

Abstract
We have asked whether p75NTR may play a role in neuronal apoptosis by producing transgenic mice that express the p75NTR intracellular domain within peripheral and central neurons. These animals showed profound reductions in numbers of sympathetic and peripheral sensory neurons as well as cell loss in the neocortex, where there is normally little or no p75NTR expression. Developmental loss of facial motor neurons was not observed, but induced expression of the p75NTR intracellular domain within adult animals led to increased motor neuron death after axotomy. Biochemical analyses suggest that these effects were not attributable to a p75NTR-dependent reduction in trk activation but instead indicate that the p75NTR intracellular domain may act as a constitutive activator of signaling cascades that regulate apoptosis in both peripheral and central neurons.
Keywords: nerve growth factor, sympathetic neuron, tumor necrosis factor, cell death, NF-kB, jun kinase
Neurotrophins play critical roles in the survival and maintenance of specific neuronal populations during development and into adulthood. Their effects are mediated by trk receptors, which are classical tyrosine kinase receptors activated in response to neurotrophin binding, and by the p75NTR, a member of the superfamily of tumor necrosis factor (TNF) receptor-related molecules. Biochemical and genetic analyses have shown that the stereotypical survival and differentiative responses mediated by neurotrophins are attributable to activation of the trk receptors (for review, see Klein, 1994; Greene and Kaplan, 1995).
The physiological role played by the p75NTR is still being determined. One hypothesized function that has gained general acceptance is that p75NTR acts as an accessory receptor for trkA, increasing the ability of trkA to bind and respond to limiting levels of nerve growth factor (NGF). Biochemical data show that trkA-expressing cells become increasingly responsive to NGF when transfected with p75NTR, that numbers of high-affinity NGF binding sites are increased in membranes in which both receptors are coexpressed, and that p75NTR-specific antibodies reduce NGF-mediated activation of trkA (Hempstead et al., 1991; Barker and Shooter, 1994;Verdi et al., 1994). p75NTR−/− mice show reductions in NGF-mediated survival (Davies et al., 1993), a trkA-dependent response, consistent with the hypothesis that p75NTR plays some role in enhancing trkA activation. However, p75NTR is also capable of autonomous activation of signal transduction cascades. In p75NTR-expressing fibroblasts and in PC12 cells, p75NTR ligation results in increased sphingomyelinase activity and accumulation of the potent lipid second messenger, ceramide (Dobrowsky et al., 1994, 1995). NGF-dependent activation of the NF-kB transcriptional complex occurs in both cultured Schwann cells and in L929 fibroblasts transfected with p75NTR (Carter et al., 1996), and NGF-dependent activation of jun kinase occurs within cultured rat oligodendrocytes that express p75NTR but not trkA (Casaccia-Bonnefil et al., 1996). Both ceramide accumulation and jnk activation are correlated with apoptotic stimuli in a number of systems, and several recent results suggest that p75NTR may play a role in regulating cellular apoptosis. For example, sensory neurons that normally apoptose very rapidly when deprived of neurotrophin show reduced rates of neurotrophin withdrawal-induced cell death if p75NTR levels are reduced (Barrett and Bartlett, 1994; K.-F. Lee, personal communication). Cell death within the developing avian retina is reduced by antibodies that block NGF binding to p75NTR (Frade et al., 1996), and cultured oligodendrocytes that express p75NTR undergo increased apoptosis in response to NGF, an effect that can be reduced by using p75NTR-blocking antibodies (Casaccia-Bonnefil et al., 1996). Thus, the available data suggest that NGF working through p75NTR can have a positive or a negative influence on cell survival and that the nature of this effect may depend on the cellular coexpression or coactivation of trkA.
Despite recent advances in this area, the consequences of p75NTR signaling within neurons remain virtually unknown. This is partially because of the paucity of neuronal systems in which activation of p75NTR signaling can be studied readily and reliably. To address this, we have exploited findings made with truncated intracellular domains of TNF-R1 and fas, members of the TNF receptor superfamily that show weak intracellular homologies to p75NTR. Truncated TNF-R1 produces cellular apoptosis when overexpressed, and the fas intracellular domain can potentiate the TNF-R1 response (Boldin et al., 1995a), both apparently by acting as constitutive activators of their signaling cascades. Reasoning that a similar truncated form of p75NTR might activate signaling pathways within neurons, we created transgenic mice in which the intracellular domain of p75NTR is expressed within peripheral and central neurons in the hopes of revealing neuronal effects of p75NTR signaling. Our results indicate that the p75NTR does, indeed, act as a constitutive signaling activator; expression of p75NTR results in cell death both in neurons that normally express p75NTRas well as in neurons that do not normally express the receptor. Expression of the intracellular domain after injury results in atypical motor neuron death in adult animals, demonstrating that the effects of p75NTR are not limited to a particular developmental time span. Together, these results demonstrate that p75NTR is capable of activating signaling cascades that regulate neuronal apoptosis.
MATERIALS AND METHODS
Creation of transgenic animals, genotyping, and breeding.A Tα1 minigene cloning cassette was constructed in which the 1.1 kb Tα1 α-tubulin promoter (Gloster et al., 1994) was separated from SV40 intron and polyadenylation sites by a short polylinker. For the Tα1:intracellular domain (ICD) construct, a cDNA encoding an initiator methionine, followed by amino acids 276–425 of the rat p75NTR (Radeke et al., 1987), was subcloned into the polylinker. Completed minigenes were purified free of vector sequence and microinjected into pronuclei to produce founder transgenic animals at the Canadian NeuroScience Network Transgenic Core Facility (McGill University). Genotyping was performed on tail (postnatal animals) or hindlimb (embryos) DNA by Southern blotting with a fragment of the Tα1 α-tubulin promoter sequence or by PCR with primers derived from the Tα1 α-tubulin promoter and p75NTR sequence. Seven lines of Tα1:ICD mice were identified; four of these were characterized to a moderate degree, and two were examined in detail (4163 Tα1:ICD and 4173 Tα1:ICD). For some studies Tα1:ICD lines were crossed to mice carrying a separate transgene in which the Tα1 α-tubulin promoter drives expression of nuclear LacZ (Gloster et al., 1994; Bamji and Miller, 1996). The resultant double transgenic lines were designated 4163 Tα1:ICD × Tα1:nlacZ and 4173 Tα1:ICD × Tα1:nlacZ.
Creation of stable PC12 sublines. pRC-CMV (Invitrogen, San Diego, CA) driving expression of the truncated intracellular domain of rat p75NTR described above was transfected into PC12 cells, using Lipofectamine (Life Technologies, Gaithersburg, MD), and stable clones were selected in 400 μg/ml G418. Sixteen clones were analyzed for expression of the p75NTR-ICD by immunoblot, as described below, and six lines showing expression were retained. The results shown in Figure 7 were repeated in two separate clonal lines.
Fig. 7.

