

Abstract
The survival, differentiation, and maintenance of responsive neurons are regulated by nerve growth factor (NGF), which is secreted by the target and interacts with receptors on the axon tip. It is uncertain how the NGF signal is communicated retrogradely from distal axons to neuron cell bodies. Retrograde transport of activated receptors in endocytic vesicles could convey the signal. However, little is known about endocytosis of NGF receptors, and there is no evidence that NGF receptors continue to signal after endocytosis. We have examined early events in the membrane traffic of NGF and its receptor, gp140TrkA (TrkA), in PC12 cells. NGF induced rapid and extensive endocytosis of TrkA in these cells, and the receptor subsequently moved into small organelles located near the plasma membrane. Some of these organelles contained clathrin and α-adaptin, which implies that TrkA is internalized by clathrin-mediated endocytosis. Using mechanical permeabilization and fractionation, intracellular organelles derived from endocytosis were separated from the plasma membrane. After NGF treatment, NGF was bound to TrkA in endocytic organelles, and TrkA was tyrosine-phosphorylated and bound to PLC-γ1, suggesting that these receptors were competent to initiate signal transduction. These studies raise the possibility that NGF induces formation of signaling endosomes containing activated TrkA. They are an important first step in elucidating the molecular mechanism of NGF retrograde signaling.
Keywords: NGF, TrkA, signaling, endosome, clathrin, PLC-γ1
Nerve growth factor (NGF), a polypeptide neurotrophic factor of the neurotrophin gene family, acts physiologically to enhance the survival and differentiation of specific populations of neurons in the central (CNS) and peripheral nervous systems (PNS) (Levi-Montalcini, 1987; Yuen and Mobley, 1995). NGF actions are mediated by its receptors, p75NTR and gp140TrkA (TrkA). p75NTR, a single-transmembrane glycoprotein, is a receptor for all of the neurotrophins (Bothwell, 1995). The role that p75NTR plays in NGF signaling is not well defined (Bothwell, 1996; Carter et al., 1996); however, p75NTR modulates NGF binding and activation of TrkA (Meakin and Shooter, 1992; Davies et al., 1993; Barker and Shooter, 1994; Hantzopoulos et al., 1994; Lee et al., 1994; Mahadeo et al., 1994; Verdi et al., 1994). TrkA is a receptor tyrosine kinase whose activation has been shown in vitro to induce many of the typical neuronal responses to NGF (Loeb and Greene, 1993). NGF binding to TrkA causes dimerization of the receptor with resulting activation of its kinase domain (Kaplan et al., 1991; Klein et al., 1991; Meakin and Shooter, 1991; Jing et al., 1992). Autophosphorylation of certain tyrosine residues in the intracellular domain of the receptor creates sites for binding and activation of signaling intermediates that continue the signal transduction cascade (Stephens et al., 1994). Known intermediaries are PLC-γ1, SHC, and PI-3 kinase (Kaplan and Stephens, 1994; Stephens et al., 1994). Significantly, TrkA plays an important role in NGF signaling in vivo. Animals in which the gene for TrkA was disrupted showed marked abnormalities in the development of NGF-responsive neurons in the CNS and PNS (Smeyne et al., 1994).
An important question is how NGF signaling in axons is communicated to neuronal cell bodies. For many NGF-responsive populations, the principal source of NGF is the target field of innervation (Longo et al., 1993). Thus, NGF is available to bind and activate its receptors only on distal axons. The importance of signaling through these receptors was demonstrated by Campenot (1977), who showed that NGF present only at the tips of neurites was sufficient to maintain the viability of cell bodies. This indicates that a signal(s) created by receptor activation on distal axons must be communicated to the cell body. For neurons with long axons, the distance through which the retrograde signal must be moved may be >1000 times the width of the cell body. In earlier studies to define the NGF retrograde signal, the following were discovered. (1) NGF itself was taken up in the target in a dose-dependent, saturable, and stereospecific manner (Hendry et al., 1974a). (2) NGF was retrogradely transported to the cell body at ∼2500 μm/hr (Hendry et al., 1974b; Claude et al., 1982). (3) A response to NGF was registered in only the neurons in which it was transported (Hendry, 1977). (4) The response to NGF coincided temporally with the arrival of NGF at the cell body (Hendry and Bonyhady, 1980). (5) Retrograde movement of NGF was abolished by colchicine (Hendry et al., 1974b; Claude et al., 1982). (6) Colchicine also blunted the retrograde signal caused by NGF (Paravicini et al., 1975). Importantly, although transport of NGF marked retrograde signaling, NGF itself was not the signal. NGF injection into the cell body did not create responses, and NGF antibodies failed to suppress them (Heumann et al., 1984). We suggest that the retrograde signaling entity must be (1) present in distal axons, (2) activated by NGF, (3) retrogradely transported to the cell body, and (4) that its activity must be sufficient to initiate the NGF response in the cell body.
Our hypothesis is that activated TrkA in endocytic vesicles is a retrograde signal. In support, the following are known. (1) TrkA is present in axons in NGF-responsive neurons (Holtzman et al., 1995). (2) TrkA in axons can be activated by NGF treatment (Knüsel et al., 1994; Li et al., 1995). (3) In vitro, surface TrkA is rapidly downregulated in response to NGF binding (Hosang and Shooter, 1987; Zhou et al., 1995). (4) TrkA, including the tyrosine-phosphorylated active form of the receptor, is retrogradely transported in axons (Ehlers et al., 1995). Significantly, the amount of TrkA that accumulated distal to a sciatic nerve ligature, and the extent of its tyrosine phosphorylation, was increased by applying NGF to the foot pad, the target of sciatic nerve sensory neurons (Ehlers et al., 1995). These experiments showed that target-derived NGF regulates retrograde transport of TrkA. However, they did not examine the mechanism by which NGF accomplishes this effect or its significance for signaling. Important, as yet untested, predictions of the hypothesis are that NGF induces endocytosis of TrkA, that internalized receptors continue to be activated, and that they are moved retrogradely in axons and are capable of signaling.
We have examined TrkA receptors internalized in response to NGF binding. PC12 cells have been widely used to study NGF responses (Tischler and Greene, 1975; Stephens et al., 1994; Zhou et al., 1995) and provide a model for neuronal signal transduction and membrane traffic. In this paper, we show that NGF rapidly induced internalization of TrkA receptors, that these receptors were localized in endocytic vesicles, and that intracellular receptors were activated as judged by the presence of phosphotyrosine and association with PLC-γ1. These data suggest that by inducing activation and endocytosis of TrkA, NGF creates signaling endosomes that could be used to convey the retrograde signal.
MATERIALS AND METHODS
Materials. Bis(sulfosuccinimidyl)suberate (BS3), disuccinimidyl suberate (DSS), and NHS-SS-biotin were obtained from Pierce (Rockford, IL). Potassium hydroxide (99%) was from Aldrich (Milwaukee, WI). Type IV collagen was from Collaborative Biomedical Products (Bedford, MA). Normal goat serum and Vectastain mounting medium were from Vector Laboratories (Burlingame, CA). Except as noted, Sigma (St. Louis, MO) was the source of all other reagents and chemicals.
NGF was prepared as described previously (Mobley et al., 1986). NGF was labeled with 125I (Amersham, Arlington Heights, IL) using lactoperoxidase, as modified from Vale and Shooter (1985). Iodinated protein was separated from free 125I on a PD-10 column (Pharmacia, Uppsala, Sweden) preequilibrated with binding buffer [PBS, pH 7.4, containing 1 mg/ml glucose and 1 mg/ml bovine serum albumin (BSA)]. Final specific activity was 25–100 cpm/pg. Radioactivity was quantified using a Beckman 2000 gamma counter.
1088 is a rabbit antibody against the C terminus of human TrkA. It was purified using protein A–Sepharose (Pierce) and has been characterized previously (Zhou et al., 1995). Sc11 is another rabbit antibody to the C terminus of human TrkA (Santa Cruz Biotechnology, Santa Cruz, CA). Both antibodies recognize full-length receptors whose kinase domains can be activated. RTA is a rabbit antibody against the extracellular domain of rat TrkA (Clary et al., 1994; Lucidi-Phillipi et al., 1996). X22 is a mouse monoclonal antibody to the clathrin heavy chain (Brodsky, 1985). AP.6 is a mouse monoclonal antibody to the α-adaptin 100 kDa subunits (Chin et al., 1989). GM10 is a mouse monoclonal antibody that stains lysosomes (Grimaldi et al., 1987; Grady et al., 1995). The antibody to PLC-γ1 was a mixed monoclonal antibody; 4G10 is a mouse monoclonal antibody to phosphorylated tyrosine (both from UBI, Lake Placid, NY). 125I-labeled goat anti-mouse was prepared using Na-125I (Amersham), iodobeads (Pierce), and goat anti-mouse (Pierce) according to the manufacturer’s instructions, and desalted on a PD-10 column (Pharmacia Biotech, Piscataway, NJ).
NGF binding and cross-linking to surface receptors.PC12 cells (a gift of Lloyd A. Greene, Columbia University) were grown on collagen-coated plates in RPMI 1640 with 10% horse serum and 5% fetal calf serum (both from HyClone Laboratories, Logan, UT). Cells were washed and harvested in warm PBS (without Ca2+ and Mg2+) and resuspended in cold (4°C) binding buffer. In all experiments, equal amounts of cells were aliquoted for each condition. Cells were incubated with [125I]NGF (1 nm = 26.5 ng/ml) at 4°C for 1 or 2 hr, warmed to 37°C in the presence of [125I]NGF for 5, 10, or 30 min, and then rapidly chilled (4°C). For cross-linking [125I]NGF to surface receptors, the membrane-impermeant cross-linking reagent BS3 was added at a final concentration of 0.8 mm, while rotating at 4°C for 30 min. To correct for nonspecific binding and cross-linking, unlabeled NGF (1 μm = 26.5 μg/ml) was included during binding and cross-linking. The reactions were quenched with 1 mm lysine for 10 min. Cells were pelleted and then lysed for 20 min on ice in 1 ml of lysis buffer #1 (20 mm Tris, pH 8.0, 150 mm NaCl, 1% NP-40, 10% glycerol, and proteinase inhibitors: 1 mm PMSF, 10 μg/ml benzamidine, 1 μg/mlO-phenantholine, and 0.1 μg/ml each pepstatin, chymostatin, leupeptin, and aprotinin). After centrifuging at 16,000 × g for 30 min, the supernatant was removed and assayed for protein (BCA Assay, Pierce). Samples were normalized for protein and immunoprecipitated with 1088 (12 μg/ml), rotating overnight at 4°C. Protein A–Sepharose beads (Pierce), 120 μl of a 20% solution per ml of lysate, were added and incubated at 4°C for 2 hr. After washing two times in lysis buffer #1 and once in H2O, 50 μl of 7 m urea SDS-PAGE sample buffer (125 mm Tris, pH 6.95, 7 m urea, 2% SDS, 1 mm EDTA, 0.1% Bromphenol Blue) with 100 mm DTT was added, and the sample was heated to 65°C for 15 min. Samples were loaded onto 8–12% SDS-PAGE. Fixed gels (10% acetic acid, 10% isopropanol, 20% methanol) were dried and exposed to the PhosphorImager and then to XAR-5 film (Eastman Kodak, Rochester, NY).
