

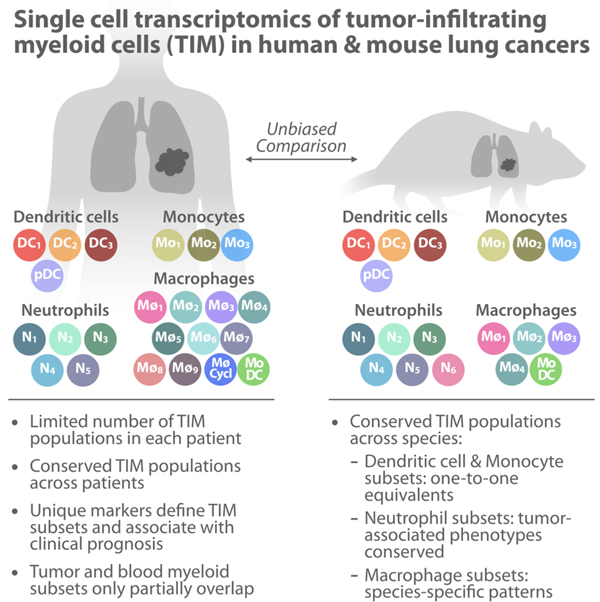
Abstract
Tumor-infiltrating myeloid cells (TIMs) comprise monocytes, macrophages, dendritic cells and neutrophils, and have emerged as key regulators of cancer growth. These cells can diversify into a spectrum of states, which may promote or limit tumor outgrowth, but remain poorly understood. Here, we used single-cell RNA sequencing to map TIMs in non-small cell lung cancer patients. We uncovered 25 TIM states, most of which were reproducibly found across patients. To facilitate translational research of these populations, we also profiled TIMs in mice. In comparing TIMs across species, we identified a near-complete congruence of population structures among dendritic cells and monocytes; conserved neutrophil subsets; and species differences among macrophages. By contrast, myeloid cell population structures in patients’ blood showed limited overlap with those of TIMs. This study determines the lung TIM landscape and sets the stage for future investigations into the potential of TIMs as immunotherapy targets.
eTOC
Tumor-infiltrating myeloid cells (TIM) have emerged as key cancer regulators and potential next-generation immunotherapy targets, yet they remain incompletely understood. Using single cell RNA-seq, Zilionis et al. map the TIM landscape in human and murine lung tumors and systematically compare cell states, revealing conserved myeloid populations across individuals and species.
Graphical Abstract
Introduction
The ability of the immune system to control tumor cells was proposed more than a century ago and recently harnessed for therapy. Therapies targeting T cell inhibitory checkpoint signaling pathways have shown unprecedented clinical benefits and are redefining cancer therapy. However, only a minority of cancer patients durably respond to current immunotherapies (Sharma and Allison, 2015). Considering that tumor microenvironments are home to diverse cell types (Binnewies et al., 2018), several efforts have begun to identify immunotherapy targets beyond T cells. Among the most compelling class of targets are tumor-infiltrating myeloid cells (TIMs), comprising of mononuclear phagocytes (monocytes, macrophages and dendritic cells) and polymorphonuclear phagocytes (granulocytes) (Engblom et al., 2016). TIMs are abundant in the stroma of a broad range of tumors but remain less studied than T cells.
At present, we have a limited understanding of the complexity of TIM subtypes, which makes them hard to study and target. TIMs consist of several distinct lineages, but each of these may further diversify into a spectrum of activation states in response to exogenous stimuli. This is most appreciated for macrophages; recently, the field has championed a more holistic analysis of these cells by considering their ontogeny, their response to environmental signals, and their transcriptional state (Ginhoux et al., 2016; Mantovani et al., 2017). Macrophages have also been catalogued as classically (M1) or alternatively (M2) activated in response to defined stimuli and are respectively associated with anti- and pro-tumor activities. Yet macrophages in vivo typically display phenotypes that vary well beyond these denominations (Ginhoux et al., 2016; Mantovani et al., 2017). In the case of granulocytes, the classification of different cell types (neutrophils, basophils, eosinophils and mast cells) has remained largely unchanged since their identification by histology, and subsets within these cell states in tumors are not as well appreciated. Tumor-infiltrating neutrophils have been ascribed both anti- and pro-tumor properties, as well as diverse molecular phenotypes (Coffelt et al., 2016; Engblom et al., 2016). Yet the full spectrum of transcriptional states of tumor-infiltrating neutrophils, particularly in patients, remains unknown. Heterogeneity among dendritic cells (DCs) is also appreciated (Broz et al., 2014), but again the complete range of DC states within tumors is not fully known.
Until recently, the complexity of TIM sub-populations meant that it was impossible to obtain a comprehensive snapshot of the tumor stroma. This is because the study of TIMs has largely relied on being able to fractionate cell sub-populations by flow cytometry and histology, using established panels of marker antigens. A drawback of these approaches is that they demand prior knowledge of TIM sub-population markers, with validated antibodies for these markers. Thus, even major TIM cell sub-states could remain unappreciated because they present no discriminating features with existing markers. It is not even clear whether each lineage of TIMs should be considered a collection of discrete sub-populations or whether they represent a complex continuum of states. Further, any change in marker gene expression (for example owing to a unique tumor microenvironment) could confound attempts at fractionation of even known cell types. Another drawback of these methods is that no single antibody panel is typically capable of capturing the full heterogeneity of TIM states. These limitations restricted the complexity of the TIM landscape that could be surveyed.
A second major challenge in studying TIMs is the possible discrepancy between the human and mouse immune systems. Mice remain the principle research model for exploring disease mechanisms, in part due to their reproducibility and scalability compared to human studies, but also due to their utility for carrying out mechanistic perturbations. Although some studies of tumorigenesis in the mouse do not model normal immune responses, syngeneic disease models and transgenic models do allow studying a more physiological immune response in laboratory animals. Yet in focusing on specific marker genes for cell populations, it has been easy to identify discrepancies between organisms. The antigen Ly6G, for example, marks mouse but not human neutrophils. Similarly, some DCs in mice are marked by CD103 but by CD141 in the human equivalent subset, and classical monocytes are marked by CD14 (human) or Ly6C (mouse). This has presented a challenge in reconciling mouse and human myeloid biology. The differences raise practical concerns about the ability to model immune responses, and immunotherapies, in mice. As we investigate TIMs, it therefore becomes critical to relate population structures in human and mice.
Addressing both challenges, single cell RNA sequencing (scRNA-Seq) offers an opportunity to sample the whole transcriptome of individual cells, and to thus organize cells into cell states independent of any prior assumptions on surface markers or species conservation, and without a strong limit on the number of marker genes that can be used for analysis. This can be done in any organism, allowing unbiased comparisons across species (Briggs et al., 2018).
In this study, we used scRNA-Seq to analyze immune populations in lung non-small cell lung cancer (NSCLC) in seven patients, as well as in a mouse model of the disease and in healthy mouse lungs. Lung cancer represents a useful case study for such a comparison because the disease is highly prevalent and often refractory to existing treatments; yet the tissue is poorly accessible in humans, making it difficult to study and requiring reliable mouse models. Though our key focus was the possible utility of mouse as a model for human TIMs, we additionally compared immune cells in patient tumors to those found in their blood. We synthesized our findings by surveying the conservation of FACS markers, transgenic mouse models, and immunotherapeutic targets between sub-populations in human and mouse. We further defined distinct marker genes for TIM subsets and their association with patient survival. The resulting resource identifies tumor-associated myeloid cell states that exist in most patients and are conserved across humans and mice. The findings may open opportunities for using defined TIM sub-populations as diagnostic tools and therapeutic targets and studying them in mice.
Results
scRNA-Seq profiling maps immune cell gene expression in human and mouse lung cancer
We set out to define TIM cell sub-populations in human NSCLC and in a mouse lung adenocarcinoma (LA) model, and to establish similarities and discrepancies between the two organisms (Fig. 1A). We first obtained fresh tumor tissue from seven human patients undergoing resection of one of two NSCLC types: LA (5 of 7) and squamous cell carcinoma (SCC; 2 of 7). The samples were from patients who had not yet received cancer treatment in all cases but one (Table S1). The patients ranged from 61 to 83 years old; the tumors were of diverse tumor stages and split between female (n=4) and male (n=3) subjects. The biopsies were dissociated, washed, and rapidly processed using the inDrop scRNA-Seq platform (Zilionis et al., 2017). After filtering scRNA-Seq data to exclude putative cell doublets (Wolock et al., 2018) and stressed or dead cells, a total of 40,362 cell transcriptomes were retained for analysis across the seven patients. These data were visualized for exploration using SPRING, a two-dimensional force-layout embedding (Weinreb et al., 2018) (Fig. 1B). Of all cells, 15% of them (n=5,912) were classified as fibroblasts, endothelial, erythroid and epithelial cell types. The population structure of these non-immune cells is analyzed in Fig. S1A–E and data on these cells are available for further exploration (see Key Resource Table). The remainder of the study focused on immune cells (Fig. 1C; n=34,450).
Figure 1. Single cell transcriptional profiling of mouse and human immune cells in non-small cell lung cancer.
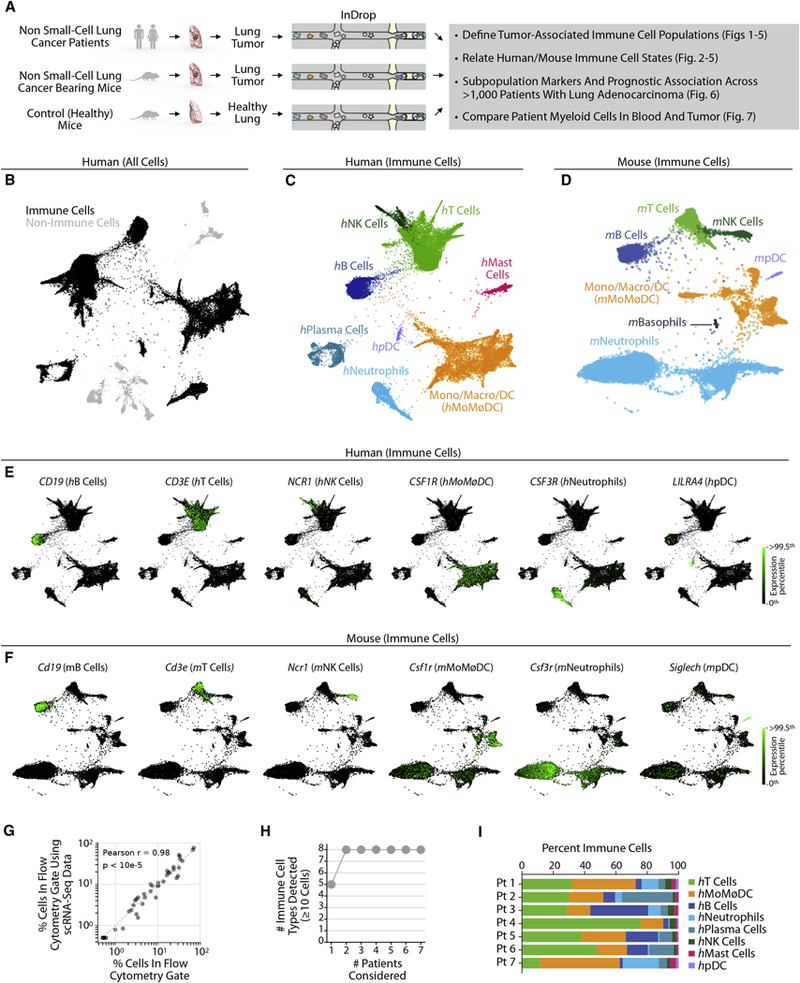
A Schematic of experimental workflow for defining and comparing immune transcriptional states in both species. Single cell suspensions for scRNAseq were prepared from patient lung tumor biopsies (n=7), murine lung tumors (n=2), and murine healthy lung tissue (n=2).
B. Two-dimensional visualization (SPRING plots) of immune and non-immune single cell transcriptomes (n= 40,362) in patient lung tumor biopsies (n=7).
C, D. SPRING plots of lung immune cells from (C) human patients (34,450 cells) and (D) mice (15,939 cells). Major cell types were defined by a Bayesian cell classifier using bulk whole-transcriptome profiles of FACS-sorted cell populations.
E, F. Single cell gene expression for representative immune cell type-enriched genes in human and mouse immune cells.
G. Comparison of mouse immune cell frequencies measured by flow cytometry to scRNA-Seq clusters satisfying the same gating scheme.
H. Cumulative plot of number of immune cell types detected with patient number. Patients were ordered by increasing number of populations detected.