Expression of the p75NTR-ICD does not affect trk receptor levels or autophosphorylation. A, B, Stable PC12 cells lines created to overexpress the p75NTR-ICD were analyzed for trkA autophosphorylation. A, p75NTR-ICD was detected by immunoblot in these lines, using αp1 directed against the p75NTRintracellular domain. B, Effects of the p75NTR-ICD on NGF-induced trkA autophosphorylation were determined in stable PC12 cell lines subjected to a 5 min treatment with either vehicle or with 4, 20, or 100 ng/ml NGF. TrkA was immunoprecipitated with anti-pan-trk 203 and subsequently was analyzed for phosphotyrosine content by immunoblot, as described in Materials and Methods. C–F, Trk receptor levels and endogenous trk tyrosine phosphorylation are similar in cortices (C, D) of neonatal animals of line 4173 and their control littermates. Cortical lysates were immunoprecipitated with pan-trk antibody 203 and analyzed on immunoblots with anti-phosphotyrosine antibody 4G10 (C). The same blots subsequently were reprobed with trkBout, which is specific for trkB (D). No difference in trk activation or levels was observed between control and transgenic animals. E, F, Trk receptor levels and endogenous trk tyrosine phosphorylation are similar in the cortex of adult animals of line 4173 and their control littermates. E, Total trk receptors immunoprecipitated from adult cortex were analyzed for phosphotyrosine content by 4G10 immunoblot. F, Anti-pan-trk immunoprecipitates from adult animals were immunoblotted to detect total trk receptor levels, using anti-pan-trk 203. G, Lysates of neonatal cortex from line 4173 and control animals were analyzed for levels of phosphotyrosine-containing proteins by immunoblot with 4G10. The arrow in Aindicates the p75NTR-ICD; the arrowsin B–F indicate trk receptors. Molecular weight standards are indicated on the left side of each panel.
Animals and surgical procedures. For biochemistry, animals were anesthetized with sodium pentobarbital (35 mg/kg), and tissues were removed immediately and processed for immunoblots. For morphometric analyses, animals were anesthetized with sodium pentobarbital (35 mg/kg) for 30 min, and ganglia were removed and immersion-fixed in 4% paraformaldehyde in phosphate buffer (PB) for 1 hr to overnight at 4°C (for morphometric analysis of peripheral ganglia) or in 1.6% glutaraldehyde in 0.1 m PB, pH 7.3 (for electron microscopy). For morphometric analysis of the cortex, animals were perfused transcardially with 4% paraformaldehyde in PB. Subsequent to transcardial perfusion, the brains were removed and post-fixed in 4% paraformaldehyde in PB. The brains subsequently were cryoprotected in graded sucrose solutions, sectioned on the cryostat, and stained with cresyl violet.
For facial nerve lesion studies, animals were anesthetized with Metophane, and the main branch of the facial nerve was resected as it exited the stylomastoid foramen, taking care not to injure the adjacent blood vessels. Seven days after axotomy, animals were anesthetized with sodium pentobarbital and transcardially perfused with 4% paraformaldehyde. Brains were cryoprotected in graded sucrose solutions and sectioned on the cryostat. In total, three control and transgenic animals from each line were analyzed.
Histological and morphometric analyses. Embryos were prepared for β-galactosidase staining essentially as described (Gloster et al., 1994). In brief, embryos were fixed for 60 min at 4°C in freshly prepared 4% paraformaldehyde in 0.1 mNaH2PO4, pH 7.3, and then rinsed three times in 0.1 m NaH2PO4, pH 7.3, 2 mm MgCl2, 0.01% sodium deoxycholate, and 0.02% Nonidet P40. The staining reaction was performed in rinse buffer supplemented with 1 mg/ml X-gal, 5 mm K3Fe(CN)6, and 5 mm K4FE(CN)6, typically for 3–5 hr at 37°C. Embryos were post-fixed up to 48 hr in 4% paraformaldehyde. For sectioning, tissues were cryopreserved and cryosectioned. For terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) analysis, embryos first were stained in X-gal, post-fixed for 24 hr in 4% paraformaldehyde, cryosectioned, and then TUNEL-labeled by an Apoptag kit (Oncor, Gaithersburg, MD) as per the manufacturer’s instructions.
Morphometric analyses of motor and sensory neurons were performed with a computer-based image analysis system that prevents double measurement of profiles (Biocom, Paris, France). For counts of facial motor neurons, cryoprotected brains were frozen and mounted for cryosectioning, and 16 μm coronal sections were collected through the extent of the facial nuclei. Slides subsequently were stained with cresyl violet to visualize nuclei, and all of the neuronal profiles with a distinct nucleolus were counted on every fifth section. Quantitative comparisons were made, in all cases, between injured facial motor neurons on one side and the contralateral, uninjured motor neurons on the other side. Similar sections through the facial nucleus were analyzed for neuronal size. For determination of sensory neuron number, L3, L4, and L5 dorsal root ganglia (DRG) were serial-sectioned at 7 μm, and neurons containing a distinct nucleolus were counted in every fourth section, as per Coggeshall (Coggeshall, 1992; Coggeshall and Lekan, 1996). This approach does not correct for split nucleoli. For comparisons of cortical neuron number, cresyl violet-stained coronal sections at the appropriate level were photographed, and the total number of neuronal profiles was counted in 530-μm-wide strips that extended from pia to corpus collosum.
For electron microscopy of the dorsal cutaneous nerve (DCN), the DCN was dissected from adult animals, immersion-fixed overnight in 1.6% glutaraldehyde in 0.1 m PB, pH 7.3, post-fixed in 1% OsO4 in sodium cacodylate, and dehydrated in a series of ascending ethanol gradients (70, 85, 95, and 100%). Samples were cleared in acetone, infiltrated, and embedded in spurr resin. Sections were collected on Formvar-coated 50 mesh copper grids, stained with lead citrate and uranyl acetate, and examined and photographed on a Hitachi H7100 transmission electron microscope. Myelinated axons were identified by their darkly stained sheaths, whereas unmyelinated axons were identified by their light appearance relative to the surrounding Schwann cell cytoplasm. These cross sections of the DCN were photographed and montaged, and the total number of myelinated and unmyelinated axons was counted. Statistical results were expressed as mean value ± SEM and were tested for significance by the one-tailed Student’s t test for paired differences.
Immunoblotting, immunoprecipitation, jnk assays, and NF-kB assays. For immunoblotting for the p75NTR-ICD, whole brains from P20 animals were homogenized in 20 mmTris, pH 8.0, 154 mm NaCl, 10% glycerol, 1% Nonidet P-40, 1 μg/ml leupeptin, 100 μm phenylmethylsulfonyl fluoride, 5 mm phenanthroline, and 1 mmorthovanadate in a polytron, spun for 30 min at 12,000 rpm in a Beckman JA-14 rotor to remove insoluble material, and then assayed for protein content by the BCA assay (Pierce, Rockford, IL). Soluble brain extract (20 μg) from NP40 lysates was separated on 12% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with αP1, a rabbit polyclonal antibody directed against the p75NTR intracellular domain (Barker et al., 1994). So that trk receptors in brain tissue could be characterized, trk receptors were immunoprecipitated from lysates prepared from neonatal or adult brains with the pan-trk antibody 203 (Hempstead et al., 1992), separated by 7.5% SDS-PAGE, and then immunoblotted either with 4G10, an anti-phosphotyrosine antibody, or with pan-trk 203, TrkBout (Kaplan et al., 1993) or TrkCin (Belliveau et al., 1997) to detect total trk, trkB, or trkC, respectively. For analysis of whole-cell lysate tyrosine phosphorylation, lysates of neural tissue were separated by 7.5% SDS-PAGE and immunoblotted with 4G10.
