

Short abstract
Blocking the negative co‐stimulatory molecule PD‐1 prevents immune suppression, blocks apoptosis, and improves survival in sepsis.
Keywords: entotoxin shock/sepsis, tolerance/suppression/anergy
Abstract
There is increasing recognition that a major pathophysiologic event in sepsis is the progression to an immunosuppressive state in which the host is unable to eradicate invading pathogens. Although there are likely numerous causes for the immunosuppression, expression of negative costimulatory molecules on immune effector cells is a likely contributing factor. PD‐1 is a recently described, negative costimulatory molecule that has potent effects to inhibit T cell activation, cytokine production, and cytotoxic functions. PD‐1 plays a critical role in the host response to specific pathogens, but relatively little work has been done on the possible effects of PD‐1 in sepsis. We hypothesized that the anti‐PD‐1 antibody would improve survival in sepsis. Mice underwent CLP, and PD‐1 expression was quantitated. Additionally, the effects of anti‐PD‐1 antibody on lymphocyte apoptosis, cytokine production, host immunity, and survival were determined. PD‐1 expression increased beginning 48 h after sepsis, and >20% of CD4 and CD8 T cells were positive by 7 days. Anti‐PD‐1 antibody administered 24 h after sepsis prevented sepsis‐induced depletion of lymphocytes and DCs, increased Bcl‐xL, blocked apoptosis, and improved survival. Anti‐PD‐1 also prevented the loss in DTH, a key indicator of immunocompetence in sepsis. Thus, delayed administration of anti‐PD‐1 antibody, an important therapeutic advantage, was effective in sepsis. Furthermore, these results add to the growing body of evidence that modulation of the positive and negative costimulatory pathways on immune cells represents a viable therapeutic approach in reversing immunosuppression and improving sepsis survival.
Abbreviations
- CLP
cecal ligation and puncture
- DC
dendritic cell
- DTH
delayed‐type hypersensitivity
- MFI
mean fluorescence intensity
- PD‐1
programmed death‐1
- PD‐L1/2
programmed death ligand 1/2
- SHP‐2
Src homology 2‐containing tyrosine phosphatase 2
- TNBS
trinitrobenzene sulfonic acid
Introduction
Sepsis, the systemic, inflammatory response syndrome that occurs during severe infection, kills more than 210,000 people in the United States annually [1]. Sepsis initiates a complex immune response that varies during the duration of the illness [2, 3, 4, 5]. Although recent studies have shown that proinflammatory and anti‐inflammatory responses occur simultaneously during the first phase of the disease, the early net result is often a potent “cytokine storm”‐mediated hyperinflammatory response [4]. With contemporary standard‐of‐care measures, the majority of patients survive the early hyperinflammatory phase and enter a stage of protracted immunosuppression, which has been termed “immunoparalysis.” This immunosuppression in sepsis is manifest by failure to clear the primary infection, reactivation of HSV‐I and cytomegalovirus, and development of new, secondary infections, often with organisms that are not particularly virulent to the immunocompetent patient [6, 7, 8]. A recent autopsy study of 235 patients who had a diagnosis of sepsis in the surgical intensive care unit showed that ∼80% had a persistent site of infection on postmortem examination, thus highlighting the significant degree of immune compromise in these patients [9].
Although a number of mechanisms of immune suppression have been shown to play a role in sepsis, the precise basis for the immunoparalysis is not well‐delineated. It is likely that a number of interacting processes are responsible, and these mechanisms include apoptotic depletion of immune effector cells, increased T regulatory cells [10], increased myeloid‐derived suppressor cells [11], and a shift from a TH1 immune phenotype to an anergic or TH2 phenotype [3, 4, 5]. Another possibility for the immunosuppression of sepsis is an up‐regulation of inhibitory proteins or a down‐regulation of costimulatory proteins [12, 13, 14, 15]. The relatively recently identified receptor, PD‐1, has been shown to be a crucial modulator of host immune responses [16, 17, 18]. PD‐1, a member of the B7‐CD28 superfamily, is a negative, costimulatory molecule that is inducibly expressed, primarily on the cell surface of activated CD4 and CD8 T cells [16, 17, 18]. Signaling through PD‐1 inhibits the ability of T cells to proliferate and produce cytokines and attenuates cytotoxic T cell function [12, 13]. The key immunomodulatory role of PD‐1 is revealed by studies showing that PD‐1 knockout mice develop autoimmune diseases, including a lupus‐like syndrome and cardiomyopathy [14, 15]. The two known agonistic ligands to PD‐1, i.e., PD‐L1 (B7‐H1) and PD‐L2 (B7‐DC), are regulated by different mechanisms and expressed by different cell types. PD‐L1 is inducibly expressed across a broad spectrum of immune and nonimmune cells, including T cells, B cells, DCs, macrophages, endothelial cells, epithelial cells, and pancreatic islet cells [17, 18]. PD‐L2 is mainly expressed on DCs and macrophages. Ligation of PD‐1 with PD‐L1, in concordance with TCR or BCR activation, leads to recruitment of SHP‐2, dephosphorylating the essential scaffolding protein Gab2 and preventing activation of PI3K, which limits downstream activation of Akt, the essential kinase in costimulatory receptor signaling [12].
PD‐1 can inhibit a robust immune response, thereby facilitating chronic viral and fungal infections [16, 19, 20, 21, 22, 23]. In a mouse model of lymphocytic choriomeningitis, functional exhaustion of virus‐specific CD8 T cells was shown to correlate with increased PD‐1 expression, and CD8+ T cells lacking PD‐1 are more resistant to tolerance than wild‐type CD8+ T cells [19]. In patients infected with HIV‐1, PD‐1 expression renders infected T cells more susceptible to apoptosis, and it is postulated that the virus down‐regulates PD‐1 to ensure its survival [24]. In a mouse model of Histoplasma capsulatum, PD‐L1 was up‐regulated on macrophages and splenocytes during infection, and PD‐1‐deficient mice had improved survival compared with wild‐type mice [23]. Treatment with a blocking antibody to PD‐1 was shown to improve survival in wild‐type mice infected with Histoplasma [23]. Blockade of PD‐1/ligand interactions has been shown to reverse T cell tolerance and exhaustion and is associated with resolution of chronic infection models [12, 13].
