

Abstract
Selenoproteins are a diverse group of proteins containing selenocysteine (Sec), the 21st amino acid, incorporated during translation via a unique recoding mechanism1,2. Selenoproteins fulfil essential roles in many organisms1, yet are not ubiquitous across the tree of life3–7. In particular, fungi were deemed devoid of selenoproteins4,5,8. However, we show here that Sec is utilized by nine species belonging to diverse early-branching fungal phyla, as evidenced by genomic presence of both Sec machinery and selenoproteins. Most fungal selenoproteins lack consensus Sec recoding signals (SECIS elements9) but exhibit other RNA structures, suggesting altered mechanisms of Sec insertion in fungi. Phylogenetic analyses support a scenario of vertical inheritance of the Sec trait within eukaryotes and fungi. Sec was then lost in numerous independent events in various fungal lineages. Notably, Sec was lost at the base of Dikarya, resulting in the absence of selenoproteins in Saccharomyces cerevisiae and other well studied fungi. Our results indicate that, despite scattered occurrence, selenoproteins are found in all kingdoms of life.
Selenocysteine (Sec), the 21st amino acid, is co-translationally inserted via an unusual recoding mechanism, wherein UGA, normally a stop codon, is translated as Sec1. Sec insertion occurs specifically in selenoprotein genes, due to cis-acting RNA structures known as SECIS elements9. Sec machinery genes (tRNASec, EFsec, PSTK, SBP2, SecS, SPS) are trans-factors necessary and sufficient for eukaryotic Sec synthesis and insertion1,2,10. Sec is believed to confer catalytic advantage over Cys, its sulphur-containing analogue11,12, for specific oxidoreductase functions. Nevertheless, selenoproteins are not found in all organisms. Sec usage is scattered across bacteria3,4,13 and archaea14. Within eukaryotes, selenoproteins are present in most metazoans (including all vertebrates15), some protists and certain algae4,5,16. They are absent in many insects6, few nematodes7, plants5, and various protists4. Notably, fungi were considered the only kingdom of life entirely devoid of Sec4,5,8. However, here we provide conclusive genomic evidence for Sec utilization by nine fungal species belonging to three early-branching phyla.
We downloaded all available fungal genomes from NCBI (1201 species, Supplementary Table 1), and searched them for the presence of eukaryotic Sec machinery genes (Methods) using Selenoprofiles17 and Secmarker18. These automatically generated predictions (Supplementary Figure 1) were analysed for two potential confounders: the occurrence of protein families with similarity to those of interest, and contaminant sequences in fungal genome assemblies. For this, we reconstructed gene trees of candidate proteins together with their most similar annotated sequences (Methods) and inspected them to distinguish protein families (Supplementary Figures 2–5). This procedure led to the dismissal of several candidates. After filtering, Sec machinery proteins (Supplementary Data 1) localized only in a handful of genomes, and co-occurred with tRNASec (Fig. 1). After extensive analysis, we filtered out three species with Sec machinery which we reckoned as resulting from genome contamination from Sec-utilizing bacteria (Supplementary Note 1). On the other hand, we concluded that Bifiguratus adelaidae (Mucoromycota), Gonapodya prolifera (Chytridiomycota), Capniomyces stellatus, Zancudomyces culisetae, Smittium culicis, Smittium simulii, Smittium megazygosporum, Smittium angustum, and Furculomyces boomerangus (Zoopagomycota) were Sec-utilizing fungi (Fig. 2). These species formed distinct clades in three early-branching fungal phyla. The order of Harpellales was particularly well represented: 7 of the 8 species analysed had Sec.
Figure 1.