NF-kB and jun kinase activity assays were performed on nuclear and cytoplasmic fractions, respectively, of embryonic day 16 (E16), E17, and E18 transgenic and wild-type littermates. NF-kB levels were determined by electrophoretic mobility shift assays, using HIV-LTR as a probe, essentially as described (Singh and Aggarwal, 1995). Jun kinase assays were performed by using a GST-jun fusion protein encoding amino acids 1–91 of human c-jun, as described in Westwick and Brenner (1995).
RESULTS
Recent studies suggest a role for p75NTR in the regulation of cellular apoptosis (Barrett and Bartlett, 1994;Casaccia-Bonnefil et al., 1996). To determine whether p75NTR is capable of promoting neuronal apoptosis and to begin to examine molecular mechanisms underlying such effects, we expressed the cytoplasmic domain of the p75NTR in neurons of transgenic mice, using a minigene composed of the 1.1 kb Tα1 α-tubulin promoter, an open reading frame encoding the intracellular domain of rat p75NTR (amino acids 276–425), and an SV40 intron and polyadenylation signal (Fig.1A). The Tα1 α-tubulin promoter produces robust expression concomitant with or shortly after neuronal terminal mitosis and is maximally active during periods of process extension (Gloster et al., 1994). Tα1-driven expression decreases to lower levels in the mature nervous system (Bamji and Miller, 1996) and is reinduced after axonal injury (Gloster et al., 1994; Wu et al., 1997).
Fig. 1.

Expression of the Tα1:ICD in the brains of transgenic mice. A, A Tα1 minigene cloning cassette was constructed in which the 1.1 kb Tα1 α-tubulin promoter was separated from an SV40 intron and polyadenylation sites by a cDNA encoding amino acids 276–425 of the rat p75NTR. The resultant minigene was purified free from vector sequence and microinjected to produce founder transgenic animals at the Canadian NeuroScience Network Transgenic Core Facility (McGill University).B, Expression of the p75NTR-ICD in the brains of various lines of Tα1:p75NTR-ICD mice detected by immunoblot. The position of the p75NTR-ICD is indicated by an arrow(right), and molecular weight standards are indicated (left).
Genotyping revealed that Tα1:ICD animals were under-represented in litters, suggesting some embryonic lethality of the transgenic animals. Nonetheless, many animals survived to term and developed into fertile adults. Immunoblot analysis showed p75NTR-ICD expression in neonatal brains of various Tα1:p75NTR-ICD lines, with levels of expression varying considerably (Fig. 1B). Transgene-positive animals from these lines exhibited a phenotype indicative of nervous system deficits, including a general lack of coordination. By the third postnatal week Tα1:ICD mice exhibited ptosis, and several animals displayed self-mutilation, consistent with defects in sympathetic and sensory innervation, respectively. To examine the cellular basis for these defects, we chose the 4163 and 4173 Tα1:p75NTR-ICD lines for detailed analysis.
p75NTR-ICD mice have deficits in sympathetic and sensory neurons
Because ptosis indicates a deficit in sympathetic innervation, we first examined the sympathetic superior cervical ganglia (SCG) from transgenic and nontransgenic littermates. To characterize the sympathetic deficit, we mated Tα1:ICD mice with homozygous Tα1:nlacZ mice in which a β-galactosidase marker gene is expressed pan-neuronally (Gloster et al., 1994; Bamji and Miller, 1996). Figure2A shows SCG from p75NTR-ICD-positive and negative littermates that were removed at postnatal day 1 (P1), which precedes the period of naturally occurring cell death in the murine SCG (Crowley et al., 1994). The SCG from p75NTR-ICD-expressing animals were greatly reduced in size, and X-gal staining indicated that these SCG contained many fewer β-galactosidase positive nuclei; in several Tα1:p75NTR-ICD animals, distinct SCG could not be identified for dissection. To confirm this apparent neuronal loss within the SCG, we dissected SCG from adult Tα1:ICD animals and sectioned and stained them with cresyl violet. The transgenic adult SCG were greatly reduced in size, as compared with their wild-type counterparts, because of a dramatic loss of neurons from these ganglia (Fig. 2B,C).
Fig. 2.
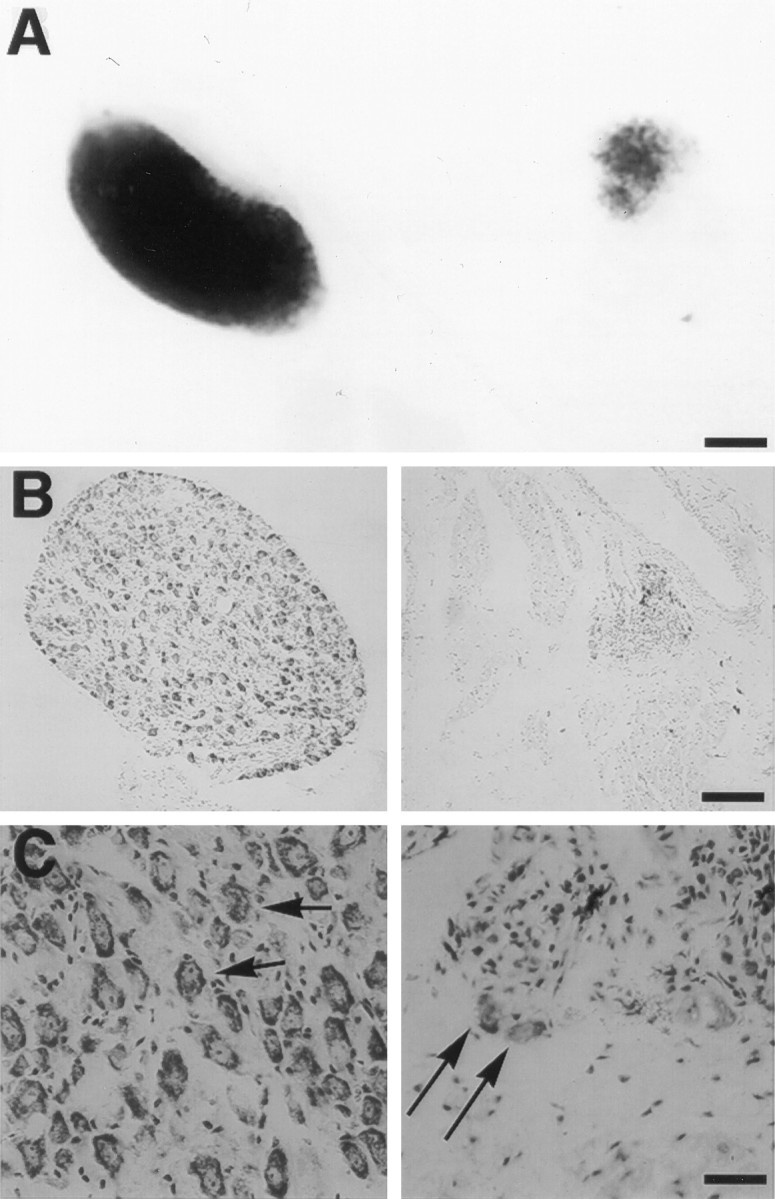
Expression of the p75NTR-ICD in developing neurons leads to loss of sympathetic neurons.A, Sympathetic superior cervical ganglion (SCG) from a postnatal day 1 Tα1:nlacZ control mouse (left) and from its Tα1:ICD × Tα1:nlacZ littermate (line 4173,right) were stained with X-gal to visualize Tα1:nlacZ-expressing neurons. Scale bar, 167 μm. B, Photomicrograph of SCG sections from an adult control (left) or from a line 4173 Tα1:ICD mouse (right) stained with cresyl violet. Scale bar, 84 μm.C, Higher magnification of sections of sympathetic cervical ganglia from a wild-type (left) or from a 4173 Tα1:ICD mouse (right). Arrows inC indicate sympathetic neurons. Scale bar, 21 μm.