Relatively little work has been performed on the role of PD‐1 in the severe bacterial infections characterizing sepsis. Huang et al. [25] showed recently that mice deficient in PD‐1 have increased bacterial clearance and improved survival in the CLP model of sepsis. Their work showed that PD‐1 contributes to macrophage dysfunction during sepsis and concluded that PD‐1 could be an important therapeutic target. Our previous studies have shown that PD‐1 mRNA is highly induced in CD4 T and B cells, beginning at 8 h and persisting at 18 h after sepsis [26].
The purpose of this study was to determine if the survival advantage observed in the PD‐1 null mice in sepsis could be recapitulated therapeutically with an antagonistic antibody to PD‐1. A potential problem with using any knockout mice to examine particular pathways and therapies is that compensatory pathways have been shown to occur with genetic deletions, thereby resulting in misleading findings about the true nature of the gene in question. Thus, the results in null mice may not reflect the effects of acute inhibition of the gene in a clinical setting. Second, the results in PD‐1 null mice may not reflect the effects of blocking PD‐1 after sepsis has already occurred. In addition, this study examined potential mechanistic insights into the putative, beneficial effect of PD‐1 antagonism in sepsis.
MATERIALS AND METHODS
CLP model of sepsis
Eight‐ to 10‐week‐old male CD1 mice (22–28 g body weight) were purchased from Charles River Laboratories (Wilmington, MA, USA) and underwent CLP according to the protocol approved by the Washington University Animal Studies Committee (St. Louis, MO, USA) and as described previously [27, 28]. Mice were anesthetized with isofluorane, and a midline abdominal incision was made. The cecum was mobilized, ligated below the ileocecal valve, and punctured twice with a 27‐gauge needle, resulting in a polymicrobial peritonitis. The abdominal wall was closed in two layers, and the mice received 1 mL 0.9% saline s.c. for volume resuscitation. Each sham‐operated mouse had its cecum externalized but not ligated or punctured. All mice were allowed free access to food and water throughout the study.
Antibodies and reagents
Antibodies were purchased from the following companies, and the company protocols were followed in all applications: BD PharMingen (San Diego, CA, USA): CD4‐FITC (Cat. #553729), CD4‐PE‐Cy5 (Cat. #553050), CD8‐PE‐Cy5 (Cat. #553034), B220‐PE‐Cy5 (Cat. #553091), and CD11c‐FITC (553801); eBioscience (San Diego, CA, USA): NK cells and DX5‐FITC (Cat. #11‐5971‐85); Cell Signaling Technologies (Danvers, MA, USA): Bcl‐xL (Cat. #2764) and active caspase‐3 (Cat. #9661); and BioLegend (San Diego, Ca, USA): PD‐1 antagonistic mAb (clone RMP1‐14, isotype IgG2α, ‐κ) and its isotype control antibody. Antibodies (200 μg in 200 μL saline) were administered i.v. by tail vein or by i.p. injection. Preliminary studies demonstrated no differences in the effects of the route of administration of the anti‐PD‐1 antibodies on the various parameters, e.g., apoptosis or Bcl‐xL formation, and after initial studies using i.v. injections of anti‐PD‐1, additional anti‐PD‐1 antibodies were administered subsequently via i.p. injections.
Determination of viable splenic immune cell counts
Approximately 24 h after treatment, mice were killed, and spleens were harvested for analysis. Splenocytes were dissociated by gently pressing through a 70‐μm filter and then washed. The total number of viable cells was determined using a Beckman‐Coulter (Fullerton, CA, USA) cell counter as described previously [29, 30, 31]. Splenocytes were also stained with fluorochrome‐conjugated antibodies (BD PharMingen) to cell subset‐specific surface markers (CD4 and CD8 for T cells, B220 for B cells, DX5 for NK cells, and CD11c for DCs), and the percentage of the different cell subsets was determined by flow cytometric analysis (FACScan, Becton Dickinson, San Jose, CA, USA), as described previously [31].
PD‐1 expression in splenic immune cells
Mice underwent CLP or sham surgery, and spleens were harvested at 1, 2, 4, or 7 days postsurgery. Splenocytes were stained with fluorochrome‐conjugated antibodies to CD4 T, CD8 T, and PD‐1. Flow cytometric analysis (50,000 events/sample) was performed on FACScan (Becton Dickinson), and CellQuest software was used to analyze the data.
Quantitation of apoptosis
Cells were fixed in 1% paraformaldehyde for 30 min at room temperature, washed, and permeabilized with 90% methanol on ice for 30 min. Apoptosis was quantified by flow cytometry using the APO‐BrdU™ TUNEL assay kit, following the manufacturer's instructions, and by fluorescent staining for active caspase‐3 (Cell Signaling Technologies) as described previously [31].
Quantitation of intracellular Bcl‐xL via flow cytometry
Harvested splenocytes were fixed in 1% paraformaldehyde and washed with Perm/Wash solution (BD Biosciences, San Jose, CA, USA). Primary antibody staining for Bcl‐xL was done overnight at 4°C as described previously [32]. Cells were then washed and stained with the donkey anti‐rabbit secondary antibody and cell surface antibodies.
Cytokine studies: plasma and stimulated splenocytes
Plasma and spleens were harvested from septic and sham‐operated mice 24 h after treatment with anti‐PD‐1, isotype control antibody, or saline. Plasma IL‐6, IL‐10, TNF‐α, and IFN‐γ were measured using the Bio‐Plex Pro cytokine assay (Bio‐Rad, Hercules, CA, USA). Splenocytes (1×106) were cultured and stimulated with anti‐CD3 and anti‐CD28 for 24 h, and supernatants were harvested for cytokine determination as described previously.