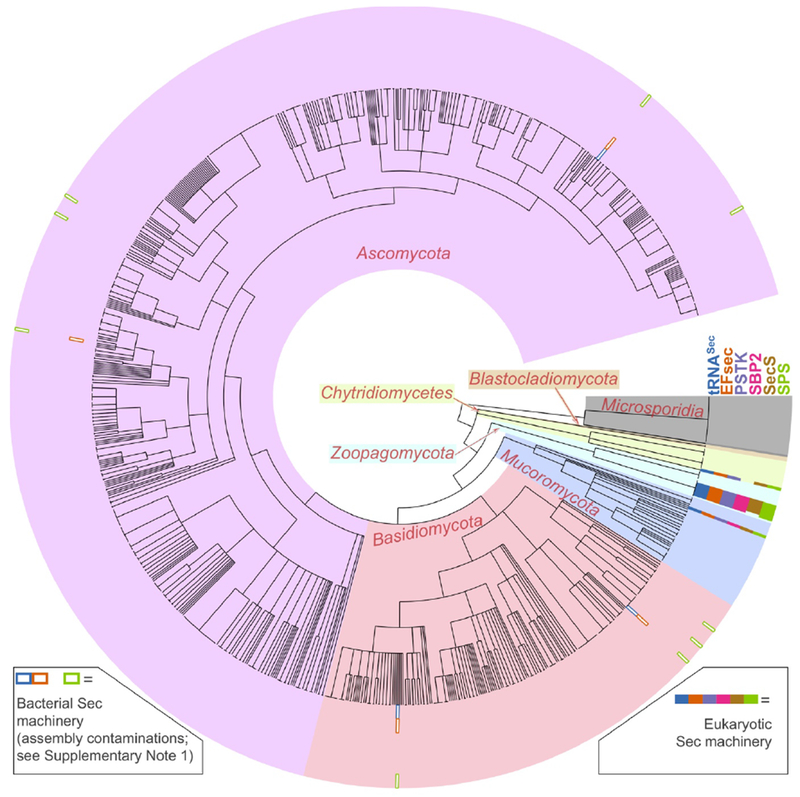
Sec machinery genes in fungal genome assemblies. The figure shows the results of genomic searches for Sec machinery genes (tRNASec, EFsec, PSTK, SBP2, SecS, SPS) in 1201 species of fungi. The tree in the center shows the phylogenetic relationships of species according to NCBI taxonomy. Gene presence is represented by colored rectangles in the outermost section. White-filled rectangles (legend at bottom left) represent genes that were attributed to assembly contamination from Sec-utilizing bacteria (Supplementary Note 1), and thus dismissed. An extended version of this figure is available as Supplementary Figure 1.
Figure 2.
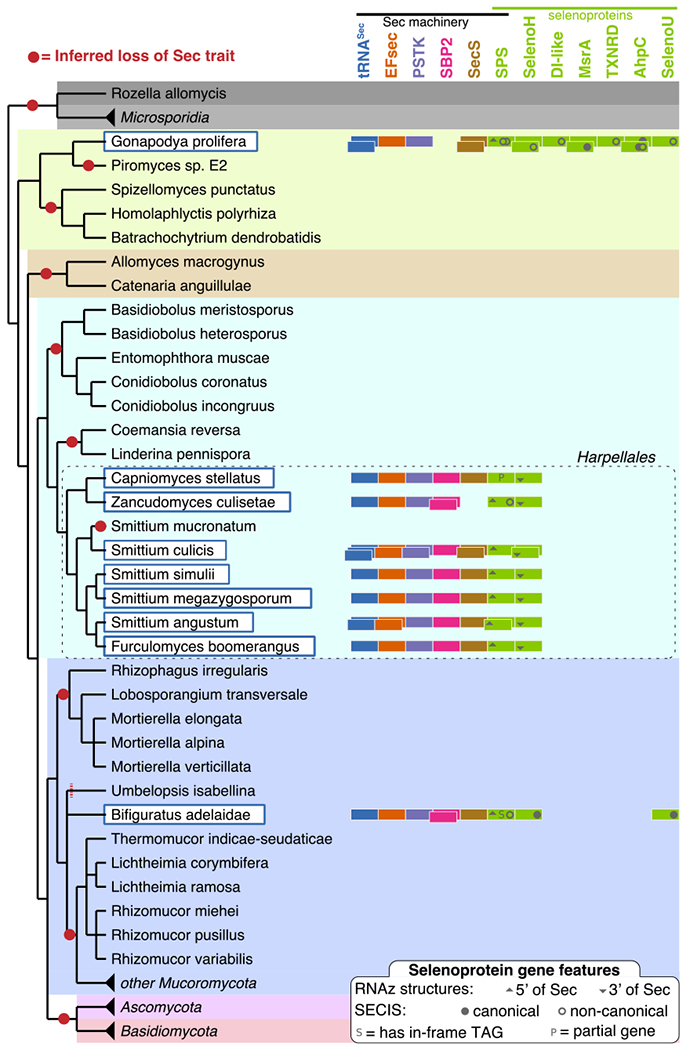
Selenoproteins and Sec machinery in Sec-utilizing fungi. On the left, a reference tree of fungi analyzed in this study (Methods) includes Sec-utilizing species highlighted in dark-blue, white-filled boxes. Each species is annotated with colored rectangles on the right, representing the Sec machinery and selenoprotein genes found in their genome. Multiple occurrences for a gene family are indicated as stacked rectangles. For selenoproteins, the presence of SECIS elements and other conserved RNA structures are indicated within each rectangle (see legend at the bottom). Losses of Sec encoding capacity, inferred by maximum parsimony, are shown as red circles along the tree. Unresolved phylogeny does not allow to determine whether the absence of Sec in Umbelopsis isabellina should be ascribed to yet another independent Sec loss event (hence the red dashed line). Microsporidia, Ascomycota, Basidiomycota and some Mucoromycota were compressed in this figure; these do not contain Sec machinery or selenoproteins.
