

Abstract
Mutations in the seizure (sei) locus cause temperature-induced hyperactivity, followed by paralysis. Gene cloning studies have established that the seizuregene product is the Drosophila homolog ofHERG, a member of the eag family of K+ channels implicated in one form of hereditary long QT syndrome in humans. A series of five null alleles with premature stop codons are all recessive, but viable. A missense mutation in thesei gene, which changes the charge at a conserved glutamate residue near the outer mouth of the pore, has a semidominant phenotype, suggesting that the mutant seizure protein acts as a poison in a multimeric complex. Transformation rescue of a null allele with a cDNA under the control of an inducible promoter demonstrates that induced expression of seizure potassium channels in adults rescues the paralytic phenotype. This rescue decays with at1/2 of ∼1-1.5 d after gene induction is discontinued, providing the first estimate of ion channel stability in an intact, multicellular animal.
Keywords: cloned potassium channel, Drosophila melanogaster, gene mapping, seizure gene, hyperexcitability, temperature-induced paralysis, sodium channel regulation, transformation, transgenic animals, HERG, IKr, eag gene family, ion channel turn over
The excitability properties of neurons are determined by the types, distribution, and density of ion channels they express. Changes in expressed ion channels can affect cell resting potentials, action potential threshold and duration, repetitive firing, and neurotransmitter release. For example, reduction of sodium channel function results in a less excitable cell, whereas reduction in potassium channel function results in hyperexcitability.
Temperature-sensitive paralytic mutations in Drosophila melanogaster, which affect sodium channel density or properties, provide a means to identify interesting gene products involved in ion channel structure or regulation. Four mutations have been implicated in sodium channel changes:parats,napts, tipE, andseits. The para(paralytic temperature-sensitive) gene encodes the α subunit of a voltage-gated sodium channel (Loughney et al., 1989). The nap (no action potential temperature-sensitive) locus encodes a nucleic acid binding protein likely to be involved in the expression or splicing of X-linked genes, including para (Kernan et al., 1991). Recent cloning of the tipE locus identified a novel membrane component that dramatically stimulates the expression ofpara sodium channels but structurally is unrelated to previously identified sodium channel subunits (Feng et al., 1995a). Theseizure (sei) gene is the last of these genes to be cloned.
The sei mutants have been an anomaly amongDrosophila mutations proposed to affect sodium channels, because the biochemical and electrophysiological phenotypes do not match the behavioral phenotypes. For example, bothseits1 (ts1) andseits2 (ts2) homozygotes are hyperactive even at the permissive temperature (Jackson et al., 1984, 1985). Hyperactivity also is seen in electrophysiological recordings from dorsal longitudinal muscles (Kasbekar et al., 1987) and from the adult cervical giant fiber pathway (Elkins and Ganetzky, 1990). In addition, there is a reduction in membrane-bound sodium channels in the seits1 allele, but not in seits2 (Jackson et al., 1984,1985). Similarly, whole-cell patch-clamp studies show reduced sodium currents in ts1, but not in ts2 (O’Dowd and Aldrich, 1988). This creates a paradox, because reduction in sodium channel density or activity should lead to hypoexcitability rather than the observed hyperexcitability. Furthermore, it is difficult to understand why the ts1 and ts2 alleles have similar effects on behavior and on cell excitability and yet have different effects on sodium channel density.
To resolve these inconsistencies, we used positional cloning to demonstrate that the sei gene encodes a channel subunit most closely related to the human HERG potassium channel involved in theIKr current in cardiac myocytes (Sanguinetti et al., 1995; Trudeau et al., 1995). Sequencing mutant seialleles shows that recessive hyperexcitability and temperature-induced paralysis are the null phenotypes for this locus. A missense mutation that forms a defective subunit has a semidominant phenotype, presumably because it interacts with wild-type subunits to form a poisoned channel. We use an inducible transgene in a null mutant background to estimate functional channel half-life in the intact animal.
MATERIALS AND METHODS
Drosophila culture. Drosophila stocks were grown at 21°C on standard cornmeal medium (Lewis, 1960). The wild-type Canton-S strain was obtained from J. C. Hall (Brandeis University, Waltham, MA). The deficiency stocks(Df(2R)orBR6,Df(2R)orBR11,Df(2R)G10BR27,Df(2R)bw-S46, and Df(2R)bw-D23) were provided by B. Reed (M.I.T., Cambridge, MA).Df(2R)Px4, Df(2R)Px,seits1, andseits2 were described previously (Lindsley and Zimm, 1992; Jackson et al., 1984, 1985). The bw ba and or49h stocks were provided by P. Bryant (University of California, Irvine, CA).Df(2R)spMP was induced by γ-ray mutagenesis (M. Parisi and L. M. Hall, unpublished data).
Mutagenesis. The bw ba males were mutagenized with 4000 rads of γ-irradiation and mated in batches of 15 males and 30 seits1 virgin females. The 106,457 F1 progeny were screened at 39°C for temperature-sensitive paralysis (Feng et al., 1995b), and eight newsei alleles were isolated. After recovery from paralysis at 21°C, individual flies were mated to CyO, cn bw/BlL. Then brown-eyed (bw) offspring were mated to establish new sei lines over the CyO, cn bw chromosome.
Chromosome walk. Fragments of yeast artificial chromosome (YAC) clone DYN13-25 (Cai et al., 1994) were used to initiate a chromosome walk by screening two Drosophila genomic cosmid libraries under high stringency conditions (Sambrook et al., 1989). Clones with an “M” as the first letter were from the KT3 cosmid library (a generous gift of Max Scott and John Lucchesi, Emory University, Atlanta, GA). Clones with an “S” in the first letter were from the iso-1 cosmid library in the Not-Bam-Not-CoSpeR vector (a generous gift of J. W. Tamkun, University of California, Santa Cruz, CA) (Tamkun et al., 1992).
