

Abstract
Numerous lysosomal enzymes and membrane proteins are essential for the degradation of proteins, lipids, oligosaccharides, and nucleic acids. The CLN3 gene encodes a lysosomal membrane protein of unknown function, and CLN3 mutations cause the fatal neurodegenerative lysosomal storage disorder CLN3 (Batten disease) by mechanisms that are poorly understood. To define components critical for lysosomal homeostasis that are affected by this disease, here we quantified the lysosomal proteome in cerebellar cell lines derived from a CLN3 knock-in mouse model of human Batten disease and control cells. We purified lysosomes from SILAC-labeled, and magnetite-loaded cerebellar cells by magnetic separation and analyzed them by MS. This analysis identified 70 proteins assigned to the lysosomal compartment and 3 lysosomal cargo receptors, of which most exhibited a significant differential abundance between control and CLN3-defective cells. Among these, 28 soluble lysosomal proteins catalyzing the degradation of various macromolecules had reduced levels in CLN3-defective cells. We confirmed these results by immunoblotting and selected protease and glycosidase activities. The reduction of 11 lipid-degrading lysosomal enzymes correlated with reduced capacity for lipid droplet degradation and several alterations in the distribution and composition of membrane lipids. In particular, levels of lactosylceramides and glycosphingolipids were decreased in CLN3-defective cells, which were also impaired in the recycling pathway of the exocytic transferrin receptor. Our findings suggest that CLN3 has a crucial role in regulating lysosome composition and their function, particularly in degrading of sphingolipids, and, as a consequence, in membrane transport along the recycling endosome pathway.
Keywords: protein sorting, trafficking, lysosome, neurodegenerative disease, lipid droplet, lipid metabolism, endocytosis, electron microscopy (EM), mannose 6-phosphate receptors, pH regulation, proteomics, membrane protein
Introduction
More than 60 soluble acid hydrolases and numerous lysosomal membrane proteins are essential for the degradation of proteins, lipids, oligosaccharides, and nucleic acids reaching lysosomes via endocytic and autophagic pathways, and for the transport of metabolites and ions across the membrane driven by a proton gradient that is maintained by the vacuolar H+ ATPase (vATPase).3 In addition to these functions, lysosomes are involved in fusion processes with the plasma membrane and other organelles, nutrient sensing, and gene regulation (1–3). The physiological importance of lysosomal homeostasis is demonstrated by the existence of numerous diseases caused by mutations in genes encoding lysosomal proteins, which result in general lysosomal dysfunction associated with accumulation of nondegraded materials in lysosomes (4).
Mutations in the CLN3 gene coding for a glycosylated multispanning lysosomal membrane protein of 438 amino acids, result in the fatal neurodegenerative lysosomal storage disorder CLN3 (also called juvenile neuronal ceroid lipofuscinosis (JNCL) or Batten disease). The world-wide most common mutation causes a 1-kb deletion in CLN3 gene that leads to the loss of exons 7 and 8, and is predicted to produce a truncated protein (5) that is retained in the endoplasmic reticulum (ER). The accumulation of heterogeneous autofluorescent ceroid lipopigment aggregates in lysosomes is not clearly disease-specific (6). The mechanisms underlying neuronal degeneration in CLN3 disease and the function of CLN3 are still unknown. It has been proposed that CLN3 contributes to the regulation of lysosomal size, pH, arginine, lipid, and Ca2+ homeostasis (7–11). Furthermore, CLN3 has been implicated in various intracellular membrane transport processes such as anterograde and retrograde transport between trans-Golgi network (TGN) and endosomes, formation of autophagolysosomes, late steps of the endocytic pathway, and vesicular trafficking along axons (5, 12–15).
To gain insight into mechanisms underlying neuronal degeneration, and to define components critical for lysosomal homeostasis in CLN3 disease, we performed a SILAC-based comparative mass spectrometric analysis of isolated lysosomes from cerebellar cell lines of WT and Cln3 knock-in mice (Cln3Δex7/8) exhibiting the 1-kb deletion in the Cln3 gene (12). We found that the protein concentration of 28 soluble lysosomal enzymes was significantly reduced in Cln3Δex7/8 lysosomes associated with various changes in the lipid composition of Cln3Δex7/8 cerebellar cells. These were biochemically evaluated and found to contribute to differentially affected transport routes of endocytic cargo receptors.
Results
Lysosomal proteomics identifies differential abundance of acid hydrolases in Cln3Δex7/8 cerebellar cells
We performed SILAC-based comparative proteomics and quantified the relative amounts of lysosomal proteins at steady state in lysosomal fractions isolated from WT and Cln3Δex7/8 cerebellar cells (PXD004548; lysosomal proteome analysis) exhibiting storage material (Fig. S1) by means of internalized dextran-stabilized magnetite transported to LysoTracker-positive organelles (Fig. S2). Gene ontology (GO) enrichment analysis revealed that 197 and 170 of the identified proteins were annotated to vacuole and lysosome annotation in the database, containing 502 and 411 mouse proteins, respectively. We found in the isolated lysosomal fractions 104 of 185 experimentally confirmed lysosomal proteins comprising 47 soluble and 23 core membrane proteins (Table S1), and 34 associated proteins on the cytoplasmic lysosomal membrane. The majority exhibited a statistically significant differential abundance. In addition, the concentrations of three cargo receptors, 300-kDa mannose 6-phosphate receptor (Mpr300), LDL receptor-related protein 1 (Lrp1), and Lrp2 (also known as megalin), involved in trafficking of M6P-containing and nonphosphorylated lysosomal enzymes (16–19) were significantly changed in Cln3Δex7/8 cells (Table S1). Furthermore, numerous peripheral membrane proteins on the cytoplasmic side of lysosomes involved in vesicular targeting, positioning, and signaling have been covered by our proteomic analysis. Six lysosomal enzymes were increased 1.5- to 2.3-fold in lysosomal fractions of Cln3Δex7/8 cerebellar cells compared with WT controls (Fig. 1A): dipeptidylpeptidase 2 (Dpp7), tripeptidyl-peptidase 1 (Tpp1), RNase T2 (Rnaset2), lysosomal α-glucosidase (Gaa), α-galactosidase A (Gla), and palmitoyl-protein thioesterase 1 (Ppt1). In contrast, the amounts of 28 proteins were clearly decreased. Among these are enzymes involved in the degradation of glycans (arylsulfatase B (Arsb), β-gal (Glb1), β-hexosaminidase subunit α (Hexa) and β (Hexb), GalNAc-6-sulfatase (Galns), β-mannosidase (Manba), β-glucuronidase (Gusb), and α-l-fucosidase (Fuca1)), or lipids (Glb1, N-acylethanolamine–hydrolyzing acid amidase (Naaa), Hexa, Hexb, prosaposin (Psap), putative phospholipase B-like 2 (Pldb2), galactosylcerebrosidase (Galc), arylsulfatase A (Arsa), acid ceramidase (Asah1), group XV phospholipase A2 (Pla2g15), and lysosomal acid lipase (Lipa)). These proteins, as well as the following were decreased by 16 to 80% in Cln3Δex7/8 lysosomal fractions compared with WT: neuronal ceroid-lipofuscin protein 5 (Cln5), Niemann Pick type C2 protein (Npc2), cathepsin D (Ctsd), DNase-2-α (Dnase2), cathepsin Z (Ctsz), lysosomal Pro-X carboxypeptidase (Prpc), Creg1, legumain (Lgmn), carboxypeptidase Q (Cpq), sialate O-acetylesterase (Siae), and mammalian ependymin-related protein 1 (Epdr1).
Figure 1.

SILAC-based quantitative analysis of lysosomal soluble proteins in Cln3Δex7/8 and WT cerebellar cells. A, cerebellar cells were grown in light (Cln3Δex7/8) and heavy isotope (WT)–labeled medium and incubated with 10% magnetite particles coupled to dextran for 24 h followed by a chase for 36 h in labeled medium. Equal amounts of both cell populations were combined, and a postnuclear fraction was loaded onto magnetic column placed in a MACS Separator. After removal of the column from Separator, lysosomes were eluted and analyzed by LC-MS. Comparative proteomic data of known soluble lysosomal proteins were plotted against Cln3Δex7/8/WT(light/heavy) ratio (mean ± S.D., n = 3, p values are given in Table S1). B, whole cell extracts from three individual samples of WT and Cln3Δex7/8 cells were analyzed by Ctsd, Ctsz, and Creg1 Western blotting. Endogenous α-tubulin was used as loading control. The positions of molecular mass marker proteins in kDa are indicated. C, relative enzyme activity of Ppt1, Gla, Hexb, Arsa, Manba, and Gusb in extracts of WT and Cln3Δex7/8 cerebellar cells. Activities in WT cells were set as 1. Statistical significance was determined using one-way ANOVA followed by Dunnett's multiple comparison test. Data represent mean ± S.D., n = 3 (Ppt1, Gla, Arsa), n = 4 (Manba), n = 9 (Hexb, Gusb), ***, p ≤ 0.001.