We identified selenoproteins in all Sec-utilizing fungi (Supplementary Data 1), which belonged to seven known selenoprotein families (gene trees provided in Supplementary Figures 6–10). Two of them were found in all Sec-utilizing fungi: SelenoH, a nuclear oxidoreductase possibly involved in redox homeostasis19, and SPS (Fig. 3), Sec machinery component and also selenoprotein itself in many prokaryotes and eukaryotes4. Other fungal selenoproteins included SelenoU, an uncharacterized oxidoreductase20; AhpC (alkyl hydroperoxide reductase C), found as selenoprotein in certain bacteria, protists and porifera21; MsrA (methionine sulfoxide reductase A)22, identified as selenoprotein in algae, protists, and various non-vertebrate metazoa; DI-like, homologous to vertebrate iodothyronine deiodinases23 and present as selenoprotein in various invertebrates, protists and bacteria16; and finally TXNRD (thioredoxin reductase24), a selenoprotein present in most Sec-utilizing eukaryotes. Notably, this constitutes the first case of animal-like TXNRD described in fungi, since this kingdom uses a shorter and Sec-independent form of TXNRD24. G. prolifera was the species with most selenoproteins, covering all selenoprotein families discussed above. Analysis of a publicly available transcriptome for this species confirmed the expression of all selenoproteins and Sec machinery factors, except for tRNASec (Methods). Selenoprotein transcripts appear to occur at high levels in G. prolifera (Supplementary Figure 11). SPS mRNA was particularly abundant, ranking in the top 1-6% (depending on the background distribution used) transcripts.
Figure 3.