I. Inter-patient heterogeneity revealed by plotting per-patient immune cell type distribution. See also Figure S1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD45 | Biolegend | Cat# 103126, cloneID 30-F11, RRID:AB_493535 |
| anti-CD11b | BD | Cat# 557397, cloneID M1/70, RRID:AB_396680 |
| anti-Ly-6G | BD | Cat# 551461, 560599, cloneID 1A8, RRID:AB_394208, RRID:AB_1727560 |
| anti-SiglecF | BD | Cat# 564514, cloneID E50–2440, RRID:AB_2738833 |
| anti-CD73 | Biolegend | Cat# 127205, cloneID TY/11.8, RRID:AB_1089065 |
| anti-Clec5a | R&D Systems | Cat# FAB1639P, cloneID 226402, RRID:AB_2081645 |
| anti-MPO | Dako | Cat# A0398, polyclonal, RRID:AB_2335676 |
| ImmPRESS Horse radish peroxidase (HRP) labelled Anti-Rabbit IgG | Vector Laboratories | Cat# MP-7401, RRID:AB_2336529 |
| Biological Samples | ||
| Human primary NSCLC samples | Dana-Farber Brigham and Women’s Cancer Center | See Table S1 for details. |
| Human peripheral blood from NSCLC patients (same as above) | Dana-Farber Brigham and Women’s Cancer Center | See Table S1 for details. |
| Mouse lungs from lung tumor-bearing or tumor free mice | Pittet lab | See Experimental Models for details |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 10% Formalin | Sigma | Cat# HT501128 |
| 3,3’-diaminobenzidine (dab) substrate chromogen system | Dako | Cat# K3468 |
| Hematoxylin | Dako | Cat# S2302 |
| OptiPrep | Sigma-Aldrich | Cat# D1556 |
| ACK lysis buffer | Lonza | Cat# 10–548E |
| ACK lysis buffer | Stemcell | Cat# 07800 |
| Collagenase type I | Worthington Biochemical Corporation | Cat# LS004197 |
| 7-aminoactinomycin | Sigma-Aldrich | Cat# A9400 |
| Critical Commercial Assays | ||
| Manual RNAscope® 2.5 HD Reagent Kit (RED) | ACD Bio | Cat# 322350 |
| RNAscope 2.5 HD (Red) detection Kit | ACD Bio | Cat# 322360 |
| RNAscope probe: Peptidase inhibitor 3 | ACD Bio | Cat# 534371 |
| RNAscope probe: Peptidylprolyl isomerase B (cyclophilin B, PPIB, positive control) | ACD Bio | Cat# 313901 |
| Tumor Dissociation kit | Miltenyi Biotec | Cat# 130–095-929 |
| ErythroClear kit | Stemcell Technologies | Cat# 01738 |
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| TaqMan Fast Advanced MasterMix | Thermo Fisher Scientific | Cat# 4444964 |
| TaqMan assay: b-Actin | Thermo Fisher Scientific | Cat# 4331182, AssayID Mm00607939_s1 |
| TaqMan assay: Car4 | Thermo Fisher Scientific | Cat# 4448892, AssayID Mm00483021_m1 |
| TaqMan assay: Clec4n | Thermo Fisher Scientific | Cat# 4448892, AssayID Mm00490934_m1 |
| TaqMan assay: Clec5a | Thermo Fisher Scientific | Cat# 4448892, AssayID Mm01131767_m1 |
| TaqMan assay: Csf1 | Thermo Fisher Scientific | Cat# 4453320, AssayID Mm00432686_m1 |
| TaqMan assay: Ltc4s | Thermo Fisher Scientific | Cat# 4448892, AssayID Mm00521864_m1 |
| TaqMan assay: Nt5e | Thermo Fisher Scientific | Cat# 4453320, AssayID Mm00501910_m1 |
| TaqMan assay: Runx1 | Thermo Fisher Scientific | Cat# 4448892, AssayID Mm01213404_m1 |
| TaqMan assay: Siglecf | Thermo Fisher Scientific | Cat# 4448892, AssayID Mm00523987_m1 |
| TaqMan assay: Spp1 | Thermo Fisher Scientific | Cat# 4331182, AssayID Mm00436767_m1 |
| TaqMan assay: Vegfa | Thermo Fisher Scientific | Cat# 4453320, AssayID Mm00437306_m1 |
| TaqMan assay: Xbp1 | Thermo Fisher Scientific | Cat# 4331182, AssayID Mm00457357_m1 |
| Deposited Data | ||
| Single cell RNA sequencing data | This paper | GEO: GSE127465 |
| Experimental Models: Cell Lines | ||
| Murine KP1.9 lung adenocarcinoma derived from lung tumor nodules of a C57BL/6 KrasLSL-G12D/WT;p53Flox/Flox mouse | Zippelius lab, University Hospital Basel, Switzerland | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | Jackson Laboratory | Cat# 000664, RRID:IMSR_JAX:000664 |
| Software and Algorithms | ||
| SPRING | Klein lab | https://github.com/AllonKleinLab/SPRING_dev |
| indrop.py pipeline | Klein lab | github.com/indrops |
| Scrublet for doublet removal | Klein lab | https://github.com/AllonKleinLab/scrublet |
| Color deconvolution plugin for IHC/RNAscope analysis | FIJI | https://imagej.net/Colour_Deconvolution |
| Shanbhag and Otsu thresholding for IHC/RNAscope analysis | FIJI | https://imagej.net/Auto_Threshold |
| Cell counter plugin for IHC/RNAscope analysis | FIJI | https://imagej.nih.gov/ij/plugins/cell-counter.html |
| Python 2.7.13 | Anaconda | https://www.anaconda.com/distribution/ |
| Other | ||
| Interactive explorer of human total cells from tumor | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Zilionis2019/human/NSCLC_all_cells |
| Interactive explorer of human immune-only cells from tumor | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Zilionis2019/human/NSCLC_immune |
| Interactive explorer of human tumor non-immune cells | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Zilionis2019/human/NSCLC_non_immune |
| Interactive explorer of human blood-only cells | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Zilionis2019/human/blood |
| Interactive explorer of human blood and tumor immune cells | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Zilionis2019/human/NSCLC_and_blood_immune |
| Interactive explorer of immune cells from lungs of tumor-bearing or tumor free mice | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Zilionis2019/mouse/all_CD45_cells |
A matched dataset was obtained by scRNA-Seq profiling of immune cells from healthy and tumor-bearing mice (Fig. 1D). To induce LA tumors, mice were injected with the KP1.9 tumor cell line (Pfirschke et al., 2016). We isolated and sequenced a total of ~17,000 CD45+ single cells from the lungs of healthy (i.e. tumor-free) and tumor-bearing mice (two mice per condition). After excluding cells of high mitochondrial load (an indicator of cell death) and putative doublets, 15,939 single cell transcriptomes were retained for analysis, and visualized using SPRING (Fig. 1D, Figs. S1F, G).
Both human and mouse immune cells partitioned into several major cell clusters, some with complex internal structure, as well as minor clusters representing <1% of the total cell population. To define the identity of the cells, we applied a Bayesian Classifier that assigns each single cell transcriptome to prior annotated transcriptional states (Figs. 1C,D, Fig. S1H). For human cell annotation, we used whole-transcriptome profiles of FACS-sorted sub-populations (Newman et al., 2015). For mouse, we used a comparable data set from the IMMGEN consortium (Heng et al., 2008). Our classification revealed that the clusters corresponded to almost all major known immune cell lineages. In humans, we identified various myeloid cells (mast cells, neutrophils, classical dendritic cells (DCs), plasmacytoid DCs (pDCs), monocytes, macrophages) and lymphoid cells (T cells, NK cells, B cells, plasma cells). In mouse, we identified the same lineages, except for plasma and mast cells. We detected basophils, which were not seen in the human biopsies. In neither organism did the classifier identify a distinct cell cluster as eosinophils, suggesting that these either did not represent a distinct cell state, were extremely rare or failed to survive sample preparation. Expression of genes defining major immune subsets is shown for humans and mice in Figs. 1E, F.
We make two technical notes about collecting these data. First, neutrophils showed very low transcript counts and required setting low filtering thresholds to allow their detection. These cells could be inadvertently excluded using the data filters commonly used in scRNA-Seq studies. Second, plasma cells in patient tumors expressed very low transcript counts for PTPRC, the gene encoding the antigen CD45 (Table S2). This may explain why plasma cells were not detected in the CD45-sorted mouse immune repertoire.
To establish confidence in the ability of scRNA-Seq to report on known cell states, we compared the abundances of cell types identified by scRNA-Seq to conventional gates used in flow cytometric analysis of mouse tissue. This comparison showed strong quantitative agreement between the two methods (Fig. 1G, p<10−5, Pearson’s R=0.98). We observed that all eight major cell types were represented by 6 of 7 patients (Figs. 1H,I and Fig. S1I), albeit in different proportions that may reflect differences in disease etiology or progression. These findings suggest that the patient cohort represented major aspects of the lung tumor immune microenvironment.
Unbiased comparison of human and mouse immune cells reveals a broad conservation of major gene expression programs
We generated a resource on the gene expression state of immune cells in mouse and human tumors, by cell type. In total, over 4,896 genes showed significant differences in expression in the major cell lineages in human, and a comparable number (4,545) in mouse (Figs. 2A,B, Tables S2,3). Although each immune cell type defined here contains further sub-structure, the gene expression profiles defined at the level of classical cell types reflect many known markers (Figs. 2A,B).
Figure 2. Immune cell types show orthologous gene expression between mouse and human at the level of major lineages.
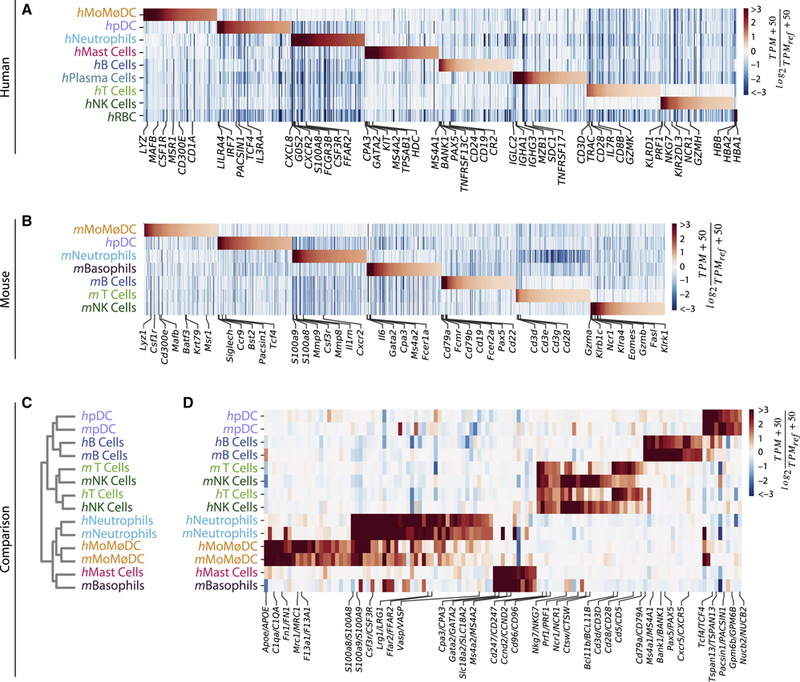
A, B. Enriched genes within major immune cell types in human and mouse samples. TPMREF = second-highest expression value per gene, transcripts per million.
C. Hierarchical clustering of major cell lineages by correlation of gene expression groups cells by cell type, not organism. Cell type labels as in Fig. 1C–D.
D. Heat map showing gene orthologs similarly enriched within mouse and human immune cell types. TPMREF = median expression value per gene, transcripts per million. See also Figure S2.
To systematically compare human and mouse immune cells, we quantified the similarity between the average transcriptome of the immune cell types identified in human and mouse (orthology method and gene list in Table S4). This ‘low resolution’ analysis ignored sub-structure within each lineage, and served as a starting point for a detailed analysis within each lineage. We observed that cell type identity dictated the similarity between gene expression profiles, rather than the source organism. This could be seen by clustering on gene expression correlations (Fig. 2C) and by inspecting the expression of cell type-specific genes across cell types in both species (Fig. 2D). With the exception of T and NK cells, each major cell lineage clustered with its ortholog. T and NK cells were more similar to each other within organisms than across organisms, reflecting their proximity in gene expression observed in SPRING visualization (Figs. 1C,D) and expression heatmaps (Figs. 2D, Table S3). The similarities in gene expression also reflected ontogeny, with myeloid cells separating from lymphoid cells. An apparent exception was the pDCs, which are considered a myeloid cell type, but associated here with lymphoid lineages. This observation is consistent with recent observations that pDCs are distinct from conventional DCs (See et al., 2017; Villani et al., 2017).
This correspondence of cell type orthologs reaffirmed that there exist coherent immune programs conserved between human and mouse, and it justified an examination of similarities and differences within each immune lineage.
Mouse and human lung tumors contain common and distinct neutrophil subsets
We analyzed the substructure of each major cluster, focusing on neutrophils, DCs, monocytes and macrophages (see Figs. S2A–R for T, NK, mast, and plasma cell sub-states). We present first data for neutrophils, which showcase examples of conserved and divergent gene expression patterns between mouse and human.
In patient tumor samples, neutrophils formed a continuum of states, which resolved into five subsets by spectral clustering (human N1–5 or hN1–5, Figs. 3A–C). The representation of these subsets varied among patients (Figs. 3D,E, Fig. S3A, Table S1). Neutrophils in mouse KP1.9 tumors also formed a continuum of states, which resolved into six subsets (mouse N1–6 or mN1–6, Figs. 3F–H). The proportion of these subsets markedly differed in tumor-free and tumor-bearing lungs, with mN1,2 enriched ~ten-fold in healthy lungs, mN4,5 increasing 10 to 20-fold in tumor tissue, and mN3,6 exclusively present in tumor tissue (Fig. 3I, Fig. S3B).
Figure 3. Human and mouse lung tumors contain a conserved axis of neutrophil phenotypes and a distinct neutrophil subset showing type I interferon response.
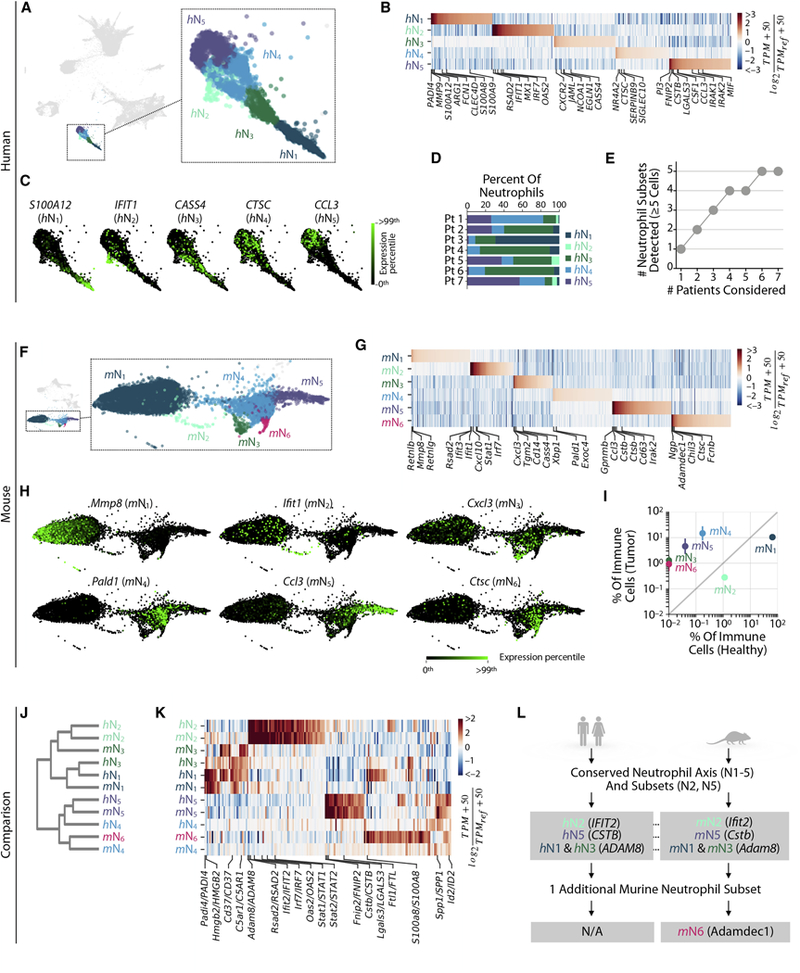
Lung neutrophil subsets defined in A-E human patient tumors and F-I mouse tumor and healthy tissues.