The Tα1 α-tubulin promoter becomes active concomitant with or immediately after terminal mitosis, suggesting that neuronal loss in Tα1:ICD mice reflects a deficit in neuronal survival rather than defects in the developmental progression of neuronal precursors to postmitotic neurons. To confirm this, we examined the development of sensory neurons of the DRG in transgenic versus control mice. DRG neurons are normally born at E10, and detectable Tα1 α-tubulin promoter activity is first observed within a subset of the developing DRG by E10.5 and within all DRG neurons by E11.5 (A. Gloster, H. El-Bizri, S. Bamji, and F. D. Miller, unpublished data). At E11.5, there were no apparent differences in the pattern or intensity of X-gal staining of line 4173 Tα1:ICD × Tα1:nlacZ DRGs relative to their control Tα1:nlacZ littermates (data not shown). In contrast, at E13.5, the DRGs of Tα1:ICD × Tα1:nlacZ were reduced considerably in size, as indicated by X-gal staining (Fig.3A), indicating a loss of neurons within days of transgene induction. To confirm that this decrease in X-gal staining corresponds to a loss of sensory neurons, we determined the number of neurons in the L3, L4, and L5 DRG of control versus line 4173 Tα1:ICD animals (Fig. 3B). To perform this analysis, we serially sectioned L3, L4, and L5 DRGs at 7 μm, and we counted neurons containing a distinct nucleolus on every fourth section. This analysis demonstrated a loss of 39% of the DRG neurons within 4173 Tα1:ICD animals relative to controls (Tα1:ICD = 6417 ± 1359; controls = 10497 ± 306; n= 3 animals each). Thus, the decrease in X-gal staining in the DRG of Tα1:ICD embryos corresponds to a decrease in sensory neuron number.
Fig. 3.

Peripheral sensory neurons are lost in p75NTR-ICD transgenic mice. A, Whole-mount E13.5 embryos derived from a Tα1:ICD × Tα1:nLacZ cross were stained with X-gal. Arrows indicate DRG within a control embryo (left) and within an embryo carrying the Tα1:ICD transgene (right). This example shows a severely affected animal. Scale bar, 400 μm.B, Counts of neuronal profiles in L3, L4, and L5 DRG of adult wild-type or line 4173 Tα1:ICD animals revealed a significant loss of sensory neurons (*p < 0.05).
To define precisely the population of sensory neurons lost in the adult Tα1:ICD mice, we characterized the distribution of axons within cross sections of the DCN of adult animals of lines 4163 and 4173. The DCN was chosen for these analyses because it is composed almost exclusively of DRG-derived sensory axons (supplying hairy skin of the back) and because shifts in size distributions of axons present within this nerve accurately reflect neuronal losses within the parental DRG. Electron micrographs of the DCN revealed fewer unmyelinated axons within the transgenic versus wild-type DCN (Fig.4A–D); counts of axonal profiles revealed that ∼50% of the unmyelinated axons were lost in the p75NTR-ICD-expressing animals (Fig.4E: wt, 1644 ± 292; line 4173, 656 ± 147; line 4163, 830 ± 246, p < 0.05;n = 4 animals each). The numbers of myelinated axons did not differ significantly between transgenic and wild-type DCN, although there was a nonsignificant trend toward fewer myelinated axons in the Tα1:ICD animals (wt, 287 ± 60; line 4173, 167 ± 34; line 4163, 188 ± 32, p > 0.05;n = 4 animals each). The small-caliber unmyelinated axons are derived from the trkA-positive population of sensory neurons (Carroll et al., 1992; Ruit et al., 1992), suggesting a selective loss of NGF-responsive sensory neurons in the Tα1:ICD mice.
Fig. 4.
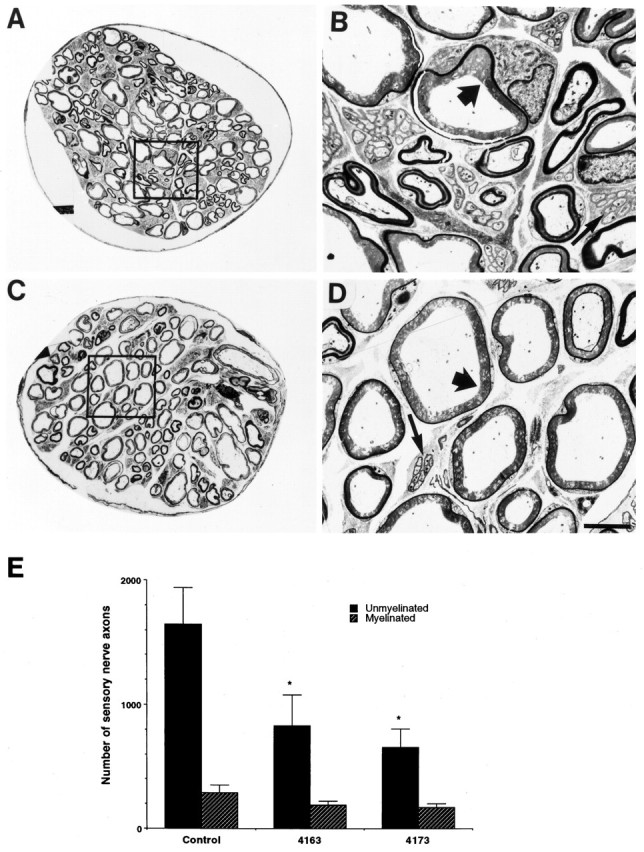
Expression of the p75NTR-ICD leads to loss of unmyelinated sensory axons of the dorsal cutaneous nerve. A–D, Cross sections of the dorsal cutaneous nerve of adult control mice (A, B) and adult Tα1:ICD mice of line 4163 (C, D), as visualized by electron microscopy. B, D, Higher magnifications of the boxes outlined in A andC, respectively. Note the selective loss of the smaller fiber unmyelinated axons (thin arrows) relative to the large fiber myelinated sensory fibers (thick arrows). Scale bar, 6 μm. E, Quantitation of axonal loss demonstrates a selective loss of small unmyelinated sensory fibers in the DCN of Tα1:ICD mice of lines 4163 and 4173 relative to controls (*p < 0.05). The myelinated population measured in this analysis includes both the small- and large-caliber myelinated sensory axons.
p75NTR-ICD expression results in neuronal loss within the neocortex
Sympathetic and sensory neurons lost in the Tα1:ICD animals coexpress p75NTR and trkA during development and in maturity, raising the possibility that the effects of the p75NTR-ICD could be attributable to interference with normal signaling through these receptors. If true, this dominant-negative effect should not be observed in neurons that do not express either p75NTR or trkA. To test this, we examined the neocortex, which lacks trkA expression and in which p75NTR is expressed only transiently within the subplate (Allendoerfer et al., 1990).