Determination of DTH
Mice underwent CLP or sham surgery as described previously [27]. A group of sham and CLP‐operated mice received 200 μg anti‐PD‐1 via i.p. injection 24 h after surgery, and other groups of mice served as controls. At 4 days postsurgery, all mice were immunized with injections of 100 μl 10 mM TNBS s.c. At Day 8, all mice had antigenic challenge with 10 mM TNBS in the right footpad. PBS was injected in the left footpad as a control. Measurements (micrometers±se) were taken 24 h later and represent the difference between the right footpad (antigen challenge) versus the left footpad (PBS challenge).
Survival studies
Survival was followed for 7 days on mice that had undergone CLP and subsequently randomized to receive anti‐PD‐1 antibody, isotype control antibody, or saline. The antibodies or saline were administered via i.v. injection at 24 and 48 h after surgery. Mice were given 1 cc normal saline s.c. within 30 min of surgery and allowed free access to food and water.
Statistical analysis
All studies involving two or more groups were analyzed using a one‐way ANOVA with a Dunnett's post‐test. A log rank test was used to evaluate statistical significance for survival studies. All statistical tests were performed using GraphPad Prism 4.00 software (GraphPad, La Jolla, CA, USA). Significance was set at P < 0.05.
RESULTS
Septic peritonitis increases expression of PD‐1 on CD4 and CD8 T cells
PD‐1 expression on CD4 and CD8 T cells was measured at various time‐points after surgery. At 24 h postsurgery, CD4 and CD8 T cell surface expression of PD‐1 did not differ significantly between CLP and sham‐operated animals ( Fig. 1 ). However, by 48 h after surgery, PD‐1 expression increased significantly in septic animals compared with sham‐operated controls on CD4 and CD8 T cells. In CLP‐operated mice, the percentage of CD4 and CD8 T cells that expressed PD‐1 continued to increase, and by 7 days after surgery, >20% of splenic CD4 and CD8 T cells expressed this receptor. A representative example of PD‐1 expression in CD4 T cells in sham and CLP mice determined via flow cytometry is presented ( Fig. 2 ).
Figure 1.
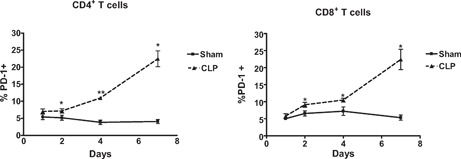
PD‐1 expression temporally increases in CD4+ and CD8+ T cells in sepsis. PD‐1 expression was measured in isolated splenocytes via flow cytometry at various time‐points. PD‐1 expression increases on splenic CD4 and CD8 T cells 2 days after sepsis when compared with sham and remains elevated at 4 and 7 days. Number of mice for sham, n = 5–12; CLP, n = 6–17 (*, P < 0.01; **, P < 0.001). Graphs are aggregates of multiple experiments.
Figure 2.
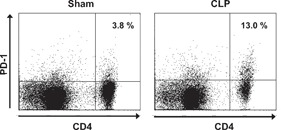
PD‐1 expression increases in sepsis–flow cytometric detection. Mice underwent sham or CLP surgery, and PD‐1 expression on CD4 T cells was quantitated via flow cytometry 4 days later. The cells located in the upper‐right quadrant of each panel are CD4+ T cells that express PD‐1. Note, the increase in PD‐1 expression in CD4 T cells from septic versus sham mouse. Flow diagram representative of data expressed in Figure 1.
PD‐1 blockade prevents loss of viable splenic immune cells by inhibiting lymphocyte apoptosis
Total splenocytes and immune cell subsets were quantitated 48 h after surgery in sham‐operated mice and septic mice that had been treated with anti‐PD‐1 antibody, isotype control antibody, or saline 24 h after surgery ( Fig. 3 ). Compared with sham‐operated mice, sepsis caused a 57.1%, 42.9%, 49.6%, 66.5%, and 50.3% loss in absolute cell counts for CD4 T, CD8 T, B, and NK cells and DCs, respectively, P < 0.01. There was no difference in total viable splenocytes in septic mice treated with saline or isotype control antibody. Treatment with anti‐PD‐1 prevented the loss of total splenocytes, CD4 T, CD8 T, and B cells, and DCs, such that the absolute values in septic mice treated with anti‐PD‐1 were not statistically different from sham‐treated mice. Anti‐PD‐1 ameliorated the loss in NK cells in sepsis, but the absolute number of NK cells in septic mice treated with anti‐PD‐1 was less than in sham‐operated mice (P<0.05). Treatment with anti‐PD‐1 prevented the loss of total viable splenocytes as a result of sepsis when compared with mice treated with isotype control antibody (58.2×106±5.0 vs. 23.4×106±3.6; **, P<0.001). For CD4 T cells, CLP mice treated with anti‐PD‐1 had increased cells compared with CLP mice treated with isotype control antibody, i.e., 9.0 ± 1.7 × 106 versus 2.7 ± 0.8 × 106, respectively; **, P < 0.01. Anti‐PD‐1 antibody provided a similar protection against cell loss for CD8 T cells (4.8±1.3×106 vs. 1.8±0.5×106), B cells (38.1±3.8×106 vs. 11.2±1.7×106; **, P<0.001), NK cells (1.9±0.2×106 vs. 0.8±0.1×106; **, P<0.01), and DCs (2.8±.4×106 vs. 0.4±0.1×106; **, P<0.01) when compared with mice treated with isotype control antibody.
Figure 3.