A. SPRING plot from Fig. 1C showing neutrophil subsets.
B. Genes enriched between neutrophil subsets. TPMREF defined as in Fig. 2A.
C. Single cell expression of representative subset-enriched genes.
D. The frequency of lung tumor neutrophils subsets varies between patients.
E. Cumulative plot of the number of neutrophil subsets detected with patient number.
F-H show mouse equivalents of A-C.
I. The tumor specificity of mouse neutrophil subsets assessed by plotting their frequency in tumor versus healthy tissue samples. Bars show the two replicate value of each condition. J-L. Comparison of mouse and human neutrophil subsets.
J. Orthologous murine and human neutrophil subsets established by hierarchal clustering.
K. Heat map showing genes similarly enriched within mouse and human neutrophil subsets. TPMREF defined as in Fig. 2D.
L. Summary of mouse/human comparison showing a conserved axis of neutrophil phenotypes ranging from N1 to N5, the latter being tumor-specific and tumor-promoting in mice. A rare, but distinct neutrophil subset with a type I interferon response expression signature is also present in both mouse and human. See also Figure S3.
We previously identified two mouse lung neutrophil subsets, characterized by variable expression of the sialic acid binding Ig-like lectin F (Siglecf). SiglecFlow cells are found in tumor-free lungs, whereas SiglecFhigh neutrophils accumulate in tumor tissues and exhibit several pro-tumor functions as (Engblom et al., 2017). Here, we identified that three neutrophil subsets (mN4–6) expressed Siglecf, and indeed these were highly tumor-enriched, whereas the remaining three subsets (mN1–3) were Siglecf-low (Fig. S3C).
To build confidence in the observed sub-population structure, we sorted SiglecFhigh and SiglecFlow cells from lung tissue (Fig. S3D) and examined predicted gene expression markers by qPCR (Fig. S3E). We expected this sorting strategy to enrich for cells in clusters mN4–6, compared to mN1–3, and indeed confirmed the predicted overexpression of the Car4, Clec4n, Clec5a, Csf1, Ltc4s, Nt5e, Runx1, Siglecf, Spp1, Vegfa and Xbp1 transcripts in SiglecFhigh cells (Figs. S3E–G). We further analyzed Nt5e and Clec5a protein expression by flow cytometry and found strong correspondence between mRNA and protein gene expression patterns (Fig. S3H).
To define how neutrophil subsets relate to each other across species, we compared human and mouse neutrophils systematically on the single cell and whole transcriptome levels (as for Fig. 2C,D). We found conservation of several aspects of neutrophil population structure between species, as can be appreciated through an unsupervised comparison (Figs. 3J–L). In both human and mouse, we observed a common ordering of neutrophil subsets, from h/mN1 neutrophils that expressed high levels of canonical neutrophil markers (MMP8,9; S100A8,9; ADAM8; synonymous lower case mouse gene symbols omitted here and below), via h/mN3,4, to h/mN5 that were tumor-specific in mouse and express cytokines CCL3 and CSF1, as well as CSTB, CTSB, and IRAK2 (Figs. 3B,G, Table S2,3). mN6 neutrophils were most closely related to their mN4 counterparts (e.g. they both expressed Hexb and Ptma) although mN6 uniquely expressed Fcnb and Ngp (Fig. 3G). In both organisms h/mN2 neutrophils formed a sub-population apart from the continuum of states; these cells were rare but exhibited a strong transcriptional signature, expressing type I interferon response genes, including IFIT1, IRF7 and RSAD2 (top 50 marker genes showed enrichment for the GO term ‘cellular response to Type I interferon’ with FDR=4×10−26). We found N2 cells in both healthy and tumor lung tissue in mice and will later show that they also appear in patients’ peripheral blood.
In summary, we identified three conserved modules of neutrophil gene expression within mouse and human neutrophils (Fig. 3L): a) neutrophils that express canonical neutrophil markers (h/mN1); these progress continuously to b) neutrophil states that are tumor-specific and promote tumor growth in mice (h/mN5); and c) a small but distinct neutrophil subset with an expression signature of type I interferon response (h/mN2).
DCs contain four distinct subsets that are conserved between mouse and human
Using the same Bayesian classifier and reference datasets, the remaining TIM populations were assigned as having gene expression profiles of monocytes, macrophages and DCs (Fig. S1H). We analyzed each subset in separation. Spectral clustering of patient tumor DCs identified four subsets: pDC (human pDC, or hpDC) and hDC1–3, which showed distinct gene expression programs across tens of genes (Fig. 4A–C, Table S1–3). The DC subsets were all present in 6/7 patients, albeit in variable proportions; one patient lacked several subsets, possibly due to the low cell numbers in this sample (Figs. 4D,E, Fig. S4A).
Figure 4. Single cell transcriptional analysis of dendritic cells reveals four distinct subsets that are conserved between mouse and humans.
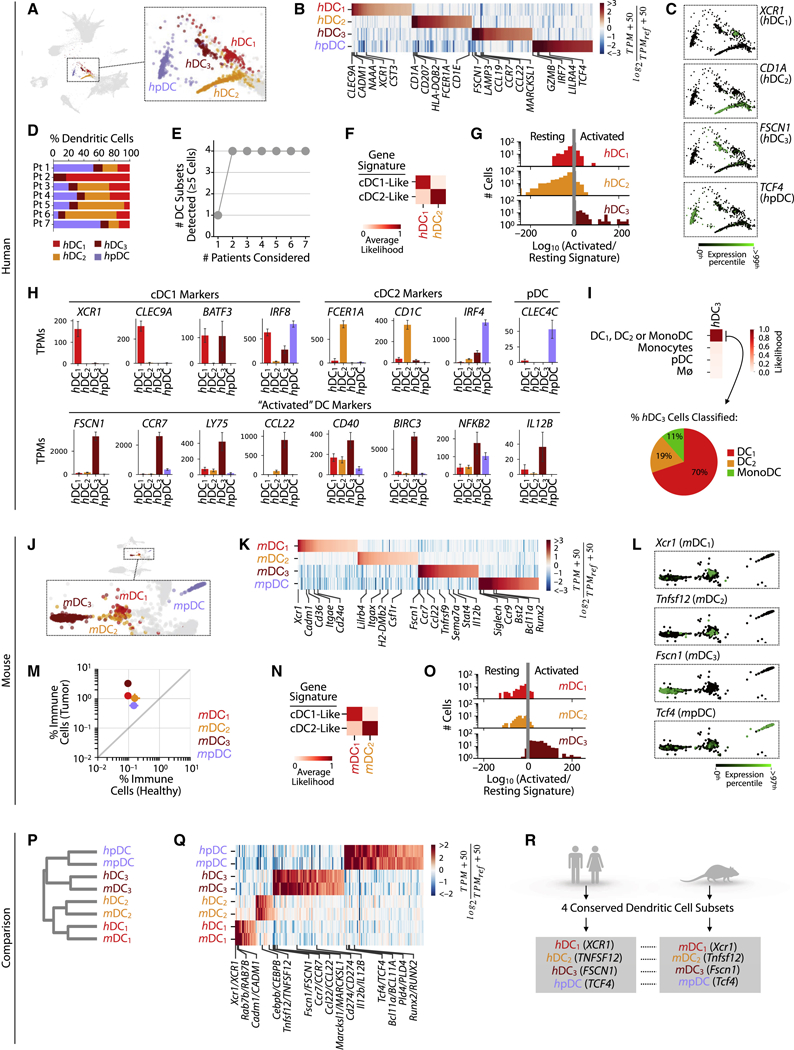
Dendritic cell (DC) subsets defined in A-G humans patients, and H-M mice.
A.SPRING plot showing DC subsets.
B. Genes enriched between DC subsets. TPMREF defined as in Fig. 2A.
C. Single cell expression of representative subset-enriched genes.
D. Distribution of lung tumor DC subsets in each patient.
E. Cumulative plot of the number DC subsets detected with patient number.
F. Classification by cDC1 or cDC2 gene signatures reveals the identity of hDC½ subsets.
G. Single cell histogram of activated vs resting likelihood ratio reveals the identity of h/mDC3.
H. Expression of canonical DC markers across DC subsets.
I. Likelihood of hDC3 cells classified as hDC1, hDC2, or MonoDC.
J-L, N-O show mouse equivalents of A-C, F-G.
M. Tumor-enrichment of all DC subsets seen from the fraction of each DC subsets in tumor and healthy tissues. Bars show the two replicate value of each condition.
P-R. Comparison of mouse and human DC subsets.
P. Orthologous murine and human DC subsets established by hierarchal clustering.
Q. Heat map showing genes similarly enriched within mouse and human DC subsets. TPMREF defined as in Fig. 2D.
R. Summary of mouse and human DC subset comparison showing a one-to-one correspondence of four distinct DC subsets (DC1–3, and pDC). See also Figure S4.
To assess hDC1–3, we compared their gene expression profiles to those of bulk-sorted classical DCs, which comprise cDC1 (defined as XCR1+ CADM1+ by flow cytometry) and cDC2 (CD1A+ CD172A+) (Eisenbarth, 2019; Guilliams et al., 2016). cDC1 are efficient antigen cross-presenters to CD8+ T cells, whereas cDC2 preferentially interact with CD4+ T cells. The hDC1 and hDC2 subsets expressed gene signatures that mapped well to these DC populations (Fig. 4F). Accordingly, hDC1 uniquely expressed the cDC1 markers XCR1, CLEC9A, TBHD (the gene encoding for cell surface protein CD141) and CADM1, whereas hDC2 expressed cDC2 markers, including CD1A, CD1C, CD1E, CD207 (encoding for langerin) and FCER1A (Fig. 4B, Table S2).
hDC3 showed an “activated” DC phenotype based on comparisons to previously published microarray datasets of in vitro LPS-stimulated DCs (Fig. 4G) (Zanoni et al., 2009) but lacked expression of key cDC1/2 and pDC genes. For example, hDC3 expressed the cDC1-associated genes BATF3 and IRF8 but neither XCR1 nor CLEC9A (Fig. 4H). Individual hDC3 cells also failed to consistently associate with a single DC signature (Fig. 4I). Many appeared similar to hDC1, however, and hDC3 were much closer in expression to DCs than to monocytes or macrophages.
Six human blood DC subsets have been reported (Villani et al., 2017), most of which showed a correspondence to our data with some exceptions (Fig S4B–D). One of the blood clusters (denoted DC5 in that paper) was absent from our tumor data, while the tumor hDC3 subset was absent in their blood data. These differences likely reflect variation in DC states between healthy blood and tumor tissue.
In mouse (Figs. 4J–O), we again found four distinct DC subsets, all of which were significantly increased in tumor-bearing lungs (Fig. 4M). These four subsets could be related one-to-one to those found in human, first by inspecting marker genes for each cell cluster (Figs. 4K,L, Table S2), and then by unsupervised hierarchical clustering using orthologous variable genes (Figs. 4P–R, Table S2,3). mDC1–3 mirrored hDC1–3 while mpDCs mirrored hpDCs.
The gene expression profiles of mDC1 and mDC3 respectively resembled those identified for Ccr7–and Ccr7+ cDC1 subsets in the thymus and peripheral lymph nodes of mice (Ardouin et al., 2016) (Fig. S4E, F). These data suggest that Ccr7– mDC1 can give rise to Ccr7+ mDC3 cells. mDC3 also resembled tumor-infiltrating Ccr7+ DCs that secrete interleukin-12 and have anti-tumor activity (Garris et al., 2018) (Fig. S4G, H). CCR7 expression may enable mDC3 to migrate to draining lymph nodes (Roberts et al., 2016) but some of these cells can remain within tumors (Garris et al. 2018).
In summary, the conserved structure of both the mouse and human DC populations, and its recurrence in other studies, supports the view that DCs can be consistently classified and compared between species (Dutertre et al., 2014). Tumors in both organisms decomposed into plasmacytoid and cDC1–2 subsets, and into resting and activated DCs, and we could identify no others. In both organisms, pDCs appeared to represent a distinct cell state, suggestive of alternate ontogeny to other DCs. Both organisms also presented monocytes showing DC-like signatures, which are described below.
Monocyte subsets are well conserved between mouse and human, while macrophage subsets show species-specific patterns
In human, we defined 14 transcriptional states of cells classified as monocytes and macrophages. Three showed gene signatures of monocytes (hMono1–3), nine of macrophages (hMø1–9), one of monocytes and DCs (hMonoDC), and one characterized by cell cycle gene expression (hMøCycl) (Figs. 5A–D, Fig. S5A, Tables S2,3). We assigned these identities to the clusters by comparing their gene expression to that of annotated reference gene expression profiles (Gentles et al., 2015; Newman et al., 2015) (Fig. 5D). Even though spectral clustering identified a relatively large number of transcriptional states, all of them could be detected using any 4/7 patients (Fig. 5C, Figs. S5B,C, Table S1). These results indicate substantial monocyte and macrophage heterogeneity in lung tumors defined by multiple gene expression programs, which are reproducible across individuals.
Figure 5. Monocyte subsets are well conserved between mouse and human, whereas macrophage subsets show inter-species heterogeneity.