Restriction fragment-length polymorphism (RFLP) mapping.Male or49h and femaleseits1 flies were mated to generate double heterozygote femalesor49h/seits1. Recombinants between or and sei were recovered by mating these females toDf(2R)orBR11/SM6a, because this deletion uncovers both recessive or andsei genes. A total of 24 or sei recombinants were recovered and mated individually toDf(2R)orBR11/SM6ato establish lines. Four lines failed to survive. RFLPs betweenor49h andseits1 were identified by extracting genomic DNA from each line (Jowett, 1986), digesting it with restriction enzymes, and comparing the resulting genomic Southern blots probed with labeled clones from the walk (Feng et al., 1995b). Each of 20 recombinant lines was analyzed by Southern blotting to determine the isoform at each RFLP site.
Northern blotting. mRNA preparation from whole adults and Northern blot conditions were as described previously (Zheng et al., 1995). To standardize for RNA recovery differences, we reprobed Northern blots with a 0.6 kb cDNA fragment encoding a widely expressed ribosomal protein (rp49) (O’Connell and Rosbash, 1984). Northern blots were quantitated with a Molecular Dynamics PhosphorImager model 400, using ImageQuant version 4.2 software.
cDNA cloning and DNA sequencing. A Drosophilahead cDNA library in λgt11 (generously provided by P. Salvaterra, Beckman Research Institute, Duarte, CA) (Itoh et al., 1986) was probed with an 0.8 kb genomic DNA segment [bases +127 to +867 of theseizure open reading frame (ORF) plus a 56 base pair (bp) intron], which was interrupted by the insertion in theseiG50 allele. Two overlapping clones (sei-w1 and sei-w2) were isolated in a screen of 8 × 105 pfu. Nested deletions (Henikoff, 1987) were prepared for the cDNA inserts subcloned into the EcoRI site of pBluescript II SK(−) (Stratagene, La Jolla, CA). Double-stranded DNA sequencing (at least twice in both directions) was done on an Applied Biosystems Sequencer Model 373A (Foster City, CA) using cycle sequencing and the dideoxy chain termination method with either fluorescent dye-tagged primers or dye-tagged terminators. Rapid amplification of cDNA ends (RACE) (Frohman et al., 1988; Hofmann and Brian, 1991) with primers from the 5′ end of the sei-w1 clone was used to isolate the 5′ end of the message. Genomic DNA fragments were sequenced after PCR amplification with primers derived from the cDNA sequence (Zheng et al., 1995).
Transformation rescue. The complete seizure ORF (see Fig. 3A) plus 24 bp of the 5′ untranslated region and 100 bp of the 3′ untranslated region was subcloned into theStuI site of the pCaSpeR-hs vector under the control of a heat–shock promoter (Thummel and Pirrotta, 1992). Germline transformation was done as described previously (Feng et al., 1995b) using w seits1 as the host.
Fig. 3.

Structural features of the seizure protein.A, The sei (S) amino acid sequence was compared with the HERG (H) potassium channel subunit. The alignment was generated using the GAP program in GCG software (Devereux et al., 1984). Vertical lines connect identical amino acids; two vertical dots represent conservative substitutions; dashes indicate gaps introduced to facilitate alignment. Conserved groups were defined as (M, I, L, V); (A, G); (S, T); (Q, N); (K, R); (E, D); and (F, Y, W). The six hydrophobic domains (S1–S6), the pore (P), and the cyclic nucleotide binding domain (cNBD) are overlined. Predicted N-glycosylation sites (Asn 515 in sei, Asn 598 in HERG) are marked with Ψ. The position of the premature stop codon inseits1 is indicated by aclosed circle; the missense mutation inseits2 is indicated by an open circle. Potential phosphorylation sites are indicated as follows: CaM kinase,downward-pointing arrows; protein kinase C, inverted open triangles; protein kinase A,upside down caret; and casein kinase, filled diamonds. The C terminus (1045–1159) of HERG is not shown.B, The hydropathy plot for the deduced sei protein was determined by the method of Kyte and Doolittle (1982). Regionsabove the line are hydrophobic.
RESULTS
Identification of the seizure transcript
Because the molecular nature of the seizure gene product was unknown, positional cloning was the method of choice to identify the transcript. As summarized in Figure1A, the seizure gene was mapped first to the cytogenetic region 60A8–B10. Then a YAC clone that mapped between 60A10–A13 and 60B12–B13 (Cai et al., 1994) was used to initiate a chromosome walk (summarized in Fig. 1D) through this region.
Fig. 1.