Cell surface biotinylation. Cells were incubated with or without NGF (1 nm) for 30 min at 4°C in PBS with 1 mg/ml glucose, and then NHS-SS-biotin (0.5 mg/ml) was added. The mixture was incubated with gentle rocking for 90 min at 4°C. Cells were pelleted (1000 rpm for 5 min) and then washed three times in cold PBS containing 1 mm lysine. Samples were resuspended in binding buffer, and equal amounts were aliquoted for three different treatments. One sample (designated 100%) was held at 4°C. Another (bkg = background) was treated at 4°C with 50 mm reduced glutathione in 50 mm Tris, pH 8.6, 100 mm NaCl, 1 mg/ml glucose, and 1 mg/ml BSA for 30 min. This treatment was repeated twice. The third sample (int = internalized) was warmed to 37°C for either 10 or 20 min to allow endocytosis, and then treated with glutathione as above. All samples were then incubated 1 hr on ice in 0.2 ml of lysis buffer #2 (20 mm Tris, pH 8.0, 150 mm NaCl, 1% NP-40, 1 mm EDTA) containing 1% BSA and 1 mg/ml iodoacetamide. The lysates were centrifuged 10 min at 10,000 × g. SDS (final concentration 0.5%) was added to the supernatant, and the lysates were boiled 5 min. Lysis buffer #2 (0.8 ml) was then added, and TrkA was immunoprecipitated with 1088 (12 μg). Each lysate was divided into two aliquots. The first was used to detect the amount of biotinylated TrkA. Proteins were separated on nonreducing 7.5% SDS-PAGE, transferred to nitrocellulose (Hoefer Pharmacia Biotech, San Francisco, CA), and blotted with [125I]streptavidin (Amersham). Biotinylated TrkA was quantified by PhosphorImager (Molecular Dynamics, Sunnyvale, CA). The second aliquot was submitted to 7.5% SDS-PAGE in a reducing environment, transferred to nitrocellulose, and blotted with 1088 (1:500). The signal was developed using [125I]protein A (Amersham) and quantified using the PhosphorImager. The signal for biotinylated TrkA was normalized to the amount of TrkA protein. The TrkA available for internalization was (100% − bkg). TrkA internalized during warming was not susceptible to reduction with glutathione. The amount of TrkA internalized was (int − bkg). The percent of TrkA internalized was (int − bkg)/(100% − bkg) × 100.
Immunostaining and confocal microscopy. Cells were plated on 8-well chamber slides (Nunc, Naperville, IL) that had been coated with Matrigel (Collaborative Biomedical Products) using a 1:200 dilution in PBS (Ca2+- and Mg2+-free) overnight at 4°C. Wells were washed three times with cold PBS. PC12 cells were plated in DME H-21 medium with 10% horse serum and 5% fetal calf serum 1–2 d before experiments. After aspirating the medium, NGF (2 nm = 53 ng/ml) was added to cells for 30 sec, 2 min, or 60 min in 300 μl of DME H-21 containing 0.5 mg/ml BSA and 10 mm HEPES at 37°C. This medium, without NGF, was added to controls.
Cells were fixed on ice with 1% paraformaldehyde in PBS for 15 min. Cells were permeabilized in PSS (PBS with 10% normal goat serum and saponin at 1 mg/ml) at room temperature for 30 min, changing the solution every 10 min. Primary antibodies were diluted in PSS (sc11 at 1 μg/ml; X22 at 3.1 μg/ml; AP.6 at 8 μg/ml; GM10 at 1:6000) and were incubated with cells overnight at 4°C. After three 10 min washes with PSS, secondary antibodies diluted in PSS (FITC-conjugated goat anti-rabbit IgG at 1:100; Texas Red-conjugated goat anti-mouse immunoglobulins, 1:200; both from Cappel Research Products, Durham, NC) were applied for 45 min at room temperature. After three washes in PBS, coverslips were mounted using Vectashield mounting solution. No staining was evident when primary antibodies were excluded. For Sc11, preliminary incubation (overnight, 4°C) with the peptide immunogen (10 μg/ml) eliminated staining.
Cells were observed with an MRC 1000 Laser Scanning Confocal Microscope (Bio-Rad, Hercules, CA) equipped with a krypton/argon laser and attached to a Zeiss Axiovert microscope. A Zeiss Neofluor ×100 oil-immersion objective with a numerical aperture of 1.3 (0.17°) was used, and images were collected using an aperture of 3–4 mm and a zoom of 2–3. Typically, 10–20 optical sections were taken at 0.5 μm intervals through the cells. The resolution of the confocal microscope in the x–y-axis was 170–200 nm, and in thez-axis was 230–400 nm. Images of 768 × 520 pixels were obtained. Images were processed using Adobe Photoshop 3.0 (Adobe Systems, Mountain View, CA) and printed using a Techtronix Printer. In experiments in which markers were colocalized, colocalization was confirmed by examining individual organelles at higher magnification. In addition, we ensured that colocalization was eliminated by merging overlaying but noncoincident sections. The images shown correspond to optical sections through cells at mid-height.
To count TrkA-positive vesicles near the surface of cells, images of individual optical sections were examined. The edge of the cell was defined as the outermost limit of staining for the clathrin heavy chain, and this was marked with a line. A second line was drawn 0.5 μm interior to the first, and all bright, punctate TrkA-positive vesicles between the lines were counted. The number of these vesicles was then divided by the perimeter of the surface of the cell to yield a value for the number per micrometer cell perimeter.
Cell fractionation. Figure 4 depicts the cell fractionation strategy. Cells grown and harvested as above were first incubated with or without NGF (1 nm) in binding buffer at 4°C for 1 hr. They were then either washed briefly in binding buffer, or not washed and warmed in binding buffer for 10 min at 37°C. Cells were then chilled (4°C) and washed in PBS with 1 mm EDTA and 1 mm EGTA, and then in a cytoplasm-like buffer (buffer B; 38 mm each of the potassium salts of aspartic, gluconic, and glutamic acids, 20 mm MOPS, 5 mm reduced glutathione, 10 mm potassium bicarbonate, 0.5 mm magnesium carbonate, 1 mm EGTA, 1 mm EDTA, pH-adjusted to 7.1 at 37°C with potassium hydroxide). Cells were resuspended in 0.5 ml of buffer B containing proteinase inhibitors (as for lysis buffer #1). Sodium orthovanadate (1 mm) was included to inhibit phosphatase activity for the experiments shown in Figures 8, 9, 10. To permeabilize cells, we used a ball homogenizer obtained from the European Molecular Biology Laboratory (Heidelberg, Germany) and tungsten carbide grade-25 balls obtained from Industrial Tectonics (Ann Arbor, MI). The cell suspension was passed through the homogenizer (8.020 mm cylinder with an 8.0186 mm ball). More than 98% of the cells stained with trypan blue after this procedure. By centrifuging at 1000 × g for 10 min, cell ghosts (P1) were pelleted, thus separating them from the cytosol and the released vesicles. Membranes in the supernatant were isolated using two different protocols. In the first, they were layered over a 0.4 ml pad of 10% sucrose in buffer B (with inclusions, as above) and then centrifuged at 100,000 × g for 1 hr in a Beckman Ti SW50.1 rotor. The membrane pellet was referred to as P2′, and the supernatant (S2′) was cytosol. In the second protocol, the 1000 ×g supernatant was centrifuged at 8000 × gfor 35 min in a TiSW50.1 rotor. The pellet (P2) contained large organelles. The supernatant (S2) was layered over a 0.4 ml pad of 10% sucrose in buffer B (with inclusions, as above) and centrifuged at 100,000 × g for 1 hr to produce the pellet, P3. S3 was the cytosol.
Fig. 4.

Strategy for cell fractionation experiments. NGF (1 nm) was bound to PC12 cells at 4°C for 1 hr. Cells were then either briefly washed in binding buffer at 4°C or not washed, and warmed at 37°C for 10 min. Cells were then chilled (4°C), washed, resuspended in a cytoplasm-like buffer, and permeabilized by passage through a ball homogenizer. The cell ghosts (P1) were separated from cytosol and organelles released from the cells by centrifugation at 1000 × g. Two alternative strategies were used to fractionate the membranes in the supernatant of the 1000 × g centrifugation. (1) They were layered over a 0.4 ml pad of 10% buffered sucrose and centrifuged at 100,000 × g (1 hr), forming a pellet (P2′) and the cytosol (S2′). (2) To separate large and small vesicles, membranes were centrifuged at 8000 × g for 35 min to pellet large vesicles (P2). The supernatant of the 8000 ×g spin was then layered over a 0.4 ml pad of 10% buffered sucrose and centrifuged at 100,000 × gfor 1 hr, which separated small vesicles (P3) from cytosol (S3).
Fig. 8.

TrkA and tyrosine-phosphorylated TrkA were detected in intracellular organelles. Top, PC12 cells incubated with or without NGF (1 nm) at 4°C were washed, warmed 10 min (37°C), chilled (4°C), permeabilized, and fractionated as in Figure 4. Equal amounts of cells were used to compare conditions. TrkA was immunoprecipitated with RTA from one-fifth of P1, one-half of P2, one-half ofP3, and one-tenth of S3. Shown is a Western blot of immunoprecipitates probed with RTA followed by HRP-conjugated anti-rabbit IgG. Chemiluminescence was used for detection. The bands for gp140TrkA and gp110TrkA are noted. Bottom, TrkA immunoprecipitates (as above) were Western-blotted and probed with anti-phosphotyrosine antibody (4G10) followed by125I-labeled goat anti-mouse IgG. Data were taken directly from the PhosphorImager. The position of the tyrosine-phosphorylated 140 kDa band comigrated exactly with TrkA. Tyrosine-phosphorylated TrkA was present in P1, P2, andP3 in NGF-treated cells.
Fig. 9.