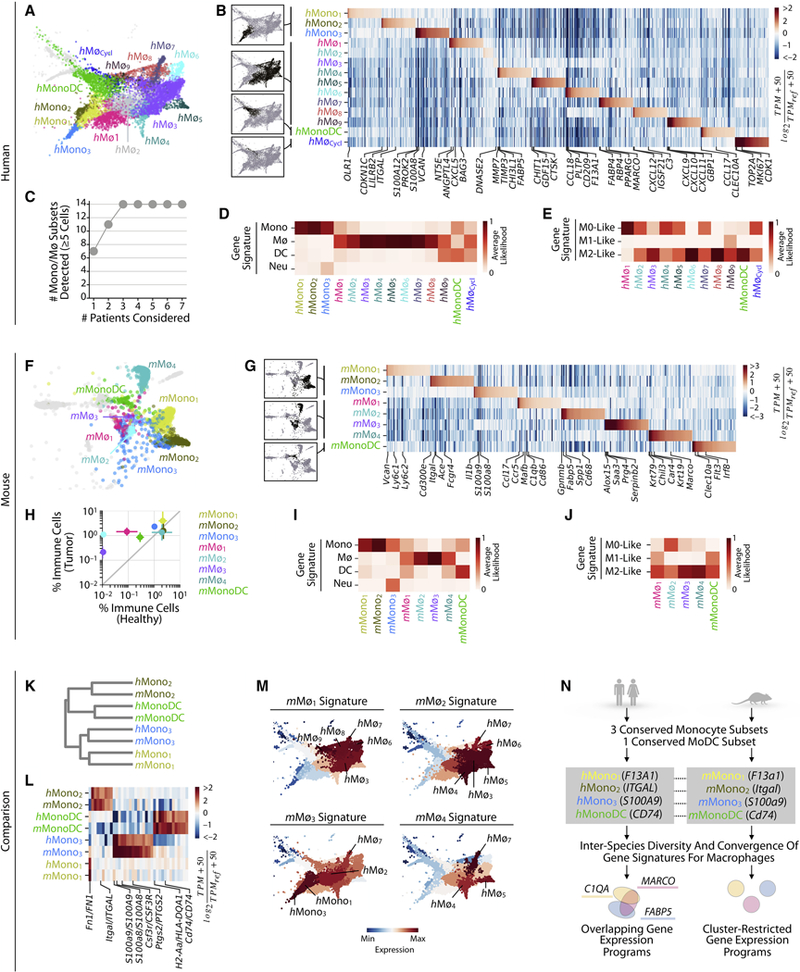
Monocyte (Mono) and macrophage (Mø) subsets defined in A-E human patients, and F-J mice.
A. SPRING plot showing Mono/Mø subsets.
B. Genes enriched between Mono/Mø subsets. TPMREF defined as in Fig. 2A.
C. Cumulative plot of the number of Mono/Mø/MonoDC subsets detected with patient number.
D. Classification of Mono/Mø/MonoDC subclusters by Mono, DC, Mø and neutrophil gene signatures.
E. Classification of macrophage subsets by M0/M1/M2-like gene signatures.
F-G, I-J show mouse equivalents of A-B and D-E.
H. Tumor-enrichment of Mø, MonoDC but not Mono subsets, seen from the fraction of each each Mono/Mø/MonoDC subset in the tumor vs healthy tissues. Bars show the two replicate value of each condition.
K-N. Comparison of mouse and human Mono/Mø/MonoDC subsets.
K. Orthologous murine and human monocyte subsets established by hierarchal clustering.
L. Heat map showing genes similarly enriched within mouse and human Mono/MonoDC subsets. TPMREF defined as in Fig. 2D.
M. Expression of mouse Mø gene signatures within human Mono/Mø/MonoDC subsets shows partially conserved patterns in macrophage transcriptional programs between species.
N. Summary of mouse and human Mono/Mø/MonoDC subset comparisons showing the correspondence between Mono and MonoDC subsets. Some gene expression patterns of murine and human Mø subsets were conserved, but Mø subsets exhibited the greatest inter-species variation overall. See also Figure S5.
We examined gene signatures to assess whether any of the states observed correspond to canonical M1 and M2 states, which have been used to define anti-inflammatory and pro-inflammatory macrophages, respectively. In humans, no macrophage cluster exhibited only an M1-like phenotype, whereas hMø3,6,8, and hMonoDC exhibited an M2 gene signature (Fig. 5E). Enrichment for the M2 signature can be expected for macrophages found in growing tumors, but these data show that M2 is not a distinct state.
In mouse, we defined a total of eight transcriptional states corresponding to monocytes (mMono1–3), macrophages (mMø1–4), and both monocytes and DCs (mMonoDC) (Figs. 5F–J, Fig. S5D, Tables S2,3). The monocytic states were well represented in healthy tumor-free tissue, as was a single cluster (mMø4) identified as a resident alveolar lung macrophage by its expression of Krt79, Krt19 and Car4 (Gautier et al., 2012). The remaining mouse macrophage states were strongly tumor-enriched (Fig. 5H). As with human, we assigned the cluster identities using annotated reference gene expression profiles (Heng et al., 2008) (Fig. 5I). Similarly to our human data, we found that previously annotated M0/½ gene expression signatures (Hou et al., 2018) did not clearly associate with murine macrophage clusters, and that all macrophages exhibited an M2-like phenotype (Fig. 5J).
The complexity of these populations makes it a challenge to identify similarities in population structure between organisms by inspection alone. Nevertheless, a systematic unbiased comparison (Figs. 5K,L, Table S3) identified that hMono1–3 corresponded respectively to mMono1–3, with each of the populations sharing distinct gene expression patterns. The MonoDC subset was likewise similar between patient and murine tumors, and was characterized by high gene expression of DC-related genes, including CLEC10A, MHC class II genes and associated genes (e.g. CD74) (Figs. 5B,G,K, L). Both the human and murine MonoDC subsets were distinct from the seemingly more mature DC populations DC1–3 and pDCs, e.g. human MonoDCs lacked or expressed lower levels of DC marker genes, including XCR1, CD1A, CD1E, FSCN1, and TCF4.
The correspondences noted above reflected canonical distinctions between monocyte subsets, as well as a less appreciated monocyte state. Historically, two types of monocytes have been described in both humans and mice albeit with different markers. In humans, these are identified in blood as CD14+ (so called ‘classical’) and CD14int CD16+ (‘non-classical’) (Ginhoux and Jung, 2014). Corresponding ontogenically and functionally related subsets in mice are defined as Ly6Chigh CCR2+ CX3CR1int and Ly6Clow CCR2– CX3CR1high. Ly6Chigh monocytes can extravasate into tissues and give rise to macrophages and DCs, whereas Ly6Clow monocytes can remain in the vasculature and patrol vessel walls. In our murine data, mMono1 expressed the Ly6Chigh monocyte markers Ly6c1, Ly6c2 and Ccr2, whereas mMono2 expressed the Ly6Clow monocyte markers Cx3cr1 and Fcgr4 (Fig. S5E, Table S2), in agreement with previous work (Ingersoll et al., 2010; Mildner et al., 2017). Similarly, hMono1 expressed classical monocyte-associated genes including CD14 and FCN1, and hMono2 expressed non-classical markers CDKN1C, LILRB2, and ITGAL (Fig. S5F, Table S2). Our genome-wide comparison of h/mMono subsets (Figs. 5K,L), confirmed the correspondence of the classical and non-classical monocyte types between organisms.
These canonical descriptions left the third cluster, h/mMono3, unaccounted for. Among all monocytes and macrophages, h/mMono3 uniquely expressed a set of neutrophil-associated genes (Fig. 5B, D, G, I), including S100A8, S100A9, and CSF3R (Fig. S5G), consistent with previous reports of both bulk (Ingersoll et al., 2010; Schmidl et al., 2014) and single cell analysis of sorted CD14+ blood monocytes (Villani et al., 2017). In humans, hMono3 also expressed markers of CD14+ CD16– monocytes suggesting that this population may be an unappreciated subtype of ‘classical’ monocytes (Fig. S5F). In mice, mMono3 did not express a clear Ly6Chigh or Ly6Clow gene signature; instead, this cluster contained both Ly6Chigh and Ly6Clow-like cells. It is therefore plausible that, in mice, the neutrophil-associated genes define a program orthogonal to that of classical and non-classical identity. A fourth monocyte subset was previously identified in peripheral blood (Villani et al., 2017). Many genes expressed by this subset were not present in any of our monocytes and other populations and we suspect that this subset might include physical doublets with NK cells (Fig. S5H).
Macrophages showed both conserved and divergent phenotypes between mouse and human. In both organisms, key macrophage-associated genes, such as MRC1, APOE, complement genes (C1QA,B,C) and cathepsins (CTSB,D) were enriched in macrophages compared to other TIMs and broadly expressed across clusters in continuous gradients (Figs. S5I, J). To relate macrophage heterogeneity across species, we asked whether gene signatures of mouse macrophage subsets varied across human macrophage clusters (Fig. 5M). We found that mMø1 and mMø2 gene signatures mapped to a coherent gradient across human macrophage subsets, especially subsets hMø8,9 and hMø3,4,5, respectively. Similarly, the mMø4 (alveolar macrophage) gene signature mapped mainly to hMø7. In contrast, the mMø3 gene signature did not map well to human macrophage subsets, suggestive of a subset unique to the murine tumors. Conversely, hMø1–2 did not appear well represented by murine macrophages. These complex relationships reinforce the difficulty in relating macrophage subsets between species. However, our analyses provide evidence that transcriptional programs broadly distinguishing tumor-associated macrophages from monocytes are well-conserved between mouse and human and that features of distinct murine tumor-infiltrating macrophage subsets may correspond to several cell subsets in patient tumors. We summarize the key similarities and differences in Fig. 5N.
Among the human macrophage subsets, we identified distinct chemokine expression (Fig. S5K, Tables S2,3) including hMø1 expressing the neutrophil chemoattractant CXCL5, hMø8 expressing the CXCR4 ligand CXCL12, and hMø9 expressing the T cell recruiting chemokines CXCL9,10,11. The respective receptors for these macrophage-expressed chemokines displayed heterogeneous expression patterns across other immune cell types. For example, CXCR2, encoding the receptor for CXCL5, is expressed exclusively by neutrophils, whereas CXCR3, encoding the receptor for CXCL9,10,11 is mainly expressed by T cells, NK cells and pDCs. In mice, we did not observe the same pattern; instead, murine mMø (mMø1 and mMø3 in particular) expressed mainly monocyte and neutrophil chemoattractants, while expressing negligible levels of the T cell chemokines CXCL9,10,11 (Fig. S5L, Tables S2,3). These findings suggest that macrophage diversity might be explained in part by their migration cues directed at cell populations with distinct chemokine receptor expression.
Defining distinct marker genes for TIM subsets and their association with patient survival
By analyzing seven patients, a question was whether the observed states were representative of a wider population. In examining non-immune proliferating epithelial cell states, which likely includes tumor cells within our samples (Fig. 6A), we found that each patient exhibited distinct cell states, indicating that our sample size is not adequate to comment on the diversity of patient epithelial phenotypes. By contrast, TIM states overlapped between patients (Fig. 6B), indicating that the myeloid microenvironment is much more stereotyped than the tumor tissue that it infiltrates. This observation gives confidence that we captured reproducible subsets within the myeloid lineages.
Figure 6. Unique marker genes for TIM subsets and their association with patient survival.
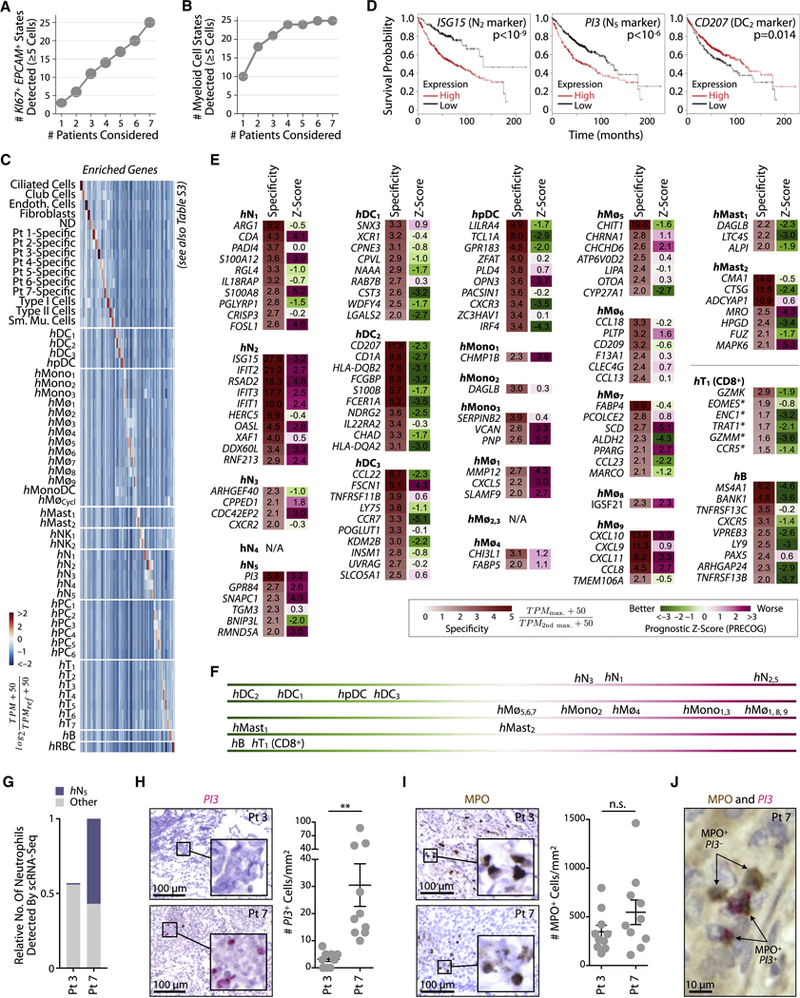
A, B. Plots of state diversity as a function of sampled patient count shows that patient tumor epithelial cell states are far from saturation while myeloid subsets approach saturation.
C. Identification of genes enriched in expression in each cell subset as compared to all others in the human tumor microenvironment. Color bar as in Fig. 2A.
D. Kaplan-Meier plots of showing differences in survival amongst lung adenocarcinoma patients (n=720) stratified by expression of selected markers (Gyorffy et al., 2013) for hN2 (LSG15), hN5 (PI3), and hDC2 (CD207). P-values from univariate cox regression.
E. Prognostic z-scores derived from lung adenocarcinoma patient data (n=1,127) (Gentles et al., 2015) for genes most specific to unique cell-states as determined by scRNAseq and shown in panel C. Negative and positive prognostic z-scores respectively associate with favorable and adverse prognosis.
F.Summary of trends in immune cell subset association with patient survival shown in E.
G-J. Detection of hN5 PI3+ and all neutrophils (MPO+) in lung adenocarcinoma in situ. G, scRNAseq prediction for the relative number of total and hN5 neutrophils detected in patients 3 and 7 (1=total neutrophil count in patient 7).
H.In situ hybridization for PI3 transcripts on tumor sections supports a significant enrichment of hN5 neutrophils in patient 7 as predicted by scRNAseq. Quantification data points correspond to distinct fields of view (n=10), two-tailed t-test p-value < 0.01.
I. Detection of the pan-neutrophil marker MPO by immunohistochemistry reveals a comparable fraction of neutrophils in patient 7 relative to patient 3 (quantification as in H).