Neuronal counts were performed on two separate regions of the forebrain of wild-type and line 4173 Tα1:ICD animals. One region was a rostral area defined by the position of the lateral ventricles and the medial septum; the other was more caudal, located at the anterior portion of the hippocampal formation. Cresyl violet-stained sections from both of these regions demonstrated that cortical thickness was decreased in Tα1:ICD mice relative to controls (Fig.5). For quantitation, neuronal counts were performed on 530-μm-wide strips extending from corpus collosum to pia; this analysis revealed that the decrease in cortical thickness within Tα1:p75NTR-ICD mice reflected a highly significant decrease in neuronal number, with 22% fewer neurons within the rostral region (wt, 3015 ± 58.3; line 4173, 2355 ± 67.7, p = 0.0005; n = 3 animals each) and 26% neuronal loss in the caudal region (wt, 3133 ± 145; line 4173, 2376 + 15.5, p = 0.003; n = 4 Tα1:ICD animals and 3 control animals). This decrease in cortical neuron number in the Tα1:ICD mice indicates that the neuronal loss is not limited to trkA- or p75NTR-expressing neurons.
Fig. 5.

Neuronal expression of the p75NTR-ICD leads to the loss of neurons within the neocortex. Shown are photomicrographs of Nissl-stained coronal sections of the neocortex of control and transgenic p75NTR-ICD animals. Right andfar left photographic panels are from control animals, whereas the three inner panels are all from transgenic animals of the 4173 line. The area examined is indicated in the schematic drawings shown at left, with the top panels representing the rostral level of the neocortex and the bottom panels representing the caudal level. Each vertically aligned pair of photomicrographs is derived from the same animal. Brackets in the far left panel indicate approximate boundaries of the different cortical layers. Scale bar, 75 μm. Neuronal counts reveal a highly significant loss of cortical neurons in the p75NTR-ICD (p < 0.001; see Results).
Inducible expression of the p75NTR-ICD leads to death of injured facial motor neurons
To determine whether the p75NTR-ICD had an effect on motor neurons, we examined the facial motor nucleus in wild-type and Tα1:ICD adult mice for deficits in neuronal number or size. Quantitation of cresyl violet-stained sections throughout the extent of the facial nucleus revealed that facial motor neuron size was not altered significantly within Tα1:p75NTR-ICD mice (wt, 317.7 ± 13.8; line 4173, 329.5 ± 8.2; line 4163, 355.3 ± 13.9, p > 0.05; n = 3 animals each) (Fig. 6A) and that numbers of facial motor neurons were similar in adult controls and transgenic animals of lines 4163 and 4173 (p= 0.3; n = 3 animals each) (Fig. 6B). Thus, unlike peripheral sympathetic and sensory neurons, developing motor neurons are resistant to deleterious effects of p75NTR-ICD expression.
Fig. 6.

Inducible expression of the p75NTR-ICD results in loss of mature adult facial motor neurons after nerve injury. Coronal sections of facial nuclei were prepared from adult animals subjected to a unilateral facial nerve lesion 7 d earlier. Sections were stained with cresyl violet, and neuronal counts were performed on every fifth section, as described in Materials and Methods. Neither the size (A; in μm2) nor the number of facial motor neurons (B) is affected significantly by developmental expression of the p75NTR-ICD in uninjured neurons, but lesion of the facial nerve results in loss of facial motor neurons in ICD-expressing animals (*p< 0.05).
Our results show that the intracellular domain of the p75NTR induces cell death of selected developing neuronal populations of neurons, but our results do not address the possibility that acute ICD expression might affect the survival of mature neurons. To examine this possibility, we took advantage of the finding that expression of the Tα1 α-tubulin promoter is induced in mature facial motor neurons after facial nerve lesion; promoter activity is maximal 1–3 d after axotomy and thereafter is maintained at high levels until neurons successfully regenerate (Gloster et al., 1994; Wu et al., 1997). To test whether induced expression of the p75NTR intracellular domain affected survival of mature neurons, we unilaterally resected the facial nerve of adult animals; 1 week later, survival of facial motor neurons was compared between lesioned and unlesioned sides. Consistent with previous findings (Tetzlaff et al., 1991), lesioned and unlesioned facial motor nuclei in control animals showed no significant decrease in relative neuron number (wt uninjured side, 2505 ± 340; wt injured side, 2267 ± 305, p > 0.05; n = 3). However, within both the 4163 and 4173 transgenic lines, facial nerve lesion resulted in motor neuron loss of ∼40% (4163 uninjured side, 2675 ± 140; 4163 injured side, 1573 ± 66, p< 0.05; n = 3; 4173 uninjured side, 2318 ± 108; 4173 injured side, 1447 ± 190, p < 0.05;n = 3) (Fig. 6B). Thus although facial motor neurons were not lost because of developmental expression of the p75NTR-ICD, the injury-induced expression of this protein resulted in the death of injured adult motor neurons.
p75NTR-ICD expression does not inhibit activity of intracellular signaling cascades
The p75NTR-ICD could signal autonomously to mediate apoptosis; alternatively, the ICD could interfere with Trk receptor activation and thereby inhibit a trk-mediated survival signal. To distinguish between these possibilities, we first asked whether the p75NTR intracellular domain has any effect on trk receptor autophosphorylation. TrkA-expressing sympathetic and sensory neurons were, in large part, lost from the transgenic animals and, as an alternative, we examined effects of the p75NTRICD on trkA using transfected PC12 cells in which the p75NTR-ICD is stably expressed (Fig.7A). PC12 cells tolerate the p75NTR-ICD well, and there was no difficulty in producing clones that stably expressed the receptor fragment (data not shown). To test whether the expression of the p75NTR-ICD alters proximal NGF-mediated signaling events, we examined trkA autophosphorylation levels over a range of NGF concentrations; Figure 7B shows that the expression of the p75NTR-ICD had no apparent effect on levels of NGF-mediated trkA phosphorylation in PC12 cells.
To determine whether the p75NTR-ICD perturbed activation of trkB or trkC, we examined levels of endogenous trk receptor activation in the cortex of neonatal (Fig. 7C,D) and adult (Fig. 7E,F) transgenic and control animals. Trk receptors were immunoprecipitated from control or transgenic cortex from both age groups and immunoblotted with antibodies directed against phosphotyrosine or with antibodies specific to either trkB or trkC (data not shown). These studies revealed no difference in endogenous receptor levels or receptor activation between wild-type and transgenic animals, although deficits in neuronal number were observed in these same cortical regions (Fig. 7E,F). To determine whether neuronal expression of the ICD affected acute activation of trkB, we divided neonatal cortex symmetrically and triturated it, and aliquots were left untreated or exposed to 100 ng/ml BDNF for 10 min. Trk receptors were immunoprecipitated with pan-trk antibody 203 and analyzed for phosphotyrosine content by immunoblot. BDNF-mediated stimulation of trkB was equivalent in both transgenic and wild-type animals (data not shown). Finally, we analyzed lysates of neonatal line 4173 cortex for tyrosine-phosphorylated proteins by immunoblot analysis. These studies revealed no apparent differences between the pattern of proteins bearing tyrosine phosphorylation in the cortex of Tα1:ICD versus control animals (Fig. 7G). Together, these studies demonstrate that the ability of the p75NTR-ICD to mediate neuronal death is unlikely to be attributable to the inhibition of trk receptor activation.