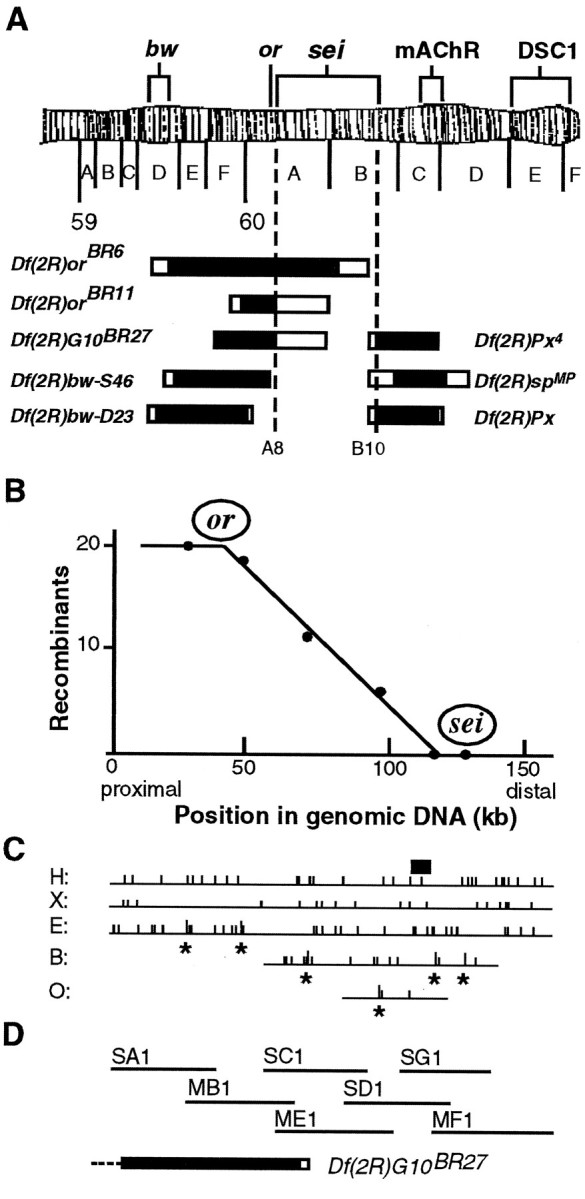
Localization of the seizuregene. A, Deletion mapping. Heterozygotes forseits1 and each deficiency (Df) were tested for temperature-induced paralysis at 38°C. OnlyDf(2R)orBR6and Df(2R)orBR11 failed to complement sei, locating the gene between 60A8 and B10. Deleted regions are indicated as black bars with regions of uncertainty shown as open bars. Other genes shown are eye color genes (bw, or), cloned genes for a muscarinic acetylcholine receptor (mAChR) (Onai et al., 1989), and a sodium channel homolog (DSC1) (Salkoff et al., 1987). The regions deleted are as follows:Df(2R)orBR6, 59D5–D10 to 60B3–B8;Df(2R)orBR11, 59F6–F8 to 60A8–A16; andDf(2R)G10BR27, 59F3 to 60A8–A16 (breakpoints defined in Reed, 1992);Df(2R)bw-S46, 59D8–D11 to 60A7 (Simpson, 1983);Df(2R)bw-D23, 59D4–D5 to 60A1–A2;Df(2R)Px, 60B8–B10 to 60D1–D2; andDf(2R)Px4, 60B8–B10 to 60D1 (breakpoints defined in Lindsley and Zimm, 1992); andDf(2R)spMP, 60B8–B13 to 60D3–D8 (Parisi and Hall, unpublished results). B–D, RFLP mapping and chromosome walking. Twenty recombinants betweenor49h andseits1 were analyzed for six RFLPs that distinguish theor49h chromosome from theseits1 chromosome. The positions of these RFLPs are indicated by an asterisk on the genomic DNA restriction maps (C). H,HpaI; X, XbaI;E, EcoRI; B,BglII; O, XhoI. The number of recombinants with the or49hRFLP is plotted versus RFLP location on genomic DNA inB. The X intercept, determined by linear regression analysis, shows the approximate physical location ofseizure. The small black bar inC shows the region in which genomic DNA aberrations were detected in seizure alleles. The cosmid clones from the chromosome walk are shown in D, relative to the end of deletion Df(2R)G10BR27. The heavy black bar represents the known end of this deletion, whereas the open bar shows a region of uncertainty. The dotted line indicates that the deletion extends to the left beyond the limits of this figure.
Because many regions of the Drosophila genome are rich with transcripts (see, for example, Feng et al., 1995b), we used two approaches to identify the seizure transcript: RFLP mapping and analysis of γ-ray-induced chromosome aberrations that disruptseizure function. Two γ-ray-induced seizurealleles (G50 and G87) showed altered restriction patterns within a 10 kb segment of the chromosome walk (indicated by a small black rectangle in Fig.1C). Thus, this 10 kb region was a candidate for at least part of the seizure gene.
RFLP mapping (Fig. 1B) was used to confirm, independently, the genomic location of seizure before cDNA cloning. The closer an RFLP is to seizure, the lower the frequency of recombination will be between them. The X intercept at position 118 kb of the chromosome walk (Fig. 1B) gives the approximate location of the seizure gene. Because the horizontal axes of Figures 1B–D are on the same scale, it can be seen that the position of seizure as defined by RFLP mapping coincides with the 10 kb region shown to contain either a small insertion or deletion in γ-ray-inducedseizure alleles.
A genomic fragment from the region proposed to contain theseizure gene detected two candidate transcripts (3.4 and 3.0 kb in wild type) that were altered in size or amount in sevenseizure alleles (Fig. 2). Some alleles (ts1 and G64) showed a dramatic reduction in both transcripts, as compared with wild-type (CS) (Fig.2, Table 1). In others, there was a change in transcript size. For example, in G87 both transcripts are 0.8 kb smaller than wild type. In G50 two transcripts are 0.5 kb larger, plus there is an additional small transcript at 2.2 kb. Bothts2 and G85 show an increase in intensity of the larger 3.4 kb transcript relative to the smaller 3.0 kb one. Northern blots probed with single-stranded riboprobes show that the 3.4 and 3.0 kb messages are transcribed in the same direction and are substantially overlapping (X. J. Wang and L. M. Hall, unpublished results). Changes in transcript size and/or intensity in so many independently isolatedseizure alleles provide compelling evidence that these cDNAs represent the seizure gene product.
Fig. 2.

Northern blot of seizure alleles. Poly(A+) RNA (∼10 μg) from wild-type (CS) and sei mutant adult flies was loaded into each lane, as indicated. The blot was probed with aseizure cDNA, including bases +127 to +2668 of the ORF. (Northern blots of wild type probed with single-stranded riboprobes or with the 0.8 kb genomic fragment originally used to probe the cDNA library each identify only the same 3.4 and 3.0 kb transcripts shown in this blot.) Later the blot was reprobed with Drosophilaribosomal protein 49 (rp49) cDNA (O’Connell and Rosbash, 1984) to standardize for mRNA recovery (bottom panel).
Table 1.
Effects of seizure alleles on transcript abundance
| Band | Wild-type | ts1 | ts2 | G43 | G50 | G64 | G85 | G87 |
|---|---|---|---|---|---|---|---|---|
| 3.9 kb | 230 | |||||||
| (44.2)a | ||||||||
| 3.5 kb | 441 | |||||||
| (20.9)b | ||||||||
| 3.4 kb | 520 | 61.2 | 818 | 456 | ND | 193 | 872 | ND |
| (100)a | (11.8)a | (157)a | (87.7)a | (37.1)a | (168)a | |||
| 3.0 kb | 2115 | 314 | 1469 | 1284 | ND | 366 | 878 | ND |
| (100)b | (14.8)b | (69.5)b | (60.7)b | (17.3)b | (41.5)b | |||
| 2.6 kb | 1075 | |||||||
| (207)a | ||||||||
| 2.2 kb | 418 | 2239 | ||||||
| (19.8)b | (106)b |
Band intensity relative to rp49 for each allele tested (cpm in indicated band)/(cpm in rp49 band) × 10−4.