J. Co-staining of MPO and PI3 supports the prediction that PI3+ cells are MPO+. See also Figure S6.
The overlap of TIM states between patients motivated us to assess their association with patient survival, by examining the expression of genes specific to each sub-population in survival studies. For this, we determined which genes were enriched within each state relative to all other cells in human tumors, taking all tumor-associated cells (non-immune and immune) into account (Figs. 6C, Tables S2,3). Some specific genes showed clear association with patient survival: for example, both the hN2 marker ISG15 and the hN5 marker PI3 associated negatively with patient survival, whereas the hDC2 marker CD207 associated positively with survival (Fig. 6D). We systematically scored marker gene prognostic association over multiple studies (n=1,127 patients) by PRECOG (Gentles et al., 2015) (Fig. 6E) and summarized the resulting associations in Fig. 6F. This analysis confirmed that genes enriched in CD8+ T cell and B cell subsets associated consistently with better patient survival, as expected (Fig. 6E, F). For some sub-populations, no single gene was expressed with sufficient specificity to directly relate its expression to population abundance (Fig. 6E). Among neutrophils, DCs and monocyte/macrophage clusters, many subsets showed specific gene expression that provided a remarkably consistent view of the prognostic association of each sub-population. The neutrophil subsets hN2 and hN5 both showed an abundance of marker genes associated with poor patient survival, as did hMø1 and hMø9, while among myeloid cells, hDC2 showed the most consistent positive association with survival, with the other DC subsets and a mast cell subset also showing positive associations. These results support findings of previous association studies and extend them to resolution of subsets within each major cell lineage.
Access to marker genes (Figs. 6C,E) allows developing strategies to detect immune cell subsets in histological sections, opening an opportunity for future large-scale cytometric analysis, e.g. using existing patient sample banks. We tested feasibility of such analyses by examining markers specific to hN5 neutrophils in situ. This subset represented one end of a continuum of neutrophil states (Fig. 3A), was associated most strongly with negative patient survival and was conserved between mouse and human. Within the patient cohort, we identified two patients (Pt 3 and Pt 7) that showed extreme differences in the abundance of hN5 cells, with Pt 3 hosting few of them and Pt 7 hosting many (Fig. 6G, Table S1). We stained tissue sections for transcripts of PI3, which marks hN5. We additionally stained for a pan-neutrophil marker, the primary granule protein myeloperoxidase, or MPO (Eruslanov et al., 2014). As predicted, Pt 7 hosted an abundance of PI3+ cells while Pt 3 hosted almost none (Fig. 6H). As predicted, both patients had detectable numbers of MPO+ cells (Fig. 6I) and PI3+ cells were MPO+ (Fig. 6J). These results validate the trends seen by scRNA-Seq, and demonstrate the utility of the latter for designing histology-based studies.
Genes targeted by mouse models, immunotherapies and immunophenotyping across murine and patient tumor-infiltrating immune cells
Defining the TIM landscape offered an opportunity to ask how genetic reagents, drugs and antibodies might target different myeloid cell populations in mice and humans. We cataloged 34 genes targeted in genetically engineered mouse models, e.g. used for fluorescent cell tracing, cell ablation, or other transgene expression (Figs. S6A–F, Table S5). We also examined 26 genes that encode proteins targeted by immunotherapeutic agents (Fig. S6G, Table S5) and 36 genes whose products encode surface antigens used in immunophenotyping (Fig. S6H, Table S5). For each of these gene sets, we assessed how well gene expression agreed between mouse and human across immune cells, and how specific gene expression was to defined cell types. Though many of the genes targeted in genetically engineered mouse models and by drugs showed a good correspondence in expression between organisms, there were notable exceptions that highlight inter-species differences. For surface antigens, as well as identifying similarities and differences between organisms, it was notable that many genes canonically used to identify specific cell types showed expression over multiple cell types. For all of these cases, we refer to Fig. S6 for notable examples. Overall, these results highlight the need to benchmark specific mouse immunotherapy models against human as performed here. The findings also indicate the potential shortcomings of quantifying or purifying cell types by immunocytometry, which may group distinct cell states together even with some of the best-established markers.
Tumor infiltrates only partially overlap with states of the peripheral blood
We asked whether TIM cell states can be appreciated by sampling peripheral blood (PB), which is a more easily accessible tissue and could be useful for diagnostic purposes. We began to address myeloid cell states’ tumor specificity by comparing them to PB myeloid cells of the same patients (we profiled 6 patients). Our goal was to define which tumor myeloid states had a circulating counterpart.
Several scRNA-Seq studies have previously examined blood immune populations focusing on PB mononuclear cells (PBMCs), which are fractionated by their density [e.g. (Zheng et al., 2017)]. PBMC preparations are deliberately depleted for polymorphonuclear cells, which include all granulocytes, and thus provide an incomplete view of myeloid cells. Therefore, we prepared blood for analysis by red blood cell depletion to obtain all other immune cells types. After filtering we collected 14,411 cell transcriptomes by scRNA-Seq.
SPRING embedding of cells from PB and tumors across all patients revealed that a few tumor and PB immune cells overlapped in state, although most did not (Fig. 7A). This separation did not reflect library batch effects, because all patient samples overlapped within the blood and tumor clusters. Therefore, PB cells were transcriptionally separable from those in tumors, but could still reflect specific tumor cell states. Applying the cell type classifier (as in Fig. 1C), we identified the expected array of blood leukocytes, including monocytes, neutrophils, pDC, T, NK and B cells (Fig. 7B). Average gene expression profiles for these blood populations are provided as a resource in Table S2 and Fig. S7A. The proportions of each cell type differed between the two tissues and some populations were only detected in the tumor (Fig. S7B).
Figure 7. Tumor infiltrates only partially overlap with states of the peripheral blood.
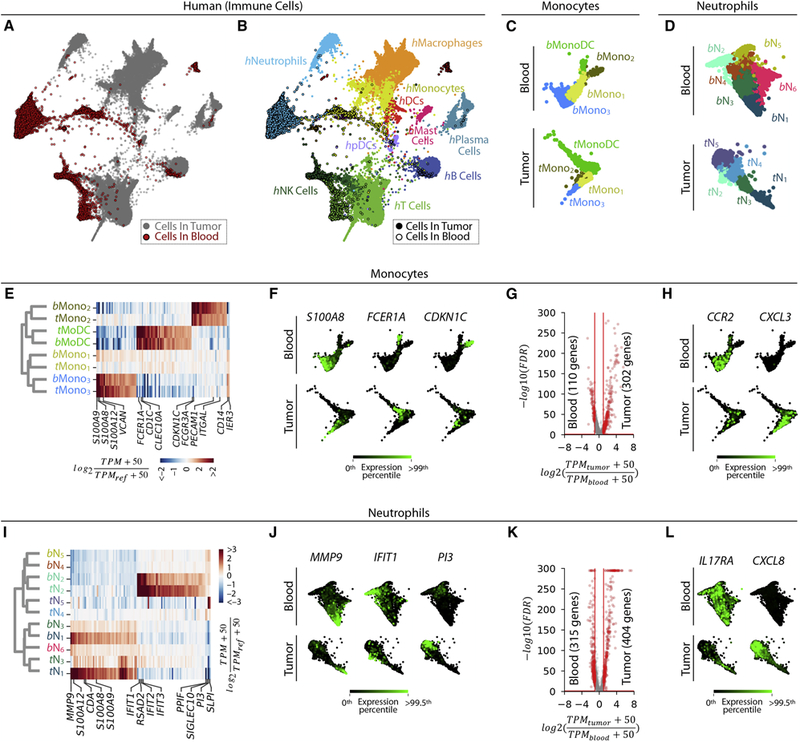
A, B. Two-dimensional visualization (SPRING plots) of immune cell transcriptomes from patient blood (n=6) and tumor samples (n=7). Cells colored by (A) sample origin (tumor= grey; blood= red) and (B) inferred immune cell type.
C, D. Spectral clustering of Mono/MonoDC and Neutrophil subsets in blood, shown alongside tumor clusters from Figs. 4, 5.
E. Homologous tumor and blood monocyte subsets established by hierarchal clustering; heat map shows similarly enriched genes. TPMREF defined as in Fig. 2D.
F. Selected examples of genes showing conserved patterns of expression between blood and tumor monocyte populations. G. Volcano plot identifying differentially expressed genes between tumor and blood monocytes.
H. Examples of genes enriched in blood (CCR2) and tumor (CXCL3) monocytes.
I-L. show Neutrophils equivalents for E-H. See also Figure S7.
We asked whether myeloid cell sub-clusters were similar across tumor and blood tissue. Clustering of PB monocyte populations revealed three monocyte states (hbMono1–3) and one monocyte-like state expressing concomitant dendritic-cell associated genes (hbMonoDC) (Fig. 7C, Figs. S7C–F). PB neutrophils formed a continuum, which we divided into six subsets (hbN1-6) by spectral clustering (Fig. 7D, Figs. S7G–J). For monocytes, hierarchical clustering revealed a one-to-one match between tumor and blood populations (Figs. 7E, F, Table S3), although the tumor and blood Mono1 subsets shared few marker genes. Part of this correspondence was anticipated, considering that monocytes are defined as circulating cells. However, blood and tumor monocytes differed (409 genes >2-fold differentially expressed at 5% FDR, Fig. 7G, Table S2). PB monocytes, for example, expressed significantly more transcripts for the chemokine receptor CCR2 while tumor monocytes expressed CXCL3, a cognate ligand for the CXCR2 receptor expressed by neutrophils (Fig. 7H). For reference, we also compared the patient blood monocyte subsets to those from healthy donors described in (Villani et al., 2017) [here denoted Mono (Vil.)] (Fig. S4B, C). We found a close correspondence between Mono1,2,3 (Vil.) and hbMono1,2,3. Mono4 (Vil.) expressing NK-associated genes, as previously defined by Villani et al., was not detected in our dataset. Additionally, one subset thought to be DCs in that study (DC4) showed a correspondence to hbMono2, consistent with a previous report (Calzetti et al., 2018).
Among neutrophils, the blood subset hbN2 corresponded strongly to the tumor subset hN2, with a similar Type I interferon signature (IFIT1,2,3, RSAD2), and the blood subsets hbN1,3,6 were related to tumor clusters hN1,3 and shared the expression of elevated levels of the canonical neutrophil-associated genes MMP9 and S100A8,9,12 (Figs. 7I,J, Table S3). Yet there was no strong PB correspondence to the CCL3+ tumor subset hN5. Overall, blood and tumor neutrophils strongly differed in their gene expression (719 genes >2-fold differentially expressed at 5% FDR Fig. 7K, Table S2). For example, tumor neutrophils expressed far higher levels of the cytokine CXCL8, and much lower levels of the IL17 receptor, IL17RA, than their blood counterparts (Figs. 7L).
PB cells also contained hbN6 neutrophils, which were undetected or very rare in the tumor (Figs. 7D,I). The hbN6 subset was found in all patients and represented 6.0 ± 0.4% (mean±SEM) of blood neutrophils (Fig. S7H). It expressed unique markers, including TNFRSF13B, which encodes the receptor for the cytokines APRIL and BAFF, as well as PDE10A and ADRA1A. TNFRSF13B is associated with B cell lineage differentiation, class switching and antibody production (Rickert et al., 2011), and was expressed by tumor, but not PB, B cells. The function of circulating TNFRSF13B+ neutrophils is unknown, though mutations in TNFRSF13B are implicated in a common variable immunodeficiency that can in rare cases present neutropenia with poorly understood etiology (Bogaert et al., 2016; Guffroy et al., 2017). The gene encoding BAFF, which binds TNFRSF13B, was expressed by other blood neutrophils (bN3–4), suggesting possible signaling between PB neutrophils. In summary, PB neutrophils show some reflection of population structure found in the tumor, but both the tumor and PB host non-overlapping and unappreciated neutrophil subsets. We conclude that to understand TIMs it is necessary to directly assess tumor tissues, as the PB does not recapitulate the tumor myeloid cell landscape.
Discussion
Here we compared TIM population structures between mice and humans by scRNA-Seq, unbiased to pre-defined markers and notions of cell state. In doing so, we defined the TIM transcriptional landscape in each organism revealing both known and unappreciated, yet reproducible, TIM phenotypes. In discussing this work, we begin with its limitations and follow with our findings regarding TIM heterogeneity at the whole-genome level; the uniqueness of tumor versus blood myeloid cells; and the reproducibility and conservation of TIM phenotypes between patients and between organisms.
We note three limitations to this study. First, profiling more patients could reveal additional TIM subsets. This may be the case for neutrophils, since identification of all five subtypes reported here required six of the seven patients. By contrast, the four DC and fourteen monocyte/macrophage subsets reported here were all represented by even three patients, indicating that human lung TIM repertoires are complex but conserved. Furthermore, even rare subsets were detected in all patients. For example, the hDC3 subset constitutes 0.25% of all cells but was found reproducibly. Second, analyzing more cells could further refine TIM heterogeneity. Nonetheless we analyzed enough cells to detect TIM states as rare as 0.05% with at least 10 cells in total across at least 3 patients, with 98% confidence. We therefore believe we cover the major TIM transcriptional programs. Third, in comparing to the mouse, one model system of LA was used. Thus, where differences occur between mouse and human, we do not yet know whether these are due to species-specific differences. However, the degree of commonality between species was considerable and argues for the relevance of using animal models to study various TIM states. Additionally, the approaches developed here can be repeated for other mouse models, which will facilitate choosing and/or justifying future models with respect to human biology.
Despite these caveats, our analysis serves to clarify TIM heterogeneity. We focus on two general lessons.
First, we see coherence of TIM heterogeneity. Unsupervised clustering revealed a landscape that is readily understandable, with each myeloid cell type showing several stereotyped gene expression programs that are either distinct, or form a simple continuum. Further, though patients showed variable proportions of TIMs in each cell state, the number of states remained limited. This contrasted strongly with tumor cells from the same patients, which were patient-specific. We conclude that we likely have a complete description of the common TIM gene expression programs. For neutrophils in particular, the h/mN2 type I interferon response signature stands apart from a continuum of states from h/mN1 to tumor-specific h/mN5, in both human and mouse. Even for macrophages, which showed the most complex phenotypes and highest variability between patients, the human and mouse macrophages expressed many of the same genes, albeit in different combinations. Among DCs, pDCs and cDC1–3 should represent a fairly complete description of tissue DCs, showing striking coherence between organisms. Similarly, monocytes are often decomposed into classical and non-classical subsets in both human and mice, and we indeed observed these two subsets with a one-to-one correspondence between organisms. However, we also observed two monocyte subsets that are characterized by neutrophil or dendritic cell gene expression, respectively. These monocytic states emerged independently in both mouse and human tumors, and in blood samples. Generally, the appearance of congruent expression programs in two different organisms provide confidence in the results and establish handles for relating TIM biology between laboratory and clinic.