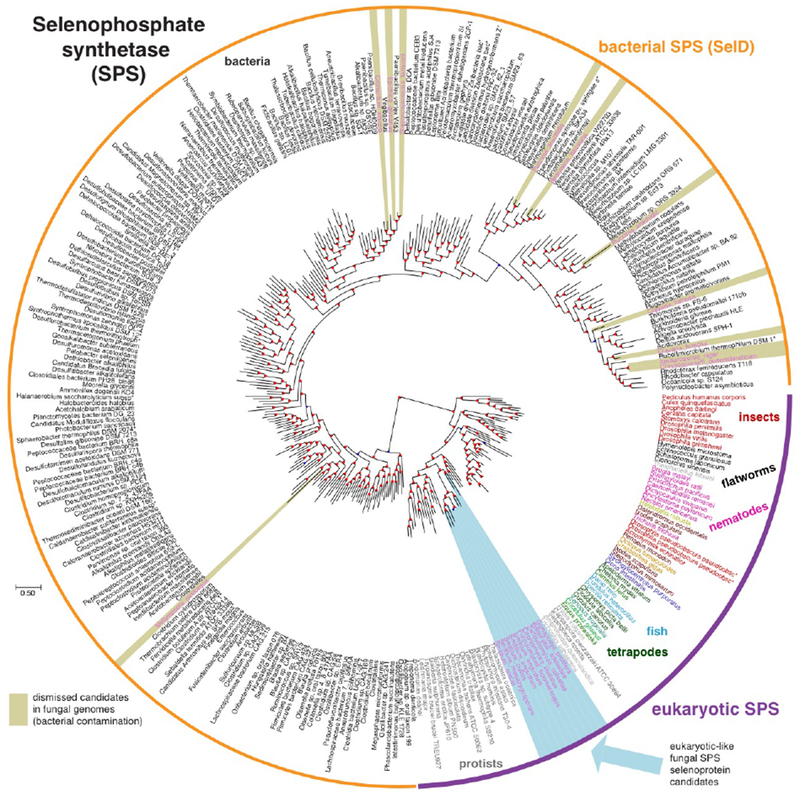
Reconstructed phylogenetic tree of SPS proteins. The tree was built based on the sequences of 21 SPS candidates in fungal genomes, aligned with 300 similar proteins annotated in NCBI NR (Methods). The source species is shown for each protein, and it is colored according to its taxonomic group. Fungal SPS candidates are highlighted in light blue (bona-fide eukaryotic SPS) and khaki (bacterial-like SPS, attributed to assembly contamination; Supplementary Note 1). Analogous trees for the rest of Sec machinery and selenoprotein genes are available as Supplementary Figures 2–10. Branch support values are provided in Supplementary Data 2.
We searched fungal selenoprotein genes for occurrence of eukaryotic SECIS elements. Surprisingly, we found canonical SECISes only in 5 of 29 selenoproteins (Fig. 2). We then broadened our searches (Methods) and detected additional SECIS-like structures in 8 genes. These contained the SECIS core motif and typically additional extended paired regions (Supplementary Figure 12). All candidate structures (except one) were located downstream of coding sequences, as expected of eukaryotic SECISes (Supplementary Figure 13). Strikingly, no SECIS candidates were predicted in any Harpellales selenoproteins (with one possible exception). Despite this, we are confident that they represent bona-fide selenoprotein genes: they show homology to known selenoproteins and exhibit UGA aligned precisely with the known Sec position. This observation suggests utilization of non-standard Sec recoding signals. We further performed de novo searches for RNA structures. First, we applied RNAz25 (Methods) to identify evolutionarily conserved structures in SelenoH and SPS genes of Harpellales. This approach identified two structured regions, one per gene, both within coding sequences. The SelenoH CDS structure consisted of two stems, including the Sec UGA and downstream region (Supplementary Figure 14). While all Harpellales sequences support this structure, the majority of pairings break down in the other two Sec-utilizing fungi, suggesting that it may be order-specific. The SPS CDS structure (Supplementary Figure 15) appears conserved across all species. It is located immediately upstream of the Sec UGA and consists of a ~30 pairs stem. Due to the lack of closely related sequences of non-Harpellales Sec-utilizing species, we could not apply RNAz to the rest of fungal selenoproteins. We thus ran RNALfold26 to predict all locally stable RNA structures. All these structures (Supplementary Figure 13) constitute Sec recoding signal candidates.