Italicized number in parenthesis = band intensity relative to the wild-type equivalent.c
Band intensity calculated relative to the 3.4 kb wild-type band.
Band intensity calculated relative to the 3.0 kb wild-type band.
After correction for loading differences.
The seizure gene encodes a potassium channel subunit of the eag family
Sequencing wild-type cDNA corresponding to the disrupted transcripts revealed a long ORF encoding a deduced protein of 855 amino acids (Fig. 3A, “S”lines) with a predicted molecular mass of 97.5 kDa. The dendrogram (Fig. 4) derived from database comparisons shows that sei is a member of the eag class of potassium channels, which also have similarities to cyclic nucleotide-gated cation channels (Guy et al., 1991). The complete sei protein shows a striking similarity (58% similarity; 47% identity) to HERG, a member of the eag family recently cloned from humans (Warmke and Ganetzky, 1994). The similarity and identity are even higher (84 and 72%, respectively) if the comparison is limited to the hydrophobic core. Identification of this new Drosophila potassium channel subunit, which is closer to a human potassium channel than to other Drosophilachannels, establishes sei and HERG as a distinct potassium channel subfamily, as predicted previously (Warmke and Ganetzky, 1994).
Fig. 4.

Dendrogram of the potassium channel family. This tree shows the relationship between sei and other potassium channel family members, using the hydrophobic cores for comparison. Similarity is inversely proportional to the horizontal distance of any two sequences from a branch point. The GrowTree program in the Wisconsin Genetics Computer Group (GCG) sequence analysis software (version 8.0) was used to produce this diagram from a distance matrix created by Distances, using UPGMA. The potassium channels (and their GenBank accession numbers) are the Drosophila Shaker family of voltage-gated channels Shab (M32659),Shaker (M17211), Shaw (M32661),Shal (M32660); Drosophilacalcium-activated channel Slo (M96840); inwardly rectifying channels IRK1 (X73052), ROMK1(X72341) GIRK1 (L25264); cyclic nucleotide-gated channels cAMP (X55519) and cGMP (X51604); plant inward rectifiers AKT1 (X62907) andKAT1 (M86990); and eag family of channels mouseM-Eag (U04294), rat R-Eag (Z34264),Drosophila Eag (M61157), Elk (U04246), and human HERG (U04270).
Structural features of the seizure protein
The hydropathy plot (Fig. 3B) and the alignment of sei- and HERG-deduced proteins (Fig. 3A) illustrates structural features that they have in common. First, each has six predicted transmembrane α-helices (Fig. 3, S1–S6) plus a pore region (P) between S5 and S6. The amphipathic S4 domain with positive amino acids (K, R) every third residue is important in voltage-sensing. The complete conservation of this domain between sei and HERG suggests they may have similar voltage-dependent properties. The sequence of the sei protein in the P region (SLTSVGFGN) is very similar to the potassium channel consensus sequence (tmttvG[y/f]Gd) (Chandy and Gutman, 1995), providing strong evidence that sei is a subunit of potassium-permeable channels.
There is also a striking identity in the proposed cyclic nucleotide binding domain (cNBD) in the C-terminal cytoplasmic region. However, the rest of the C terminus and most of the N terminus of the sei protein have very little in common with HERG or with other members of the eag potassium channel gene family. Both the N and C termini of the sei protein are significantly shorter than HERG.
There is a consensus site for N-glycosylation 12 or 14 amino acids upstream of the P region, which is conserved in all eag family members (Warmke and Ganetzky, 1994). In addition, three potential phosphorylation sites just downstream of S6 are conserved in sei and in HERG as is a phosphorylation site within the cyclic nucleotide binding domain. In total, the sei protein has 9 possible protein kinase C phosphorylation sites, 6 CaM kinase phosphorylation sites, 1 protein kinase A phosphorylation site, and 11 casein kinase phosphorylation sites on predicted cytoplasmic domains. These sites, especially those conserved between sei and HERG, suggest the potential for regulation of this channel by phosphorylation.
Mutations in seizure alleles
To determine the molecular basis for the recessive versus dominant phenotypes of different seizure alleles, we sequenced genomic DNA corresponding to the entire coding region from wild-type and homozygous mutant lines. We identified mutant changes in six different alleles. The alkylating agent ethyl methanesulfonate (EMS) generally induces point mutations, and that is what we found for the EMS-induced ts1 and ts2 alleles. Theseits1 allele has a T to A change, which converts K282 (Fig. 3A) into a premature stop codon. The resulting truncated protein completely lacks the hydrophobic core that forms the transmembrane channel (Fig. 5). Barring read-through of the stop codon, this nonsense allele should be a functional null.
Fig. 5.

Location of mutations in the sei protein. The predicted membrane topology of the sei protein is shown. Theheavy black vertical lines indicate the six transmembrane domains. The postulated pore-forming domain dips into the membrane between transmembrane domains 5 and 6. A potential cyclic nucleotide binding domain (cNBD) is shown as ahatched box in the C terminus. Ψ shows the position of a predicted N-glycosylation site. The filled trianglerepresents the site of a 0.5 kb insertion containing an in-frame stop codon in seiG50. Thescissors indicate the 779 bp deletion inseiG87. This causes a +1 frameshift leading to a premature stop codon that follows the seven altered amino acids after N548. The positions of changes in the other four mutant alleles are shown by open circles.G64 has a single nucleotide deletion leading to five changed amino acids that follow P363 and end with a premature stop codon at amino acid 369. G43 has a TG to ATTT change leading to a +2 frameshift. The 49 amino acids that follow I473 are changed, and there is a premature stop codon in place of I522. The nature of the ts1 and ts2 point mutations is discussed in the text.