Recent studies also have demonstrated that a direct signaling cascade mediated by p75NTR may result in the activation of the transcription factor NF-kB and of jun kinase (Carter et al., 1996; Casaccia-Bonnefil et al., 1996). We therefore measured these activities in lysates of Tα1:ICD brains from E16 and E18 embryos in a total of eight litters; although both jnk activity and NF-kB DNA binding activity were detected readily in brain lysates prepared from control or transgenic animals, neither activity was affected by neuronal expression of the p75NTR-ICD (Fig. 8A,B).
Fig. 8.
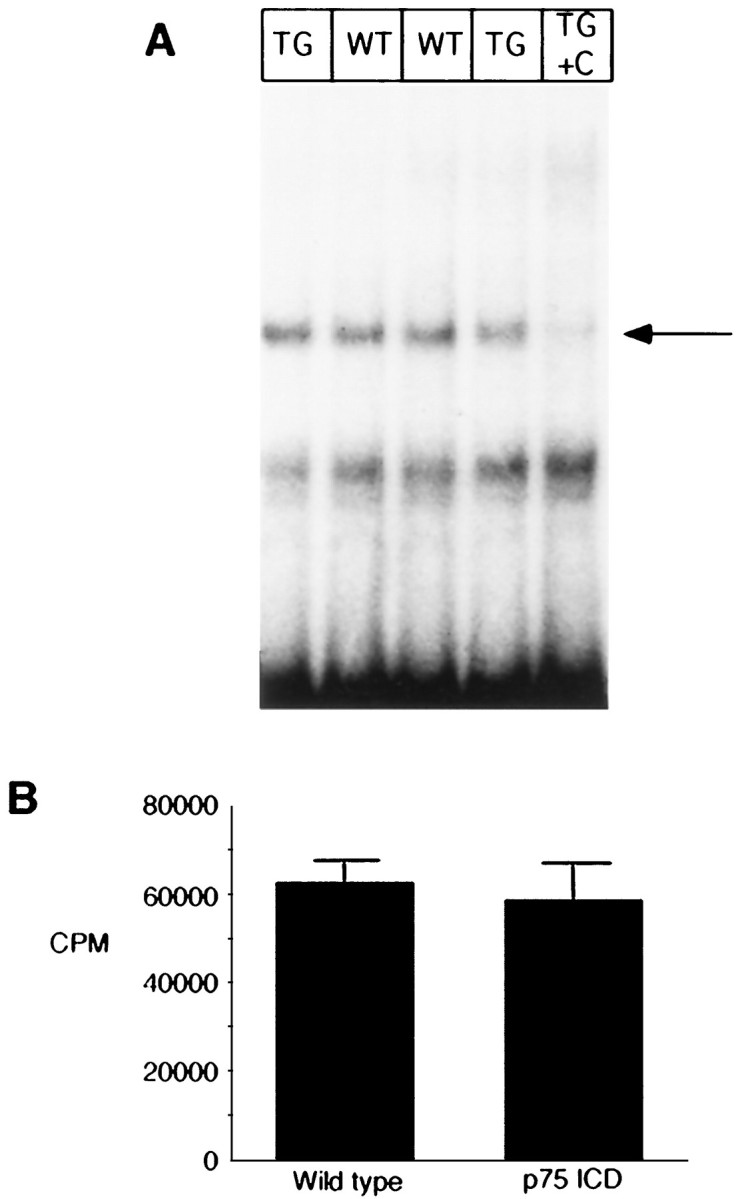
Levels of NF-kB and jun kinase activity are not altered in brain lysates of embryonic transgenic animals.A, Nuclear extracts from E16 brain lysates were analyzed for NF-kB activity by electrophoretic mobility shift, using an end-labeled 32 bp fragment derived from an HIV-LTR as probe. Inlane 5 (TG + C), 50 ng of an unlabeled NF-kB element was included with the lane 4 sample to determine specific NF-kB binding complexes (indicated byarrow). Shifted patterns are similar in wild-type (lanes 2 and 3) and in transgenic line 4173 (lanes 1 and 4) brains. This is typical of four similar experiments. B, Brain lysates from a litter of E16 animals were analyzed for jnk activity, as described in Materials and Methods. Results shown represent mean of assays of individual complete litter (n = 5 wild-type and 4 for Tα1:ICD mice) and represent one of three similar experiments.
DISCUSSION
Although p75NTR was cloned over a decade ago, its physiological role remains unclear. Results presented in this paper indicate that the p75NTR, like other members of this receptor family, is capable of autonomously signaling to mediate the death of selected populations of developing and injured neurons. Specifically, we have demonstrated that neuronal expression of the truncated intracellular domain (ICD) of p75NTR is sufficient to cause developmental apoptosis of developing sensory and sympathetic neurons in the PNS but not of facial motor neurons. This deficit in neurons was not limited to TrkA- or p75NTR-expressing neurons because there are decreased numbers of neurons in the neocortex, where TrkA is not expressed and where p75NTR is expressed only transiently. Moreover, our data indicate that neurons are differentially vulnerable to the ICD at different points in their lifetime; inducible expression of the ICD in injured adult facial motor neurons led to increased neuronal death, whereas the ICD did not affect survival of these same neurons during development. This effect on neuronal apoptosis is not attributable to a direct effect on trk receptors, because trk protein levels and endogenous and induced activation levels all remained the same regardless of the presence or absence of the p75NTR-ICD. On the basis of these data, we believe that the intracellular domain of the p75NTR is a constitutively active autonomous signaling protein that, in the appropriate neuronal context, will mediate neuronal death.
The structural motif that defines the TNF receptor superfamily is a tandemly repeated extracellular cysteine-rich domain (Banner et al., 1993; Bazan, 1993); many of the receptors within this family play important roles in regulation of proliferation and apoptosis, particularly within the immune system (for review, see Baker and Reddy, 1996). The intracellular domains of these proteins contain no enzymatic activity, and, until recently, the signaling paths that they activated were poorly characterized. The only intracellular homology identified among members of the TNFR superfamily is a region of 80 amino acids that is found in TNF-R1, fas, DR3, and p75NTR(Boldin et al., 1995b; Chapman, 1995; Chinnaiyan et al., 1996). In TNF-R1 and fas, this domain is required for ligand-mediated activation of apoptotic pathways, and it therefore has been termed the “death domain” (Tartaglia et al., 1993). A major advance in understanding signaling through this class of receptor has come with the identification of proteins, including TRADD, FADD, and RIP, that interact with TNF-R1 and fas intracellular domains (Chinnaiyan et al., 1995; Hsu et al., 1996). It is now clear that death domain interactions play a critical role in mediating the mobilization of these signaling molecules and the activation of a proteolytic apoptotic cascade (for review, see Nagata, 1997). The fact that a soluble cytoplasmic fragment of TNF-R1 can provoke cellular apoptosis indicates that overexpressed receptor fragments are capable of activating these signaling proteins (Boldin et al., 1995a).