Second, we find that blood myeloid cells poorly reflect TIM states. We asked whether the complexity of TIM cells is reflected in blood myeloid cell populations, which would offer practical advantages in their accessibility for diagnostics and clinical research. ScRNA-Seq is particularly valuable for this question since cell surface marker expression can often change as cells exit the blood. From our study, we conclude that the blood provides a limited view of TIMs, and it also hosts myeloid cell states not seen at the tumor site. This does not rule out that liquid biopsies could predict disease outcomes, but it does establish a poor relationship between blood myeloid cell states to those at the tumor site.
With the population structure and marker genes defined here for multiple cell populations, in the future we can test the functional relevance of distinct subsets to tumor progression; correlate the abundances of cell types to clinical outcomes and therapeutic response in humans and in animals; and establish the developmental relationships between TIM subsets and their dependence on signaling. Knowledge of the human-mouse correspondence of TIMs will play a critical role in achieving these objectives with the ultimate goal of gaining insight into myeloid biology and myeloid immunotherapy.
STAR methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Allon M. Klein (allon_klein@hms.harvard.edu).
Raw RNA sequence data from human samples are not available publicly due to patient privacy concerns.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human sample acquisition.
In this study we analyzed 7 human primary NSCLC samples (Table S1). Fresh samples used in the study were obtained from patients undergoing surgical resection. Blood was drawn off of patients’ arterial or intravenous line in the operating room during their surgery, while the patient was under anesthesia. This study was conducted with approval of the Dana-Farber Brigham and Women’s Cancer Center IRB and written informed consent from subjects. The protocol allows collection of discarded tissue samples as well as blood. De-identified genomic information will be deposited in protected public repositories for subjects explicitly allowing it on the consent form. Human tissue samples were de-identified before transfer to the Klein laboratory, and analysis is not considered human subject research under the US Department of Human and Health Services regulations and related guidance (45 CFR part 46). Tumor lung samples were dissociated for single cell RNA sequencing as described below. Perpendicular sections immediately flanking 1-to 3-mm thick fragments of all tumor tissues were reviewed by a pulmonary pathologist to confirm the diagnosis and tumor content. Where available, basic demographic information for the patient samples in the study is included in Table S1.
Mouse tumor model.
Murine KP1.9 lung adenocarcinoma tumor cells were injected into 10 weeks old C57BL/6 male mice intravenously (2.5×105 cells in 100µl PBS) to develop orthotopic tumors in the lung. The tumor cell line was derived from lung tumor nodules of a C57BL/6 KrasLSL-G12D/WT;p53Flox/Flox (KP) mouse and was kindly provided by Dr. Zippelius (University Hospital Basel, Switzerland). Mice were analyzed for tumor phenotypes 4 weeks post-tumor initiation. Tumor burden was scored by measuring postmortem lung weight as proxy for tumor burden as previously described (Pfirschke et al., 2016). Age and sex matched mice (14 weeks old male mice) were used as tumor free, healthy controls. C57BL/6 wild type mice were obtained from the Jackson Laboratory. All animals were housed under specific pathogen free conditions at the Massachusetts General Hospital (MGH). Experiments were approved by the MGH Institutional Animal Care and Use Committee (IACUC) and were performed in accordance with MGH IACUC regulations.
METHOD DETAILS
RNA in situ hybridization (RNA scope) and Immunohistochemistry (IHC)
Tissues were fixed in 10% Formalin overnight (HT501128 Sigma) while shaking, paraffin embedded and cut in 5 µm sections. RNA in situ hybridization was performed with Manual RNAscope® 2.5 HD Reagent Kit (RED) (ACD Bio, # 322350) following manufacturer’s instructions as previously described (Wang et al., 2012). The tissue sections were hybridized with pre-designed probes spanning peptidase inhibitor 3 (PI3) (NM_002638.3) or peptidylprolyl isomerase B (cyclophilin B, PPIB, positive control) (NM_000942.4) mRNA (ACD bio, # 534371 and #313901) in the HybEZ hybridization oven (ACD Bio) for 2 hours at 40°C, following a ser ies of pretreatment steps (deparaffinization in xylene, dehydration, peroxidase blocking, and heat-induced epitope retrieval by the target retrieval and protease plus reagents; ACD Bio). The slides were then processed for standard signal amplification steps and the red chromogen development was performed by the RNAscope 2.5 HD (Red) detection Kit (ACD Bio, #322360). For sections that were double labeled with PI3 and Myeloperoxidase, the tissues were blocked in 2% bovine serum albumin (BSA) in Tris-buffered saline (TBS) for an hour, and then incubated with rabbit polyclonal anti-MPO antibody (Dako, #A0398, 1:800) at 4o overnight. Horse radish peroxidase (HRP) labelled Anti-Rabbit IgG (ImmPRESS, #MP-7401) was applied for 30 minutes after washes in TBS (3X10 minutes). The HRP signal was detected by 3,3’-diaminobenzidine (dab) substrate chromogen system (Dako, #K3468). The slides were then counterstained in 50% hematoxylin (Dako, #S2302) for 2 minutes, air-dryed and coverslipped with EcoMount. For sections stained with MPO only, the tissues were rehydrated in EtOH series after deparaffinization in xylene. Antigen retrieval was performed in parboiling 1X Target Retrieval solution (RNA scope) for 20 minutes, the slides were cooled at room temperature, followed by peroxidase (3% H202) (10 minutes) and BSA (%2 in TBS, 30 minutes) blocking steps. The tissues were incubated with MPO antibody (#A0398, Dako, 1:800) for two hours at room temperature. ImmPRESS™ HRP Anti-Rabbit IgG (#MP-7401) and Dako DAB substrate chromogen (#K3468) systems were used for detection. The slides were then counterstained in 50% hematoxylin for 2 minutes, air-dryed and coverslipped with EcoMount. Images were taken with the Keyence BZ-X800 microscope at 20x resolution. Approximately 100 images were obtained for each section. Ten non-overlapping 20x images for each patient and staining were analyzed in FIJI. For PI3 analysis: The Color deconvolution plugin (Ruifrok and Johnston, 2001) was used to extract the PI3 signal (Fast red). Images were thresholded using Shanbhag thresholding (Shanbhag, 1994): lack of signal in the negative control sample and strong positive signal in the RNAscope positive control were confirmed (PPIB provided by ACD bio). The same threshold was applied across samples and PI3+ cells (>1 positive dot) were counted using the Cell counter plugin (https://imagej.nih.gov/ij/plugins/cell-counter.html). The results are expressed as PI3+ cells/mm2. For MPO analysis: the DAB signal was extracted using the Color Deconvolution plugin and thresholded images (Otsu, 1979) were quantified using the Analyze Particle tool (using watershedding to separate nearby particles). An MPO+ particle was defined as ≥20 pixels2, or ~3 µm2. The results are expressed as MPO+ cells/mm2.
Patient lung tumor sample dissociation.
Human lung tumor tissue was dissociated for single cell RNA sequencing using the Tumor Dissociation kit by Miltenyi Biotec (cat. no. 130–095-929). Briefly, the tissue was placed in a petri dish on ice and cut into small pieces of 2–4 mm. The pieces were infused with the RPMI/enzyme mix (Miltenyi Biotec), transferred to a gentleMACS C tube containing RPMI/enzyme mix, attached to the sleeve of the gentleMACS Octo Dissociator and run using the “37_h-TDK2” program. After termination of the program, the cells were spun down at 300 x g for 10 min at 4°C, resusp ended in RPMI-2% FBS, passed through a 70µm strainer and centrifugation was repeated. The cell pellet was treated with 1 ml ACK solution for 7 min at room temperature, the lysis stopped with 4 ml RPMI-2% FBS. After centrifugation, the cells were resuspended in 1 ml of RPMI-2 % FBS and passed through a cell strainer to obtain a single-cell suspension. Immediately before transcriptome barcoding using the inDrop platform, cells were pelleted at 300 x g for 5 min at 4°C in a swing-bucket rotor, gently resuspended in 1x PBS with 0.1% w/v BSA, manually counted on a hemocytometer and diluted to 100 k cells/ml. The final cell suspension included 15% v/v OptiPrep (Sigma-Aldrich, cat. no. D1556).
Whole blood preparation for single cell RNA sequencing (scRNAseq).
3–4 ml of blood were withdrawn in a K2EDTA vacutainer and stored on ice. Upon delivery to the Klein laboratory where scRNAseq was performed, 10µl of 0.5M EDTA were added per ml of blood and cells were resuspended by inverting the tube 3–5 times. Blood was kept on ice at all times prior to red blood cell (RBC) removal by one of the following protocols.
For patients 1 and 2, RBC removal was performed by ACK lysis. 2 ml of blood were transferred into a 50-ml tube prefilled with 20 ml of ACK lysis buffer (Lonza, cat. no. 10–548E or Stemcell, cat. no. 07800) and the tube was rotated for 10 min, protected from light. ACK lysis was performed at room temperature, all other steps were performed on ice. After stopping lysis by topping the tube with ice-cold 1xPBS, the cells were pelleted by centrifugation at 450 x g for 5 min at 4 °C in a swing-bucked rotor and the superna tant decanted. The cells were then resuspended in 1x PBS with 0.1% w/v BSA and transferred into a 15-ml tube. The cell suspension was underlaid with two layers of OptiPrep solution in 1xPBS, 5 ml of 15% v/v and 2 ml of 47% v/v, and centrifuged for 15 min at 800 x g in a swing-bucket rotor with the break turned off. RBC-depleted blood cells were retrieved from the interface between the two layers of OptiPrep, counted, and diluted to a final concentration of 100k/ml in 1xPBS with 0.1% w/v BSA and 15% v/v OptiPrep.
For the remaining patients, RBC removal was performed using the ErythroClear kit (Stemcell Technologies, cat. no. 01738). In the final scRNAseq data we did not find evidence of biases specific to the RBC-removal protocol used and we prefer this second protocol for allowing a shorter sample processing time. 100 ul of blood were transferred to a 1.5-ml tube prefilled with 500 ul of 1xPBS with 0.1% w/v BSA and mixed by pipetting up-and-down. 170 ul of ErythroClear bead suspension were transferred to a separate 1.5-ml tube, placed on a magnet for 1 min, and the unbound storage solution discarded. Beads were resuspended in 100 ul of 1xPBS with 0.1% w/v BSA by vortexing vigorously, then gently mixed with the cell suspension and left at room temperature for 1 min. The tube was placed on a magnet for 3 min. The RBC-depleted unbound fraction was carefully pipetted out and into another tube, pelleted by centrifugation at 450 x g for 5 min at 4 °C, and resuspended to a final concentra tion of 100k/ml in 1xPBS with 0.1% w/v BSA and 15% v/v OptiPrep.
Flow cytometry-based sorting of CD45+ cells from lung tissues.
Single cell suspensions were obtained from lung tumor or tumor-free (i.e. healthy) lung tissue of age and sex matched C57BL/6 male mice which were perfused with PBS. Small tissue pieces were generated from perfused lungs using scissors and digested (RPMI containing 0.2 mg/ml collagenase type I, Worthington Biochemical Corporation) for 15min at 37°C while shaking (700rpm). Digested lung tissue was gently meshed through 70µM cell strainers using a plunger. Single cell suspensions were then stained with a fluorescent conjugated antibody specific to CD45 (clone 30-F11, Biolegend) for 45min at 4°C. The cells were washed with staining buffer (PBS containing 0.5% BSA and 2 mM EDTA) and CD45+ live cells were sorted on a FACS Aria cell sorter (BD) into FBS containing tubes which were kept on ice until the cells were further processed for scRNASeq. 7-aminoactinomycin (7AAD, Sigma) positivity was used to exclude dead cells.
Immunophenotyping by flow cytometry.
Single cell suspensions from lung tumor or tumor-free tissues were obtained as described above and were incubated with FcBlock (clone 93, Biolegend) for 15min before staining with fluorescent conjugated antibodies for 45min at 4ºC. The cells were washed with staining buffer (PBS containing 0.5% BSA and 2 mM EDTA) and analyzed on a LSRII flow cytometer (BD). 7AAD was used to exclude dead cells and doublet cells were removed based on their forward/side scatter properties. Based on cell surface marker expression, H-SiglecFlow neutrophils (CD45+CD11b+Ly-6G+SiglecFlow) from healthy lung tissue and T-SiglecFhigh neutrophils (CD45+CD11b+Ly-6G+SiglecFhigh) as well as T-SiglecFlow neutrophils (CD45+CD11b+Ly-6G+SiglecFlow) from lung tumor tissue were identified by flow cytometry and analyzed for further marker expression identified by scRNASeq. Following Abs were purchased from BD: CD11b (557397, clone M1/70); Ly-6G (551461, 560599, clone 1A8); SiglecF (564514, clone E50–2440), Biolegend: CD45 (103126, clone 30-F11); CD73 (127205, clone TY/11.8) or R&D Systems: Clec5a (FAB1639P, clone 226402).
Gene expression quantification by quantitative PCR.
Neutrophils from lungs of KP tumor-bearing or tumor-free mice were investigated in order to validate whether these cells exhibited transcriptional characteristics identified by scRNASeq. Neutrophils were FACS sorted based on surface marker expression (CD45+CD11b+Ly-6G+SiglecFlow or high) as described above. RNA was isolated from the sorted cells using the RNeasy Micro Kit (Qiagen) according to manufactures procedures and cDNA was generated utilizing the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Afterwards Real-time PCR assays were performed using the TaqMan Fast Advanced MasterMix together with TaqMan probes at the 7500 Fast Real-Time PCR System (Applied Biosystems). b-Actin was used as a housekeeping gene.
Single cell RNA sequencing (scRNAseq) and read processing.
scRNAseq was performed using inDrop as described previously (Zilionis et al., 2017), with modifications to DNA primers indicated in Table S6. Briefly, a microfluidic device was used to co-encapsulate individual cells and hydrogel beads carrying barcoding reverse transcription (RT) primers into 2–3 nl droplets. After a RT reaction, droplets were broken, and the resulting bulk material was taken through 1) second strand synthesis, 2) in vitro transcription providing linear amplification of the material, 3) fragmentation of the amplified RNA, 4) a second reverse transcription, and 5) PCR, yielding in a sequencing-ready library. Libraries were sequenced on the NextSeq Illumina platform, paired-end mode, with read lengths summarize in Table S6. Gene expression counts for individual cells were generated from raw fastq files using the indrop.py pipeline (Zilionis et al., 2017), (github.com/indrops), using human and mouse genome assemblies GRCh38/81 and GRCm38/91 (genome assembly/ENSEMBL release).