To verify the proteomic data, the expression levels of nine soluble lysosomal proteins were determined by Western blotting and enzymatic activity in whole cell lysates (Fig. 1, B and C). The amounts of immunoreactive Ctsd, Ctsz, and Creg1 were strongly reduced or almost absent in Cln3Δex7/8 cells (Fig. 1B). In agreement with the comparative proteomic data, the activities of Ppt1 and Gla, representing the most enriched proteins in lysosomal fractions of Cln3Δex7/8 cells, were significantly increased, whereas the activities of Hexb, Arsa, Manba, and Gusb were reduced in Cln3Δex7/8 cells (Fig. 1C). Next, we examined whether changes in the amounts of lysosomal membrane proteins in Cln3Δex7/8 cells affect other lysosomal functions. Although three subunits of the vATPase (Atp6d1, Atp6g1, and Atp6v1h) and the accessory protein 1 (Atp6ap1) were significantly changed in their abundance in lysosomal fractions of Cln3Δex7/8 cerebellar cells (Fig. 2A and Table S1), ratiometric pH measurements showed unchanged lysosomal H+ concentrations in mutant cells (pH 4.53 ± 0.08 (WT) versus 4.49 ± 0.09 (Cln3Δex7/8); Fig. 2B). Therefore, our data do not support a role of CLN3 in lysosomal pH maintenance of neuronal cells, although previous studies reported on changes in vacuolar pH in yeast cells lacking the CLN3 homologue btn1 (8, 20).
Figure 2.

Lysosomal pH in relation to altered vATPase compositionin Cln3Δex7/8 cerebellar cells. A, comparative proteomic data of known lysosomal membrane proteins (mean ± S.D., n = 3, p values given in Table S1). B, ratiometric imaging of lysosomal pH in WT and Cln3Δex7/8 cerebellar cells using dextran-coupled pH-sensitive Oregon Green® 488. Mean ± S.E. measuring the lysosomes of at least 10 different cells per genotype; n = 6.
Clonal effects responsible for the observed phenotype in Cln3Δex7/8 cerebellar cells are unlikely because we found similar reductions in Ctsd and Ctsz protease or Lrp1 level in primary embryonic fibroblasts isolated from Cln3Δex7/8 mice (Fig. S3)
Increased Mpr300-mediated uptake of 125I-arylsulfatase B in Cln3Δex7/8 cells
The proteomic analysis of lysosomal fractions revealed a 2.2-fold increased concentration of Mpr300 in Cln3Δex7/8 cerebellar cells compared with WT controls (Fig. 3A) which was confirmed by Western blotting (Fig. 3B). Furthermore, the relative number of Mpr300 was also determined by ligand-binding experiments on saponin-treated cerebellar cells (representing the total number of Mpr300) or on untreated cells (representing Mpr300 localized at the cell surface) using recombinant human 125I-labeled arylsulfatase B (125I-ARSB) as lysosomal indicator ligand. Binding studies with 125I-ARSB at 4 °C revealed about doubled Mpr300 sites at the surface of Cln3Δex7/8 cells compared with WT cells, which corresponded to the 2.2-fold higher expression in Cln3Δex7/8 cerebellar cells determined in the presence of saponin (Fig. 3C).
Figure 3.
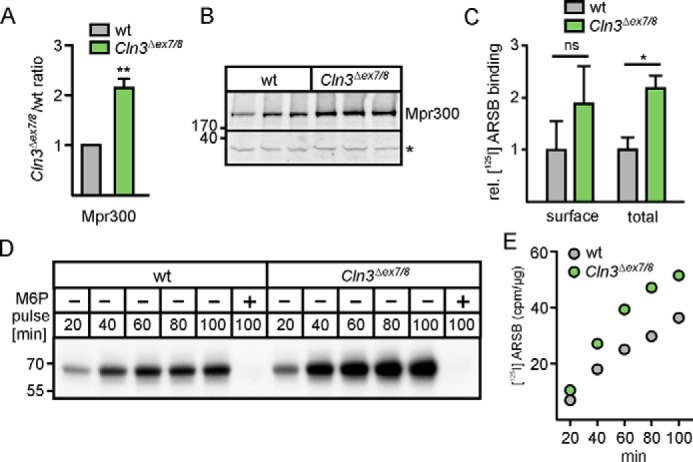
Mpr300 expression and Mpr300-mediated uptake of ligands into Cln3Δex7/8 cerebellar cells. A, relative amounts of Mpr300 were determined by quantitative proteomics in lysosomal fractions of Cln3Δex7/8 and WT cells represented as Cln3Δex7/8/WT ratio (mean of three individual SILAC experiments; **, p ≤ 0.01). B, Western blotting of Mpr300 of three individual samples of WT and Cln3Δex7/8 cells. The positions of molecular mass marker proteins are indicated in kDa. The unspecific band (*) from the same blot was used as loading control. C, 125I-ARSB binding to Mpr300 at 4 °C in the absence of saponin (cell surface binding; n = 3) or in the presence of saponin (total binding; n = 3). The values were corrected by binding in the presence of 10 mm M6P. Statistical significance was determined using one-way ANOVA followed by Bonferroni's multiple comparison test. Data represent mean ± S.D., n = 3 (*, p ≤ 0.05; ns = not significant). D, increased M6P-dependent internalization of recombinant 125I-ARSB in cerebellar cells at 33 °C over time. E, absolute radioactivity values of 125I-ARSB (in cpm/μg) were determined by γ-counting of excised 125I gel bands corrected by the radioactivity in the presence of M6P.
To examine the functional significance of increased amounts of Mpr300 in Cln3Δex7/8 cerebellar cells, we performed Mpr300-dependent internalization assays using 125I-ARSB as Mpr300 ligand. Because there is a continuous and constitutive exchange between Mpr300 localized at the cell surface and Mpr300 in all intracellular membranes with the same kinetics (21), the uptake is proportional to the total number of Mpr300. Cerebellar cells were incubated with 125I-ARSB for 20 to 100 min. The specificity of Mpr300-mediated uptake of 125I-ARSB was demonstrated by the complete inhibition by M6P (Fig. 3D, lanes 6 and 12). In both WT and Cln3Δex7/8 cerebellar cells, the amounts of 125I-ARSB precursors increased with incubation time. Quantification of both cell-associated radioactivity and measurement of radioactivity in excised 125I-ARSB bands from the gel demonstrated 2-fold higher amounts of 125I-ARSB in Cln3Δex7/8 cerebellar cells compared with WT controls (Fig. 3E).
Lipidome analysis reveals alterations of glycosphingolipids in Cln3Δex7/8 cerebellar cells
Because the amounts of 11 soluble lysosomal proteins involved in lipid degradation are significantly reduced in lysosomes of Cln3Δex7/8 cerebellar cells, we analyzed the lipidome of WT and Cln3Δex7/8 cerebellar cells by electrospray ionization MS (ESI-MS). To increase the sensitivity of MS detection, cerebellar cells of three independent preparations were combined, and anionic and neutral lipids were analyzed separately (Tables S2 and S3). The eight anionic lipid species which showed the most pronounced change in their relative amount included several GM3 and GD1 ganglioside species with a sphingosine (d18:1) backbone and variable acyl chains. The levels of GM3 species were increased 1.4- to 2.8-fold in Cln3Δex7/8 cerebellar cells compared with the WT cells, whereas GD1 species were reduced by 50%. Particular phosphatidylinositol (PI) and phosphatidylserine (PS) species (38:3) had 1.6-fold elevated values in Cln3Δex7/8 cerebellar cells (Table S2). In addition, numerous alterations in the relative amounts of neutral lipid species were detected in Cln3Δex7/8 cells. Lactosylceramides (LacCer) 24:0 and 16:0 showed the strongest increase (5.0- and 4.7-fold, respectively) in Cln3Δex7/8 cells. The elevated levels of LacCer were confirmed by BODIPY® FL C5-Lactosylceramide (Bodipy-LacCer) fluorescence live cell microscopy. Following binding at 4 °C and a switch to 33 °C, in WT cells, the majority of Bodipy-LacCer co-localized with Golgi-RFP used to stain the Golgi apparatus (Fig. 4, A and B). In contrast, in Cln3Δex7/8 cells, the total Bodipy fluorescence was higher than in WT cells and mostly co-localized in LysoTracker-positive organelles (Fig. 4, C and D). Other lipid species that showed increased levels (1.5- to 2.1-fold) for these cells included lysophosphatidyl-ethanolamine (LPE) 20:3, sphingomyelin (SM) 24:1, ceramides (Cer) 24:0 and 24:1, and phosphatidylethanolamines (PE) 38:3 and 40:3. At the same time SM14:0 and 15:0, PE40:4, phosphatidylcholine (PC) 38:4, lysophosphatidylcholine (LPC) 20:4, and hexose cerebrosides (HexCer) 18:0, 20:0, 22:0, and 23:0 were reduced to 30–60% of the WT levels (Table S3). GC with mass selective detection (GC-MS) revealed losses of essential arachidonic acid, 20:4n-6 (in PE40:4, PC38:4) compensated by increases of its precursor 20:3n-6 and nonessential 20:3n-9 (in LPE20:3, PE38:3, PE40:3) (Table S4).
Figure 4.