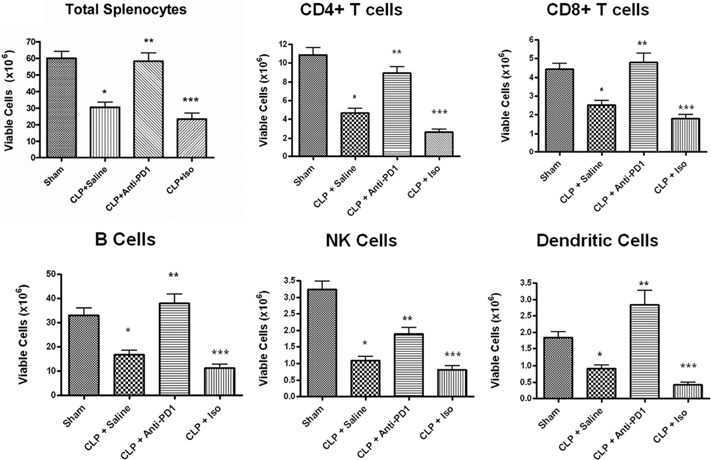
PD‐1 blockade prevents loss of viable splenic immune cells. Viable splenocyte subsets were quantitated 48 h after surgery in sham and septic mice that had been treated with anti‐PD‐1 antibody, isotype control antibody (Iso), or saline 24 h after surgery. Treatment with anti‐PD‐1 prevented the loss of total viable splenocytes as a result of sepsis when compared with mice treated with isotype controls. There was no difference in total viable splenocytes in septic mice treated with saline or isotype control antibody. Anti‐PD‐1 antibody also prevented the loss in absolute cell counts for CD4 T, CD8 T, B, and NK cells and DCs when compared with mice treated with isotype control antibody. Each panel is an aggregate of two to three experiments; sham, n = 10; CLP + saline, n = 16; CLP + anti‐PD‐1, n = 7; CLP + isotype control antibody, n = 7; *, P < 0.01, for sham versus CLP + saline; **, P < 0.01, for CLP + saline versus CLP + anti‐PD‐1; ***, P < 0.01, for CLP + anti‐PD‐1 versus CLP + isotype control.
Flow cytometric analysis of active caspase‐3 and TUNEL in splenic CD3 T cell populations after sham or CLP surgery was performed to quantify lymphocyte apoptosis. Although active caspase‐3 and TUNEL are used to detect apoptosis, the two tests differ in sensitivity, specificity, and time of positivity. Thus, these two tests of apoptosis considered together are a reliable, nonoverlapping measure of apoptotic cell death. Anti‐PD‐1 treatment prevented the sepsis‐induced increase in apoptosis, as detected by TUNEL and active caspase‐3 for T and B cells ( Fig. 4 ). TUNEL and caspase‐3 activity were not different between sham‐operated animals that received saline versus sham mice that received anti‐PD‐1 antibody or the isotype control (data not shown).
Figure 4.
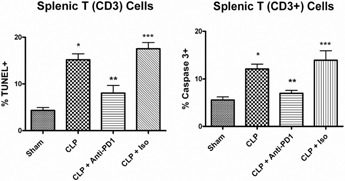
PD‐1 blockade inhibits splenic T cell apoptosis. Mice underwent sham or CLP surgery and 24 h later, had administration of anti‐PD‐1 antibody, isotype control antibody, or saline. At 48 h after surgery, splenocytes were harvested. Apoptosis was quantitated by TUNEL or active caspase‐3. Compared with sham, CLP causes significant increases in apoptosis as evaluated by TUNEL and active caspase‐3, which was prevented by treatment with anti‐PD‐1 antibody. Each figure is an aggregate of two to three experiments; sham, n = 10; CLP (CLP + saline), n = 16; CLP + anti‐PD‐1, n = 7; isotype (CLP + isotope control antibody), n = 7; *, P < 0.01, for sham versus CLP + saline; **, P < 0.01, for CLP + saline versus CLP + anti‐PD‐1; ***, P < 0.01, for CLP + anti‐PD‐1 versus CLP + isotype control.
Anti‐PD‐1 antibody prevents the sepsis‐induced decrease in Bcl‐xL
The level of pro‐ and antiapoptotic Bcl‐2 family members determines the life or death of immune effector cells [33, 34]. Given that anti‐PD‐1 prevented sepsis‐induced apoptosis of lymphocytes, we examined the effect of blocking PD‐1 on the intracellular abundance of Bcl‐xL in CD4 and CD8 T cells. Mice received anti‐PD‐1, saline, or the isotype control antibody 24 h after sham or CLP surgery, and spleens were harvested 48 h postsurgery. Bcl‐xL was quantitated using intracellular staining and expression of MFI. Sepsis caused a significant loss in the MFI of Bcl‐xL in septic versus sham CD4 and CD8 T cells ( Fig. 5 ). In contrast, there was no difference in the MFI for Bcl‐xL in CD4 or CD8 T cells from septic mice that were treated with anti‐PD‐1 versus sham‐operated mice.
Figure 5.
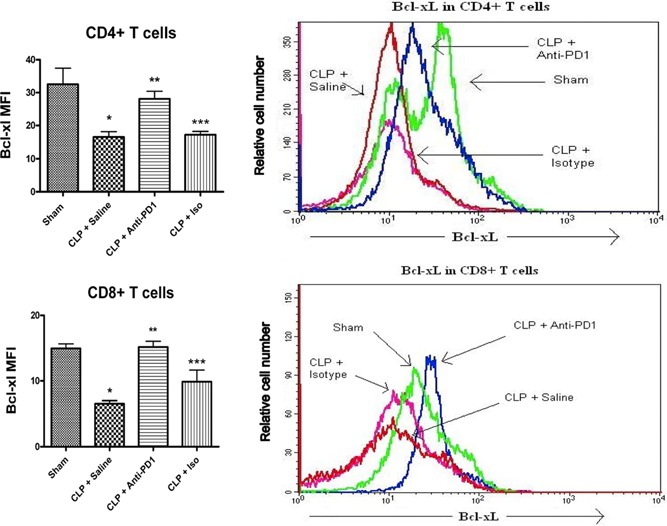
PD‐1 blockade prevents the sepsis‐induced loss in Bcl‐xL. Mice underwent sham or CLP surgery and 24 h later, had administration of anti‐PD‐1 antibody or isotype control antibody. At 48 h after surgery, splenocytes were harvested. Mice undergoing CLP have decreased intracellular Bcl‐xL (indicated by decreased MFI) in CD4 and CD8 T cells when compared with sham‐operated animals, which was prevented by treatment with anti‐PD‐1 antibody. *, P < 0.01, for sham versus CLP + saline; **, P < 0.01, for CLP + saline versus CLP + anti‐PD‐1; ***, P < 0.05, for CLP + anti‐PD‐1 versus CLP + isotype control. Sham, n = 2; CLP + saline, n = 4; CLP + anti‐PD‐1, n = 5; and CLP + isotype control, n = 5. Histogram overlays represent one mouse from each group and show that treatment with the anti‐PD‐1 antibody increases MFI of Bcl‐xL.