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell data filtering and normalization.
To retain high quality transcriptomes, total count and a mitochondrial count filters were applied. For mouse, transcriptomes with more than 600 total counts and less than 15% of counts coming from mitochondrial genes were retained. For human, these thresholds were 300 total counts and 20%, respectively. A more permissive filtering was necessary to i) avoid filtering out human neutrophils, that naturally have a lower mRNA content; ii) retain non-hematopoietic cells which are more sensitive to dissociation and show a higher mitochondrial gene fraction, possibly indicating premature lysis. After this initial cleanup, data was normalized by total counts as described (Klein et al., 2015).
Cell multiplet removal.
We applied Scrublet (Wolock et al., 2018) to remove putative hybrid transcriptomes occurring when two or more cells enter the same microfluidic droplet and receive the same barcode. Scrublet assigns each measured transcriptome a ‘doublet score’, which indicates the likelihood of being a hybrid transcriptome. For mouse samples, we removed the cells with the top 2.5% highest doublet scores. For human samples, a cluster-level approach was applied. First, doublet scores were determined for each patient and source (tumor vs blood) individually, and 1.2–5.4% of highest scoring cells were flagged as potential doublets after visual inspection of doublet score distributions. Next, for tumor and blood separately, a kNN graph of cells from all patients was built using SPRING (see below), divided into 500 spectral clusters, and clusters with a large fraction of potential doublets were removed.
Dimensionality reduction of single cell data.
Single cell data was pre-processed for visualization and clustering by dimensionality reduction as described in (Klein et al., 2015). In brief, highly-variable genes were identified using the V-statistic, a modified Fano Factor corrected for variation in sequencing depth and cell size (Klein et al., 2015). Genes with V<mode(V) were excluded, and genes were further filtered based on minimal expression level (minimum 5 UMI filtered counts in a minimum of 5 cells), and based on minimum correlation to other genes (see table below). A principal component analysis was then carried out using gene correlation, and the state vector of each cell was defined as the first m principal components (PCs). For human samples, m was set to be the number of non-random PCs, as in (Klein et al., 2015). In brief, PCA was performed on a shuffled gene correlation matrix, and the largest eigenvalue for shuffled data, λ*, over 10 permutations, was defined as the threshold eigenvalue. m was then set to include all eigenvalues λ>λ*. The number of resulting highly-variable genes, and the number of resulting PCs is shown for each of the visualizations in the paper.
| Data set | Gene correlation cutoff* | Resulting number variable genes** | Number PCs |
|---|---|---|---|
| Human total cells from tumor (Fig. 1B) | N=2,R=0.1 | 4056 | 370 |
| Human tumor immune-only (Fig. 1C) | N=2,R=0.1 | 3095 | 211 |
| Human tumor non-immune-only (Fig. S1A) | N=50,R=0.1 | 1235 | 89 |
| Human blood-only (Fig. 7C) | N=1,R=0.2 | 1649 | 139 |
| Human blood and tumor immune cells (Fig. 7A) | N=2,R=0.1 | 3534 | 306 |
| Mouse total cells (Fig. 1D) | N=50,R=0.05 | 4965 | 60*** |
Genes correlated at >R (Pearson’s) to ≥ N genes are retained
Specific genes available in Table S2, Excel sheet “genes_used_SPRING”
For mouse, no randomization was carried out
Batch effect correction.
We observed a technical batch effect in mouse libraries, which were prepared for sequencing in several rounds. To prevent batch effects from distorting subsequent data analysis, we used one batch to select variable genes and to calculate principal component (PC) gene loadings. Cells from all batches were then projected into the reduced space as described in the previous section, and all subsequent analysis was performed on the reduced PC space. The batch selected to generate the PC embedding included libraries from all biological mouse samples used in this study.
Partitioning of immune and non-immune transcriptomes from human tumor samples.
For human tumor tissues, cell transcriptomes were computationally annotated as “immune” (main figures) and “non-immune” (Fig. S1) as follows. Cells from tumor samples from all patients were first embedded in PC sub-space as described above. Cells were then used to define a k-nearest neighbor graph (k=5 nearest neighbors), which was partitioned into 200 spectral clusters for the sole purpose of this partitioning. Average gene expression was defined for each cluster. Cells were defined as non-immune if belonging to a cluster low for PTPRC (gene for CD45) and high for any of the following genes: MUC5A (goblet cell marker), KRT5 (basal epithelial cell marker), SFTPD (secretory cell marker), EPCAM (pan-epithelial cell marker), CDH5 (endothelial cell marker), COL1A2 and ACTA2 (fibroblast markers). Fig. 1B shows the labels assigned to each of the cells.
Bayesian classifier of immune cell types and comparison cell states to published data sets.
To annotate the cell type of each single cell transcriptome (Figs. 1C,D), we used a Bayesian cell type classifier as reported in (Zemmour et al., 2018) and adapted from the approach in (Jaitin et al., 2014). The classifier defines a set of labeled reference transcriptomes using publicly available bulk transcriptional profiles of annotated and sorted cell populations. The classifier returns a maximum likelihood (ML) cell type based on a multinomial model (reproduced below from the above references for completeness), as well as the likelihoods themselves. Both ML cell types and underlying likelihoods are reported at different points in the paper. ML cell types are shown for:
All human immune cells (Fig. 1C, Fig. S1H) using as a reference the RNA-Seq data from (Newman et al., 2015) (“LM22” dataset).
All mouse immune cells in Fig. 1D. The reference data set used is microarray data from phase 1 and 2 of the Immunological Genome Project by the IMMGEN consortium (Heng et al., 2008).
Cells of the human DC3 population described in this study classified by hDC1, hDC2 or hMonoDC subsets (Fig. S4C, bar chart).
For microarray data, linear (i.e. non-log transformed) microarray data was used. Probes were collapsed to unique gene symbols by selecting the probe with the maximum mean expression. For all data (microarray and RNA-Seq), the data was quantile normalized, replicates averaged (when available). For RNA-Seq data, a pseudovalue of 50 TPM was added. Only genes symbols shared between with single cell RNA-seq data were retained.
For each analysis, the reference data set consists of a gene expression matrix Xi,j (cell type i=1,..,M; gene j=1,…,N), and M cell type labels. A single cell RNA-Seq profile defines raw expression counts uj for gene j=1,..N. The classifier returns the likelihood of cell type i given P(i|u), defined as
, where Z is a normalization constant (ensuring ,P0(i) is a prior expectation on the abundance of cell type i, and is the fraction of transcripts from gene j in cell type i. Except where stated otherwise in Table S7, we used a naïve prior . In the cases summarized in Table S7, several labels for the same cell type were included in the classification to reflect different experimental conditions. In this case, we adjusted Po(i) to ensure that each major cell type had equal prior likelihood.
Table S7 shows the reference data sets and priors for the likelihood calculations in the paper. Heat maps in Figs. 4F,L, Figs. 5D,E,I,J, and Figs. S4C, D, F, H show the average likelihood of cells per cluster.
Comparison of DC and Monocytes subsets to previously reported populations (supporting Fig S4).
To relate DC and Monocyte subsets described in this study with populations previously reported in (Villani et al., 2017), (Ardouin et al., 2016), and (Garris et al., 2018), we applied the Bayesian classifier described in the previous section, and treated the resulting likelihoods as a convenient distance measure. For all three comparisons in Figs. S4D,F,H the same strategy was applied. First, we classified the previously described populations by all of the immune human (Fig. S4D) or mouse (Figs. S4F,H) subsets described in this study. The classification was performed on either single cells (Figs. S4D,H) or bulk microarray replicates (Fig. S4F). For heat maps showing classification to major cell types, the priors of detailed subsets Po(i) were adjusted to ensure that each major cell type had equal prior likelihood (see previous section and Table S7). For heat maps showing likelihoods of previously reported populations being DCs, pDCs, Monocytes and Macrophages, the classification included only subsets that fall in these myeloid cell categories. Heatmaps showing ‘reciprocal likelihood products’ show the product of Likelihoods obtained by applying the Bayesian classifier in both directions. Using this approach, only subset pairs with mutual cross-correspondence show non-vanishing scores.
For Fig. S4E, the gene set shown is the union of top 5 most enriched genes per cell population identified separately within each of the two data sets (i.e. Villani et al. and this study). Only genes present in both datasets were considered. For each study separately, genes were selected as follows:
Genes were pre-filtered to be expressed at at least 250 TPM in at least 3 cells.
For each population i, a two-tailed Mann-Whitney U test was performed to only retain genes that show a significantly (FDR<0.05) different expression level between cells inside i vs all other cells.
The top 5 genes with the highest fold-change in average expression in population i vs outside i were selected. A pseudovalue of 50 TPM was added before calculating fold-changes.
The selection of genes shown in Figs. S4G and I was analogous, with the exception that step (1) was omitted for bulk microarray data by Ardouin et al. (Fig. S4G). This dataset is composed of an equal number of replicates (n=3) per DC population.
Processed previously published datasets used in this analysis are provided in Table S7.
SPRING plots.
The single cell transcriptomes embedded in a low-dimensional PC space were visualized using SPRING (Weinreb et al., 2018). SPRING builds a k-nearest neighbor (kNN) graph of cells and represents it in 2D through a force-directed layout. Shown in the paper are graphs with k=5. The SPRING plots are available for interactive exploration at the web addresses listed under DATA AVAILABILITY and in the Key Resource Table.
Clustering of transcriptomes.
Spectral clustering was used to define cell subsets, specifically k-means clustering on k graph spectral coordinates after L2 normalization (Wagner et al., 2018). To determine the number of clusters, we first over-clustered the data (large k), and then merged adjacent spectral clusters that showed only small differences in gene expression (see Differential gene expression analysis below).
Comparing abundances of FACS gates to scRNA-Seq.
For Fig. 1G, each cell in the scRNA-Seq data set was assigned to a FACS gate based on mRNA expression of surface antigens. For this, we assumed that transcript levels detected by RNA-Seq can be related to the protein levels that are quantified by FACS. Inspection of bulk transcriptomic data on IMMGEN confirmed that this is broadly true for all antigens (Heng et al., 2008). As a first step, we calculated for each cell transcriptome a binary vector of FACS markers as follows: (1) we clustered the scRNA-Seq data as described, and (2) as a denoising step, all cells in each cluster were assigned the average gene expression of the cluster. (3) For each gene corresponding to a FACS marker described in the “Flow cytometry-based sorting of CD45+ cells from lung tissues” section above, we discretized expression (1=expressed, 0 not expressed) by picking a threshold below which the marker would be considered low at the protein level. (4) The threshold parameters were chosen manually to generally delineate a defined second mode (present in most markers) or a right heavy tail (Cd4). Gr-1, which is known to react with both Ly6c and Ly6g, was modeled as the average of the two.
Each gate was encoded as a set of markers and a “gate logic” expression, which specified whether a cluster should express the marker to fall into the gate. The gate logic reflects the precise order in which gates were applied to FACS data. We quantified the gate abundance as the fraction of all cells which fell into the clusters assigned to that gate.
Differential gene expression analysis and marker gene identification.
We define cell subset marker genes and variable genes as follows. Let C be the set of all cells considered, partitioned into n disjoint subsets c1, …,cn. Define as the average expression of gene j in subset i, and as the values of for gene j sorted in descending order. Define as the ratio of the k-th highest average subset expression of gene j to the (k+1)-th expression value, after adding a pseudovalue of 50TPM to all average gene expression values.
Cell subset marker genes:
A gene j is a marker gene for cell subset ci if:
Gene j is detected in at least 3 cells at least 3 counts across all cells in C.
Gene j is statistically significantly higher in expression in state ci compared to the complement set (all cells C not in ci). To establish significance, we used a two-tailed Mann-Whitney U test with multiple hypothesis correction, FDR<5%.
Gene j has maximal average expression in subset i, i.e. .
Gene j satisfies , i.e. the max-to-second-max ratio is at least 1.1x.
Prior to marker gene identification, a denoising step was carried out to remove rare outliers which may be due to cell doublets that survived data filtering, or low-quality transcriptomes clustering with transcriptionally unrelated cells. We defined a gene j in population ci as outlier if it was expressed at non-zero value in only one cell in ci and the expression of gene j in that cell was more than 10 counts. The average expression value of outliers in population ci was set to 0 for the purpose of DGE analysis and heatmap normalization.
Heatmaps in Figs. 2A–B, 3B, 3G, 4B, 4I, and Figs. S1B, 2B, 2G, 2J, 7A, 7C and 7G show the expression of the top 20–200 most specific genes for each cell population, ordered by decreasing (max-to-second-max ratio). Full lists of enriched genes and their Whitney-U p values, FDRs, and max-to-second-max ratios are available in Table S2.
Variable genes:
The approach described above for identifying genes enriched in a cell subset will exclude genes that are specific to more than one state, i.e. genes that are variable in average expression level across subsets. Variable genes are used for unsupervised comparison and mouse and human immune cells as described in the following results section. Defining then a gene j was considered a non-exclusive marker gene for subsets with . The fold-change threshold T was set to T=1.5.
Mouse-human and blood-tumor comparison dendrograms and heatmaps
Mouse-human comparisons:
The mouse-human ortholog table was downloaded from the Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org/downloads/reports/HMD_HumanPhenotype.rpt), and only genes with a one-to-one translation between human and mouse were considered (see Table S4).
For an unsupervised comparison of a set of mouse cell states cM1,…,cMn and human cell states cH1, …,cHm the following steps were performed. We define pseudocount-adjusted median-normalized average gene expression of subset i as .
Normalization is performed within each species separately. Then:
Identify the set of variable genes (see previous section) across states cM1, …,cMn (mouse).
Identify set of variable genes across cH1,…,cHm (human).
Retain the intersection of the two gene sets, called set h. Gene set h is used for cross-species comparisons.
Combine median-normalized average subset expression counts mouse and human for gene set h into a single table with i=1,…,n,…,m+n cell states, labeled h-1,…,h-n,m-1…,m-m.
Log-transform , and hierarchically cluster rows and columns (genes and cell states). For hierarchical clustering, Ward linkage and correlation distance (defined as d = (1-r)½) were used (r=Pearson correlation).
Full expression tables underlying dendrograms are provided in Table S2. Heat maps in Figs. 2D, 3K, 4O, 5L show a subset of all genes used for hierarchical clustering.