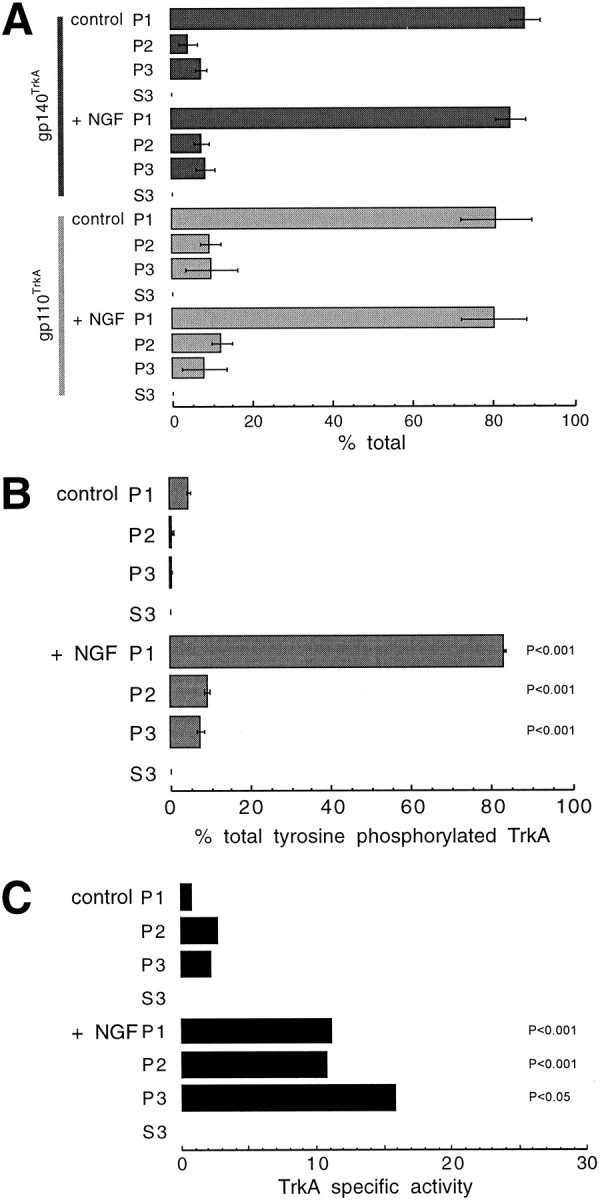
The “specific activity” of TrkA tyrosine phosphorylation increased after NGF treatment. A, Data for gp140TrkA and gp110TrkA from three experiments as in the top of Figure 8 were quantified by densitometry and plotted with error bars (±SEM). The proteins, conditions, and fractions are labeled at the left. B, Data for tyrosine-phosphorylated TrkA from three experiments as in the bottom of Figure 8 were quantified by PhosphorImaging or densitometry and plotted as in A. The values are reported as a percentage of total tyrosine-phosphorylated TrkA in NGF-treated cells.C, The specific activity of TrkA tyrosine phosphorylation was the ratio of the amount of tyrosine-phosphorylated TrkA to the amount of gp140TrkA, plotted in arbitrary units. The average specific activity was calculated using data from four individual experiments. Differences between the P2and P3 fractions within a treatment group were not significant. When comparing fractions from control and NGF-treated cells (for example, P1-control vs P1-NGF-treated), significant differences, calculated using Student’s t test, are indicated by the probability value (P).
Fig. 10.

PLC-γ1 was bound to TrkA in intracellular organelles. PC12 cells incubated at 4°C for 1 hr with NGF (1 nm; lanes 2–4,6–8, 10) or without NGF (lanes 1, 5, 9) were warmed 10 min at 37°C. Cells were chilled (4°C), permeabilized, and fractionated as in Figure 4. P1, P2′, andS2′ were lysed and immunoprecipitated with anti-PLC-γ-1 (lanes 1, 2,5, 6, 9,10) or anti-TrkA (1088; lanes 4,8). In lanes 3 and 7, TrkA-immunoprecipitated lysates were subsequently immunoprecipitated with anti-PLC-γ1. Immunoprecipitates were Western-blotted with anti-phosphotyrosine antibody (4G10; A) and then stripped and reprobed with anti-PLC-γ1 (B) and, finally, with anti-TrkA (RTA; C). Chemiluminescence was used for detection: P1 and S2′, 1 min exposure; P2′, 2 hr exposure. The positions for PLC-γ1 and TrkA are indicated. NGF treatment resulted in association of tyrosine-phosphorylated TrkA and tyrosine-phosphorylated PLC-γ1 inP1 and P2′ (see text). Although anti-PLC-γ1 brought down TrkA in P1 andP2′ after NGF treatment (A,C, lanes 2, 6), PLC-γ1 was not reproducibly found on blots of TrkA immunoprecipitates (lanes 4, 8, A,B). Because TrkA immunoprecipitation clearly brought down PLC-γ1 in both P1 and P2′, the complexes present in the TrkA immunoprecipitates may have been unstable.
To determine whether fragments of plasma membrane contaminated released intracellular organelles, we biotinylated cell surface proteins and determined whether amyloid precursor protein (APP), a relatively abundant protein (Haass et al., 1992), was detected in P2 and P3. PC12 cells were harvested, and cell surface proteins were biotinylated, as indicated above, for 20 min at 4°C. Cells were washed three times with 1 mm lysine in PBS and then permeabilized and fractionated. P1, P2, and P3 were lysed in lysis buffer #2 and immunoprecipitated with α-C7, an antibody to the C terminus of APP (Podlisny et al., 1991). Immunoprecipitates were submitted to 7.5% SDS-PAGE and blotted. [125I]streptavidin was used to probe the blots, and the PhosphorImager was used for detection.
Electron microscopy of cell fractionations. PC12 cells treated with NGF, as just described, were permeabilized and fractionated. The P1, P2, and P3 pellets were fixed in 2% glutaraldehyde in 100 mm sodium cacodylate buffer, pH 7.4, for 90 min at room temperature. Each pellet was washed sequentially in the sodium cacodylate buffer, pH 7.4, and in 50 mm veronyl acetate buffer, pH 7.4, before fixation in 1% osmium tetroxide in the veronyl acetate buffer at 0°C for 45 min. After a final wash in the same veronyl acetate buffer, the pellets were dehydrated and embedded in Epon–Araldite. Thin sections were cut and examined in a Zeiss EM 10CA microscope. Micrographs were taken at ∼20,000×. Several areas, each 20.5 μm2, were chosen for vesicle measurements.
Fractionation of internalized NGF. To quantify [125I]NGF in intracellular organelles, cells incubated with [125I]NGF (1 nm) for 1 hr were washed, warmed at 37°C for 10 min, chilled, permeabilized, and fractionated. Fractions were assayed as above for [125I]NGF. In some cases, we determined the amount of [125I]NGF associated with the cytoskeleton, which was defined as the NP-40-insoluble pellet of P1 (Vale and Shooter, 1985). For these studies, P1 was resuspended in 0.45 ml of PBS, 1 mm EGTA, 1 mm EDTA containing the protease inhibitors listed above. NP-40 (1%) was then added, and the suspension was incubated on ice for 1 hr before centrifuging for 10 min at 10,000 × g in a microcentrifuge. In some experiments, acid washing was used to measure surface-bound [125I]NGF (Bernd and Greene, 1984). To examine the distribution of [125I]NGF in intracellular organelles, P2′ was resuspended in 10% sucrose in buffer B (with inclusions) using a 25 G needle and then applied to a 10–40% (w/w) sucrose gradient with a 0.4 ml 50% sucrose pad. The gradient was centrifuged at 100,000 × g for 1 hr in a Beckman Ti SW50.1 rotor. Gradient fractions (4 drops each) were collected from the bottom of the tube. Each fraction was surveyed for radiolabeled NGF using the gamma counter.
NGF cross-linking to intracellular TrkA. Cells incubated with [125I]NGF (1 nm) in binding buffer for 1 hr at 4°C were washed and then warmed for 10 min at 37°C before permeabilization, as above. Unlabeled NGF (1 μm) was added to control for nonspecific binding and cross-linking. DSS (2 mm) was added to the permeabilized cells and released membranes, and the mixture was incubated while rotating for 30 min (4°C). The reaction was quenched with lysine (10 mm) for 10 min. P1, P2, and P3 were then prepared, as above. P1 was solubilized by extracting in 1% NP-40 in buffer B for 1 hr (4°C). After centrifuging at 1000 × g (10 min), SDS was added to bring the final concentration to 0.5%. P2 and P3 were resuspended in H2O with 0.5% SDS. S3 was brought to the same SDS concentration. All samples were boiled for 5 min, chilled (4°C), and then brought to a volume of 1 ml and a final concentration of 0.1% SDS by diluting with immunoprecipitation (IP) buffer (20 mmHEPES, pH 7.4, 0.15 m NaCl, 1% NP-40, 0.5% DOC, 1 mm EDTA). To this was added 1088 (17 μg/ml). After incubating at 4°C overnight, one-tenth of the volume of 20% protein A–Sepharose beads (Pierce) was added for 1 hr, with rotation. The beads were washed twice with IP buffer and once with water before resuspending in 7 m urea SDS-PAGE sample buffer with 20 mm DTT. Samples were heated to 65°C for 10 min and run on a 5–12% SDS-PAGE. Dried gels were exposed to XAR film for 1–3 weeks.
Immunoprecipitation and blotting. Immunoprecipitation of cell fractions was performed by dissolving P1, P2, P3, or S3 in 1 ml of lysis buffer #1 with 1 mm Na-orthovanadate. To this was added 12 μg of 1088, 12 μg of RTA, or 5 μg of anti-PLCγ1. The mixture was incubated overnight at 4°C, and one-tenth of the volume of protein A– or protein A/G–Sepharose beads was added for 2 hr at 4°C. Sepharose beads were washed three times in lysis buffer #1 and once with water, then treated with 50 μl of 7 m urea sample buffer and heated (55°C, 15–30 min). Samples were loaded on 7.5% SDS-PAGE. After transferring to nitrocellulose, blots were probed with anti-phosphotyrosine (4G10), RTA, or anti-PLCγ1. Immune complexes were detected with horseradish peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG and chemiluminescence (Amersham) or with 125I-labeled goat anti-mouse IgG. Data were quantified from multiple chemiluminescent exposures using National Institutes of Health Image or using a Molecular Dynamics PhosphorImager.
RESULTS
TrkA was internalized in response to NGF
Two studies were used to define how NGF binding regulates trafficking of TrkA receptors. In the first, TrkA at the surface of PC12 cells was marked by cross-linking to radiolabeled NGF using BS3, a membrane-impermeable cross-linker (Hartman et al., 1992). PC12 cells were incubated with [125I]NGF (1 nm) at 4°C to allow for binding at a temperature that inhibits membrane traffic. The mixture was then warmed at 37°C for 0, 5, 10, or 30 min to permit endocytosis. After chilling (4°C) and cross-linking, immunoprecipitated TrkA was analyzed by SDS-PAGE. Two major radiolabeled cross-linked species were found (Fig.1A). Each was specific for [125I]NGF, because cross-linking done in the presence of excess unlabeled NGF eliminated the bands (data not shown). One band migrated at ∼150 kDa, the position expected for a TrkA monomer–NGF monomer complex. A more slowly migrating band represents NGF bound to TrkA in an undefined oligomeric complex. There was a marked decrease in the amount of TrkA that could be cross-linked to NGF after warming. Relative to that present without warming, the level of the 150 kDa complex decreased by 15 ± 4% (SEM; n = 2) at 5 min and by 45 ± 3% (n = 2) at 30 min. Similar decreases were seen in the more slowly migrating species. We reproducibly detected a small increase in surface cross-linking between 10 and 30 min, which may be caused by receptors recycling back to the plasma membrane. These data indicate that NGF caused rapid downregulation of surface TrkA.