In seits2 there is a G to A change, which changes a negatively charged group to a positively charged one (E490K in Fig. 3A) in a region close to the extracellular side of the S5 domain. This charge change is near the channel pore (Fig. 5) and could disrupt channel function. Because this is a missense mutation, seits2 should still make a full-length subunit.
The four γ-ray-induced alleles diagrammed in Figure 5 are all recessive and show hyperactivity and temperature-induced paralysis phenotypes indistinguishable from the ts1 allele. Three (G50, G43, and G64) are frameshift mutations leading to a premature stop codon in the approximate positions shown in Figure 5. The G87 allele deletes a large region from just before the start of transmembrane 6 through to a point in the C terminus past the cyclic nucleotide binding domain (Fig. 5). Although they differ in the sites of change, none of the γ-ray-induced alleles would produce a functional channel, because they lack part or all of important transmembrane domains. Thus,seits1, plus the γ-ray-induced alleles, defines the null phenotype of this gene and illustrates the consequences to the animal of complete absence of theseizure gene product.
The entire ORF also was sequenced for a fifth γ-ray-induced allele (G85, transcripts shown in Fig. 2), but no changes were identified despite the failure of this allele to complement otherseizure alleles. This allele may have alterations in the 5′ or 3′ untranslated regions or in an alternative exon not yet identified. Interestingly, G85 and ts2 both show an increase in the 3.4 kb transcript relative to the 3.0 kb transcript (Table 1). The reason for this shift remains to be determined.
Expression of a seizure+ transgene: paralysis rescue and estimation of channel half-life
Mutant strains that completely lack the seizure potassium channel provide a unique opportunity to define when expression of the gene product is needed to rescue the paralytic phenotype. To address this question, we made transgenic flies in a null mutant background. The transgene was wild-type seizure cDNA encoding the protein shown in Figure 3A and was under the control of an inducible heat–shock promoter. In the absence of induction (Fig.6, circles), the transgenic flies show a paralytic phenotype indistinguishable from the null mutant without the transgene. This suggests that uninduced expression of the transgene is not at physiologically relevant levels, at least with respect to the paralysis phenotype. In contrast, repeated induction of this transgene in adults (Fig. 6, squares) over a 5 d period rescues the paralytic phenotype in most flies by the sixth day. This experiment suggests that synthesis of this channel subunit in adults only is sufficient to restore normal locomotor function.
Fig. 6.

Paralysis rescue (A) and estimation of apparent channel half-life (B) using an induciblesei+ transgene. Adult flies homozygous for the seits1 null allele and carrying the wild-type sei transgene under the control of a heat–shock promoter were grown at 21°C and collected within 18 hr of adult eclosion. The transgene was induced by heat treatments at 35°C for 1 hr per day. A, Induction began within 2 hr after collection and continued for a total of 5 d. Each day, ∼24 hr after the last induction, a group of flies was scored for percentage standing (not paralyzed) after 2 min at 38°C. Under these conditions, 100% of wild-type flies would be standing (data not shown). Flies were discarded after a single paralysis test. Data from multiple experiments have been combined. Filled squaresrepresent transgenic flies given the inducing heat treatment (109–226 flies per point); filled circles show their uninduced sibs (102–204 flies per point). B, Flies tested on day 6 (after 5 d of induction) showed 94% rescue of the paralytic phenotype. After day 5 no further inducing heat pulses were given, but paralysis testing on different batches of flies continued until day 7.
When induction of the wild-type transgene is stopped, the return of the paralytic phenotype should reflect the net rate of loss of functional channels from membranes and thus will provide an estimate of apparent half-life of functional channels in membranes in an intact animal. To estimate this half-life, just before reaching 100% rescue, we stopped induction of the transgene and monitored the return of the paralytic phenotype. As shown in Figure 6B, return of paralytic phenotype was rather rapid. The time to return to 50% paralyzed flies is ∼1-1.5 d after 94% rescue is achieved.
DISCUSSION
Potassium channel mutations are the direct cause ofseizure hyperexcitability and paralysis
RFLP mapping, identification of mutant changes in the coding region of genomic DNA from six independently isolatedseizure alleles, and transformation rescue with cDNA under an inducible promoter provide conclusive evidence that the potassium channel sequence presented here is the seizure gene product. The sequence conservation between seizure and other ion channels shows that it is a potassium channel of the eag subfamily and is related most closely to the HERG inwardly rectifying potassium channel. Althoughsei has not yet been expressed in Xenopusoocytes, the virtually complete conservation of the ion selective pore region (P) leaves little doubt that sei encodes a potassium channel α-type subunit. These results provide an explanation for the hyperexcitable phenotype seen with all sei mutant alleles. In general, decreases in potassium channel activity cause hyperexcitability because of prolonged depolarization during an action potential and/or failure to maintain a normally negative resting potential. Decrease in activity of this sei potassium channel subunit leads to hyperexcitability similar to that observed for other potassium channel subunit loss of function mutants, includingSh, eag, and Hyperkinetic (Wu and Ganetzky, 1992).
A recent report suggests that a “set” of cloned potassium channels (including Sh, Shab, Shal, and Shaw) accounts for most embryonic potassium currents in Drosophila (Tsunoda and Salkoff, 1995). Given the dramatic effects that sei mutants have on behavior, sei also should be included in the list of physiologically important potassium channel components inDrosophila.
Genetic evidence that the seizure potassium channel is a multimer
Null alleles of sei are completely recessive, indicating that the 50% reduction of gene product generally expected in a heterozygous null is without consequence to the behavior of the organism. This suggests that excess sei gene product is normally available in wild type. In contrast, the missense alleleseits2 has a partially dominant phenotype, although its overall hyperexcitability phenotype as a homozygote is less extreme than the null alleles (Kasbekar et al., 1987). This suggests that the potassium channel that contains the sei protein is a multimer, as are other potassium channels (Isacoff et al., 1990). The presence of one or more copies of a mutant subunit in a multimer could act as a dominant “poison” to disrupt the functional properties of the remaining wild-type subunits. It is interesting to note that missense mutations in HERG, the human homolog ofsei, also cause a dominant phenotype, in this case, of cardiac arrhythmia (Curran et al., 1995).