Intracellular localization of Bodipy lactosylceramide in cerebellar cells. A, WT and Cln3Δex7/8 cells were incubated with CellLight® Golgi-RFP for 16 h followed by BODIPY® FL C5-Lactosylceramide (Bodipy-LacCer) loading for 30 min at 33 °C. Cells were analyzed by confocal fluorescence life cell microscopy. Scale bars = 10 μm. B, Bodipy-LacCer fluorescence intensity was quantified in Golgi-GFP-positive compartments. C, WT and Cln3Δex7/8cells were incubated with Bodipy-LacCer for 30 min followed by LysoTracker® Deep Red loading for 30 min at 33 °C, and confocal fluorescence live cell microscopy. Scale bars = 10 μm. D, quantification of Bodipy-LacCer fluorescence intensities in LysoTracker® Deep Red positive structures. Data are presented as mean ± S.D. of three independent experiments (25 cells were analyzed per condition) (***, p ≤ 0.001).
In addition, immunofluorescence intensity of perilipin-2 (Plin2), an envelope protein of lipid droplets (22), was increased in Cln3Δex7/8 compared with WT cells (Fig. 5, A and B). Furthermore, loading of the cells with oleate revealed a stronger expansion in cytosolic Plin2-associated lipid droplets in Cln3Δex7/8 cells, suggesting impaired degradative capabilities.
Figure 5.

Accumulation of Plin2-positive lipid droplets. A, WT and Cln3Δex7/8 cells were incubated with or without 400 μm BSA-coupled oleate overnight at 33 °C followed by co-immunofluorescence staining of the lipid droplet marker Plin2 (red) and the lysosomal membrane protein Lamp1 (green). Nuclei were stained with DAPI (blue). Scale bars: 10 μm. B, quantification of Plin2 fluorescence intensities in WT and Cln3Δex7/8cells with (+) or without (−) oleate treatment. Data are presented as mean ± S.D. of three independent experiments (25 cells were analyzed per condition). Statistical significance was determined using one-way ANOVA (*, p ≤ 0.05, ***, p ≤ 0.001).
Taken together, the comparative lipidome analyses of cerebellar cells revealed distinct alterations, in particular in the relative contents of glycosphingolipids in Cln3Δex7/8 cells, which can be partially explained by changes in the steady-state expression levels of lysosomal sphingolipid-degrading hydrolases.
To determine whether re-expression of CLN3 could reverse alterations in lysosomal homeostasis, we infected WT and Cln3Δex7/8 cerebellar cells with lentiviruses expressing human CLN3 N-terminally tagged with GFP (GFP-CLN3) or GFP alone under a CMV promotor driving high transgene expression. After antibiotics selection for GFP- and GFP-CLN3–positive cells (Fig. S4), in both cell lines GFP-CLN3 was found primarily localized in the ER and to some extent in lysosomes (Fig. S5). However, re-expression of GFP-CLN3 neither affected the reduced cathepsin D and Z levels and decreased activities of lysosomal glycosidases, nor the oleate-induced formation/accumulation of Plin2-positive lipid droplets in Cln3Δex7/8 cerebellar cells (Fig. S6).
Impaired recycling of transferrin receptor in Cln3Δex7/8 cells
Because the intracellular distribution and transport of membrane proteins between different subcellular compartments, in particular along the endocytic and autophagic pathways, depend on the lipid composition of the membranes (23–25), we examined the endocytosis of fluorophore-labeled transferrin (Trf-Alexa Fluor® 546) mediated by transferrin receptor (TrfR). The TrfR protein level was not changed in Cln3Δex7/8 cerebellar cells (Fig. S7). However, the amount of Trf-Alexa Fluor® 546 detected intracellularly after 30-min incubation was significantly increased in Cln3Δex7/8 cerebellar cells compared with WT cells (Fig. 6, A and D). The continuously internalized Trf-Alexa Fluor® 546 is detectable beneath the plasma membrane, in small vesicles evenly distributed over the whole cell, and in larger, more dilated structures in perinuclear regions partially co-localizing with Rab11a and the Golgi marker protein Gm130 characteristic for the transferrin-recycling pathway (Fig. S8) (26, 27). The identity of organelles mainly retaining Trf in Cln3Δex7/8 cerebellar cells needs to be studied.
Figure 6.

Alteration in transferrin receptor recycling in Cln3Δex7/8 cells. A–C, confocal fluorescence images of WT and Cln3Δex7/8 cells after incubation with Trf-Alexa Fluor® 546 (red) for 30 min at 33 °C (A), and subsequent incubation with Trf-Alexa Fluor® 488 (green) for 15 min at 33 °C (B). After removal of the medium, WT and Cln3Δex7/8cells were further incubated with fresh medium in the absence of Trf-Alexa Fluor® (chase) for 15 min at 33 °C (C). Confocal immunofluorescence images were captured with identical exposure settings. Nuclei were visualized using DAPI (blue). Scale bars = 10 μm. D, quantification of Trf-Alexa Fluor® 546 and Trf-Alexa Fluor® 488 fluorescence intensities at each time point. Statistical significance was determined using one-way ANOVA followed by Dunnett's multiple comparison test. Data are presented as mean ± S.E. of three independent experiments (15 cells were analyzed per experiment), (ns, not significant; *, p ≤ 0.05, ***, p ≤ 0.001).
To examine whether the increased accumulation of Trf-Alexa Fluor® 546 in Cln3Δex7/8 cells is because of elevated uptake or reduced recycling and exocytosis, the cells were incubated for 30 min with Trf-Alexa Fluor® 546, washed, and further incubated for 15 min in the presence of Trf-Alexa Fluor® 488 (Fig. 6B). As expected, the Alexa Fluor® 546 fluorescence completely disappeared in WT cells because of the release of Apo-Trf into the medium accompanied by the accumulation of Trf-Alexa Fluor® 488 (Fig. 6, B and D, upper row). In contrast, in Cln3Δex7/8 cells both Trf-coupled fluorophores partially co-localized in the same intracellular membrane structures (Fig. 6, B and D, lower row). During a further 15-min chase in medium lacking Trf-Alexa Fluor® ligands, almost no ligand-related fluorescence was observed in WT cells (Fig. 6, C and D, upper row), whereas both Trf-Alexa Fluor® 546 and Trf-Alexa Fluor® 488 were still detectable in Cln3Δex7/8 cells (Fig. 6, C and D, lower row). These data demonstrate that the exocytic pathway of Trf from the recycling endosome to the plasma membrane is strongly disturbed in Cln3Δex7/8 cells.
Discussion
The homeostasis of lysosomes relies on the balance between the biosynthetic replenishment of lysosomal components, degradation, and recycling of macromolecules obtained by endocytosis and autophagy. Our analysis of the lysosomal proteome of Cln3Δex7/8 cerebellar cells provides novel insight into complex regulatory mechanisms linking altered composition of lysosomes and dysregulated membrane transport. We found that the amounts of only few enzymes are increased, whereas the majority is decreased in Cln3Δex7/8 lysosomes (Fig. 1A and Table S1). Therefore, we predicted impaired capabilities for degradation of multiple classes of substrates in Cln3Δex7/8 lysosomes, which was supported by transmission EM revealing heterogeneous storage material in enlarged lysosomes composed by dense and multilamellar bodies, electron-lucent floccular bodies and curvilinear and fingerprint-like material in Cln3Δex7/8 cerebellar cells characteristic for accumulation of proteins, glycans, and glycolipids (Fig. S1). Recently, Sabatini and colleagues (28) applied an immunoprecipitation approach for the rapid purification of lysosomes (LysoIP) to determine the mTORC1-regulated lysosomal proteomes. Although both LysoIP and our magnetite-loading protocol differ in recovery of lysosomes, cell types (lysosomal membrane protein–overexpressing HEK 293T cells versus SILAC-labeled mouse-derived cerebellar cells), the total number of identified proteins in the lysosomal fractions, the composition of soluble and integral lysosomal membrane proteins, and peripheral proteins on the cytoplasmic side of lysosomes are relatively similar. Furthermore, the high sensitivity of MS leads to the identification of a considerable number of nonlysosomal “contaminating” proteins. Because the role of lysosomes as purely degradative organelles changed in the past years toward central players in autophagy/mitophagy/ribophagy, energy metabolism, nucleus signaling, physical contacts with other organelles, or adaptive intracellular positioning (28–31), numerous co-purified contaminating proteins may be functionally linked to diverse functions of lysosomes.
The altered lysosomal protein levels in Cln3Δex7/8 cells might be caused by changes in their mRNA level, differences in lysosomal stability, and/or intracellular transport efficiency. Although it is rather unlikely that the low expression of cathepsin D and Z in Cln3Δex7/8 lysosomes results in stabilization of few selected proteins, it was shown that CLN3 is necessary for normal protein trafficking along the secretory, TGN-endosome, and endocytic pathway (12, 32, 33). Consistent with these observations, we show here that in Cln3Δex7/8 cells the transport efficiency of two endocytic cargo receptors, the Mpr300 and TrfR, is affected by two different mechanisms: alteration of the steady-state receptor protein concentration (Mpr300) (Fig. 3, B and C), and changes in the recycling efficiency (TrfR) (Fig. 6). The altered levels of Mpr300 correspond to alterations in ligand uptake and lysosome delivery (Fig. 3, D and E). Our ratiometric pH measurements (Fig. 2B) exclude that changes in H+ concentration are responsible for impaired dissociation processes of receptor-ligand complexes or lysosomal dysfunction in Cln3Δex7/8 cells and provide no evidence for a role of CLN3 in lysosomal pH maintenance of neuronal cells as reported previously for the vacuolar pH in yeast cells lacking the CLN3 homologue btn1 (8, 20).