Notes:
* In steps 1,2, changing the threshold value T for highly variable genes by 20% did not affect outcome of clustering, with the exception of Monocytes and MonoDCs. For Fig. 5K we therefore used a precise threshold value of T=1.6. Changing T for this figure clusters hMono1 with hMono3, and mMono1 with mMono3, suggesting that differences between these states within species may be less pronounced that trans-species differences.
* For Fig. 2A, steps 1 and 2 were omitted, i.e. no selection on variability was performed and all 1-to-1 orthologs detected in at least one mouse and one human subset were included. The 1-to-1 match of DCs and the main trends for Neutrophils (summarized in Fig. 3J) remained unchanged with the inclusion of all genes.
Blood-tumor comparisons
For comparing blood to tumor immune cell populations, the analysis was as above but replacing mouse/human with blood/tumor, and without needing to consider orthologs.
Mouse macrophage signature expression in human macrophages.
Given a list of genes L, a subset of cells C, and an expression matrix Xij, (cell i, gene j) we define a gene expression signature as a function σi(L,C,X) that returns a scalar signature score for each cell i in C:
where ranki(XCj) is the dense ranking for gene j of cell i considering only cells in C.
To establish the expression of mouse macrophage gene expression programs in human as shown in Fig. 5M, we calculated signatures σC(L, C, X) through the following steps.
The human cells subset C for signature analysis was taken to be all human monocytes, macrophages, MMDCs and DCs.
One signature gene list LS was defined for each mouse macrophage subset S as 50 genes that (a) have one-to-one orthology between mouse and human (see Table S4), and (b) rank highest in specificity for the subset, as defined above in the “Marker gene” methods section (shown in Fig 5G).
Calculate for each subset S on human cells.
Average across each of the human cell subsets hMono/hMø.
Visualization of cell density changes between healthy and tumor states.
To visualize which immune cell populations are enriched relative to healthy tissues (Fig. S1F), the local density of tumor cells, 0<c<1, was calculated at each node of the k-nearest neighbor graph G defined above (see SPRING plots above). The plot then visualizes the log-transformed ratio of tumor and healthy densities, log2[c/(1-c)].
To calculate c, every node i corresponding to a tumor cell was assigned an initial value , and every node corresponding to a healthy cell was assigned initial value . The vector c′ was then averaged over a local neighborhood to generate c using a graph diffusion approach on the graph G. G is an unweighted undirected graph. The smoothing operation specifically corresponds to the matrix operation c = OSc′, where the smoothing operator is OS = eLβ and L is the random walk graph Laplacian of G. The smoothing operator accepts a single parameter, β, which determines the kernel size, i.e., the extent of smoothing. β=1 was used for Fig. S1F; no data imputation was performed for the rest of the analyses
Kaplan-Meier plots (Fig. 6D).
Kaplan-Meier plots and univariate cox regression p-values were generated using the database and tools described in (Gyorffy et al., 2013), using the associated online tool kmplot.com/analysis/index.php?p=service&cancer=lung. This tool combines data on 1,928 lung cancer patients from multiple studies, of which survival and gene expression data on 720 patients were retained for survival analysis after filtering based on histology (lung adenocarcinoma) and on valid probe sets. For each gene, all patients were included in either a “high expressing” or a “low expressing” group, with the high/low expression threshold defined to maximize statistical significance in differential survival between the two populations. For PI3, the threshold corresponded to the 61st percentile; for ISG15 and CD207 the threshold corresponded to the 30th percentile. Given the small number of hypotheses tested, no multiple hypothesis correction was performed for this figure panel.
Differential gene expression (DGE) analysis between tumor and blood monocytes (Fig. 7G) and neutrophils (Fig. 7K).
Genes differentially expressed between two sets of cells (blood monocytes vs. tumor monocytes, and blood neutrophils vs. tumor neutrophils) were identified using a two-tailed Mann-Whitney U test, with multiple hypothesis correction using the Benjamini-Hochberg procedure. Prior to analysis genes with average expression level of <10TPM in both populations were excluded. Genes identified as significantly different at at 5% false discovery rate, and with an absolute fold-change > 2, were considered as differentially expressed. A pseudovalue of 50 TPM was added before calculating fold-changes. As the Mann-Whitney U-test can give p-values equal to 0, to allow representation in a volcano plot these genes were given the lowest non-zero FDR-corrected p-value.
Embedding of additional transcriptomes into the mouse SPRING plot
Late in the manuscript production process we had the opportunity to include additional mouse scRNA-Seq libraries into the mouse SPRING plot, representing 6% of the cells. To make this data available, we embedded the cells into the SPRING representation using the following approach:
the additional 1088 transcriptomes from tumor-bearing mouse #2 underwent data cleanup steps described above leaving 1047 high-quality transcriptomes. Cleanup included filtering on total counts and percentage of mitochondrial counts, and multiplet removal.
A kNN graph was build using the same parameters as for the rest of mouse cells (see sections Dimensionality reduction of single cell data and SPRING plots).
Edge information from the additional 1047 cells was added to the original kNN graph of the rest of mouse cells.
A force-directed layout was used to obtain the xy coordinates of the additional 1047. The remaining 14,892 transcriptomes were fixed at their original xy coordinates.
The additional transcriptomes were assigned cell subset identities using the Bayesian classifier described above.
Supplementary Material
Table S6. Summary of modifications to published inDrop protocol (Zilionis et al., 2017)(supporting STAR methods).
Table S1. Human non-small cell lung cancer patient sample information and cell numbers for myeloid subsets (supporting Figure 1).
Table S2. Whole-genome average gene expression of major immune cell types in human and mouse lung cancer from scRNAseq data, as well as results of differential gene expression analysis (supporting Figures 1 and 7).
Table S3. Data underlying heatmaps shown in figures of the paper (supporting all figures).
Table S4. Lookup table of one-to-one gene orthologs used for human-mouse comparisons (see methods; supporting all figures).
Table S5. Average expression for selected genes targeted by mouse models, immunotherapy drugs, IHC/FACS across mouse and human subsets (supporting Figure 6).
Table S7. Processed single cell RNA-seq and microarray datasets and priors used for Bayesian classification (supporting all figures).
Highlights.
Human dendritic cell and monocyte subsets show one-to-one equivalence in mouse
Neutrophils exhibit tumor-associated phenotypes that are conserved across species
Myeloid subsets in patient blood only partially overlap with those in their tumors
Unique markers define myeloid cell subsets and associate with clinical prognosis
Acknowledgements
We thank the Harvard Stem Cell Institute for help with FACS sorting; the Single Cell Core Facility at Harvard Medical School for inDrop reagents; the Bauer Core Facility for sequencing; Lucian Chirieac, M.D. for reviewing the H&E slides for the patient tumor samples; Klein and Pittet lab members for helpful discussions; David Pepin for expertise and support with ISH/IHC. This work was supported in part by the Samana Cay MGH (Massachusetts General Hospital) Research Scholar Fund, the Robert Wenner Award from the Swiss Cancer League, and NIH grants R01-AI084880, R01-CA206890, and P50-CA86355 (to M.J.P.); NIH grant R33-CA212697 and an Edward J. Mallinckrodt Jr. Fellowship (to A.M.K.); NIH grant R01-CA218579 (to A.M.K and M.J.P); Boehringer Ingelheim Fonds PhD fellowships (to C.E. and D.Z.); Deutsche Forschungsgemeinschaft PF809/1–1 and MGH ECOR (Executive Committee on Research) Tosteson Postdoctoral Fellowship (to C.P.); the Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research (STaR) Investigator Award, NCIS Yong Siew Yoon Research Grant through donations from the Yong Loo Lin Trust, and NIH grant P01 HL131477–01A1 (to D.G.T.); A.M.K. is a founder of 1CellBio. V.S. is an employee of Sanofi US.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
Single cell RNAseq data generated in this study is available at the NCBI GEO depository under the accession number GSE127465 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE127465). It includes genes counts pre- and post-normalization, and per-cell meta for both mouse and human. The raw fastq files are only provided for mouse due to human patient privacy concerns.
- Human total cells from tumor (Fig. 1B) https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Zilionis2019/human/NSCLC_all_cells
- Human tumor non-immune-only (Fig. S1A) https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Zilionis2019/human/NSCLC_non_immune
- Human blood and tumor immune cells (Fig. 7A) https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Zilionis2019/human/NSCLC_and_blood_immune
Declaration of Interests
M.J.P. has served as a consultant for Aileron Therapeutics, Cygnal Therapeutics, Elstar Therapeutics, KSQ Therapeutics and Siamab Therapeutics; these commercial relationships are unrelated to the current study. AMK is a founder and shareholder in 1CellBio, Inc.
References
- Ardouin L, Luche H, Chelbi R, Carpentier S, Shawket A, Montanana Sanchis F, Santa Maria C, Grenot P, Alexandre Y, Gregoire C, et al. (2016). Broad and Largely Concordant Molecular Changes Characterize Tolerogenic and Immunogenic Dendritic Cell Maturation in Thymus and Periphery. Immunity 45, 305–318. [DOI] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert DJ, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, and Haerynck F (2016). Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet 53, 575–590. [DOI] [PubMed] [Google Scholar]
- Briggs JA, Weinreb C, Wagner DE, Megason S, Peshkin L, Kirschner MW, and Klein AM (2018). The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science 360, eaar5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzetti F, Tamassia N, Micheletti A, Finotti G, Bianchetto-Aguilera F, and Cassatella MA (2018). Human dendritic cell subset 4 (DC4) correlates to a subset of CD14(dim/-)CD16(++) monocytes. J Allergy Clin Immunol 141, 2276–2279 e2273. [DOI] [PubMed] [Google Scholar]
- Coffelt SB, Wellenstein MD, and de Visser KE (2016). Neutrophils in cancer: neutral no more. Nat Rev Cancer 16, 431–446. [DOI] [PubMed] [Google Scholar]
- Dutertre CA, Wang LF, and Ginhoux F (2014). Aligning bona fide dendritic cell populations across species. Cell Immunol 291, 3–10. [DOI] [PubMed] [Google Scholar]
- Eisenbarth SC (2019). Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol 19, 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom C, Pfirschke C, and Pittet MJ (2016). The role of myeloid cells in cancer therapies. Nat Rev Cancer 16, 447–462. [DOI] [PubMed] [Google Scholar]
- Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, Courties G, Rickelt S, Severe N, Baryawno N, Faget J, et al. (2017). Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science 358, eaal5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, Akimova T, Vachani A, Litzky L, Hancock WW, et al. (2014). Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest 124, 5466–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 49, 1148–1161 e1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. (2012). Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13, 1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. (2015). The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, and Jung S (2014). Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14, 392–404. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Schultze JL, Murray PJ, Ochando J, and Biswas SK (2016). New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 17, 34–40. [DOI] [PubMed] [Google Scholar]
- Guffroy A, Mourot-Cottet R, Gerard L, Gies V, Lagresle C, Pouliet A, Nitschke P, Hanein S, Bienvenu B, Chanet V, et al. (2017). Neutropenia in Patients with Common Variable Immunodeficiency: a Rare Event Associated with Severe Outcome. J Clin Immunol 37, 715–726. [DOI] [PubMed] [Google Scholar]
- Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, Van Gassen S, Chen J, Poidinger M, De Prijck S, et al. (2016). Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 45, 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Surowiak P, Budczies J, and Lanczky A (2013). Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 8, e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TS, Painter MW, and Immunological Genome Project, C. (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Hou Y, Zhu L, Tian H, Sun HX, Wang R, Zhang L, and Zhao Y (2018). IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell 9, 1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, et al. (2010). Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 115, e10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, and Amit I (2014). Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna A, Tallapragada N, Veres A, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single cell transcriptomics applied to embryonic stem cells. Cell, accepted for publication. [DOI] [PMC free article] [PubMed]
- Mantovani A, Marchesi F, Malesci A, Laghi L, and Allavena P (2017). Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14, 399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nature methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu N (1979). A Threshold Selection Method from Gray-Level Histograms. IEEE Transactions on Systems, Man, and Cybernetics 9, 62–66. [Google Scholar]
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. (2016). Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 44, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber P, Ochoa AC, and Rodriguez PC (2012). Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Invest 41, 614–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert RC, Jellusova J, and Miletic AV (2011). Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol Rev 244, 115–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruifrok AC, and Johnston DA (2001). Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 23, 291–299. [PubMed] [Google Scholar]
- Schmidl C, Renner K, Peter K, Eder R, Lassmann T, Balwierz PJ, Itoh M, Nagao-Sato S, Kawaji H, Carninci P, et al. (2014). Transcription and enhancer profiling in human monocyte subsets. Blood 123, e90–99. [DOI] [PubMed] [Google Scholar]
- See P, Dutertre CA, Chen J, Gunther P, McGovern N, Irac SE, Gunawan M, Beyer M, Handler K, Duan K, et al. (2017). Mapping the human DC lineage through the integration of high-dimensional techniques. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag AG (1994). Utilization of Information Measure as a Means of Image Thresholding. CVGIP: Graphical Models and Image Processing 56, 414–419. [Google Scholar]
- Sharma P, and Allison JP (2015). The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, et al. (2017). Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, and Klein AM (2018). Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 360, 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang L-C, Bui S, Nielson A, Wu X, Vo H-T, Ma X-J, and Luo Y (2012). RNAscope: A Novel in Situ RNA Analysis Platform for Formalin-Fixed, Paraffin-Embedded Tissues. The Journal of Molecular Diagnostics 14, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C, Wolock S, and Klein AM (2018). SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2018). Scrublet: computational identification of cell doublets in single-cell transcriptomic data. bioRxiv [DOI] [PMC free article] [PubMed]
- Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, and Benoist C (2018). Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol 19, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R, Nainys J, Veres A, Savova V, Zemmour D, Klein AM, and Mazutis L (2017). Single-cell barcoding and sequencing using droplet microfluidics. Nat Protoc 12, 44–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S6. Summary of modifications to published inDrop protocol (Zilionis et al., 2017)(supporting STAR methods).
Table S1. Human non-small cell lung cancer patient sample information and cell numbers for myeloid subsets (supporting Figure 1).
Table S2. Whole-genome average gene expression of major immune cell types in human and mouse lung cancer from scRNAseq data, as well as results of differential gene expression analysis (supporting Figures 1 and 7).
Table S3. Data underlying heatmaps shown in figures of the paper (supporting all figures).
Table S4. Lookup table of one-to-one gene orthologs used for human-mouse comparisons (see methods; supporting all figures).
Table S5. Average expression for selected genes targeted by mouse models, immunotherapy drugs, IHC/FACS across mouse and human subsets (supporting Figure 6).
Table S7. Processed single cell RNA-seq and microarray datasets and priors used for Bayesian classification (supporting all figures).