This genetic analysis cannot predict whether the sei potassium channel is a homo- or heteromultimer. There is, however, indirect evidence that suggests an additional component is required. cRNA from several different sei cDNA constructs fails to make functional channels when injected into Xenopus oocytes. Mistakes or an incomplete sequence cannot be the cause for this failure, because the same cDNA sequence rescues the sei behavioral phenotype in transgenic animals. This suggests that there is a factor required forsei expression that is present in the fly and absent inXenopus oocytes. This factor may be another pore-forming subunit, or it may be an auxiliary subunit like tipE, which stimulates expression of a Drosophila sodium channel (Feng et al., 1995a). Candidates for a component needed for seiexpression include a potassium channel β subunit, such as theHyperkinetic gene product (Chouinard et al., 1995), or an unidentified component, such as the enhancer of seizure gene product (Kasbekar et al., 1987).
Premature nonsense codons affect seizureRNA abundance
As reviewed by Maquat (1995), mRNAs containing a nonsense codon as a result of either frameshift or nonsense mutations often are reduced in abundance. The severity of the reduction is correlated with the closeness of the nonsense mutation to the normal initiation codon. The different sei nonsense alleles described here provide a unique opportunity to determine whether this relationship holds true for mRNAs that encode integral membrane proteins and are associated with the endoplasmic reticulum during translation. In general,sei transcripts show a similar effect of premature nonsense codons, as reported in other systems. Northern analysis ofsei alleles (Fig. 2, Table 1) shows that the presence of a premature nonsense codon in the first half of the message (ts1 and G64) causes a marked decrease in both sei transcripts, as compared with wild type. The case of G50 is complicated by the appearance of an extra transcript, which may be the result of a cryptic transcription start site. However, even if all three transcripts are considered, there is an overall reduction in total transcript level, as compared with wild type (after correcting for loading differences).
Rescue of paralytic phenotype with an inducible wild-typeseizure transgene
Expression of the seizure potassium channel subunit in adults only is sufficient to rescue paralysis. This stands in contrast to the requirement for the tipE gene product during pupal development to rescue adult paralysis (Feng et al., 1995a). Although mutations in both the tipE gene and the sei gene cause a temperature-sensitive paralysis, they seem to do so by different mechanisms that are distinct in time.
Potassium channel turnover in the whole animal
To our knowledge there has been no previous estimate of stability of sei/HERG potassium channels in an intact, multicellular organism. By inducing expression of the seizure+transgene and monitoring the paralysis phenotype, we were able to titrate expression to the point at which 94% rescue of the mutant phenotype was attained. Then induction was discontinued. The rate of return of the paralytic phenotype should reflect the net loss of functional channels from the membrane. With this method we estimate that the apparent half-life of functional sei channels is ∼1–1.5 d. This is consistent with metabolic labeling studies of sodium channels that estimated the apparent half-lives of the α subunit and the αβ2 complex to be 30 and 50 hr, respectively, in cultured embryonic rat brain neurons (Schmidt and Catterall, 1986). It is also consistent with estimates of calcium channel lifetime of 5–8 d inParamecium (Schein, 1976). There is one report (Zhao et al., 1995) suggesting that Shaker potassium channels in cultured “giant”Drosophila neurons in some genetic backgrounds turn over more rapidly than we report here. This could indicate differences in stability of different potassium channel types or could reflect differences in different mutant backgrounds.
Relationship between seizure and sodium channel regulation
How can the fact that seizure encodes a potassium channel subunit be reconciled with observations that the ts1allele is associated with changes in levels of functional sodium channels (Jackson et al., 1985; O’Dowd and Aldrich, 1988) and that thets2 allele has an altered pH dependence and alteredKD for saxitoxin binding (Jackson et al., 1984)? We propose that the reduction in channel levels in ts1 is a consequence of sei effects on cell excitability. There are previous reports that link electrical activity to density of sodium channels (Dargent and Couraud, 1990; Dargent et al., 1994) (for review, see Catterall, 1992), including a report of downregulation of sodium channels in nerve terminals of epileptic mice (Willow et al., 1986). The effects of the ts2 allele on saxitoxin binding sites are more difficult to understand. They may be an experimental artifact because of the high incubation temperatures used in these studies. Alternatively, they may reflect a ts2-induced shift in expression of sodium channels with different binding properties as a result of alternative splicing (Thackeray and Ganetzky, 1994, 1995) or expression of different α-subunit genes [para, Loughney et al. (1989) vs DSC1, Salkoff et al. (1987)].
There are multiple mechanisms by which sodium channel density is regulated by electrical activity of the cell. In cultured rat muscle fibers, spontaneous electrical activity causes a feedback downregulation of sodium channel density by changing levels of mRNA encoding sodium channel α subunits (Offord and Catterall, 1989). Treatments that mimic increased electrical activity, such as elevation of cytosolic calcium with ryanodine or the ionophore A23187, also cause sodium channel downregulation (Sherman and Catterall, 1984; Brodie et al., 1989; Offord and Catterall, 1989).
Sodium channel downregulation in response to hyperexcitability also has been observed in neurons treated with sodium channel activators, including α-scorpion toxin, batrachotoxin, and veratridine (Dargent and Couraud, 1990). In these studies, the mechanism of regulation is by sodium channel internalization (Dargent et al., 1994) rather than by channel synthesis, as reported for muscle (Offord and Catterall, 1989).
Although the mechanisms for sodium channel downregulation differ for the two cases summarized above, they have in common that the response to hyperexcitability is a decrease in the density of functional sodium channels in the plasma membrane. We propose that the decrease in functional sodium channels associated with theseits1 allele inDrosophila may be the consequence of hyperexcitability. To explain the normal sodium channel levels found in the ts2missense allele, we postulate that there is a minimum hyperexcitability level required to induce the downregulation. This level is reached in the null ts1 allele, but not in the missense ts2allele. The different sei alleles described here, plus mutations in other potassium channel components (Wu and Ganetzky, 1992), provide a unique opportunity to dissect genetically the role of potassium channel activity in sodium channel regulation.