Fig. 1.
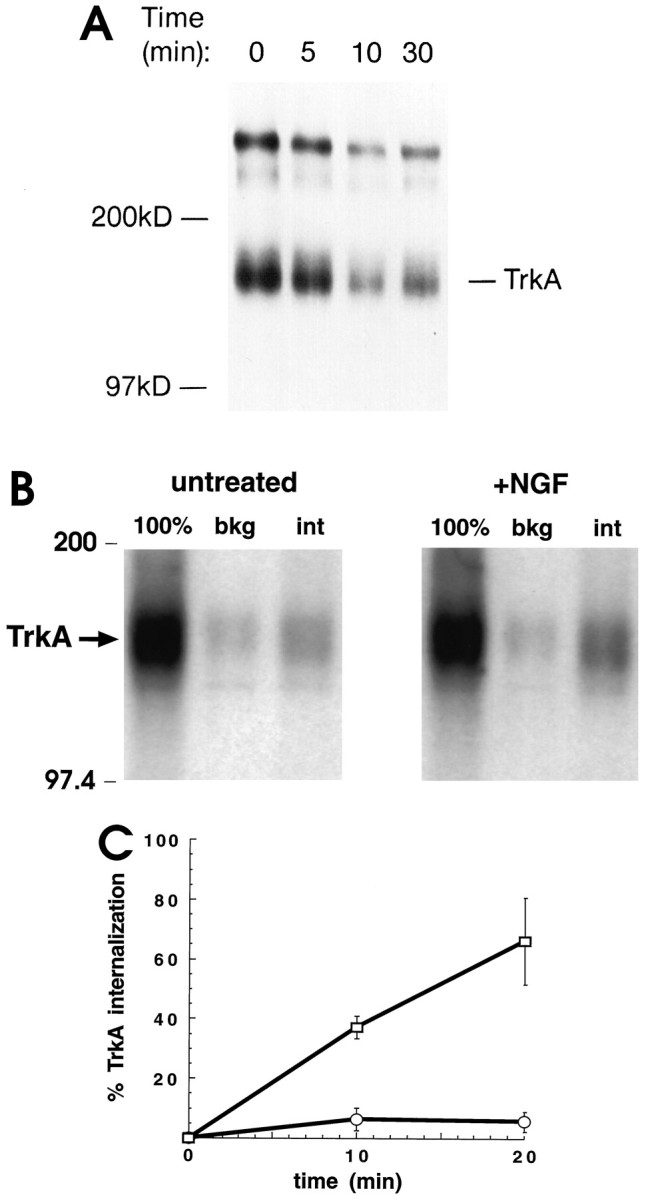
NGF induced trkA internalization.A, NGF treatment decreased cross-linking to surface TrkA receptors. PC12 cells, an equal number for each condition tested, were incubated with [125I]NGF (1 nm) at 4°C for 2 hr and then warmed to 37°C for 0, 5, 10, or 30 min. Cells were chilled, and the membrane-impermeant cross-linker BS3 was added for 30 min at 4°C. Cell lysates were immunoprecipitated with 1088, an anti-Trk C-terminal antibody, before SDS-PAGE. The dried gel was exposed to x-ray film. The positions of molecular weight markers are indicated, as is a band corresponding to a complex containing an [125I]NGF monomer cross-linked to a TrkA monomer. The more slowly migrating band marks a higher-molecular-weight complex containing [125I]NGF and TrkA. There was no [125I]NGF cross-linking in experiments in which unlabeled NGF (1 μm) was present during binding and cross-linking.B, Constitutive and NGF-induced internalization of TrkA: internalization of TrkA increased after warming in the presence of NGF (+NGF). Cells were incubated with unlabeled NGF (1 nm) for 30 min at 4°C (untreated). Control samples were handled identically except that no NGF was present. NHS-SS-biotin was added at 4°C to biotinylate cell surface proteins; cells were then either kept on ice or warmed 10 min at 37°C to allow for endocytosis. Glutathione was added to some samples to remove biotin on cell surface proteins. After samples were lysed and boiled in 0.5% SDS, TrkA was immunoprecipitated with 1088. After SDS-PAGE, proteins were transferred to nitrocellulose and blotted with [125I]streptavidin. TrkA is marked by anarrow. Biotinylated TrkA was analyzed without warming and without glutathione (100%), without warming but with glutathione (bkg), or with warming and with glutathione (int). Biotin on TrkA receptors internalized during warming was protected from glutathione reduction.C, Quantitation of TrkA internalization. Experiments were performed as in B. The signals for biotinylated TrkA were normalized for TrkA protein. The percent internalization of TrkA was computed as described in Materials and Methods. Values are mean ± SEM from three separate experiments at 10 min and two at 20 min (open circles, untreated; open squares, NGF-treated). There was a low level of constitutive internalization. NGF markedly increased TrkA internalization.
We next determined whether NGF downregulation of surface TrkA was caused by enhanced endocytosis of these receptors. Surface biotinylation has been used to examine trafficking of membrane proteins (Schmid and Carter, 1990). Using this method, constitutive and NGF-induced TrkA internalization were evaluated. Cells were incubated at 4°C either in the presence of NGF (1 nm) or in its absence. Surface proteins were then biotinylated using NHS-SS-biotin. After removing unreacted NHS-SS-biotin, cells were warmed to 37°C for 10 min to allow endocytosis, or not warmed. Samples were then chilled (4°C), and glutathione was added to release biotin on cell surface proteins (Schmid and Carter, 1990). TrkA immunoprecipitated from cell lysates was submitted to SDS-PAGE and blotting; [125I]streptavidin was used to detect biotinylated TrkA. In Figure 1B, the lanes labeled (100%) show TrkA labeling under conditions in which there was neither warming nor addition of glutathione. This corresponds to TrkA present at the surface of cells before initiation of membrane traffic. The signals were large for both NGF-treated and untreated cells. The lanes labeled bkg (for background) show the extent to which the biotin label resisted reduction with glutathione when cells were not warmed. The signals were small for both NGF-treated and untreated cells. The TrkA available for internalization was 100% − bkg. The lanes labeled int (for internalized) show the signal when cells were warmed and then treated with glutathione. Whether or not cells were treated with NGF, the amount of labeled TrkA, representing internalized receptors, was increased by warming. In the absence of NGF, 6% of surface TrkA was internalized after 10 min. This finding points to constitutive endocytosis of TrkA receptors. Remarkably, ∼37% of TrkA was internalized after 10 min warming with NGF. More than 66% of labeled TrkA was internalized after 20 min warming with NGF (Fig.1C). These data show that NGF induced extensive and rapid internalization of TrkA receptors present at the cell surface.
TrkA was present in intracellular organelles
Internalization of TrkA by NGF suggested that it would be possible to detect TrkA in the endocytic pathway. Immunofluorescence and confocal microscopy were used to localize TrkA. In the first series of experiments, PC12 cells were incubated with or without NGF (2 nm) at 37°C. Cells were then fixed, permeabilized, and incubated with sc11, an antibody to the C terminus of Trk that binds to full-length receptors. TrkA antibody binding was detected using fluoresceinated goat anti-rabbit IgG. The confocal micrographs of cells not exposed to NGF showed that TrkA staining was predominantly intracellular (Fig. 2A). Some immunopositivity was found near the nucleus; much was present as small punctate accumulations distributed throughout the cytoplasm. Surprisingly little staining was noted at the cell surface. NGF treatment induced marked changes. At early times, there was dispersion of staining such that the boundaries of cells were better defined (Fig.2B–D). There was also the appearance of densely stained punctate structures, many of which were present near the cell surface. The number of bright punctate organelles located within 0.5 μm of the cell surface was counted. After 30 sec NGF treatment, the number increased threefold from 0.08 ± 0.01 per μm of cell perimeter (n = 12 cells) in untreated cells to 0.21 ± 0.02 per μm (n = 12) in NGF-treated cells (Student’s t test, p < 0.05). At later times (Fig. 2D), there was marked redistribution of TrkA staining to the perinuclear region. In part, this staining corresponded to TrkA in lysosomes, because a lysosomal marker (GM10) colocalized with some of the TrkA staining (data not shown). The presence of TrkA immunostaining in lysosomes at 60 min suggests that endocytosed TrkA was destined for degradation, a finding consistent with earlier observations (Zhou et al., 1995). Thus, by confocal microscopy, NGF treatment markedly influenced TrkA trafficking.
Fig. 2.

NGF changed the distribution of TrkA immunostaining in PC12 cells. Cells were exposed to media at 37°C without NGF (a) or with NGF (2 nm) for 30 sec (b), 2 min (c), and 60 min (d). A Trk-specific antibody, sc11, was used to examine the distribution of TrkA. Most TrkA staining was intracellular. With NGF treatment there was an increase in bright punctate staining near the plasma membrane (e.g., small arrows inb). Note the marked increase with NGF treatment of TrkA staining in the juxtanuclear region at 60 min (large arrows in d).
To characterize further the punctate TrkA-positive organelles produced with NGF treatment, we attempted to colocalize TrkA with markers of the clathrin-coated pit endocytic pathway. Cells were first incubated with or without NGF at 37°C for 30 sec and then chilled and fixed. Clathrin vesicles were visualized with antibodies to the clathrin heavy chain (X22) and to α-adaptin (AP.6). Figure3 shows that in the absence of NGF there was a small amount of TrkA and clathrin colocalization in an area near the nucleus (Fig. 3D–F). The perinuclear colocalization of TrkA and clathrin was probably attributable to the presence of these proteins in the biosynthetic pathway. Only rarely were TrkA and clathrin colocalized near the plasma membrane. In the presence of NGF, an increased number of clathrin and TrkA-positive organelles were located near the cell surface (Fig.3A,B). Significantly, TrkA and clathrin could be colocalized in these organelles (Fig.3C,G,H). Figure 3 (A–C, G,H) demonstrates a cell in which TrkA and clathrin colocalization was particularly marked. To quantitate the change with NGF, we determined the number of puncta showing colocalization in randomly selected control and NGF-treated cells. Puncta were counted if they fell within 0.5 μm of the plasma membrane. In control cells, such puncta ranged from 0 to 2 per section; they averaged 0.68 ± 0.10 puncta/section (n = 40 cells). With NGF, the number increased nearly fivefold. It ranged from 1 to 18 per section and averaged 3.25 ± 0.56 puncta/section (n = 31). The difference between NGF-treated and control cells was highly significant (p < 0.001). TrkA could also be colocalized with α-adaptin in NGF-treated cells (data not shown). These data show that TrkA is found in endocytic vesicles produced via the clathrin-coated pit pathway.
Fig. 3.