The reconstructed gene trees for Sec machinery and selenoproteins roughly recapitulate the species tree. These results are illustrated for SPS in Fig. 3, with the rest of protein trees provided in Supplementary Figures 2–10. SPS sequences from Sec-utilizing fungi form a monophyletic cluster, as expected from species phylogeny. This supports the scenario of common descent, with the Sec-containing SPS gene inherited from the ancestor of fungi to these extant species. The same clustering pattern was observed for SBP2. In the case of EFsec and SecS, fungal sequences also form a monophyletic cluster, with the exception of G. prolifera. Noteworthily, G. prolifera possesses two SecS paralogs, both containing similar C-terminal extensions. In these regions, we identified RNA recognition motifs (PFAM family RRM_1). In the PSTK tree, Harpellales form a unique cluster, while the other two Sec-utilizing fungi branch out of a cluster including protists and algae. The position of the fungal clusters relative to the homologs in other lineages is also informative: they are located in the expected position as an outgroup of metazoa, sometimes within protist lineages. Beyond fungal species, Sec machinery trees are in general accordance with the known species phylogeny, with some exceptions. In particular, nematodes and insects are typically placed in trees in more basal positions than expected. This may be caused by diversification of divergence rates across eukaryotic groups, causing long-branch attraction of genes of fast evolving lineages. In a previous study4, we concluded that the Sec encoding trait has been likely directly inherited from the root of eukaryotes to extant Sec-utilizing species, while the various non-Sec-utilizing groups underwent a number of parallel Sec losses. The phylogenetic trees presented here are consistent with this scenario, and thus support direct and continuous inheritance of Sec from the root of eukaryotes to Sec-utilizing fungi.
Given the remarkable diversity spanned by fungal genomes, we hypothesized that phylogenetic profiling could be profitably taken to discover fungal genes related to the Sec trait. This technique27 exploits gene co-occurrence across genomes to link genes to pathways. Aiming to uncover potential undiscovered selenoproteins and proteins involved in the Sec pathway, we developed a custom phylogenetic profiling procedure (Methods) to identify genes present in Sec-utilizing fungal genomes but absent in selenoproteinless species. Our procedure resulted in a list of 57 candidate proteins clustered in 27 homologous groups (Supplementary Table 2). PSTK, EFsec, SecS, SPS and SelenoH emerged as top scoring candidates, supporting robustness of the procedure. The rest of candidates had diverse annotated functions, and included oxidoreductases, transporters, uncharacterized proteins and others. We further examined candidates as potential selenoproteins, seeking conserved Sec-UGA codons, but found none (Methods). These genes may be involved in Sec transport, metabolism, or regulation, or represent false positives. Future experiments will be necessary to validate candidates and clarify their role in Sec biology.
In summary, we report here that Sec is genetically encoded by nine species of fungi belonging to three early-branching phyla (Fig. 2). Each phyla includes several other species with no trace of Sec usage in their genomes. We estimated the completeness of the genome assemblies (Methods) and dismissed it as explanation of the scattered pattern of Sec presence (Supplementary Figure 16). We found complete Sec machinery in each Sec-utilizing genome, with two exceptions: SecS in Z. culisetae and SBP2 in G. prolifera. The absence of the former is likely due to incomplete assembly, since we detected SecS in all other Harpellales. In contrast, SBP2 may be truly absent in G. prolifera (or diverged beyond recognition). In fact, while there are no genomes available from closely related species, there is a public G. prolifera transcriptome, and although we found all other machinery proteins transcribed at detectable levels, we still could not identify SBP2. Since this species possesses two SecS paralogs with unique RNA-binding domains, it is tempting (yet far-fetched, from current data) to speculate that one of these may have acquired the SBP2 function.
The scattered distribution of Sec across fungi, together with phylogenetic signal in machinery genes, supports a scenario of vertical inheritance followed by multiple independent Sec losses in this kingdom (Supplementary Note 2). Even in the most parsimonious scenario (Fig. 2), we infer at least 10 different complete losses of Sec encoding capacity in fungi. One of these losses was mapped prior to the split of Dikarya (Ascomycota and Basidiomycota, diverged around 700 mya28), resulting in Sec absence in Saccharomyces cerevisiae and related fungi. The most recent loss occurred in S. mucronatum, as the rest of Harpellales all utilize Sec. We analysed stop codon usage across fungi, and saw no obvious correlation with Sec usage (Supplementary Figure 17). The high incidence of parallel losses may seem surprising; however, this was previously well documented for the Sec trait at various evolutionary scales. Parallel Sec losses were frequently observed in prokaryotes 3,4,13,14, in various protist groups4,16 and even within animals, with at least 5 independent Sec loss events meticulously traced within insects6. Selenoprotein losses can occur either through whole gene loss or through conversion of Sec codons to Cys, the latter partially retaining enzymatic functions. With few exceptions, Sec-devoid fungi do not contain Cys orthologs of fungal selenoproteins (Supplementary Note 3), suggesting gene losses as primary mechanism of selenoprotein depletion in fungi.