GenBank accession number
The GenBank accession number for the seizure cDNA reported here is U36925.
Footnotes
This work was supported by National Institutes of Health Jacob Javits Neuroscience Investigator Award NS16204 to L.M.H. E.R.R. was supported, in part, by National Institutes of Health postdoctoral fellowship NS08442. We thank Bruce Reed for supplying deletions important for the cytogenetic studies, Ian Duncan for providing the YAC clone, and Qian Yan for invaluable help in producing transgenic flies. We also thank Diane Hodges, Diane O’Dowd, Dejian Ren, Ted Shih, and Michael Pugsley for helpful comments on this manuscript. We especially thank Barry Ganetzky and coworkers for their cooperative spirit in the joint publication of our independent results.
Correspondence should be addressed to Dr. Linda Hall, Department of Biochemical Pharmacology, State University of New York at Buffalo, 329 Hochstetter Hall, Buffalo, NY 14260-1200.
Dr. Reynolds’s present address: Department of Environmental Science, Entomology Division, University of California–Berkeley, 201 Wellman Hall, Berkeley, CA 94720.
Dr. Déak’s present address: Department of Genetics, Attila József University, 6726 Szeged, Közepfasor 52, Hungary.
REFERENCES
- 1.Brodie C, Brody M, Sampson SR. Characterization of the relation between sodium channels and electrical activity in cultured rat skeletal myotubes: regulatory aspects. Brain Res. 1989;488:186–194. doi: 10.1016/0006-8993(89)90708-7. [DOI] [PubMed] [Google Scholar]
- 2.Cai H, Kiefel P, Yee J, Duncan I. A yeast artificial chromosome clone map of the Drosophila genome. Genetics. 1994;136:1385–1399. doi: 10.1093/genetics/136.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catterall WA. Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev. 1992;72:s15–s48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- 4.Chandy KG, Gutman GA. Voltage-gated potassium channel genes. In: North RA, editor. Ligand- and voltage-gated ion channels. CRC; Boca Raton, FL: 1995. pp. 1–71. [Google Scholar]
- 5.Chouinard SW, Wilson GF, Schlimgen AK, Ganetzky B. A potassium channel beta subunit related to the aldo-keto reductase superfamily is encoded by the Drosophila Hyperkinetic locus. Proc Natl Acad Sci USA. 1995;92:6763–6767. doi: 10.1073/pnas.92.15.6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 7.Dargent B, Couraud F. Down-regulation of voltage-dependent sodium channels initiated by sodium influx in developing neurons. Proc Natl Acad Sci USA. 1990;87:5907–5911. doi: 10.1073/pnas.87.15.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dargent B, Paillart C, Carlier E, Alcaraz G, Martin-Eauclaire MF, Couraud F. Sodium channel internalization in developing neurons. Neuron. 1994;13:683–690. doi: 10.1016/0896-6273(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 9.Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elkins T, Ganetzky B. Conduction in the giant nerve fiber pathway in temperature-sensitive paralytic mutants of Drosophila. J Neurogenet. 1990;6:207–219. doi: 10.3109/01677069009107111. [DOI] [PubMed] [Google Scholar]
- 11.Feng G, Deák P, Chopra M, Hall LM. Cloning and functional analysis of tipE, a novel membrane protein that enhances Drosophila para sodium channel function. Cell. 1995a;82:1001–1011. doi: 10.1016/0092-8674(95)90279-1. [DOI] [PubMed] [Google Scholar]
- 12.Feng G, Deák P, Kasbekar DP, Gil DW, Hall LM. Cytogenetic and molecular localization of tipE: a gene affecting sodium channels in Drosophila melanogaster. Genetics. 1995b;139:1679–1688. doi: 10.1093/genetics/139.4.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frohman MA, Dush MK, Martin GR. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guy HR, Durell SR, Warmke J, Drysdale R, Ganetzky B. Similarities in amino acid sequences in Drosophila eag and cyclic nucleotide-gated channels. Science. 1991;254:730. doi: 10.1126/science.1658932. [DOI] [PubMed] [Google Scholar]
- 15.Henikoff S. Unidirectional digestion with exonuclease III in DNA sequence analysis. Methods Enzymol. 1987;155:156–165. doi: 10.1016/0076-6879(87)55014-5. [DOI] [PubMed] [Google Scholar]
- 16.Hofmann MA, Brian DA. A PCR-enhanced method for determining the 5′ end sequence of mRNAs. PCR Methods Appl. 1991;1:43–45. doi: 10.1101/gr.1.1.43. [DOI] [PubMed] [Google Scholar]
- 17.Isacoff EY, Jan YN, Jan LY. Evidence for the formation of heteromultimeric potassium channels in Xenopus oocytes. Nature. 1990;345:530–534. doi: 10.1038/345530a0. [DOI] [PubMed] [Google Scholar]
- 18.Itoh N, Slemmon JR, Hawke DH, Williamson R, Morita E, Itakura K, Roberts E, Shively JE, Crawford GD, Salvaterra PM. Cloning of Drosophila choline acetyltransferase cDNA. Proc Natl Acad Sci USA. 1986;83:4081–4085. doi: 10.1073/pnas.83.11.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson FR, Wilson SD, Strichartz GR, Hall LM. Two types of mutants affecting voltage-sensitive sodium channels in Drosophila melanogaster. Nature. 1984;308:189–191. doi: 10.1038/308189a0. [DOI] [PubMed] [Google Scholar]
- 20.Jackson FR, Gitschier J, Strichartz GR, Hall LM. Genetic modifications of voltage-sensitive sodium channels in Drosophila: gene dosage studies of the seizure locus. J Neurosci. 1985;5:1144–1151. doi: 10.1523/JNEUROSCI.05-05-01144.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jowett T. Preparation of nucleic acids. In: Roberts DB, editor. Drosophila: a practical approach. IRL; Oxford: 1986. pp. 275–286. [Google Scholar]