PD‐1 blockade improves IL‐6 production in isolated splenocytes
Mice received anti‐PD‐1 or the isotype control antibody 24 h after CLP surgery, and spleens were harvested 24 h later. Isolated splenocytes from septic mice receiving anti‐PD‐1 produced more IL‐6 after stimulation with CD3 and CD28 than splenocytes taken from animals receiving isotype controls (372.2 pg/ml±23.8 vs. 196.1 pg/ml±43.0, respectively; P<0.05). There was no difference between groups in levels of IL‐10, TNF‐α, or IFN‐γ production. Analysis of cytokine levels from plasma taken 24 h after treatment did not reveal any differences between CLP‐operated mice that received anti‐PD‐1 antibody or CLP mice that received saline (data not shown).
Anti‐PD‐1 preserves the DTH response in septic mice
The DTH response requires a coordinated interaction of cells of the innate and adaptive immune system. A hallmark of patients with sepsis is a loss of the DTH response to positive control antigens, i.e., antigens to which they had previous exposure [35]. We tested whether anti‐PD‐1 could reverse the loss in DTH that occurs in sepsis. Mice with sepsis failed to mount an effective DTH response to antigenic challenge ( Fig. 6 ). Importantly, the DTH response in septic mice that received anti‐PD‐1 was greater than the DTH response in untreated CLP mice (P<0.05) and was not statistically different than the response in the sham‐positive control mice (Fig. 6).
Figure 6.
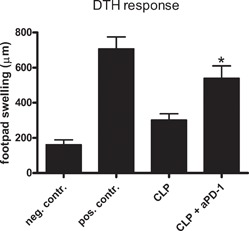
Anti‐PD‐1 prevents the sepsis‐induced loss in DTH. Mice underwent sham or CLP surgery, and selected groups were treated with anti‐PD‐1 at 24 h after surgery. Four days postsurgery, mice were sensitized with TNP, and 4 days after sensitization, mice had rechallenge with TNP (footpad injection). At 24 h after rechallenge, the degree of footpad swelling was quantitated versus PBS injection, which functioned as the negative control (neg. contr.). The footpad swelling in the foot injected with TNP is the positive control (pos. contr.). The footpad swelling response of CLP mice treated with anti‐PD‐1 (aPD‐1) and then injected with TNP was significantly greater than the footpad swelling in the CLP mice that were injected with TNP and that did not receive anti‐PD‐1; *, P < 0.05; n = 10–12 mice/group.
Anti‐PD‐1 improves survival in intra‐abdominal sepsis
Mice treated 24 and 48 h after CLP with anti‐PD‐1 antibodies had an improved 7‐day survival (70.6%) when compared with mice treated with saline (33.3%; P<0.05) or with an isotype control antibody (28.6%; P<0.05; Fig. 7 ). The majority of deaths in the control groups occurred by 3 days after surgery. There was no statistical difference in survival between mice treated with the isotype controls or with saline. These data show that PD‐1 blockade, initiated at a delayed time‐point of 24 h after the onset of polymicrobial sepsis, improves survival in mice.
Figure 7.
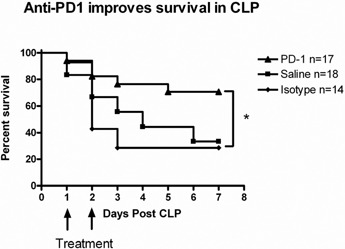
PD‐1 blockade improves survival in CLP. Mice underwent CLP and 24 h later, were injected with anti‐PD‐1 antibody, saline, or isotype control antibody. A second injection of the anti‐PD‐1 or isotype control antibody was administered at 48 h after surgery. Survival was recorded for 78 days. Mice treated with blocking antibodies to PD‐1 had an improved 7‐day survival (70.6%) compared with mice treated with saline (33.3%; P<.05) or the isotype control (28.6%; *, P<0.05).
DISCUSSION
PD‐1 is an important modulator of T cell function. It mediates peripheral tolerance, induces T cell anergy and exhaustion in chronic infections, and renders T cells more susceptible to apoptosis [12, 13]. PD‐1 deficiency or blockade has been shown to improve survival in a small number of viral, bacterial, and fungal infections [16, 23, 24, 36]. We hypothesized that blockade of PD‐1 with a mAb would improve survival in a mouse model of intra‐abdominal sepsis. Data herein show that PD‐1 blockade improves survival in the widely used and clinically relevant CLP model of sepsis. It is important to note that the administration of anti‐PD‐1 did not occur until 24 h after the onset of sepsis, during the time‐point when PD‐1 receptor expression was beginning to increase. If these beneficial effects in animal models of sepsis translate into clinical studies, it will mean that delayed administration of the therapy can still be effective in patients with sepsis, a major benefit in treatment, as patients with sepsis may not present to the hospital until many hours after sepsis onset.