The analysis of p75NTR signaling in physiologically relevant settings is complicated by the fact that many p75NTR-expressing cells also express trk receptors that activate distinct signaling cascades in response to the same ligand(s). p75NTR functionally interacts with trk receptors (Ip et al., 1993; Barker and Shooter, 1994; Verdi et al., 1994), and trk receptor activation may inhibit signaling by p75NTR (Dobrowsky et al., 1995), further complicating the analysis of autonomous p75NTRsignaling cascades. We therefore took a transgenic expression approach to allow us to examine the effects of p75NTR-dependent signaling in physiologically relevant settings irrespective of ligand binding. The neuronal apoptosis observed in the Tα1:ICD mice is consistent with the hypothesis that the truncated p75NTR fragment does, indeed, act as a constitutive activator of signaling pathways within neurons and suggests that a p75NTR-derived signal is capable of mediating apoptosis in a manner similar to other members of this receptor family. Like the intracellular domains of fas and TNF-R1, our evidence suggests that the p75NTR-ICD constitutively may activate a cell death cascade. Mechanisms that may underlie this signaling path include activation of sphingomyelinase, JNK, NF-kB, or members of the TRAF/TRAD family. Our examination of NF-kB or JNK activity in embryonic brains from Tα1:ICD mice did not reveal differences from nontransgenic littermates, but this does not rule out the possibility that activation of these pathways contributes to the transgenic phenotype in subtle cell-specific manners not detected in our biochemical assays. It is possible that changes in neuronal jnk or NF-kB activation that may result from p75NTR-ICD expression are obscured by high levels of activity from unaffected cell types or, alternatively, that high jnk or NF-kB activity occurs in a short developmental window that precedes cell loss. In this regard, the effects of the p75NTR-ICD show an intriguing cellular specificity, with developmental loss of sympathetic and peripheral sensory neurons and of central neurons of the neocortex. One possible explanation for this is that only these susceptible populations express the signaling partners necessary for mediation of a p75NTRapoptotic cascade. As p75NTR signaling partners are identified and their expression patterns are determined, the basis of the cell specificity of the p75NTR-ICD may become apparent.
Our data do not support the hypothesis that the intracellular p75NTR fragment acts in a dominant-negative manner, either inhibiting p75NTR-dependent survival signal or interfering with the normal action of trkA. If the p75NTR intracellular domain had a dominant-negative effect on p75NTR signaling, the transgenic phenotype should mimic that of the p75NTR−/− mice. However, whereas p75NTR-ICD transgenics show almost complete loss of sympathetic neurons of the SCG and increased motor neuron loss after injury, in p75NTR−/− mice sympathetic neuron number is not reduced (Lee et al., 1992; Brennan et al., 1996), and reduced motor neuron death is observed after facial nerve lesion (K.-F. Lee, personal communication). Expression of the p75NTR-ICD has no demonstrable effect on ligand-dependent activation of trkA (in stably transfected PC12 cell lines) or trkB (in transgenic brain). It is possible that negative selection has occurred within the transgenic mice such that only those neurons resistant to the effects of the p75NTR-ICD are present at the time of our analysis. However, results from the PC12 cell sublines suggest that a direct effect of the p75NTR-ICD on proximal events in trkA activation is unlikely. Rather, the observation that p75NTR-ICD expression results in a highly significant loss of neurons from multiple cortical layers, which normally do not express trkA or p75NTR (Allendoerfer et al., 1990), suggests that the receptor fragment can have a dominant effect, possibly by constitutively activating an apoptotic signaling cascade.
The findings reported here are consistent with several recent reports indicating that p75NTR may, in some circumstances, play a role in cell death. Supernumerary basal forebrain cholinergic neurons are observed in the p75NTR−/− mice, and their normal developmental loss in wild-type animals is reduced by blocking ligand binding to p75NTR(Vanderzee et al., 1996). Similarly, cell loss that normally occurs in the developing retina is reduced by reagents that block NGF access to p75NTR (Frade et al., 1996). So far, only NGF appears to be capable of mediating this effect; in the avian isthmo-optic nucleus (von Bartheld et al., 1994) and on cultured oligodendrocytes that express p75NTR but not trkA (Casaccia-Bonnefil et al., 1996), only NGF treatment results in cell death. Together, these results suggest that a normal role of p75NTR may be to aid in the process of developmental apoptosis that occurs within the maturing nervous system. The use ofin vivo models such as the Tα1:p75NTR-ICD mice described here should, in the future, allow us to identify functionally relevant p75NTR signaling pathways that contribute to neuronal apoptosis.
Footnotes
This work was supported by grants from the Canadian Neurosciences Network Program (to P.A.B. and F.D.M.), from the Medical Research Council (MRC) of Canada (to P.A.B. and F.D.M.), and from the Fond de la Recherche en Santé du Québec (to P.A.B.). C.L. was supported by a fellowship from the Fond de la Recherche en Santédu Québec, A.G. by a Canadian Parkinson’s Foundation Fellowship, R.A. by a Canadian Neurosciences Network Fellowship, S.B. by an MRC Studentship, A.B. by a studentship from the Rick Hansen Man in Motion Foundation, and D.B. by a joint MRC–Genentech Fellowship. P.B. and F.M. are Scholars of the Killam Foundation, and P.B. is an MRC Scholar. We are grateful to Audrey Speelman for excellent technical assistance, to Dr. John Hiscott for advice on NF-kB assays, and to Dr. David Kaplan for supplying antibodies and for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Freda D. Miller or Dr. Philip A. Barker, Center for Neuronal Survival, Montreal Neurological Institute, McGill University, 3801 University Avenue, Montréal, Québec, Canada H3A 2B4.
Dr. Belliveau’s present address: NeuroVir Research #100-2386 East Mall, Vancouver, BC, Canada V6T 1Z3.