NGF treatment resulted in TrkA and clathrin colocalization. PC12 cells were treated with NGF for 30 sec (A–C, G,H) or with vehicle alone for the same interval (D–F). After fixation and permeabilization, cells were immunostained using antibodies to Trk (sc11) and the clathrin heavy chain (X22). TrkA staining is shown ingreen; clathrin heavy chain staining is inred. In the absence of NGF, TrkA (D) and clathrin (E) staining is present diffusely in the cytosol and in the juxtanuclear region. There is little overlap in their distribution (F) except in the juxtanuclear region. In the presence of NGF, TrkA (A) and clathrin (B) staining is widely distributed in the cytosol; some TrkA staining is seen near the plasma membrane. Clathrin staining appears to be concentrated at the plasma membrane. C (at lower power) and G and H (at higher power; scale bar, 2 μm) show colocalization of TrkA and clathrin staining near the plasma membrane (arrows;yellow denotes colocalization). The organelles showing colocalization had the same distribution and size as those seen with increased frequency after NGF treatment (Fig. 2).
Large and small endocytic organelles contained NGF
To characterize further the endocytic organelles that contain TrkA, we first examined those that could be labeled with [125I]NGF. This approach was suggested by studies showing that TrkA expression confers the ability to internalize NGF on mutant PC12 cells that lack TrkA (Loeb et al., 1992). To prepare intracellular organelles, we took advantage of the fact that mechanically permeabilized cells release untethered organelles (Grimes and Kelly, 1992a,b). PC12 cells were permeabilized by passage through a Balch homogenizer (Balch and Rothman, 1985; Martin and Walent, 1989), and released membranes were harvested. PC12 cells incubated with NGF (1 nm) at 4°C for 1 hr were washed briefly to enrich for binding to slowly dissociating (i.e., high-affinity) receptors (Zhou et al., 1995) and to minimize fluid-phase endocytosis of free ligand. Cells were then warmed for 10 min, chilled (4°C), and permeabilized. Cytosol and released organelles were separated from the cell ghosts by centrifugation at 1000 × g (10 min). Organelles were then separated from cytosol using one of two alternative fractionation strategies (Fig. 4).
Electron microscopic examination of PC12 cells before and after permeabilization revealed several alterations (Fig. 5A,B). The cytoplasm of intact cells was typically dense with a normal complement of subcellular organelles, including ribosomes, endoplasmic reticulum, and mitochondria. The plasma membrane and nuclear envelope were intact. Although many of the same organelles could be identified in the permeabilized cells, they were less frequent and were dispersed in a less electron-dense cytoplasm. More significantly, the plasma membrane of permeabilized cells was interrupted (arrow). In the P2 fraction, the organelles were heterogeneous (Fig. 5C). P2 contained mitochondria, dense bodies, ribosomes, and many clear uncoated vesicles with a mean diameter of 180 ± 71 nm (SD; n = 57). In contrast, the P3 fraction contained a more homogeneous population of organelles (Fig. 5D). Most of the organelles were small vesicles of one of three types. Small, uncoated vesicles with a dense core were the most frequent; they were 86 ± 15 nm in diameter on average (n = 56). A second class consisted of clear, coated vesicles that were 63 ± 15 nm in diameter on average (n = 22). Infrequently, we found clear, uncoated vesicles that were variable in diameter (mean 94 ± 24 nm;n = 8) and small mitochondria and dense bodies.
Fig. 5.
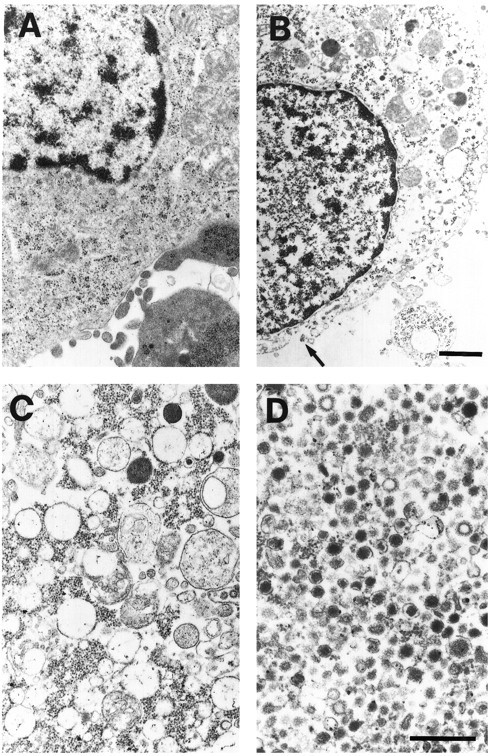
Electron micrographs of PC12 cells and vesicles released from these cells. Cell fractions were processed as indicated in Materials and Methods. A, A cell not permeabilized.B, P1: a cell after permeabilization. Note the marked decrease in the electron density of the cytoplasm. Numerous discontinuities were seen in the plasma membrane (arrow). C, Organelles in the P2 fraction. D, Organelles in the P3 fraction. Scale bars: A, B, 0.5 μm; C, D, 0.4 μm.
The electron microscopy studies suggested that this method of very gentle homogenization tears the plasma membrane and allows some intracellular organelles to leak out of the cells. Two additional observations showed that intracellular organelles could be separated from plasma membrane using this fractionation scheme. First, we carried out experiments at 4°C in which cells were incubated with [125I]NGF and washed, and the cross-linker BS3 was added before permeabilization and fractionation. Under these conditions, the NGF–TrkA complex was not detected in either P2 or P3. Second, we asked whether surface APP could be detected in P2 or P3. At 4°C, APP was biotinylated at the cell surface and cells were then permeabilized and fractionated. Surface-labeled APP in P2 was 2.9% (n = 2) of that in P1; the corresponding value for P3 was 0.6% (n = 2) and for S3 was 0.1% (n = 2). Thus, although many intracellular organelles remained with the cell ghosts (Fig. 5B), those that emerged were virtually free of plasma membrane.
We quantified the amount of internalized [125I]NGF that was released and recovered in membrane fractions. When cells were washed and then warmed 10 min at 37°C, 6.9 ± 0.6% (SEM;n = 9) of the total counts were recovered in P2 and 3.1 ± 0.4% (n = 9) were present in P3 (Student’s t test, p < 0.05). Interestingly, a substantial fraction (32 ± 4%,n = 9) of the labeled NGF that remained associated with the cell ghosts was in the detergent-insoluble fraction, which was shown to be the cytoskeleton (Vale et al., 1985). Acid washing experiments (Bernd and Greene, 1984) indicated that 49.8 ± 4.9% (n = 3) of the total bound [125I]NGF was on the cell surface under these conditions. This means that P2 + P3 together contained about one-fifth (20%) of total intracellular [125I]NGF and about half of that which was not bound to the cytoskeleton. These studies show that the fractionation scheme used allowed us to recover a substantial fraction of intracellular organelles that contain internalized NGF.
Sucrose velocity gradients were used to analyze the intracellular organelles that emerged from permeabilized cells (Fig.6). In this case, all organelles were concentrated and applied to the gradients (P2′, see Fig. 4). [125I]NGF was found in organelles that migrated near the bottom of the gradient (fractions 2–9) as well as in lighter vesicles that were heterogeneous. Thus, NGF was present in both large vesicle- and small vesicle-containing fractions.
Fig. 6.

Sucrose gradient fractionation of internalized NGF. Cells incubated with [125I]NGF (1 nm) for 1 hr at 4°C were washed, warmed 10 min, chilled (4°C), and then permeabilized. P2′ was applied to 10–40% sucrose gradients over a 50% sucrose pad and centrifuged at 100,000 × gfor 1 hr. Gradient fractions were collected from the bottom of the tube. [125I]NGF was quantified in each fraction. Data are representative of two experiments.
TrkA in intracellular organelles was bound to NGF and tyrosine-phosphorylated
Our data indicated that after binding, both NGF and TrkA were internalized. We next asked whether Trk was bound to NGF in intracellular organelles. Cells were incubated with [125I]NGF (1 nm) at 4°C, washed, warmed 10 min as above, then chilled. After permeabilization, DSS, a membrane-permeable cross-linking reagent that has been used to cross-link NGF to TrkA (Radeke and Feinstein, 1991), was added to the cell suspension before fractionation. TrkA was immunoprecipitated from cell fraction lysates before SDS-PAGE and autoradiography. A radiolabeled band that migrated at the position expected for a complex containing [125I]NGF cross-linked to TrkA was seen in the membrane fractions (P1, P2, and P3) but not the cytosol (S3, Fig. 7). P2 contained 8 ± 2% (n = 3) of the total TrkA cross-linked to [125I]NGF and P3 contained 10 ± 2% (n = 3; p > 0.05). These data roughly correlate with the amount of [125I]NGF in these fractions in the experiments above. However, more [125I]NGF was found in the P2. Either a smaller fraction of NGF was bound to TrkA in P2, or the cross-linking efficiency was lower in this fraction (or both). NGF cross-linked to TrkA was also seen when the cross-linker was added after fractionation (data not shown). These data are evidence that (1) Trk and NGF were present together in the same compartments after internalization and (2) NGF remained bound to TrkA after endocytosis.
Fig. 7.

TrkA was cross-linked to NGF in intracellular organelles. Cells were incubated with [125I]NGF (1 nm), washed, warmed 10 min (37°C), chilled (4°C), and then permeabilized and fractionated as in Figure 4. The membrane-permeable cross-linking reagent DSS was added before fractionation. One-fifth of the cell ghost membranes (P1), the entire 8000 × g pellet (P2), the entire 100,000 × g pellet (P3), and one-tenth of the 100,000 ×g supernatant (S3) were immunoprecipitated with 1088 and analyzed by SDS-PAGE and autoradiography. The arrow marks the cross-linked complex containing TrkA and [125I]NGF in P1, P2, and P3. There was no cross-linking when [125I]NGF binding was carried out in the presence of unlabeled NGF (1 μm). The amount of [125I]NGF cross-linked to TrkA was quantified by PhosphorImager.
We next sought to determine whether organelles derived from endocytosis contained activated TrkA. Cells were warmed for 10 min in the presence or absence of bound NGF and then submitted to fractionation. The phosphatase inhibitor sodium orthovanadate was added to the cell suspension during permeabilization at 4°C. The presence of TrkA was assessed by immunoprecipitation, followed by Western blotting, with RTA (Clary et al., 1994). Two proteins were identified, gp140TrkA and gp110TrkA (Fig. 8,top). The latter is a high-mannose precursor to the mature form, gp140TrkA, which acts as a cell surface receptor (Martin-Zanca et al., 1989; Zhou et al., 1995). Neither form of TrkA was detected in the 100,000 × g supernatant (S3, Fig. 8). The amount of gp140TrkA in P2 and P3 fractions together increased from 12% of the total in control cells to 16% after NGF treatment. The increase in gp140TrkA was mostly in the P2 fraction (Fig. 9A). The amount of gp110TrkA was ∼20% of total, without or with NGF (Figs. 8, 9A). The presence of gp110TrkAsuggests that up to one-fifth of the endoplasmic reticulum, or vesicles derived from it, emerge upon permeabilization.