The present studies complement the work of Huang and associates [25], who noted that PD‐1 null mice had improved survival in sepsis. The present study does significantly strengthen the hypothesis that inhibiting PD‐1 in sepsis is a viable therapeutic approach. Studies involving knockout mice, in which the gene has been deleted, are hostage to the fact that compensatory pathways may have occurred in response to the genetic deletion. These compensatory changes may confound the proper interpretation of the nature of the gene of interest. Thus, the findings in these gene knockout mice, with the exception of conditional knockout mice, may not reflect the results of acute inhibition of the gene. Most significantly, the results in knockout mice do not allow for investigation of the effects of blocking the gene after the particular insult in question, i.e., sepsis. Despite the methodological differences between the two studies, there is broad agreement on a number of points. The present study showing increased PD‐1 expression on CD4 and CD8 T cells is consistent with the work from Huang et al. [25], who reported a time‐dependent increase in PD‐1 expression on macrophages from septic mice. The studies by Huang et al. [25] pointed to a key role of PD‐1 expression in macrophages in modulating microbial clearance and the inflammatory response. These investigators also determined that there was decreased tissue destruction in jejunum and kidney and less evidence of compacted and fragmented nuclei (indicators of cellular apoptosis) in spleens and thymi from PD‐1 null mice. Collectively, the two studies suggest that PD‐1 is involved in the innate and adaptive immune responses to infection and emphasize the important “crosstalk” between the innate and adaptive immune system in sepsis.
An important effect of anti‐PD‐1 therapy was its action to prevent the sepsis‐induced loss in the DTH response. A key feature of patients with sepsis is their loss of the DTH response to recall antigens [35]. This finding is emblematic of the profound immunosuppression that characterizes patients with sepsis. PD‐1 is recognized to be an important factor in the “exhaustion” of T cells during chronic viral infection. Work by several laboratories has demonstrated that blockade of the PD‐1 inhibitory pathway leads to restoration of T cell function, including enhancing their ability to proliferate and produce appropriate cytokines with stimulation [12, 13, 37]. The ability of the septic mice that were treated with anti‐PD‐1 to mount an appropriate DTH response in the present study is consistent with the re‐establishment of effective immunity. In patients with sepsis, this re‐establishment of immune function may allow the host to eradicate the primary infection and resist new secondary infections.
An interesting and highly important action of blocking PD‐1 was the effect to markedly diminish sepsis‐induced apoptosis. A key determinant of life or death of immune cells is the relationship between the pro‐ and antiapoptotic Bcl‐2 family members. Few studies have examined the relationship of PD‐1 on Bcl‐2 family members [38]. Lin et al. [38] transferred splenocytes from mice infected with lymphocytic choriomeningitis virus into lymphopenic T cell‐deficient hosts and showed that a PD‐1‐positive and a PD‐1‐negative population of T cells resulted. The population of cells that were positive for PD‐1 was dysfunctional, had lower Bcl‐2 levels, and disappeared from the host by Day 70, presumably because of apoptosis. Petrovas et al. [20] examined CD8 T cells from patients with HIV‐1‐positive donors and reported that high expression of PD‐1 correlated with increased ex vivo, spontaneous apoptosis, low expression of Bcl‐2, and decreased IL‐7R expression. The most likely explanation for the inter‐relationship between PD‐1 and Bcl‐xL is a result of the inhibitory action of PD‐1 on CD28 signaling [12, 39], which plays an essential role in the translation of Bcl‐xL via a PI3K/Akt‐dependent pathway [39]. PD‐1 acts to increase SHP‐2, which inhibits PI3K signaling, thereby preventing phosphorylation of AKT [17], and inhibiting production of BcL‐xL [12, 39]. Work presented herein showing that the anti‐PD‐1 antibody markedly increased Bcl‐xL is particularly important, given previous studies from our group showing that transgenic mice that overexpress Bcl‐xL have improved survival in sepsis [32].
The present results add to the increasing body of evidence demonstrating that immune modulation represents a viable therapeutic approach to sepsis. Although there are a number of possible methods to enhancing the immune response in sepsis, one such approach involves manipulation of the costimulatory molecules that have diverse functions on cells of the innate and adaptive immune system. Animal studies have now shown that modulation of the positive and negative costimulatory receptors on immune cells using agonistic antibodies to CD40, anti‐PD‐1, or anti‐CTLA‐4 can ameliorate adverse effects of sepsis and improve survival ([25, 40] and unpublished data). Collectively, these diverse studies make a compelling argument for a new approach to sepsis that involves manipulation of the host immune response. Anti‐PD‐1‐ and anti‐PD‐L1 antibody‐based therapy is currently in several Phase 2 and 3 clinical trials in patients with cancer (http://clinicaltrials.gov). Although there are some concerns about the potential for autoimmune reactions in patients who are on long‐term anti‐PD‐1/anti‐PD‐L1 therapy, these antibodies appear to be well‐tolerated to date. Given the fact that patients with sepsis would not need prolonged therapy with anti‐PD‐1/anti‐PD‐L1 antibodies, the concerns for development of autoimmunity would be much diminished. If additional animal studies confirm its efficacy, anti‐PD‐1 should be strongly considered for clinical trials in sepsis.
AUTHORSHIP
P.M. performed many experiments, analyzed data, and helped write the manuscript. S.I. helped perform experiments. J.U. helped perform experiments and analyzed data. K.C.C. helped perform experiments. J.E.M. helped design experiments and write the manuscript. R.S.S. helped design experiments and write the manuscript.