REFERENCES
- 1.Allendoerfer KL, Shelton DL, Shooter EM, Shatz CJ. Nerve growth factor receptor immunoreactivity is transiently associated with the subplate neurons of the mammalian cerebral cortex. Proc Natl Acad Sci USA. 1990;87:187–190. doi: 10.1073/pnas.87.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker SJ, Reddy EP. Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene. 1996;12:1–9. [PubMed] [Google Scholar]
- 3.Bamji SX, Miller FD. Comparison of the expression of a Ta1:nlacZ transgene and Ta1 α-tubulin mRNA in the mature central nervous system. J Comp Neurol. 1996;374:52–69. doi: 10.1002/(SICI)1096-9861(19961007)374:1<52::AID-CNE4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 4.Banner DW, D’Arcy A, Janes W, Gentz R, Schoenfeld H-J, Broger C, Loetscher H, Lesslauer W. Crystal structure of the soluble human 55 kDa TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- 5.Barker PA, Shooter EM. Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to trkA on PC12 cells. Neuron. 1994;13:203–215. doi: 10.1016/0896-6273(94)90470-7. [DOI] [PubMed] [Google Scholar]
- 6.Barker PA, Barbee G, Misko TP, Shooter EM. The low affinity neurotrophin receptor, p75LNTR, is palmitoylated by thioester formation through cysteine 279. J Biol Chem. 1994;269:30645–30650. [PubMed] [Google Scholar]
- 7.Barrett GL, Bartlett PF. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc Natl Acad Sci USA. 1994;91:6501–6505. doi: 10.1073/pnas.91.14.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bazan JF. Emerging families of cytokines and receptors. Curr Biol. 1993;3:603–606. doi: 10.1016/0960-9822(93)90009-d. [DOI] [PubMed] [Google Scholar]
- 9.Belliveau DJ, Krivko I, Kohn J, Lachance C, Pozniak C, Rusakov D, Kaplan DR, Miller FD. NGF and NT-3 both activate trkA on sympathetic neurons but differentially regulate survival and neuritogenesis. J Cell Biol. 1997;136:375–388. doi: 10.1083/jcb.136.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boldin M, Mett I, Varfolomeev E, Chumakov I, Shemeravni Y, Camonis J, Wallach D. Self-association of the death domains of the p55 tumor necrosis factor (TNF) receptor and fas/APO1 prompts signaling for TNF and fas/APO1 effects. J Biol Chem. 1995a;270:387–391. doi: 10.1074/jbc.270.1.387. [DOI] [PubMed] [Google Scholar]
- 11.Boldin M, Varfolomeev E, Pancer Z, Mett I, Camonis J, Wallach D. A novel protein that interacts with the death domain of fas/apoI contains a sequence motif related to the death domain. J Biol Chem. 1995b;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- 12.Brennan C, Phillips HS, Landis SC. p75 expression affects the NGF dependency of sympathetic neuron survival in transgenic mice. Soc Neurosci Abstr. 1996;22:300.20. [Google Scholar]
- 13.Carroll SL, Silos-Santiago I, Frese SE, Ruit KG, Milbrandt J, Snider WD. Dorsal root ganglion neurons expressing trk are selectively sensitive to NGF deprivation in utero. Neuron. 1992;9:779–788. doi: 10.1016/0896-6273(92)90040-k. [DOI] [PubMed] [Google Scholar]
- 14.Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohmmatthaei R, Baeuerle PA, Barde YA. Selective activation of nf-kappa B by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- 15.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- 16.Chapman BS. A region of the 75 kDa neurotrophin receptor homologous to the death domains of TNFR-I1 and Fas. FEBS Lett. 1995;374:216–220. doi: 10.1016/0014-5793(95)01113-s. [DOI] [PubMed] [Google Scholar]
- 17.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 18.Chinnaiyan AM, O’Rourke K, Yu GL, Lyons RH, Garg M, Duan DR, Xing L, Gentz R, Ni J, Dixit VM. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–992. doi: 10.1126/science.274.5289.990. [DOI] [PubMed] [Google Scholar]
- 19.Coggeshall RE. A consideration of neuronal counting methods. Trends Neurosci. 1992;15:9–13. doi: 10.1016/0166-2236(92)90339-a. [DOI] [PubMed] [Google Scholar]
- 20.Coggeshall RE, Lekan HA. Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol. 1996;364:6–15. doi: 10.1002/(SICI)1096-9861(19960101)364:1<6::AID-CNE2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 21.Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts MS, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD, Phillips HS. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- 22.Davies AM, Lee KF, Jaenisch R. p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron. 1993;11:565–574. doi: 10.1016/0896-6273(93)90069-4. [DOI] [PubMed] [Google Scholar]
- 23.Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low affinity neurotrophin receptor. Science. 1994;265:1596–1598. doi: 10.1126/science.8079174. [DOI] [PubMed] [Google Scholar]
- 24.Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysis—modulation by co-expression of p75ntr with trk receptors. J Biol Chem. 1995;270:22135–22142. doi: 10.1074/jbc.270.38.22135. [DOI] [PubMed] [Google Scholar]
- 25.Frade JM, Rodriguez-Tebar A, Barde Y-A. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature. 1996;383:166–168. doi: 10.1038/383166a0. [DOI] [PubMed] [Google Scholar]
- 26.Gloster A, Wu W, Speelman A, Weiss S, Causing C, Pozniak C, Reynolds B, Chang E, Toma JG, Miller FD. The Tα1 α-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J Neurosci. 1994;14:7319–7330. doi: 10.1523/JNEUROSCI.14-12-07319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greene LA, Kaplan DR. Early events in neurotrophin signaling via Trk and p75 receptors. Curr Opin Neurobiol. 1995;5:579–587. doi: 10.1016/0959-4388(95)80062-x. [DOI] [PubMed] [Google Scholar]
- 28.Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires co-expression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- 29.Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR. Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron. 1992;9:883–896. doi: 10.1016/0896-6273(92)90241-5. [DOI] [PubMed] [Google Scholar]
- 30.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 31.Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD. Similarities and differences in the way neurotrophins interact with the trks in neuronal and non-neuronal cells. Neuron. 1993;10:137–149. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan DR, Matsumoto K, Lucarelli E, Thiele CJ. Induction of trkB by retinoic acid mediates biologic responsiveness to BDNF and differentiation of human neuroblastoma cells. Neuron. 1993;11:321–331. doi: 10.1016/0896-6273(93)90187-v. [DOI] [PubMed] [Google Scholar]
- 33.Klein R. Role of neurotrophins in mouse development. FASEB J. 1994;8:738–744. doi: 10.1096/fasebj.8.10.8050673. [DOI] [PubMed] [Google Scholar]
- 34.Lee K-F, Li E, Huber J, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- 35.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–366. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 36.Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature. 1987;325:593–597. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- 37.Ruit KG, Elliott JL, Osborne PA, Yan Q, Snider WD. Selective dependence of mammalian dorsal root ganglion neurons on nerve growth factor during embryonic development. Neuron. 1992;8:573–587. doi: 10.1016/0896-6273(92)90284-k. [DOI] [PubMed] [Google Scholar]
- 38.Singh S, Aggarwal BB. Protein-tyrosine phosphatase inhibitors block tumor necrosis-dependent activation of the nuclear transcription factor NF-kappa B. J Biol Chem. 1995;270:10631–10639. doi: 10.1074/jbc.270.18.10631. [DOI] [PubMed] [Google Scholar]
- 39.Tartaglia LA, Ayres TM, Wong G, Goeddel DV. A novel domain within the 55 kd TNF receptor signals cell death. Cell. 1993;74:845–853. doi: 10.1016/0092-8674(93)90464-2. [DOI] [PubMed] [Google Scholar]
- 40.Tetzlaff W, Alexander SW, Miller FD, Bisby MA. Response of facial and rubrospinal neurons to axotomy: changes in mRNA expression for cytoskeletal proteins and GAP-43. J Neurosci. 1991;11:2528–2544. doi: 10.1523/JNEUROSCI.11-08-02528.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vanderzee CEEM, Ross GM, Riopelle R, Hagg T. Survival of cholinergic forebrain neurons in developing p75(NGFR)-deficient mice. Science. 1996;274:1729–1732. doi: 10.1126/science.274.5293.1729. [DOI] [PubMed] [Google Scholar]
- 42.Verdi JM, Birren SJ, Ibanez CF, Persson H, Kaplan DR, Benedetti M, Chao MV, Anderson DJ. p75LNGFR regulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron. 1994;12:733–745. doi: 10.1016/0896-6273(94)90327-1. [DOI] [PubMed] [Google Scholar]
- 43.von Bartheld CS, Kinoshita Y, Prevette D, Yin QW, Oppenheim RW, Bothwell M. Positive and negative effects of neurotrophins on the isthmo-optic nucleus in chick embryos. Neuron. 1994;12:639–654. doi: 10.1016/0896-6273(94)90219-4. [DOI] [PubMed] [Google Scholar]
- 44.Westwick JW, Brenner DA. Methods for analyzing c-jun kinase. Methods Enzymol. 1995;255:342–359. doi: 10.1016/s0076-6879(95)55037-2. [DOI] [PubMed] [Google Scholar]
- 45.Wu W, Gloster A, Miller FD. Transcriptional repression of the growth-associated Ta1 α-tubulin gene by target contact. J Neurosci Res. 1997;48:477–487. [PubMed] [Google Scholar]