Other vesicles that emerge from permeabilized cells include regulated and constitutive secretory vesicles (Grimes and Kelly, 1992a,b). Newly synthesized TrkA should be present in these. Receptors in the biosynthetic pathway have no direct access to NGF, and it can be assumed that they have not been activated. The presence of phosphotyrosine on TrkA is a measure of its activation (Kaplan et al., 1991; Klein et al., 1991). Thus, tyrosine-phosphorylated TrkA should comprise plasma membrane and internalized TrkA, but not that in the biosynthetic pathway. Activated TrkA was detected in P2 and P3 fractions from NGF-treated cells by blotting TrkA immunoprecipitates with an anti-phosphotyrosine antibody (Fig. 8, bottom). Total gp140TrkA was greater in P3 than P2; the reverse was true for tyrosine-phosphorylated TrkA (Figs. 8, 9). Fractions prepared from cells not exposed to NGF contained very little tyrosine-phosphorylated TrkA (Fig. 9B). In contrast, in the NGF-treated cells there was an ∼17-fold increase overall in tyrosine-phosphorylated TrkA. Seventeen percent of tyrosine-phosphorylated TrkA was recovered in the P2 and P3 fractions (Fig. 9B). Comparing these data to those from Figure1C, it appears that almost half of internalized TrkA was recovered in organelles that emerged from permeabilized cells. The data for TrkA and tyrosine-phosphorylated TrkA were used to calculate a “specific activity” for the receptor: the ratio of tyrosine-phosphorylated TrkA to total TrkA (Fig. 9C). The specific activity was about the same in intracellular organelles as in the cell ghost fraction.
TrkA in intracellular organelles was bound to PLC-γ1
Another measure of TrkA activation, one that contributes to TrkA signaling leading to differentiation, is binding of PLC-γ1 (Stephens et al., 1994). To determine whether PLC-γ1 was bound to internalized TrkA receptors, we examined immunoprecipitates from cells that were incubated with NGF, or left untreated, at 4°C and then warmed for 10 min at 37°C before chilling (4°C). After permeabilization, cells were fractionated into P1, P2′, and S2′. For these experiments, lysates were immunoprecipitated with antibodies to PLC-γ1 (Fig.10, lanes 1, 2, 5, 6, 9, and 10), with anti-Trk (1088; lanes 4 and 8), or with anti-Trk followed by anti-PLC-γ1 (lanes 3 and7). In P1, P2′, and S2′, there was a marked increase in tyrosine-phosphorylated PLC-γ1 with NGF treatment (A,lane 1 vs 2, 5 vs 6, and 9vs 10). In the membrane fraction, there was a corresponding band for PLC-γ1 protein, and the amount of this was slightly increased with NGF treatment (B, lane 1 vs2 and 5 vs 6). In the P1 and P2′ fractions, there was an additional tyrosine-phosphorylated band that migrated below PLC-γ1, whose amount was increased by NGF treatment. Evidence that this band corresponded to tyrosine-phosphorylated TrkA is as follows. (1) Tyrosine-phosphorylated TrkA was present in these fractions after NGF treatment [Figs. 8,10A (lanes 4 and 8)]. (2) The tyrosine-phosphorylated band below PLC-γ1 migrated at the position expected for TrkA (Fig. 10A, lanes 2 and 6). (3) When cells were treated with NGF, TrkA was present in the PLC-γ1 immunoprecipitates formed with antibodies to PLC-γ1 (Fig. 10C, lane 1 vs2 and 5 vs 6). (4) When lysates were first cleared by immunoprecipitating with anti-Trk, in the subsequent PLC-γ1 immunoprecipitate there was a marked decrease in the amount of tyrosine-phosphorylated PLC-γ1 (Fig.10A, lane 2 vs 3 and6 vs 7). In the P2′ fraction, the amount of PLC-γ1 was also decreased after preclearing with anti-Trk (Fig.10B, lane 6 vs 7). These data show that TrkA in intracellular organelles is associated with PLC-γ1 and are further evidence for the ability of activated TrkA in intracellular vesicles to signal.
DISCUSSION
NGF signaling must be communicated from the target of responsive neurons to their cell bodies. Our studies were aimed at exploring the hypothesis that endocytosed activated TrkA receptors serve as the retrograde message (Grimes et al., 1993). We discovered that (1) NGF induced rapid internalization of TrkA in PC12 cells, (2) NGF and TrkA were both found in intracellular vesicles, (3) NGF was bound to TrkA in these vesicles, and (4) TrkA receptors in vesicles were activated, as assessed by tyrosine phosphorylation and association with PLC-γ1. Our findings raise the possibility that it is through the creation of signaling endosomes containing activated TrkA that NGF signals retrogradely to regulate neuronal survival and differentiation.
Target-derived NGF is critical for the normal survival and differentiation of several populations of PNS and CNS neurons (Levi-Montalcini, 1987; Longo et al., 1993; Crowley et al., 1994; Li et al., 1995). Given that NGF gene expression is localized to target tissues (Longo et al., 1993), a mechanism must exist to carry the NGF signal retrogradely from the processes of neurons to their cell bodies. Studies characterizing retrograde NGF signaling showed that although NGF and the signal were similar with respect to both the time course for retrograde transport and the requirement for microtubules (Hendry et al., 1974b; Paravicini et al., 1975; Hendry and Bonyhady, 1980), NGF itself was not the signal (Heumann et al., 1984). One possibility for the NGF signal is an intracellular signaling intermediate that is distinct from the receptor. Another is an activated NGF receptor or NGF–NGF receptor complex that continues to signal after endocytosis. Whether p75NTR signals, and if so, whether it could serve as a retrograde signal for NGF, is an active area of investigation (Bothwell, 1996; Carter et al., 1996). Significantly, p75NTR is retrogradely transported in NGF-responsive CNS and PNS neurons (Taniuchi and Johnson, 1985; Johnson et al., 1987;Raivich et al., 1991; Kiss et al., 1993). However, there is no evidence that NGF induces p75 internalization and endocytosis. Indeed, in earlier studies using PC12 cells, there was little change in the amount of p75NTR at the surface of cells incubated with NGF for up to 5 hr at 37°C (Hosang and Shooter, 1987). Furthermore, Curtis et al. (1995) have shown recently that disrupting p75NTRfunction had little effect on the retrograde transport of NGF in sensory and sympathetic neurons. Thus, current data provide little support that the NGF retrograde signal is carried by p75NTR.
Internalized, activated TrkA is an attractive candidate for the NGF retrograde signal. Ehlers et al. (1995) have shown recently that NGF induced an increased accumulation of tyrosine-phosphorylated TrkA distal to a ligature on the sciatic nerve. The studies reported herein suggest that NGF-mediated induction of rapid, extensive endocytosis of TrkA in the distal processes of DRG neurons was responsible for increased retrograde transport of activated TrkA. Using two different methods to assess the disposition of surface receptors on PC12 cells, the internalization of TrkA was increased significantly after NGF addition; indeed, surface-biotinylated TrkA was decreased by >60% after 20 min. These data are consistent with earlier studies showing that NGF downregulated surface TrkA receptors (Hosang and Shooter, 1987; Zhou et al., 1995) and extends them by demonstrating that endocytosed receptors are intact, at least at the treatment times assayed. These biochemical observations were complemented by confocal microscopy studies that showed an increase in TrkA in bright, punctate structures near the cell surface after NGF addition. These organelles were evident soon after NGF treatment and persisted through 60 min. Using Sc11, an antibody to the C terminus of TrkA, there was comparatively little TrkA staining at the surface of cells. However, we know that TrkA is present at the cell surface because of our biotinylation and cross-linking studies (Zhou et al., 1995). Also, we were able to stain the surface of live cells with RTA, a TrkA extracellular domain antibody (Clary et al., 1994) (D. Hall and W. Mobley, unpublished observations), which suggests that certain epitopes are more easily detected than others at the cell surface. In some TrkA-positive organelles, staining with antibodies to TrkA colocalized with staining for the clathrin heavy chain and for α-adaptin. Colocalization of TrkA with these markers indicates that TrkA internalization is mediated, at least in part, through clathrin-coated pit-mediated endocytosis. Many of the TrkA-positive organelles of the same size and distribution that failed to stain with antibodies to clathrin and α-adaptin may also have been derived from this pathway. Taken together, our findings suggest that activation of TrkA enhances recruitment of the receptor into clathrin-coated pits. In this respect, TrkA may behave as do other receptor tyrosine kinases (Lamaze and Schmid, 1995).
Surface downregulation targets other receptor tyrosine kinases to lysosomes and is believed to serve an important role in regulating signaling (van der Geer et al., 1994). There was evidence in our studies that endocytosed TrkA was also targeted to lysosomes. In confocal microscopy, we noted marked redistribution of TrkA to the juxtanuclear region in NGF-treated cells. By 60 min, staining for TrkA at this site was quite intense. Using confocal microscopy, some juxtanuclear TrkA staining was colocalized with GM10, a lysosomal marker. This finding suggests that these TrkA molecules were destined for degradation, a view consistent with earlier studies in which NGF treatment for 60 min markedly decreased total cellular TrkA levels (Zhou et al., 1995). The significance of the perinuclear TrkA staining not present in lysosomes is uncertain. It is likely these receptors are also destined for degradation. However, earlier studies suggest an additional possibility. Using EM, Schwab (1977) detected an NGF–HRP conjugate in nonlysosomal smooth vesicles in the perinuclear cytoplasm of adrenergic neurons. Bernd and Greene (1983), using EM autoradiography to detect labeled NGF, found grains well above background at the nuclear membrane. It is possible, although not proven, that the TrkA-immunopositive organelles present in the juxtanuclear region also contained NGF. If so, they would be ideally positioned to initiate NGF signaling leading to changes in gene expression.
It is possible that activated TrkA receptors internalized at the tips of axons would be targeted to lysosomes. However, if activated TrkA is the retrograde NGF signal, it must avoid degradation in the axon. Current evidence suggests that it would. Studies on the endosomal–lysosomal pathway in neurons (Parton et al., 1992;Hollenbeck, 1993; Parton and Dotti, 1993; Nixon and Cataldo, 1995) suggest that late endosomes and lysosomes are located predominantly in the cell body and proximal dendrites. Indeed, there was no evidence for these organelles in the axons or presynaptic terminals of cultured hippocampal neurons (Parton et al., 1992). Further evidence to suggest that degradative activity in axons is limited is that a minority of endocytic organelles in axons are acidic, and those that are present have an average pH of 5.4, a value consistent with that for late endosomes (Overly et al., 1995). These observations suggest that endocytosed NGF and TrkA would remain intact during retrograde transport. Retrogradely transported NGF was shown to be intact in one study (Claude et al., 1982), and in another, using EM, an NGF–HRP conjugate was found in multivesicular bodies in cell bodies and dendrites but not in axons (Schwab, 1977). Moreover, TrkA retrogradely transported in the sciatic nerve was apparently intact (Ehlers et al., 1995). These data suggest that NGF, and the TrkA receptors endocytosed in response to NGF, resist degradation during retrograde transport.