REFERENCES
- 1. Angus, D. C. , Linde‐Zwirble, W. T. , Lidicker, J. , Clermont, G. , Carcillo, J. , Pinsky, M. R. (2001) Epidemiology of severe sepsis in the United States. Crit. Care Med. 29, 1303–1310. [DOI] [PubMed] [Google Scholar]
- 2. Monneret, G. , Venet, F. , Pachot, A. , Lepape, A. (2008) Monitoring immune dysfunctions in the septic patient: a new skin for the old ceremony. Mol. Med. 14, 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adib‐Conquy, M. , Cavaillon, J. M. (2009) Compensatory anti‐inflammatory response syndrome. Thromb. Haemost. 101, 36–47. [PubMed] [Google Scholar]
- 4. Osuchowski, M. F. , Welch, K. , Siddiqui, J. , Remick, D. G. (2006) Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J. Immunol. 177, 1967–1974. [DOI] [PubMed] [Google Scholar]
- 5. Hotchkiss, R. S. , Karl, I. E. (2003) The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348, 138–150. [DOI] [PubMed] [Google Scholar]
- 6. Limaye, A. P. , Kirby, K. A. , Rubenfeld, G. D. , Leisenring, W. M. , Bulger, E. M. , Neff, M. J. , Gibran, N. S. , Huang, M. L. , Santo Hayes, T. K. , Corey, L. , Boeckh, M. (2008) Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA 300, 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luyt, C. E. , Combes, A. , Deback, C. , Aubriot‐Lorton, M. H. , Nieszkowska, A. , Trouillet, J. L. , Capron, F. , Agut, H. , Gibert, C. , Chastre, J. (2007) Herpes simplex virus lung infection in patients undergoing prolonged mechanical ventilation. Am. J. Respir. Crit. Care Med. 175, 935–942. [DOI] [PubMed] [Google Scholar]
- 8. Hotchkiss, R. S. , Coopersmith, C. M. , McDunn, J. E. , Ferguson, T. A. (2009) The sepsis seesaw: tilting toward immunosuppression. Nat. Med. 15, 496–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Torgersen, C. , Moser, P. , Luckner, G. , Mayr, V. , Jochberger, S. , Hasibeder, W. R. , Dunser, M. W. (2009) Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth. Analg. 108, 1841–1847. [DOI] [PubMed] [Google Scholar]
- 10. Venet, F. , Chung, C. S. , Monneret, G. , Huang, X. , Horner, B. , Garber, M. , Ayala, A. (2008) Regulatory T cell populations in sepsis and trauma. J. Leukoc. Biol. 83, 523–535. [DOI] [PubMed] [Google Scholar]
- 11. Delano, M. J. , Scumpia, P. O. , Weinstein, J. S. , Coco, D. , Nagaraj, S. , Kelly‐Scumpia, K. M. , O'Malley, K. A. , Wynn, J. L. , Antonenko, S. , Al‐Quran, S. Z. , Swan, R. , Chung, C. S. , Atkinson, M. A. , Ramphal, R. , Gabrilovich, D. I. , Reeves, W. H. , Ayala, A. , Phillips, J. , Laface, D. , Heyworth, P. G. , Clare‐Salzler, M. , Moldawer, L. L. (2007) MyD88‐dependent expansion of an immature GR‐1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med. 204, 1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Keir, M. E. , Butte, M. J. , Freeman, G. J. , Sharpe, A. H. (2008) PD‐1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fife, B. T. , Bluestone, J. A. (2008) Control of peripheral T‐cell tolerance and autoimmunity via the CTLA‐4 and PD‐1 pathways. Immunol. Rev. 224, 166–182. [DOI] [PubMed] [Google Scholar]
- 14. Nishimura, H. , Nose, M. , Hiai, H. , Minato, N. , Honjo, T. (1999) Development of lupus‐like autoimmune diseases by disruption of the PD‐1 gene encoding an ITIM motif‐carrying immunoreceptor. Immunity 11, 141–151. [DOI] [PubMed] [Google Scholar]
- 15. Nishimura, H. , Okazaki, T. , Tanaka, Y. , Nakatani, K. , Hara, M. , Matsumori, A. , Sasayama, S. , Mizoguchi, A. , Hiai, H. , Minato, N. , Honjo, T. (2001) Autoimmune dilated cardiomyopathy in PD‐1 receptor‐deficient mice. Science 291, 319–322. [DOI] [PubMed] [Google Scholar]
- 16. Day, C. L. , Kaufmann, D. E. , Kiepiela, P. , Brown, J. A. , Moodley, E. S. , Reddy, S. , Mackey, E. W. , Miller, J. D. , Leslie, A. J. , DePierres, C. , Mncube, Z. , Duraiswamy, J. , Zhu, B. , Eichbaum, Q. , Altfeld, M. , Wherry, E. J. , Coovadia, H. M. , Goulder, P. J. , Klenerman, P. , Ahmed, R. , Freeman, G. J. , Walker, B. D. (2006) PD‐1 expression on HIV‐specific T cells is associated with T‐cell exhaustion and disease progression. Nature 443, 350–354. [DOI] [PubMed] [Google Scholar]
- 17. Sharpe, A. H. , Wherry, E. J. , Ahmed, R. , Freeman, G. J. (2007) The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 8, 239–245. [DOI] [PubMed] [Google Scholar]
- 18. Blattman, J. N. , Greenberg, P. D. (2006) PD‐1 blockade: rescue from a near‐death experience. Nat. Immunol. 7, 227–228. [DOI] [PubMed] [Google Scholar]
- 19. Barber, D. L. , Wherry, E. J. , Masopust, D. , Zhu, B. , Allison, J. P. , Sharpe, A. H. , Freeman, G. J. , Ahmed, R. (2006) Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687. [DOI] [PubMed] [Google Scholar]
- 20. Petrovas, C. , Casazza, J. P. , Brenchley, J. M. , Price, D. A. , Gostick, E. , Adams, W. C. , Precopio, M. L. , Schacker, T. , Roederer, M. , Douek, D. C. , Koup, R. A. (2006) PD‐1 is a regulator of virus‐specific CD8+ T cell survival in HIV infection. J. Exp. Med. 203, 2281–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trautmann, L. , Janbazian, L. , Chomont, N. , Said, E. A. , Gimmig, S. , Bessette, B. , Boulassel, M. R. , Delwart, E. , Sepulveda, H. , Balderas, R. S. , Routy, J. P. , Haddad, E. K. , Sekaly, R. P. (2006) Upregulation of PD‐1 expression on HIV‐specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 12, 1198–1202. [DOI] [PubMed] [Google Scholar]