The concentration of transferrin receptors was not changed in Cln3Δex7/8 cells, whereas the amount of internalized transferrin was increased. We show here that the accumulation of transferrin in Cln3Δex7/8 cells is caused by slowdown of the exocytic route and subsequent release of apotransferrin (Fig. 6). Although the molecular mechanism, the identity, and protein-lipid composition of vesicular structures accumulating internalized transferrin in Cln3Δex7/8 cells are unknown, it is likely that e.g. alterations in lipid composition of membranes affect the recycling route of transferrin. Indeed, our lipidomic studies based on total cell extracts show abnormal lipid compositions in Cln3Δex7/8 cerebellar membranes, in particular of gangliosides, sphingomyelin, ceramide, and cerebroside species (Table 1 and Tables S2 and S3), known to impair protein sorting, trafficking, and the autophagic pathway (25, 33–36). Therefore, it is possible that the reduction of distinct sphingomyelin species (14:0 or 15:0) in Cln3Δex7/8 cells (Table S3) which are highly enriched in microdomains of purified recycling endosomes (37) might impair the transport of transferrin to the plasma membrane. Our findings are consistent with data on the coordinate mislocalization of microdomain-associated proteins to the plasma membrane in immortalized brain endothelial cells from Cln3-null mice (33).
Table 1.
Relative intensities of anionic and neutral lipid species in wildtype and Cln3Δex7/8 cerebellar cells
PS, phosphatidylserine; PI, phosphatidylinositol; GM, monosialoganglioside; GD, disialoganglioside; LPE, lysophosphatidylethanolamine; PE, phosphatidylethanolamine; LPC, lysophosphatidylcholine; PC, phosphatidylcholine; Cer, ceramide; SM, sphingomyelin; HexCer, monohexose cerebroside; LacCer, lactosyl ceramide.
| Lipid species | WT | Cln3Δex7/8 | Ratio (Cln3Δex7/8/WT) | |
|---|---|---|---|---|
| Anionic | ||||
| GM3 | d18:1/24:1 | 0.029 | 0.080 | 2.76 |
| GM3 | d18:1/16:0 | 0.077 | 0.153 | 1.99 |
| GM3 | d18:1/24:0 | 0.019 | 0.026 | 1.37 |
| PI | 38:3 | 3.363 | 5.361 | 1.59 |
| PS | 38:3 | 2.577 | 4.043 | 1.57 |
| GD1 | d18:1/16:0 | 2.927 | 1.440 | 0.49 |
| GD1 | d18:1/22:0 | 0.622 | 0.265 | 0.43 |
| GD1 | d18:1/25:0 | 0.339 | 0.145 | 0.43 |
| Neutral | ||||
| LacCer | 24:0 | 0.001 | 0.005 | 5.00 |
| LacCer | 16:0 | 0.003 | 0.014 | 4.67 |
| PE | 38:3 | 0.354 | 0.738 | 2.08 |
| Cer | 24:0 | 0.015 | 0.029 | 1.93 |
| Cer | 24:1 | 0.034 | 0.056 | 1.65 |
| P E | 40:3 | 0.030 | 0.047 | 1.57 |
| SM | 24:1 | 0.373 | 0.579 | 1.55 |
| LPE | 20:3 | 0.111 | 0.170 | 1.53 |
| LPC | 20:4 | 0.870 | 0.527 | 0.61 |
| HexCer | 18:0 | 0.054 | 0.030 | 0.56 |
| HexCer | 22:0 | 0.102 | 0.056 | 0.55 |
| SM | 14:0 | 0.267 | 0.146 | 0.55 |
| PE | 40:4 | 0.110 | 0.058 | 0.53 |
| SM | 15:0 | 0.354 | 0.185 | 0.52 |
| PC | 38:4 | 2.606 | 1.307 | 0.50 |
| HexCer | 23:0 | 0.030 | 0.013 | 0.43 |
| HexCer | 20:0 | 0.034 | 0.010 | 0.29 |
The low abundance of the lysosomal hydrolases acid ceramidase, lipase, galactosylcerebrosidase, or the sphingolipid activator protein precursor prosaposin, which is proteolytically processed to saposins A–D in lysosomes making lipid substrates accessible to lysosomal galactosylcerebrosidase, sulfatide arylsulfatase A, glucocerebrosidase, and acid ceramidase, respectively, might partially explain the accumulation or reduction of sphingolipids in Cln3Δex7/8 cells (38). The intracellular compartments affected by the changes in sphingolipids in Cln3Δex7/8 cerebellar cells remain to be further studied.
In agreement with our data, elevated levels of GM3 gangliosides have been reported in Cln3Δex7/8 cerebellar cells, which are caused by transcriptional down-regulation of GM2 synthetase and hexosaminidase A responsible for conversion of GM3 into GM2 and the lysosomal degradation of GM2 to GM3 gangliosides, respectively (39). However, secondary accumulation of GM2 and GM3 gangliosides has been reported in various neuropathic lysosomal storage disorders such as Niemann-Pick diseases, mucopolysaccharidoses, mucolipidosis type II, or cathepsin D deficiency (40). The mechanisms, molecules, and sensors initiating the differential expression and abundance of acid hydrolases in lysosomes in the absence of Cln3, in particular lipid-degrading enzymes, are poorly defined. Different transcription factors have been described that control transcriptional expression of acid lipase (FoxO1), ceramidase (KLF6), and sphingomyelinase (Sp1, AP-2), suggesting that Cln3 deficiency and subsequent lysosomal dysfunctions impair various signaling pathways (41–43). Our own data show that the mTORC1 pathway is activated in Cln3Δex7/8 cerebellar cells (Fig. S9). The subsequent downstream targets, however, affecting the protein and lipid composition of lysosomes and membranes in Cln3Δex7/8 cerebellar cells remain to be examined.
The lentiviral-mediated re-expression of GFP-CLN3 in Cln3Δex7/8 cerebellar cells did not reverse any of the altered lysosomal components or functions tested here (Fig. S6). This might be explained by the high CMV promotor driven expression of the transgene in the functionally unrelated ER compartment in relation to the small population of CLN3 in lysosomes (Fig. S5). These data are in agreement with studies on high-dosage systemic scAAV9 delivery of CLN3 which failed to restore motor activity and glial activation in Cln3Δex7/8 mice (44). Moreover, a marginal 2.9-fold increase of CLN3 over basal level of CLN3 led to a degenerative eye phenotype in Drosophila (45). This suggests that tightly regulated levels of CLN3 are critical to maintain lysosomal functions and communication with other organelles. Therefore, antibodies suitable to determine the endogenous level of CLN3 in intracellular membranes, are a prerequisite to define conditions for re-expression experiments in CLN3-defective cells and organisms. Additionally, it cannot be excluded that the GFP-fusion interferes with the so far unknown function of CLN3.
In summary, our work indicates that Cln3 has a more generalized role in cellular and in particular lysosomal homeostasis than previously recognized. The low abundance of several lysosomal enzymes in the absence of Cln3 affecting the lysosomal proteome, the associated degradation of substrates, and secondary cellular protein trafficking processes makes Cln3 a central player that is functionally connected to many other proteins and signaling pathways in neuronal cells.
Experimental procedures
Antibodies and materials
The following antibodies were used: mouse anti-α-tubulin (Sigma-Aldrich); polyclonal goat anti-Ctsd (Santa Cruz Biotechnology), goat anti-Ctsz and goat anti-Creg1 (R&D Systems), mouse anti-Gm130 (BD Biosciences), mouse anti-TrfR (Thermo Fisher Scientific), rabbit anti-Plin2 (Abcam), mouse anti-PDI (Enzo Life Sciences), rat anti-Lamp1 (clone 1D4B, Developmental Studies Hybridoma Bank), anti-Lrp1 (Abcam), rabbit anti-Rab11a (Cell Signaling Technology), and rat anti-Mpr300 (kindly provided by Dr. K. von Figura, University of Göttingen, Germany); horseradish peroxidase (HRP)–coupled secondary antibodies (Dianova); Alexa Fluor® 546–coupled anti-mouse IgG, Alexa Fluor® 488–coupled anti rabbit IgG, and Alexa Fluor® 488–coupled anti rat IgG (Thermo Fisher Scientific). Golgi-RFP (CellLightTM BacMam 2.0 Reagents) and LysoTrackerTM Deep Red, and BODIPY® FL C5-lactosylceramide complexed to BSA were purchased from Thermo Fisher Scientific. Recombinant human ARSB was kindly provided by Dr. M. Vellard (BioMarin). Sodium 125I (75 terabecquerels/mmol) was purchased from Hartmann Analytic. M6P sodium salt, penicillin/streptomycin, BSA, protease inhibitor mixture, 4-nitrophenyl-N-acetyl-β-d-glucosaminide, p-nitrocatechol sulfate, 4-methylumbelliferyl (4MU)-6-thio-palmitate-β-d-glucopyranoside, 4MU-α-d-galactopyranoside, 4MU-β-d-mannopyranoside, 4MU-β-d-glucuronide, paraformaldehyde, nigericin, and monensin, and other common laboratory reagents were obtained from Sigma-Aldrich. FBS, l-lysine 13C6 isotopes, IODO-GEN®, DMEM, geneticin, GlutaMAXTM, Oregon Green® 488-dextran, Alexa Fluor® 546– or Alexa Fluor® 488–coupled transferrin were from Thermo Fisher Scientific. LiquidMAG® QD (dextran-stabilized magnetite nanoparticles) and FluoreMAG® A (fluorescent dextran magnetite nanoparticles) and MACS LS separation columns were purchased from Liquid Research Ltd and Miltenyi Biotech, respectively. μ-Slides (8 well) were purchased from ibidi.