Based on sequence homology, Sec location and phylogenetic signal, we are confident that all identified fungal candidates constitute bona-fide selenoproteins. However, canonical SECIS elements were identified only in a few cases. We further detected non-canonical, SECIS-like structures (with extra paired regions) downstream of various selenoproteins genes (Fig. 2, Supplementary Figures 12–13). If these truly function as Sec insertion signals, then significant alterations occurred in fungi in the SECIS consensus. In any case, none of the selenoproteins of Harpellales (except one) exhibit SECIS/SECIS-like candidates. This indicates non-canonical mechanisms of Sec recoding, at the very least in this fungal order. By analysing sequences of this lineage, we identified two evolutionary conserved structures in the coding regions of SPS and SelenoH (Supplementary Figures 14–15). The SelenoH structure is localized to the Sec TGA and immediately downstream region. In this sense, it resembles recoding-enhancing structures previously characterized in other selenoprotein genes, known as SREs29. The SPS structure locates instead just upstream of Sec TGA. In metazoans, SPS contains a SRE downstream of Sec4. It is unclear how the fungal CDS structure relates to the metazoan SRE of SPS, both in phylogeny and in function. Future experiments will be essential to pinpoint the roles of the various RNA elements identified here, as they may as well be unrelated to Sec insertion.
The different Sec-utilizing eukaryotic lineages have typically both common and private selenoprotein families. However, we identified in fungi only selenoproteins with clear homology to known metazoan families. This may be due to purely technical reasons: identification of previously undescribed selenoproteins is a difficult task and typically relies on identification of SECIS elements. Since Sec signals appear altered in fungi, it is not surprising that our SECIS-dependent searches (Methods) did not result in any such candidate. It is conceivable that additional selenoproteins are encoded in Sec-utilizing fungal genomes, possibly including families exclusive of this kingdom. The characterization of fungal Sec recoding signals will facilitate their identification.
Our discoveries may open the way for the bio-engineered production of selenoproteins in fungi. The synthesis of exogenous selenoproteins has previously been attempted in S. cerevisiae: all human Sec machinery genes were transferred and successfully expressed, but Sec insertion was never observed30. This was speculatively attributed to incompatibilities between vertebrates and fungi, due to differences in ribosome structure and translation mechanism. Possibly, the use of Sec machinery native to the fungal kingdom may lead to better results. We also propose G. prolifera as attractive model for eukaryotic selenoprotein expression. The naturally high transcript levels of selenoproteins (Supplementary Figure 11) suggest a tolerance to selenoprotein concentrations higher than most organisms.
In conclusion, we showed here genomic and transcriptomic evidences for Sec usage in fungi. While Sec synthesis machinery matches that of other eukaryotes, fungi may employ unusual mechanisms of Sec insertion involving private Sec signals, requiring further ad-hoc experiments to elucidate. In this work, we provide RNA structure candidates identified in selenoproteins, as well as a list of genes showing genomic co-occurrence with Sec machinery, to facilitate targeted experiments. Lastly, the observation of so many Sec losses within fungi opens the way to investigate the selective forces driving Sec maintenance and loss, and any possible commonalities between fungi and other lineages undergoing Sec extinctions. We anticipate that characterizing the functions of fungal selenoproteins will be key to clarify why some organisms have clung to them for so long, and, by extension, why many other species could instead lose Sec with no apparent consequence.
MATERIAL AND METHODS
Sequence data
We downloaded all NCBI genome assemblies available for fungi (Supplementary Table 1) using the script ncbi_assembly.py (https://github.com/marco-mariotti/ncbi_db), which wraps the Entrez Bio Python module. When multiple assemblies were available for the same species, only the most recent one was used. This resulted in a set of 1201 fungal genomes, each corresponding to a different species.