- 22.Kasbekar DP, Nelson JC, Hall LM. Enhancer of seizure: a new genetic locus in Drosophila melanogaster defined by interactions with temperature-sensitive paralytic mutations. Genetics. 1987;116:423–431. doi: 10.1093/genetics/116.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kernan MJ, Kuroda MI, Kreber R, Baker BS, Ganetzky B. napts, a mutation affecting sodium channel activity in Drosophila, is an allele of mle, a regulator of X chromosome transcription. Cell. 1991;66:949–959. doi: 10.1016/0092-8674(91)90440-a. [DOI] [PubMed] [Google Scholar]
- 24.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 25.Lewis EB. A new standard food medium. Dros Inf Serv. 1960;34:117–118. [Google Scholar]
- 26.Lindsley DL, Zimm GG. The genome of Drosophila melanogaster. Academic; New York: 1992. [Google Scholar]
- 27.Loughney K, Kreber R, Ganetzky B. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell. 1989;58:1143–1154. doi: 10.1016/0092-8674(89)90512-6. [DOI] [PubMed] [Google Scholar]
- 28.Maquat LE. When cells stop making sense: effects of nonsense codons on RNA metabolism in vertebrate cells. RNA. 1995;1:453–465. [PMC free article] [PubMed] [Google Scholar]
- 29.O’Connell P, Rosbash M. Sequence, structure, and codon preference of Drosophila ribosomal protein 49 gene. Nucleic Acids Res. 1984;12:5495–5513. doi: 10.1093/nar/12.13.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Dowd DK, Aldrich RW. Voltage-clamp analysis of sodium channels in wild-type and mutant Drosophila neurons. J Neurosci. 1988;8:3633–3643. doi: 10.1523/JNEUROSCI.08-10-03633.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Offord J, Catterall WA. Electrical activity, cAMP, and cytosolic calcium regulate mRNA encoding sodium channel alpha subunits in rat muscle cells. Neuron. 1989;2:1447–1452. doi: 10.1016/0896-6273(89)90190-6. [DOI] [PubMed] [Google Scholar]
- 32.Onai T, FitzGerald MG, Arakawa S, Gocayne JD, Urquhart DA, Hall LM, Fraser CM, McCombie WR, Venter JC. Cloning, sequence analysis, and chromosome localization of a Drosophila muscarinic acetylcholine receptor. FEBS Lett. 1989;255:219–225. doi: 10.1016/0014-5793(89)81095-6. [DOI] [PubMed] [Google Scholar]
- 33.Reed BH (1992) The genetic analysis of endoreduplication inDrosophila melanogaster. PhD thesis, University of Cambridge, UK.
- 34.Salkoff L, Butler A, Wei A, Scavarda N, Giffen K, Ifune C, Goodman R, Mandel G. Genomic organization and deduced amino acid sequence of a putative sodium channel gene in Drosophila. Science. 1987;237:744–749. doi: 10.1126/science.2441469. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual, 2nd Ed. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 36.Sanguinetti MC, Jiang C, Curran ME, Keating ME. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 37.Schein SJ. Calcium channel stability measured by gradual loss of excitability in pawn mutants of Paramecium aurelia. J Exp Biol. 1976;65:725–736. doi: 10.1242/jeb.65.3.725. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt JW, Catterall WA. Biosynthesis and processing of the α subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell. 1986;46:437–445. doi: 10.1016/0092-8674(86)90664-1. [DOI] [PubMed] [Google Scholar]
- 39.Sherman SJ, Catterall WA. Electrical activity and cytosolic calcium regulate levels of tetrodotoxin-sensitive sodium channels in cultured rat muscle cells. Proc Natl Acad Sci USA. 1984;81:262–266. doi: 10.1073/pnas.81.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simpson P. Maternal-zygotic gene interactions during formation of the dorsoventral pattern in Drosophila embryos. Genetics. 1983;105:615–632. doi: 10.1093/genetics/105.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC, Kennison JA. Brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell. 1992;68:561–572. doi: 10.1016/0092-8674(92)90191-e. [DOI] [PubMed] [Google Scholar]
- 42.Thackeray JR, Ganetzky B. Developmentally regulated alternative splicing generates a complex array of Drosophila para sodium channel isoforms. J Neurosci. 1994;14:2569–2578. doi: 10.1523/JNEUROSCI.14-05-02569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thackeray JR, Ganetzky B. Conserved alternative splicing patterns and splicing signals in the Drosophila sodium channel gene para. Genetics. 1995;141:203–214. doi: 10.1093/genetics/141.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thummel CS, Pirrotta V. New pCaSpeR element vectors. Dros Inf Serv. 1992;71:150–151. [Google Scholar]
- 45.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 46.Tsunoda S, Salkoff L. Genetic analysis of Drosophila neurons: Shal, Shaw, and Shab encode most embryonic potassium currents. J Neurosci. 1995;15:1741–1754. doi: 10.1523/JNEUROSCI.15-03-01741.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci USA. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Willow M, Taylor SM, Catterall WA, Finnell RH. Down-regulation of sodium channels in nerve terminals of spontaneously epileptic mice. Cell Mol Neurobiol. 1986;6:213–220. doi: 10.1007/BF00711071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu C-F, Ganetzky B. Neurogenetic studies of ion channels in Drosophila. In: Narahashi T, editor. Ion channels, Vol 3. Plenum; New York: 1992. pp. 261–314. [DOI] [PubMed] [Google Scholar]
- 50.Zhao M-L, Sable EO, Iverson LE, Wu C-F. Functional expression of Shaker K+ channels in cultured Drosophila “giant” neurons derived from Sh cDNA transformants: distinct properties, distribution, and turnover. J Neurosci. 1995;15:1406–1418. doi: 10.1523/JNEUROSCI.15-02-01406.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng W, Feng G, Ren D, Eberl DF, Hannan F, Dubald M, Hall LM. Cloning and characterization of a calcium channel alpha 1 subunit from Drosophila melanogaster with similarity to the rat brain type D isoform. J Neurosci. 1995;15:1132–1143. doi: 10.1523/JNEUROSCI.15-02-01132.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]