Cell culture
Cultured cerebellar cells derived from Cln3Δex7/8 and WT mice, respectively, were described previously (12) and maintained in DMEM containing 10% heat-inactivated FBS, 1× GlutaMAXTM, 24 mm KCl, 200 μg/ml geneticin, and penicillin/streptomycin at 33 °C and 5% CO2. All experiments have been performed with 70 to 80% confluent cerebellar cells exhibiting heterogeneous storage material ranging from dense and multilamellar bodies, electron-lucent floccular bodies and fingerprint-like structures in Cln3Δex7/8 cells (Fig. S1). Isolation and cultivation of primary embryonic mouse fibroblasts (MEF) from WT and Cln3Δex7/8 mice were performed as described (18).
Endocytosis assay of 125I-ARSB
ARSB was iodinated with sodium 125I and IODO-GEN® as described (21) to specific activities of 15–20 μCi/μg protein. Cerebellar cells grown in 3.5-cm plates were pre-incubated with DMEM containing 0.1% BSA for 1 h, followed by incubation with 125I-labeled ARSB (∼250,000 cpm/ml) in DMEM/0.1% BSA in the absence or presence of 10 mm M6P for the indicated time points. Cells were washed with PBS and either harvested or chased as indicated. Cell lysates prepared in PBS containing 0.2% Triton X-100 and protease inhibitors were separated by SDS-PAGE and visualized by autoradiography.
Binding of 125I-ARSB
Cerebellar cells were cultured on 3.5-cm plates and chilled to 4 °C. To determine the relative number of Mpr300 at the cell surface, cerebellar cells were incubated with 125I-ARSB (∼600,000 cpm/ml) in DMEM/0.1% BSA adjusted to 20 mm Hepes (pH 7.4) in the absence or presence of 10 mm M6P for 2 h at 4 °C. To measure the total number of M6P-binding sites, cells were pre-incubated at 4 °C in DMEM/0.1% BSA adjusted to 20 mm Hepes (pH 7.4) containing 0.1% saponin and M6P. Saponin was included in all cell buffers further used. After two washes with PBS, cells were incubated with 125I-ARSB for 2 h at 4 °C in the presence or absence of 10 mm M6P followed by four washes with PBS. The M6P-specific cell-associated radioactivity was determined and related to the cellular protein amount.
Endocytosis/recycling of transferrin
Cells were cultured on glass coverslips in 24-well plates and subsequently incubated in serum-free medium containing 25 μg/ml Alexa Fluor® 546– or Alexa Fluor® 488–coupled transferrin for 30 or 15 min at 33 °C, respectively, followed by a chase in serum-free medium for 15 min. Cells were then washed three times with PBS, fixed in 4% paraformaldehyde on ice for 30 min, and embedded in Aqua-Poly/Mount. Fluorescence was detected and images were obtained using a Leica DMIRE2 digital scanning confocal microscope (Leica Microsystems). Fluorescence intensities were quantified using the ImageJ 1.49v software (National Institutes of Health). Data represent the mean ± S.E. (n = 15) and significance was evaluated using one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparison test. Statistics were performed using GraphPad PRISM (GraphPad Software).
EM
Ultrastructural analysis of cells was performed as described previously (46).
Measurement of lysosomal pH
Cerebellar cells were plated on glass-bottom live-cell dishes (MatTek) and ratiometric pH measurements were performed 24 h after plating (47). Lysosomes were loaded with 0.5 mg/ml Oregon Green® 488-dextran in growth medium overnight, followed by a 2-h chase. Images were acquired using a Zeiss Axiovert 200 equipped with a 100 × 1.30 NA oil-immersion lens with excitation at 440 and 488 nm delivered by a Polychrom V monochromator system (TILL Photonics). The emitted light was selected with a 535 ± 20 nm filter and captured with a Sensicam CCD camera (PCO AG). For each dish, at least 10 different cells were measured in 10 mm Hepes (pH 7.4), 150 mm NaCl, 1 mm MgCl2, 2 mm CaCl2, and 10 mm glucose and analyzed using a Fiji plug-in, in which regions of interest were defined as areas above a defined fluorescence threshold in the acquired images at 488 nm excitation. The mean intensity ratio between 488 and 440 nm excitation was calculated for each region of interest. At the end of each experiment, in situ pH calibration curves were obtained after treatment in isotonic K-based solutions (5 mm NaCl; 115 mm KCl; 1.2 mm MgSO4; 10 mm glucose; 25 mm Hepes, MES, or acetate, ranging in pH from 3.9 through 6.45) supplemented with 10 μm of both nigericin and monensin. Cells were equilibrated for at least 2 min for each pH. The resulting fluorescence intensity ratio (488/440) as a function of pH was fit to a sigmoid and used to interpolate pH values from the experimental ratio data.
Western blotting
Preparation of whole cell extracts, SDS-PAGE and Western blot analysis were performed as described previously (18). For Mpr300 Western blotting, cells were solubilized in 10 mm Tris/HCl, pH 7.4, containing 300 mm NaCl, 1 mm EDTA, 1.5% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and protease inhibitor mixture (48). Anti-α-tubulin Western blotting was used as loading control. After incubation with secondary HRP-conjugated IgG antibody, the immunoreactive bands were visualized by enhanced chemiluminescence.
Enzyme activity measurements
The enzymatic activities of Ppt1, Hexb, and Arsa in whole cell extracts were determined as described previously (49, 50). To measure Gusb, Gla, or Manba activities, cell extracts were incubated with 1 mm 4MU-β-d-glucuronide, 0.75 mm 4MU-α-d-galactopyranoside, or 0.75 mm 4MU-β-mannopyranoside, respectively, in 0.2 m sodium-citrate (pH 4.6) containing 150 mm NaCl and incubated for 2 h (Gusb) or for 16 h (Gla, Manba) at 37 °C. The reactions were stopped by the addition of 0.4 m glycine/NaOH buffer (pH 10.4). The liberated 4MU was measured fluorimetrically with excitation at 360 nm and emission at 440 nm. The specific activities (nmol/h/mg cell protein) of Cln3Δex7/8 cells were normalized to the WT cells.
Bodipy lactosylceramide labeling
Cln3Δex7/8 and WT cerebellar cells were plated on μ-slides (8 well) in a density of 10,000 cells/well. After the cells reached 70% confluency, they were cultured for 16 h in Golgi-RFP (CellLightTM BacMam 2.0 Reagents) supplemented culture medium according to manufacturer's instructions, followed by 30-min incubation with 5 μm BODIPY® FL C5-Lactosylceramide complexed to BSA in Hanks' Balanced Salt solution at 4 °C. After three washes with ice-cold fresh medium the cells were chased for further 30 min in fresh culture medium at 33 °C. For intravital co-staining of BODIPY® FL C5-Lactosylceramide and lysosomes, 50 nm LysoTrackerTM Deep Red probe was added to fresh medium after the BODIPY® FL C5-Lactosylceramide uptake and incubated for 30 min at 33 °C. Cells were analyzed by confocal fluorescence life cell microscopy. Fluorescence intensities were quantified in defined reference regions (Golgi-RFP– or LysoTracker-positive cell compartments) using the ImageJ 1.49v software (National Institutes of Health). Data represent the mean ± S.D. of three independent experiments (25 cells were analyzed per condition) and significance was evaluated with two-tailed Student's t test. Statistics were performed using GraphPad PRISM (GraphPad Software).
Oleate treatment
Cln3Δex7/8 and WT cerebellar cells grown on glass cover slides to 70% confluency were incubated overnight in culture medium supplemented with the standard dose of 400 μm Na-oleate complexed to fatty acid–free BSA overnight, washed with PBS, and fixed in 4% PFA for 20 min at room temperature. After permeabilization with PBS containing 0.1% saponin and 5% BSA (blocking buffer) for 30 min, the cells were incubated with blocking buffer containing primary antibodies against the lipid droplet marker protein Plin2, and the lysosomal membrane protein Lamp1 for 1 h in the dark in a humidified atmosphere at room temperature, washed, and re-incubated with secondary antibodies (anti-rabbit Alexa Fluor® 546 and anti-rat Alexa Fluor® 488, respectively) in blocking buffer for 30 min. After washing, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 2 min, washed with PBS, and embedded in Fluoromount. Confocal microscopy was performed on a fluorescence microscope (Leica TCS SP8 X). Fluorescence intensities were quantified using the ImageJ 1.49v software (National Institutes of Health). Data represent the mean ± S.D. of three independent experiments (25 cells were analyzed per condition) and significance was evaluated with one-way ANOVA. Statistics were performed using GraphPad PRISM.