Cell fractionation studies were used to characterize the organelles that contained NGF and TrkA. A very gentle method of homogenization was chosen in order to avoid contaminating intracellular organelles with bits of plasma membrane (Martin and Walent, 1989; Grimes and Kelly, 1992b). Based on data for the kinetics of NGF and TrkA internalization and degradation in PC12 cells (Bernd and Greene, 1983, 1984; Layer and Shooter, 1983; Hosang and Shooter, 1987; Eveleth and Bradshaw, 1988;Zhou et al., 1995), we examined organelles produced after a brief period of internalization (10 min) so as to restrict our attention to primary endocytic vesicles, endosomes, and the vesicles derived from them. NGF and TrkA were both present in the large and small vesicle fractions. Indeed, using a membrane-permeable cross-linking reagent, we found that NGF was bound to TrkA in these fractions. Given the need for receptor dimerization to induce TrkA activation (Jing et al., 1992), it is possible that persistent NGF binding to TrkA may be required to maintain TrkA kinase activation. In recent studies using 3T3 cells expressing TrkA, we have found that most NGF bound to surface receptors at pH 7.4 remains bound at pH 5.5 (J. Zhou and W. Mobley, unpublished observations), i.e., near the average pH for acidified endocytic organelles in axons (Overly et al., 1995). Thus, some NGF could remain bound to TrkA during retrograde transport. It will be important to define further the organelles that contain NGF and TrkA, those in which NGF is bound to TrkA, and those that carry activated TrkA retrogradely. In earlier studies of NGF retrograde transport in axons, NGF was found in smooth-walled tubules (45–60 nm) and in clear vesicles ranging in diameter from 50 to 150 nm (Schwab, 1977; Claude et al., 1982). Recent data (M. Grimes, E. Beattie, and W. Mobley, unpublished observations) in which in vitro reactions were used to characterize organelles that emerged from permeabilized cells suggest that P2 contains clathrin-coated vesicles and that P3 contains uncoated primary endocytic vesicles as well as transport vesicles (Grimes and Kelly, 1992b). We do not know which organelle(s) carries the NGF signal, but small transport vesicles are an attractive possibility (Grimes et al., 1993).
NGF binding to TrkA in intracellular vesicles suggested that activation of TrkA receptors persisted after endocytosis. Evidence for this was the presence of tyrosine-phosphorylated TrkA in both P2 and P3 after NGF treatment. NGF markedly increased the specific activity of tyrosine-phosphorylated TrkA in all cell fractions. TrkA signaling is communicated through the activation of signaling intermediates, including PLC-γ1. Immunoprecipitation of TrkA in intracellular organelles of NGF-treated cells showed that activated TrkA formed a complex with tyrosine-phosphorylated PLC-γ1. In preliminary studies, we have shown that tyrosine-phosphorylated SHC is also associated with activated TrkA in intracellular organelles (J. Zhou and W. Mobley, unpublished observations). These data link activated intracellular TrkA to important signaling cascades (Stephens et al., 1994) and thereby suggest strongly that internalized activated TrkA receptors are capable of signaling. Together with the data of Ehlers et al. (1995), our observations support the hypothesis that through endocytosis of activated TrkA, NGF creates signaling endosomes that convey its retrograde signal. To test this idea, studies must be done to determine whether activated TrkA in endocytic vesicles can initiate NGF signal transduction in the neuron cell body.
As second authors, J.Z. and E.B. contributed equally to this work.
Abbreviations: APP, amyloid precursor protein; BS3, bis(sulfosuccinimidyl)suberate; DSS, disuccinimidyl suberate; HRP, horseradish peroxidase; PNS, peripheral nervous system; p75NTR, low-affinity neurotrophin receptor; TrkA, receptor tyrosine kinase activated by NGF; PLC-γ1, phospholipase C-γ1; PI-3 kinase, phosphatidylinositol 3′-kinase.
M.G. was supported by the National Alliance for Research on Schizophrenia and Depression (NARSAD), the Whitehall Foundation, the Cancer Society of New Zealand, Lottery Health and Science, the National Child Health Research Foundation, and the Palmerston North Medical Research Foundation. We also acknowledge the support of the Adler Foundation (J.Z.), National Institutes of Health (NIH) Grants T32 NS07219 (E.B.), K11 AG00649 (E.C.Y.), and EY08773 (J.H.L.), and grants to W.C.M. from the March of Dimes (FY95-0625), the McGowan Charitable Trust, and NIH (RO1 NS24054). We thank Patrick Gamp and Eileen Grady for technical assistance, Drs. Steven Morris and Frances Brodsky for helpful discussions and antibodies, and Dianne Esson for secretarial assistance.
Correspondence should be addressed to Dr. William C. Mobley, Department of Neurology, M-794, University of California, San Francisco, San Francisco, CA 94143-0114.
REFERENCES
- 1.Balch WE, Rothman JE. Characterization of protein transport between successive compartments of the Golgi apparatus: asymmetric properties of donor and acceptor activities in a cell-free system. Arch Biochem Biophys. 1985;240:413–425. doi: 10.1016/0003-9861(85)90046-3. [DOI] [PubMed] [Google Scholar]
- 2.Barker PA, Shooter EM. Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to trkA on PC12 cells. Neuron. 1994;13:203–215. doi: 10.1016/0896-6273(94)90470-7. [DOI] [PubMed] [Google Scholar]
- 3.Bernd P, Greene LA. Electron microscopic radioautographic localization of iodinated nerve growth factor bound to and internalized by PC12 cells. J Neurosci. 1983;3:631–643. doi: 10.1523/JNEUROSCI.03-03-00631.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernd P, Greene LA. Association of 125I-nerve growth factor with PC12 pheochromocytoma cells: evidence for internalization via high-affinity receptors only and for long-term regulation by nerve growth factor of both high- and low-affinity receptors. J Biol Chem. 1984;259:15509–15516. [PubMed] [Google Scholar]
- 5.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 6.Bothwell M. p75NTR: a receptor after all. Science. 1996;272:506–507. doi: 10.1126/science.272.5261.506. [DOI] [PubMed] [Google Scholar]
- 7.Brodsky FM. Clathrin structure characterized with monoclonal antibodies. I. Analysis of multiple antigenic sites. J Cell Biol. 1985;101:2047–2054. doi: 10.1083/jcb.101.6.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campenot RB. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci USA. 1977;74:4516–4519. doi: 10.1073/pnas.74.10.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhäuser N, Böhm-Matthaei R, Baeuerle PA, Barde Y-A. Selective activation of NF-kappaB by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- 10.Chin DJ, Straubinger RM, Acton S, Nathke I, Brodsky FM. 100 kDa polypeptides in peripheral clathrin-coated vesicles are require for receptor-mediated endocytosis. Proc Natl Acad Sci USA. 1989;86:9289–9293. doi: 10.1073/pnas.86.23.9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clary DO, Weskamp G, Austin LR, Reichardt LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claude P, Hawrot E, Dunis DA, Campenot RB. Binding, internalization, and retrograde transport of 125I-nerve growth factor in cultured rat sympathetic neurons. J Neurosci. 1982;2:431–442. doi: 10.1523/JNEUROSCI.02-04-00431.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts MS, Armanini MP, Ling LH, MacMahon SB, Shelton DL, Levinson AD, et al Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- 14.Curtis R, Adryan KM, Stark JL. Differential role of the low affinity neurotrophin receptor (p75) in retrograde axonal transport of the neurotrophins. Neuron. 1995;14:1201–1211. doi: 10.1016/0896-6273(95)90267-8. [DOI] [PubMed] [Google Scholar]
- 15.Davies AM, Lee K-F, Jaenisch R. p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron. 1993;11:565–574. doi: 10.1016/0896-6273(93)90069-4. [DOI] [PubMed] [Google Scholar]
- 16.Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. NGF-stimulated retrograde transport of TrkA in the mammalian nervous system. J Cell Biol. 1995;130:1–8. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eveleth DD, Bradshaw RA. Internalization and cycling of nerve growth factor in PC12 cells: interconversion of type II (fast) and type I (slow) nerve growth factor receptors. Neuron. 1988;1:929–936. doi: 10.1016/0896-6273(88)90150-x. [DOI] [PubMed] [Google Scholar]
- 18.Grady EF, Garland AM, Gamp PD, Lovett M, Payan DG, Bunnett NW. Delineation of the endocytic pathway of substance P and its seven-transmembrane domain NK1 receptor. Mol Biol Cell. 1995;6:509–524. doi: 10.1091/mbc.6.5.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grimaldi KA, Hutton JC, Siddle K. Production and characterization of monoclonal antibodies to insulin secretory granule membranes. Biochem J. 1987;245:557–566. doi: 10.1042/bj2450557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimes ML, Kelly RB. Sorting of chromogranin B into immature secretory granules in pheochromocytoma (PC12) cells. Ann NY Acad Sci. 1992a;674:38–52. doi: 10.1111/j.1749-6632.1992.tb27475.x. [DOI] [PubMed] [Google Scholar]
- 21.Grimes ML, Kelly RB. Intermediates in the constitutive and regulated secretory pathways released in vitro from semi-intact cells. J Cell Biol. 1992b;117:539–549. doi: 10.1083/jcb.117.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grimes ML, Zhou J, Li Y, Holtzman D, Mobley WC. Neurotrophin signalling in the nervous system. Semin Neurosci. 1993;5:239–247. [Google Scholar]
- 23.Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992;357:500–503. doi: 10.1038/357500a0. [DOI] [PubMed] [Google Scholar]
- 24.Hantzopoulos PA, Suri C, Glass DJ, Goldfarb MP, Yancopoulos GD. The low affinity NGF receptor, p75, can collaborate with each of the trks to potentiate functional responses to the neurotrophins. Neuron. 1994;13:187–201. doi: 10.1016/0896-6273(94)90469-3. [DOI] [PubMed] [Google Scholar]
- 25.Hartman DS, McCormack M, Schubenel R, Hertel C. Multiple trkA proteins in PC12 cells bind NGF with a slow association rate. J Biol Chem. 1992;267:24516–24522. [PubMed] [Google Scholar]
- 26.Hendry IA. The effect of the retrograde axonal transport of nerve growth factor on the morphology of adrenergic neurones. Brain Res. 1977;134:213–223. doi: 10.1016/0006-8993(77)91068-x. [DOI] [PubMed] [Google Scholar]