- 22. Urbani, S. , Amadei, B. , Tola, D. , Massari, M. , Schivazappa, S. , Missale, G. , Ferrari, C. (2006) PD‐1 expression in acute hepatitis C virus (HCV) infection is associated with HCV‐specific CD8 exhaustion. J. Virol. 80, 11398–11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lazar‐Molnar, E. , Gacser, A. , Freeman, G. J. , Almo, S. C. , Nathenson, S. G. , Nosanchuk, J. D. (2008) The PD‐1/PD‐L costimulatory pathway critically affects host resistance to the pathogenic fungus Histoplasma capsulatum . Proc. Natl. Acad. Sci. USA 105, 2658–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Venkatachari, N. J. , Buchanan, W. G. , Ayyavoo, V. (2008) Human immunodeficiency virus (HIV‐1) infection selectively downregulates PD‐1 expression in infected cells and protects the cells from early apoptosis in vitro and in vivo. Virology 376, 140–153. [DOI] [PubMed] [Google Scholar]
- 25. Huang, X. , Venet, F. , Wang, Y. L. , Lepape, A. , Yuan, Z. , Chen, Y. , Swan, R. , Kherouf, H. , Monneret, G. , Chung, C. S. , Ayala, A. (2009) PD‐1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. USA 106, 6303–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brahmamdam, P. , Watanabe, E. , Unsinger, J. , Chang, K. C. , Schierding, W. , Hoekzema, A. S. , Zhou, T. T. , McDonough, J. S. , Holemon, H. , Heidel, J. D. , Coopersmith, C. M. , McDunn, J. E. , Hotchkiss, R. S. (2009) Targeted delivery of siRNA to cell death proteins in sepsis. Shock 32, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hotchkiss, R. S. , Swanson, P. E. , Knudson, C. M. , Chang, K. C. , Cobb, J. P. , Osborne, D. F. , Zollner, K. M. , Buchman, T. G. , Korsmeyer, S. J. , Karl, I. E. (1999) Overexpression of Bcl‐2 in transgenic mice decreases apoptosis and improves survival in sepsis. J. Immunol. 162, 4148–4156. [PubMed] [Google Scholar]
- 28. Unsinger, J. , McDonough, J. S. , Shultz, L. D. , Ferguson, T. A. , Hotchkiss, R. S. (2009) Sepsis‐induced human lymphocyte apoptosis and cytokine production in “humanized” mice. J. Leukoc. Biol. 86, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Unsinger, J. , Herndon, J. M. , Davis, C. G. , Muenzer, J. T. , Hotchkiss, R. S. , Ferguson, T. A. (2006) The role of TCR engagement and activation‐induced cell death in sepsis‐induced T cell apoptosis. J. Immunol. 177, 7968–7973. [DOI] [PubMed] [Google Scholar]
- 30. Unsinger, J. , Kazama, H. , McDonough, J. S. , Hotchkiss, R. S. , Ferguson, T. A. (2009) Differential lymphopenia‐induced homeostatic proliferation for CD4+ and CD8+ T cells following septic injury. J. Leukoc. Biol. 85, 382–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang, K. C. , Unsinger, J. , Davis, C. G. , Schwulst, S. J. , Muenzer, J. T. , Strasser, A. , Hotchkiss, R. S. (2007) Multiple triggers of cell death in sepsis: death receptor and mitochondrial‐mediated apoptosis. FASEB J. 21, 708–719. [DOI] [PubMed] [Google Scholar]
- 32. Hotchkiss, R. S. , McConnell, K. W. , Bullok, K. , Davis, C. G. , Chang, K. C. , Schwulst, S. J. , Dunne, J. C. , Dietz, G. P. , Bahr, M. , McDunn, J. E. , Karl, I. E. , Wagner, T. H. , Cobb, J. P. , Coopersmith, C. M. , Piwnica‐Worms, D. (2006) TAT‐BH4 and TAT‐Bcl‐xL peptides protect against sepsis‐induced lymphocyte apoptosis in vivo. J. Immunol. 176, 5471–5477. [DOI] [PubMed] [Google Scholar]
- 33. Strasser, A. (2005) The role of BH3‐only proteins in the immune system. Nat. Rev. Immunol. 5, 189–200. [DOI] [PubMed] [Google Scholar]
- 34. Youle, R. J. , Strasser, A. (2008) The BCL‐2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59. [DOI] [PubMed] [Google Scholar]
- 35. Meakins, J. L. , Pietsch, J. B. , Bubenick, O. , Kelly, R. , Rode, H. , Gordon, J. , MacLean, L. D. (1977) Delayed hypersensitivity: indicator of acquired failure of host defenses in sepsis and trauma. Ann. Surg. 186, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yao, S. , Wang, S. , Zhu, Y. , Luo, L. , Zhu, G. , Flies, S. , Xu, H. , Ruff, W. , Broadwater, M. , Choi, I. H. , Tamada, K. , Chen, L. (2009) PD‐1 on dendritic cells impedes innate immunity against bacterial infection. Blood 113, 5811–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sester, U. , Presser, D. , Dirks, J. , Gartner, B. C. , Kohler, H. , Sester, M. (2008) PD‐1 expression and IL‐2 loss of cytomegalovirus‐specific T cells correlates with viremia and reversible functional anergy. Am. J. Transplant. 8, 1486–1497. [DOI] [PubMed] [Google Scholar]
- 38. Lin, S. J. , Peacock, C. D. , Bahl, K. , Welsh, R. M. (2007) Programmed death‐1 (PD‐1) defines a transient and dysfunctional oligoclonal T cell population in acute homeostatic proliferation. J. Exp. Med. 204, 2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chemnitz, J. M. , Parry, R. V. , Nichols, K. E. , June, C. H. , Riley, J. L. (2004) SHP‐1 and SHP‐2 associate with immunoreceptor tyrosine‐based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 173, 945–954. [DOI] [PubMed] [Google Scholar]
- 40. Schwulst, S. J. , Grayson, M. H. , DiPasco, P. J. , Davis, C. G. , Brahmbhatt, T. S. , Ferguson, T. A. , Hotchkiss, R. S. (2006) Agonistic monoclonal antibody against CD40 receptor decreases lymphocyte apoptosis and improves survival in sepsis. J. Immunol. 177, 557–565. [DOI] [PubMed] [Google Scholar]