FluoreMAG labeling
WT and Cln3Δex7/8 cerebellar cells were incubated with 10% FluoreMAG-dextran nanoparticle solution in culture medium for 24 h followed by a 36-h chase in FluoreMAG dextran-free culture medium, and subsequent LysoTracker® Deep Red loading for 30 min. Cells were analyzed by confocal fluorescence life cell microscopy using a Leica TCS SP8 X.
Mass spectrometry and data analysis
Stable isotope labeling of three biological replicates of WT (control) and Cln3Δex7/8 cerebellar cells, isolations of lysosomal fractions with magnetic beads, and sample preparation for LC-MS analysis were performed as described recently (51, 52). Cells were seeded with a density of 5 × 105 cells/cm2 in SILAC DMEM with 10% dialyzed and heat-inactivated FBS, 24 mm KCl, 200 μg/ml geneticin, 110 mg/liter sodium pyruvate, and 10% magnetite particles (Liquids Research Ltd.) for 24 h. Medium contained either light (Cln3Δex7/8) or heavy (WT) labeled lysine (Lys/Lys(13C6) 181.2 mg/liter). The magnetic medium was removed and after three washing steps with PBS, heavy and light fresh medium were added for a 36-h chase time, respectively. Afterward, cells were cooled on ice, washed twice with ice-cold PBS, and then scraped and resuspended in buffer A (10 mm HEPES, 250 mm sucrose, 15 mm KCl, 1.5 mm MgAc, 1 mm CaCl2, and 1 mm DTT). Light and heavy labeled cells were mixed and homogenized with a 7 ml Dounce homogenizer (tight pestle). Nuclei and unbroken cells were removed by centrifugation for 10 min at 1000 × g. The postnuclear supernatant (PNS) was loaded onto a pre-equilibrated (0.5% BSA in PBS) Miltenyi LS magnetic column placed in a MACS Separator. After washing, the column was treated with 10 units/ml DNase 1 in buffer A for 10 min. The column was then removed from the MACS Separator and the lysosomes were eluted with 2 × 500 μl of buffer A.
The lysosomal eluate was concentrated in Amicon Centrifugal Filters at 14,000 × g and 4-fold SDS-sample buffer (250 mm Tris/HCl, pH 6.8, 8% (w/v) SDS, 40% (v/v) glycerol, 10% (v/v) β-mercaptoethanol, 0.004% (w/v) bromphenol blue) was added. After solubilization at 95 °C for 5 min, samples were cooled to room temperature and alkylation was performed by adding 1% acrylamide for 30 min at room temperature. The sample was separated by 10% SDS-PAGE, and the gel was then washed twice in HPLC water and stained with Coomassie Blue for 1 h. After destaining the gel with distilled water, the eluate band profile was divided into 10 fragments.
The gel slices were washed consecutively with water, 50% acetonitrile (ACN), and 100% ACN. Proteins were reduced with 20 mm DTT in 50 mm ammonium bicarbonate and alkylated with 40 mm iodoacetamide (in 50 mm bicarbonate). The slices were washed again and dehydrated with ACN. Dried slices were incubated with 400 ng sequencing grade trypsin at 37 °C overnight. The peptide extract was separated and remaining peptides extracted with 50% ACN. Peptides were dried in a vacuum concentrator and stored at −20 °C.
Peptides were dissolved in 8 μl 0.1% TFA and 1.5 μl were injected onto a C18 trap column (20 mm length, 100 μm inner diameter) coupled to a C18 analytical column (200 mm length, 75 μm inner diameter), made in house with 1.9 μm ReproSil-Pur 120 C18-AQ particles (Dr. Maisch, Ammerbuch, Germany). Solvent A was 0.1% formic acid. Peptides were separated during a linear gradient from 4 to 40% solvent B (80% ACN, 0.1% formic acid) within 80 min at a flow rate of 320 nl/min. The nanoHPLC was coupled online to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Ions between 330 and 1600 m/z were scanned in the Orbitrap detector with a resolution of 30,000 (maximum fill time 400 ms, automatic gain control target 106). The 25 most intense precursor ions (threshold intensity 5000) were subjected to collision-induced dissociation and fragments analyzed in the linear ion trap. Fragmented peptide ions were excluded from repeat analysis for 15 s. Raw data processing and analysis of database searches were performed with Proteome Discoverer software 1.4.1.14 (Thermo Fisher Scientific). Peptide identification was done with an in-house Mascot server version 2.4 (Matrix Science Ltd.). MS2 data were searched against mouse sequences from SwissProt (release 2014_01). Precursor ion m/z tolerance was 8 ppm, fragment ion tolerance 0.6 Da. Tryptic peptides were searched with up to two missed cleavages. Carbamidomethylation was set as a static modification (Cys). Oxidation (Met), acetylation (protein N terminus), Gln->pyro-Glu (N-term Q), Glu->pyro-Glu (N-term E), and 13C6 (Lys) were set as dynamic modifications. Mascot results from searches against SwissProt were sent to the percolator algorithm (53) version 2.04 as implemented in Proteome Discoverer. Spectra without high confident peptide match were sent to a second round of search with semi-tryptic specificity and phosphorylation as dynamic modification (Ser, Thr, Tyr). Only proteins with two peptides (maximum posterior error probability 1%) were considered identified. The false discovery rate (FDR) calculated by Percolator and peptide identifications were accepted with a cut-off of 0.01. Data were filtered for peptide-spectrum matches assigned to only one accession number and the median intensity computed across all peptide-spectrum matches identified for the respective protein. Protein intensities of all replicates were then combined into a feature table and such identified in only one replicate were excluded. Statistical analysis of the heavy to light ratios on the protein level was performed with the R package limma (54) whose applicability on MS data is described in Ref. 55. Corrected p values were computed with the R package q value (56) for false discovery rate estimation, using the corrected p values obtained from the Benjamini-Hochberg output. All data processing steps were implemented in Perl and R scripts. The MS proteomics datasets have been deposited to the Proteome Xchange consortium with the dataset identifiers PXD004548 (lysosomal proteome analysis).
Lipidomics
Total lipids were extracted in three independent experiments from 10 10-cm plates (∼900 mg cells) and separated into a neutral and an anionic fraction by reversed-phase C18 columns (Agilent Technologies, Santa Clara, CA), as described previously (57). Aliquots of the neutral and anionic lipid fractions were analyzed by electrospray ionization-MS using Agilent 6490 Triple Quad LC/MS (Agilent Technologies) and Micromass Quattro Micro triple quadrupole mass spectrometers (Waters, Milford, MA), and lipid class-specific detection modes (57, 58).
Acyl chain alterations in the lipid fractions were studied by GC (Shimadzu GC-2010 Plus with FID, and Shimadzu GCMS-QP2010 Ultra with MSD, Kyoto, Japan) and employing standard protocols (59).
Construction of lentiviral GFP-CLN3 vectors
To express human CLN3 under control of a ubiquitous promoter together with GFP as an N-terminal fusion, a PCR was performed using PrimeStar GLX polymerase (Takara Clontech) and pEGFP-C1 GFP-CLN3 as a template. The following primer pair was employed to generate an expression cassette with the GFP-CLN3 fragment under control of the CMV promoter: 5′-ACCCAAGCTGGCTAGCACCATGGTGAGCAAGGGC and 5′-gaacccttaaacatgtTCAGGAGAGCTGGCAGAG. PCR products were inserted using the InFusion Cloning Kit (Takara Clontech) into modified versions of pSicoR (Addgene, 11579) after digestion with NheI and PciI. Resulting Expression clones were checked by restriction digest and PCR and were finally verified by sequencing. A stock of VSV-G pseudotyped viral particles was produced at the Vector Facility of the University Medical Center Eppendorf using lentiviral packaging plasmids psPAX2 (Addgene plasmid 12260) and pMD2.G (Addgene plasmid 12259). After concentration by ultracentrifugation for 2 h at 4 °C (25,000 rpm, SW32Ti rotor) on a 20% sucrose cushion, the pellet was resuspended in Dulbecco's PBS. The functional titer was determined by transduction of HEK 293T cells and quantification by flow cytometry (FACS Canto II, BD Biosciences; FITC Channel).
WT and Cln3Δex7/8 cerebellar cells were seeded in a 24-well plate at a cell density of 30,000 cells/well. For viral infection, cells were incubated in normal growth medium containing lentiviral particles (GFP: 2 μl, 9 × 108 transduction units/ml; GFP-CLN3: 10 μl, 1.82 × 108 transduction units/ml). After 4 h, medium was exchanged and after 48 h cells were subjected to puromycin selection for GFP- and GFP-CLN3–positive cells, respectively.
Statistical analysis
Data represent the mean ± standard deviation (S.D.) or standard error of the mean (S.E.), and significance was evaluated with one-way ANOVA followed by Dunnett's or Bonferroni's multiple comparison test. A result was considered statistical significant if the p value was ≤ 0.05 (*), ≤ 0.01 (**), or ≤ 0.001 (***).