- 27.Hendry I, Bonyhady R. Retrogradely transported nerve growth factor increases ornithine decarboxylase activity in rat superior cervical ganglia. Brain Res. 1980;200:39–45. doi: 10.1016/0006-8993(80)91092-6. [DOI] [PubMed] [Google Scholar]
- 28.Hendry IA, Stach R, Herrup K. Characteristics of the retrograde axonal transport system for nerve growth factor in the sympathetic nervous system. Brain Res. 1974a;82:117–128. doi: 10.1016/0006-8993(74)90897-x. [DOI] [PubMed] [Google Scholar]
- 29.Hendry IA, Stockel K, Thoenen H, Iversen LL. The retrograde axonal transport of nerve growth factor. Brain Res. 1974b;68:103–121. doi: 10.1016/0006-8993(74)90536-8. [DOI] [PubMed] [Google Scholar]
- 30.Heumann R, Schwab M, Merkl R, Thoenen H. Nerve growth factor-mediated induction of choline acetyltransferase in PC12 cells: evaluation of the site of action of nerve growth factor and the involvement of lysosomal degradation products of nerve growth factor. J Neurosci. 1984;4:3039–3050. doi: 10.1523/JNEUROSCI.04-12-03039.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollenbeck PJ. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol. 1993;121:305–315. doi: 10.1083/jcb.121.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holtzman DM, Kilbridge J, Li Y, Cunningham ET, Lenn NJ, Clary DO, Reichardt LF, Mobley WC. TrkA expression in the CNS: evidence for the existence of several novel NGF-responsive CNS neurons. J Neurosci. 1995;15:1567–1576. doi: 10.1523/JNEUROSCI.15-02-01567.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hosang M, Shooter EM. The internalization of nerve growth factor by high-affinity receptors on pheochromocytoma PC12 cells. EMBO J. 1987;6:1197–1202. doi: 10.1002/j.1460-2075.1987.tb02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jing S, Tapley P, Barbacid M. Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron. 1992;9:1067–1079. doi: 10.1016/0896-6273(92)90066-m. [DOI] [PubMed] [Google Scholar]
- 35.Johnson EMJ, Taniuchi M, Clark HB, Springer JE, Koh S, Tayrien MW, Loy R. Demonstration of the retrograde transport of nerve growth factor receptor in the peripheral and central nervous system. J Neurosci. 1987;7:923–929. doi: 10.1523/JNEUROSCI.07-03-00923.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 37.Kaplan DR, Stephens RM. Neurotrophin signal transduction by the Trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- 38.Kiss J, Shooter EM, Patel AJ. A low-affinity nerve growth factor receptor antibody is internalized and retrogradely transported selectively into cholinergic neurons of the rat basal forebrain. Neuroscience. 1993;57:297–305. doi: 10.1016/0306-4522(93)90063-l. [DOI] [PubMed] [Google Scholar]
- 39.Klein R, Jing SQ, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 40.Knüsel B, Rabin S, Hefti F, Kaplan D. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lamaze C, Schmid SL. Recruitment of epidermal growth factor receptors into coated pits requires their activated tyrosine kinase. J Cell Biol. 1995;129:47–54. doi: 10.1083/jcb.129.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Layer PG, Shooter EM. Binding and degradation of nerve growth factor by PC12 pheochromocytoma cells. J Biol Chem. 1983;258:3012–3018. [PubMed] [Google Scholar]
- 43.Lee K-F, Bachman K, Landis S, Jaenisch R. Dependence on p75 for innervation of some sympathetic targets. Science. 1994;263:1447–1449. doi: 10.1126/science.8128229. [DOI] [PubMed] [Google Scholar]
- 44.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1161. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Holtzman DM, Kromer LF, Kaplan DR, Chua-Couzens J, Clary DO, Knusel B, Mobley WC. Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci. 1995;15:2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loeb DM, Greene LA. Transfection with trk restores “slow” NGF binding, efficient NGF uptake, and multiple NGF responses to NGF-nonresponsive PC12 cell mutants. J Neurosci. 1993;13:2919–2929. doi: 10.1523/JNEUROSCI.13-07-02919.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loeb DM, Tsao H, Cobb MH, Greene LA. NGF and other growth factors induce an association between ERK1 and the NGF receptor, gp140protoTrk. Neuron. 1992;9:1053–1065. doi: 10.1016/0896-6273(92)90065-l. [DOI] [PubMed] [Google Scholar]
- 48.Longo FM, Holtzman DM, Grimes ML, Mobley WC. Nerve growth factor: actions in the peripheral and central nervous systems. In: Fallon J, Loughlin S, editors. Neurotrophic factors. Academic; New York: 1993. pp. 209–256. [Google Scholar]
- 49.Lucidi-Phillipi CA, Clary DO, Reichardt LF, Gage FH. TrkA activation is sufficient to rescue axotomized cholinergic neurons. Neuron. 1996;16:653–663. doi: 10.1016/s0896-6273(00)80084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahadeo D, Kaplan L, Chao MV, Hempstead BL. High affinity nerve growth factor binding displays a faster rate of association than p140trk binding. J Biol Chem. 1994;269:6884–6891. [PubMed] [Google Scholar]
- 51.Martin TF, Walent JH. A new method for cell permeabilization reveals a cytosolic protein requirement for Ca2+-activated secretion in GH3 pituitary cells. J Biol Chem. 1989;264:10299–10308. [PubMed] [Google Scholar]
- 52.Martin-Zanca D, Oskam R, Mitra G, Copeland T, Barbacid M. Molecular and biochemical characterization of the human trk proto-oncogene. Mol Cell Biol. 1989;9:24–33. doi: 10.1128/mcb.9.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meakin SO, Shooter EM. Tyrosine kinase activity coupled to the high-affinity nerve growth factor-receptor complex. Proc Natl Acad Sci USA. 1991;88:5862–5866. doi: 10.1073/pnas.88.13.5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meakin SO, Shooter EM. The nerve growth factor family of receptors. Trends Neurosci. 1992;15:323–331. doi: 10.1016/0166-2236(92)90047-c. [DOI] [PubMed] [Google Scholar]
- 55.Mobley WC, Rutkowski JL, Tennekoon GI, Gemski J, Buchanan K, Johnston MV. Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Mol Brain Res. 1986;387:53–62. doi: 10.1016/0169-328x(86)90020-3. [DOI] [PubMed] [Google Scholar]
- 56.Nixon RA, Cataldo AM. The endosomal-lysosomal system of neurons: new roles. Trends Neurosci. 1995;18:489–496. doi: 10.1016/0166-2236(95)92772-i. [DOI] [PubMed] [Google Scholar]
- 57.Overly CC, Lee K-D, Berthiaume E, Hollenbeck PJ. Quantitative measurement of intraorganelle pH in the endosomal-lysosomal pathway in neurons by using ratiometric imaging with pyranine. Proc Natl Acad Sci USA. 1995;92:3156–3160. doi: 10.1073/pnas.92.8.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paravicini U, Stoeckel K, Thoenen H. Biological importance of retrograde axonal transport of nerve growth factor in adrenergic neurons. Brain Res. 1975;84:279–291. doi: 10.1016/0006-8993(75)90982-8. [DOI] [PubMed] [Google Scholar]
- 59.Parton RG, Dotti CG. Cell biology of neuronal endocytosis. J Neurosci Res. 1993;36:1–9. doi: 10.1002/jnr.490360102. [DOI] [PubMed] [Google Scholar]
- 60.Parton RG, Simons K, Dotti CG. Axonal and dendritic endocytic pathways in cultured neurons. J Cell Biol. 1992;119:123–137. doi: 10.1083/jcb.119.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Podlisny MB, Tolan DR, Selkoe DJ. Homology of the amyloid beta protein precursor in monkey and human supports a primate model for beta amyloidosis in Alzheimer’s disease. Am J Pathol. 1991;138:1423–1435. [PMC free article] [PubMed] [Google Scholar]
- 62.Radeke MJ, Feinstein SC. Analytical purification of the slow, high affinity NGF receptor: identification of a novel 135 kd polypeptide. Neuron. 1991;7:141–150. doi: 10.1016/0896-6273(91)90082-b. [DOI] [PubMed] [Google Scholar]
- 63.Raivich G, Hellweg R, Kreutzberg GW. NGF receptor-mediated reduction in axonal NGF uptake and retrograde transport following sciatic nerve injury and during regeneration. Neuron. 1991;7:151–164. doi: 10.1016/0896-6273(91)90083-c. [DOI] [PubMed] [Google Scholar]
- 64.Schmid SL, Carter LL. ATP is required for receptor-mediated endocytosis in intact cells. J Cell Biol. 1990;111:2307–2318. doi: 10.1083/jcb.111.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwab ME. Ultrastructural localization of a nerve growth factor-horseradish peroxidase (NGF-HRP) coupling product after retrograde axonal transport in adrenergic neurons. Brain Res. 1977;130:190–196. doi: 10.1016/0006-8993(77)90857-5. [DOI] [PubMed] [Google Scholar]
- 66.Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–249. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- 67.Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, Kaplan DR. Trk receptors use redundant signal transduction pathways involving SHC and PLC-γ1 to mediate NGF responses. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- 68.Taniuchi M, Johnson EMJ. Characterization of the binding properties and retrograde axonal transport of a monoclonal antibody directed against the rat nerve growth factor receptor. J Cell Biol. 1985;101:1100–1106. doi: 10.1083/jcb.101.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tischler AS, Greene LA. Nerve growth factor-induced process formation by cultured rat pheochromocytoma cells. Nature. 1975;258:341–342. doi: 10.1038/258341a0. [DOI] [PubMed] [Google Scholar]
- 70.Vale RD, Shooter EM. Assaying binding of nerve growth factor to cell surface receptors. Methods Enzymol. 1985;109:21–39. doi: 10.1016/0076-6879(85)09073-5. [DOI] [PubMed] [Google Scholar]
- 71.Vale RD, Ignatius MJ, Shooter EM. Association of nerve growth factor receptors with the Triton X-100 cytoskeleton of PC12 cells. J Neurosci. 1985;5:2762–2770. doi: 10.1523/JNEUROSCI.05-10-02762.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van der Geer P, Hunter T, Lindberg RA. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol. 1994;10:251–337. doi: 10.1146/annurev.cb.10.110194.001343. [DOI] [PubMed] [Google Scholar]
- 73.Verdi JM, Birren SJ, Ibanez CF, Persson H, Kaplan DR, Benedetti M, Chao MV, Anderson DJ. p75LNGFR regulates trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron. 1994;12:733–745. doi: 10.1016/0896-6273(94)90327-1. [DOI] [PubMed] [Google Scholar]
- 74.Yuen EC, Mobley WC. Therapeutic applications of neurotrophic factors in disorders of motor neurons and peripheral nerves. Mol Med Today. 1995;1:278–286. doi: 10.1016/s1357-4310(95)91189-8. [DOI] [PubMed] [Google Scholar]
- 75.Zhou J, Valletta JS, Grimes ML, Mobley WC. Multiple levels for regulation of TrkA in PC12 cells by nerve growth factor. J Neurochem. 1995;65:1146–1156. doi: 10.1046/j.1471-4159.1995.65031146.x. [DOI] [PubMed] [Google Scholar]