Author contributions
C. S., S. T., R. K., and M. Sylvester data curation; C. S., I. B., and N. B.-A. validation; C. S., S. T., M. T., R. K., S. J., G. M., M. Sylvester, M. Schweizer, and I. B. investigation; S. T., M. T., G. M., M. Sylvester, M. Schweizer, and N. B.-A. formal analysis; S. T. and M. Schweizer visualization; M. T. and T. B. conceptualization; M. T., R. K., S. J., G. M., M. Sylvester, I. B., and S. L. C. methodology; I. B., S. L. C., and V. G. resources; N. B.-A. software; A. S., V. G., and T. B. supervision; A. S., V. G., and T. B. funding acquisition; V. G. and T. B. writing-review and editing; T. B. writing-original draft.
Supplementary Material
Acknowledgment
We thank the Microscopy Imaging Facility of the University Medical Center Hamburg-Eppendorf for support.
This work was supported by the European Union Seventh Framework Program, DEM-CHILD project Grant agreement n_281234; the European Horizon 2020 Program, BATCURE project Grant n_666918; Deutsche Forschungsgemeinschaft (FOR2625); Bundesministerium für Bildung und Forschung (NCL2TREAT), and Naechstenliebe e.V. (to C. S., G. M., S. T., A. S., V. G., and T. B.); and NINDS, National Institutes of Health, Grant R01NS073813 (to S. L. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S9 and Tables S1–S4.
The MS proteomics datasets have been deposited to the Proteome X-change consortium with the dataset identifier PXD004548 (lysosomal proteome analysis).
- vATPase
- vacuolar H+ ATPase
- ACN
- acetonitrile
- ANOVA
- analysis of variance
- ARSB
- arylsulfatase B
- buffer A
- 10 mm HEPES, 250 mm sucrose, 15 mm KCl, 1.5 mm MgAc, 1 mm CaCl2, and 1 mm DTT
- CMV
- cytomegalovirus
- ER
- endoplasmic reticulum
- LacCer
- lactosylceramides
- M6P
- mannose 6-phosphate
- MES
- 2-(N-morpholino)ethanesulfonic acid
- Mpr300
- 300-kDa mannose 6-phosphate receptor
- SILAC
- stable isotope labeling by amino acids in cell culture
- TGN
- trans-Golgi network
- Trf
- transferrin
- TrfR
- Trf receptor.
References
- 1. Lim C. Y., and Zoncu R. (2016) The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 214, 653–664 10.1083/jcb.201607005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schröder B. A., Wrocklage C., Hasilik A., and Saftig P. (2010) The proteome of lysosomes. Proteomics 10, 4053–4076 10.1002/pmic.201000196 [DOI] [PubMed] [Google Scholar]
- 3. Chapel A., Kieffer-Jaquinod S., Sagné C., Verdon Q., Ivaldi C., Mellal M., Thirion J., Jadot M., Bruley C., Garin J., Gasnier B., and Journet A. (2013) An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell. Proteomics 12, 1572–1588 10.1074/mcp.M112.021980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parenti G., Andria G., and Ballabio A. (2015) Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 66, 471–486 10.1146/annurev-med-122313-085916 [DOI] [PubMed] [Google Scholar]
- 5. Kollmann K., Uusi-Rauva K., Scifo E., Tyynelä J., Jalanko A., and Braulke T. (2013) Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim. Biophys. Acta 1832, 1866–1881 10.1016/j.bbadis.2013.01.019 [DOI] [PubMed] [Google Scholar]
- 6. Palmer D. N., Barry L. A., Tyynelä J., and Cooper J. D. (2013) NCL disease mechanisms. Biochim. Biophys. Acta 1832, 1882–1893 10.1016/j.bbadis.2013.05.014 [DOI] [PubMed] [Google Scholar]
- 7. Chandrachud U., Walker M. W., Simas A. M., Heetveld S., Petcherski A., Klein M., Oh H., Wolf P., Zhao W. N., Norton S., Haggarty S. J., Lloyd-Evans E., and Cotman S. L. (2015) Unbiased cell-based screening in a neuronal cell model of Batten disease highlights an interaction between Ca2+ homeostasis, autophagy, and CLN3 protein function. J. Biol. Chem. 290, 14361–14380 10.1074/jbc.M114.621706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gachet Y., Codlin S., Hyams J. S., and Mole S. E. (2005) btn1, the Schizosaccharomyces pombe homologue of the human Batten disease gene CLN3, regulates vacuole homeostasis. J. Cell Sci. 118, 5525–5536 10.1242/jcs.02656 [DOI] [PubMed] [Google Scholar]
- 9. Ramirez-Montealegre D., and Pearce D. A. (2005) Defective lysosomal arginine transport in juvenile Batten disease. Hum. Mol. Genet. 14, 3759–3773 10.1093/hmg/ddi406 [DOI] [PubMed] [Google Scholar]
- 10. Padilla-López S., and Pearce D. A. (2006) Saccharomyces cerevisiae lacking Btn1p modulate vacuolar ATPase activity to regulate pH imbalance in the vacuole. J. Biol. Chem. 281, 10273–10280 10.1074/jbc.M510625200 [DOI] [PubMed] [Google Scholar]
- 11. Rusyn E., Mousallem T., Persaud-Sawin D. A., Miller S., and Boustany R. M. (2008) CLN3p impacts galactosylceramide transport, raft morphology, and lipid content. Pediatr. Res. 63, 625–631 10.1203/PDR.0b013e31816fdc17 [DOI] [PubMed] [Google Scholar]
- 12. Fossale E., Wolf P., Espinola J. A., Lubicz-Nawrocka T., Teed A. M., Gao H., Rigamonti D., Cattaneo E., MacDonald M. E., and Cotman S. L. (2004) Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC Neurosci. 5, 57 10.1186/1471-2202-5-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luiro K., Yliannala K., Ahtiainen L., Maunu H., Järvelä I., Kyttälä A., and Jalanko A. (2004) Interconnections of CLN3, Hook1 and Rab proteins link Batten disease to defects in the endocytic pathway. Hum. Mol. Genet. 13, 3017–3027 10.1093/hmg/ddh321 [DOI] [PubMed] [Google Scholar]
- 14. Metcalf D. J., Calvi A. A., Seaman M. N. J., Mitchison H. M., and Cutler D. F. (2008) Loss of the Batten disease gene CLN3 prevents exit from the TGN of the mannose 6-phosphate receptor. Traffic 9, 1905–1914 10.1111/j.1600-0854.2008.00807.x [DOI] [PubMed] [Google Scholar]
- 15. Cotman S. L., and Staropoli J. F. (2012) The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clin. Lipidol. 7, 79–91 10.2217/clp.11.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hiesberger T., Hüttler S., Rohlmann A., Schneider W., Sandhoff K., and Herz J. (1998) Cellular uptake of saposin (SAP) precursor and lysosomal delivery by the low density lipoprotein receptor-related protein (LRP). EMBO J. 17, 4617–4625 10.1093/emboj/17.16.4617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nielsen R., Courtoy P. J., Jacobsen C., Dom G., Lima W. R., Jadot M., Willnow T. E., Devuyst O., and Christensen E. I. (2007) Endocytosis provides a major alternative pathway for lysosomal biogenesis in kidney proximal tubular cells. Proc. Natl. Acad. Sci. U.S.A. 104, 5407–5412 10.1073/pnas.0700330104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Markmann S., Thelen M., Cornils K., Schweizer M., Brocke-Ahmadinejad N., Willnow T., Heeren J., Gieselmann V., Braulke T., and Kollmann K. (2015) Lrp1/LDL receptor play critical roles in mannose 6-phosphate-independent lysosomal enzyme targeting. Traffic 16, 743–759 10.1111/tra.12284 [DOI] [PubMed] [Google Scholar]
- 19. Braulke T., and Bonifacino J. S. (2009) Sorting of lysosomal proteins. Biochim. Biophys. Acta 1793, 605–614 10.1016/j.bbamcr.2008.10.016 [DOI] [PubMed] [Google Scholar]
- 20. Pearce D. A., Ferea T., Nosel S. A., Das B., and Sherman F. (1999) Action of BTN1, the yeast orthologue of the gene mutated in Batten disease. Nat. Genet. 22, 55–58 10.1038/8861 [DOI] [PubMed] [Google Scholar]
- 21. Braulke T., Gartung C., Hasilik A., and von Figura K. (1987) Is movement of mannose 6-phosphate-specific receptor triggered by binding of lysosomal enzymes? J. Cell Biol. 104, 1735–1742 10.1083/jcb.104.6.1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sztalryd C., and Brasaemle D. L. (2017) The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 1221–1232 10.1016/j.bbalip.2017.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kobayashi T., Beuchat M. H., Lindsay M., Frias S., Palmiter R. D., Sakuraba H., Parton R. G., and Gruenberg J. (1999) Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1, 113–118 10.1038/10084 [DOI] [PubMed] [Google Scholar]
- 24. Takahashi M., and Kobayashi T. (2009) Cholesterol regulation of rab-mediated sphingolipid endocytosis. Glycoconj J. 26, 705–710 10.1007/s10719-008-9191-z [DOI] [PubMed] [Google Scholar]
- 25. Tommasino C., Marconi M., Ciarlo L., Matarrese P., and Malorni W. (2015) Autophagic flux and autophagosome morphogenesis require the participation of sphingolipids. Apoptosis 20, 645–657 10.1007/s10495-015-1102-8 [DOI] [PubMed] [Google Scholar]
- 26. Ullrich O., Reinsch S., Urbé S., Zerial M., and Parton R. G. (1996) Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 135, 913–924 10.1083/jcb.135.4.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamashiro D. J., Tycko B., Fluss S. R., and Maxfield F. R. (1984) Segregation of transferrin to a mildly acidic (pH 6.5) para-Golgi compartment in the recycling pathway. Cell 37, 789–800 10.1016/0092-8674(84)90414-8 [DOI] [PubMed] [Google Scholar]
- 28. Wyant G. A., Abu-Remaileh M., Frenkel E. M., Laqtom N. N., Dharamdasani V., Lewis C. A., Chan S. H., Heinze I., Ori A., and Sabatini D. M. (2018) NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360, 751–758 10.1126/science.aar2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bonifacino J. S., and Neefjes J. (2017) Moving and positioning the endolysosomal system. Curr. Opin. Cell Biol. 47, 1–8 10.1016/j.ceb.2017.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henne W. M. (2016) Organelle remodeling at membrane contact sites. J. Struct. Biol. 196, 15–19 10.1016/j.jsb.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lamb C. A., Yoshimori T., and Tooze S. A. (2013) The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 14, 759–774 10.1038/nrm3696 [DOI] [PubMed] [Google Scholar]
- 32. Codlin S., Haines R. L., Burden J. J., and Mole S. E. (2008) Btn1 affects cytokinesis and cell-wall deposition by independent mechanisms, one of which is linked to dysregulation of vacuole pH. J. Cell Sci. 121, 2860–2870 10.1242/jcs.030122 [DOI] [PubMed] [Google Scholar]
- 33. Tecedor L., Stein C. S., Schultz M. L., Farwanah H., Sandhoff K., and Davidson B. L. (2013) CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. J. Neurosci. 33, 18065–18079 10.1523/JNEUROSCI.0498-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kobayashi T., Gu F., and Gruenberg J. (1998) Lipids, lipid domains and lipid-protein interactions in endocytic membrane traffic. Semin. Cell Dev. Biol. 9, 517–526 10.1006/scdb.1998.0257 [DOI] [PubMed] [Google Scholar]
- 35. Hannun Y. A., and Obeid L. M. (2008) Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 10.1038/nrm2329 [DOI] [PubMed] [Google Scholar]
- 36. Maceyka M., and Spiegel S. (2014) Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67 10.1038/nature13475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gagescu R., Demaurex N., Parton R. G., Hunziker W., Huber L. A., and Gruenberg J. (2000) The recycling endosome of Madin-Darby canine kidney cells is a mildly acidic compartment rich in raft components. Mol. Biol. Cell 11, 2775–2791 10.1091/mbc.11.8.2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schulze H., Kolter T., and Sandhoff K. (2009) Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochim. Biophys. Acta 1793, 674–683 10.1016/j.bbamcr.2008.09.020 [DOI] [PubMed] [Google Scholar]
- 39. Somogyi A., Petcherski A., Beckert B., Huebecker M., Priestman D. A., Banning A., Cotman S. L., Platt F. M., Ruonala M. O., and Tikkanen R. (2018) Altered expression of ganglioside metabolizing enzymes results in GM3 ganglioside accumulation in cerebellar cells of a mouse model of juvenile neuronal ceroid lipofuscinosis. Int. J. Mol. Sci. 19, E625 10.3390/ijms19020625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Walkley S. U., and Vanier M. T. (2009) Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 1793, 726–736 10.1016/j.bbamcr.2008.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langmann T., Buechler C., Ries S., Schaeffler A., Aslanidis C., Schuierer M., Weiler M., Sandhoff K., de Jong P. J., and Schmitz G. (1999) Transcription factors Sp1 and AP-2 mediate induction of acid sphingomyelinase during monocytic differentiation. J. Lipid Res. 40, 870–880 [PubMed] [Google Scholar]
- 42. Park J. H., Eliyahu E., Narla G., DiFeo A., Martignetti J. A., and Schuchman E. H. (2005) KLF6 is one transcription factor involved in regulating acid ceramidase gene expression. Biochim. Biophys. Acta 1732, 82–87 10.1016/j.bbaexp.2006.01.002 [DOI] [PubMed] [Google Scholar]
- 43. Lettieri Barbato D., Tatulli G., Aquilano K., and Ciriolo M. R. (2013) FoxO1 controls lysosomal acid lipase in adipocytes: Implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis. 4, e861 10.1038/cddis.2013.404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bosch M. E., Aldrich A., Fallet R., Odvody J., Burkovetskaya M., Schuberth K., Fitzgerald J. A., Foust K. D., and Kielian T. (2016) Self-complementary AAV9 gene delivery partially corrects pathology associated with juvenile neuronal ceroid lipofuscinosis. J. Neurosci. 36, 9669–9682 10.1523/JNEUROSCI.1635-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tuxworth R. I., Vivancos V., O'Hare M. B., and Tear G. (2009) Interactions between the juvenile Batten disease gene, CLN3, and the Notch and JNK signalling pathways. Hum. Mol. Genet. 18, 667–678 10.1093/hmg/ddn396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marschner K., Kollmann K., Schweizer M., Braulke T., and Pohl S. (2011) A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science 333, 87–90 10.1126/science.1205677 [DOI] [PubMed] [Google Scholar]
- 47. Weinert S., Jabs S., Supanchart C., Schweizer M., Gimber N., Richter M., Rademann J., Stauber T., Kornak U., and Jentsch T. J. (2010) Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl− accumulation. Science 328, 1401–1403 10.1126/science.1188072 [DOI] [PubMed] [Google Scholar]
- 48. Körner C., Nürnberg B., Uhde M., and Braulke T. (1995) Mannose 6-phosphate/insulin-like growth factor II receptor fails to interact with G-proteins. Analysis of mutant cytoplasmic receptor domains. J. Biol. Chem. 270, 287–295 10.1074/jbc.270.1.287 [DOI] [PubMed] [Google Scholar]
- 49. Van Diggelen O. P., Keulemans J. L., Kleijer W. J., Thobois S., Tilikete C., and Voznyi Y. V. (2001) Pre- and postnatal enzyme analysis for infantile, late infantile and adult neuronal ceroid lipofuscinosis (CLN1 and CLN2). Eur. J. Paediatr. Neurol. 5, Suppl. A, 189–192 10.1053/ejpn.2001.0509 [DOI] [PubMed] [Google Scholar]
- 50. Kollmann K., Damme M., Markmann S., Morelle W., Schweizer M., Hermans-Borgmeyer I., Röchert A. K., Pohl S., Lübke T., Michalski J. C., Käkelä R., Walkley S. U., and Braulke T. (2012) Lysosomal dysfunction causes neurodegeneration in mucolipidosis II 'knock-in' mice. Brain 135, 2661–2675 10.1093/brain/aws209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Di Lorenzo G., Velho R. V., Winter D., Thelen M., Ahmadi S., Schweizer M., De Pace R., Cornils K., Yorgan T. A., Grüb S., Hermans-Borgmeyer I., Schinke T., Müller-Loennies S., Braulke T., and Pohl S. (2018) Lysosomal proteome and secretome analysis identifies missorted enzymes and their non-degraded substrates in mucolipidosis III mouse cells. Mol. Cell. Proteomics 17, 1612–1626 10.1074/mcp.RA118.000720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thelen M., Winter D., Braulke T., and Gieselmann V. (2017) SILAC-based comparative proteomic analysis of lysosomes from mammalian cells using LC-MS/MS. Methods Mol. Biol. 1594, 1–18 10.1007/978-1-4939-6934-0_1 [DOI] [PubMed] [Google Scholar]
- 53. Käll L., Storey J. D., MacCoss M. J., and Noble W. S. (2008) Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 7, 29–34 10.1021/pr700600n [DOI] [PubMed] [Google Scholar]
- 54. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schwämmle V., León I. R., and Jensen O. N. (2013) Assessment and improvement of statistical tools for comparative proteomics analysis of sparse data sets with few experimental replicates. J. Proteome Res. 12, 3874–3883 10.1021/pr400045u [DOI] [PubMed] [Google Scholar]
- 56. Storey J. D. (2002) A direct approach to false discovery rates. J. R. Stat. Soc. Series B. Stat. Methodol. 64, 479–498 10.1111/1467-9868.00346 [DOI] [Google Scholar]
- 57. Jabs S., Quitsch A., Käkelä R., Koch B., Tyynelä J., Brade H., Glatzel M., Walkley S., Saftig P., Vanier M. T., and Braulke T. (2008) Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J. Neurochem. 106, 1415–1425 10.1111/j.1471-4159.2008.05497.x [DOI] [PubMed] [Google Scholar]
- 58. Käkelä R., Somerharju P., and Tyynelä J. (2003) Analysis of phospholipid molecular species in brains from patients with infantile and juvenile neuronal-ceroid lipofuscinosis using liquid chromatography-electrospray ionization mass spectrometry. J. Neurochem. 84, 1051–1065 10.1046/j.1471-4159.2003.01602.x [DOI] [PubMed] [Google Scholar]
- 59. Käkelä R., Käkelä A., Kahle S., Becker P. H., Kelly A., and Furness R. W. (2005) Fatty acid signatures in plasma of captive herring gulls as indicators of demersal or pelagic fish diet. Mar. Ecol. Prog. Ser. 293, 191–200 10.3354/meps293191 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.