Gene prediction
Protein-coding gene prediction was performed using Selenoprofiles v.3.5a, a homology-based gene finder developed specifically to identify selenoproteins and related proteins17. Selenoprofiles is a pipeline based on an extended version of tblastn, which use alignments of multiple sequences, rather than single queries, to scan genomes for homologs. These genomic matches are then processed using the programs exonerate31 and genewise32 to produce complete gene predictions. tRNASec was searched using the newly developed ad-hoc tool Secmarker18 v0.2. We initially scanned all fungal genome assemblies for eukaryotic Sec machinery and inspected results (271 hits in total; Supplementary Figure 1). Then, we searched for all known selenoprotein families (eukaryotic33 and prokaryotic34) in those species with tRNASec. This resulted in a number of putative genes, both for Sec machinery and selenoproteins, which were then subjected to phylogenetic analysis (see below). We also searched Sec-utilizing species for previously undescribed selenoproteins, using the program Seblastian21, which uses SECIS identification as first step. This approach did not report any suitable selenoprotein candidate other than those predicted by Selenoprofiles. However, this may depend on the fact that fungal selenoproteins use non-canonical Sec insertion signals.
Phylogenetic analysis
In order to provide phylogenetic context, each candidate protein was searched with blastp35 v2.6.0+ against the NCBI NR database36, and the most similar proteins (e-value<1e-5) were downloaded and aligned to the fungal candidates. When more than 300 proteins were matched in NR for a certain family, these were aligned with mafft37 v7.123b, and 300 sequence representative were selected using Trimal38 v1.4.rev15. Phylogenetic reconstruction was then performed using the “build” routine of the ETE3 package39 v3.0.0b36, using the workflow “PhylomeDB”. This pipeline40 comprises the use of various aligner programs combined to a consensus alignment with M-Coffee41 v11.00.8cbe486. This is then trimmed to remove uninformative and confounding positions using Trimal. Neighbor joining phylogenetic reconstruction is then performed to assess the likelihood of 5 different evolutionary models. Lastly, the final tree is computed by maximum likelihood with PhyML42 v20160115 using the best evolutionary model. These trees were plotted using custom ETE3 scripts. The descriptions of NR proteins were then inspected to assign protein families to each phylogenetic cluster and mark them in the tree figures (Fig. 3, Supplementary Figures 2–10). Branch support values (Supplementary Data 2) were computed with approximate likelihood ratio tests, as implemented in PhyML. Our phylogenetic analyses resulted in the dismissal of a number of candidates, either attributed to bacterial contaminations of genome assemblies, or assigned to a different protein family than the one originally searched.
Species phylogeny
For most analysis (Fig. 1, Supplementary Figure 1), NCBI taxonomy was used as rough phylogenetic backbone of fungal species. Attempting to trace the events of Sec loss, we then employed a fully resolved tree of fungi including early branching lineages28. However, various species, whose genome we analysed in this study, were missing from this tree. The tree was thus merged with NCBI taxonomy and with a recently published tree of Zoopagomycota43, producing a nearly completely resolved reference tree (Fig. 2).
Prediction of SECIS elements and other RNA structures
SECIS candidates were predicted using the program SECISearch321. Our initial searches identified only five elements downstream of selenoprotein genes, which fit the canonical SECIS consensus (Supplementary Figure 12). We thus built new SECIS covariance models for Infernal44 v1.0.2 and covels: we enriched those originally used21 with the fungal sequences, and we removed 90% of vertebrate sequences to avoid overtraining on this lineage. A modified version of SECISearch3 was created to put these models to use. The built-in filter of SECISearch3, enforcing strict constraints on the various SECIS parts, was relaxed in this version. The modified filter required candidates to contain GA dinucleotides at the SECIS core (preceding stem II), a minimum length of 25 nucleotides in-between (corresponding to stem II and its apical part, possibly including any additional stems), and free energy estimated by RNAfold26 v2.4.6 smaller than −8.0 Kcal/Mol. This resulted in nine additional non-canonical SECIS-like candidates (Supplementary Figure 12). Furthermore, we performed de novo searches for any RNA structures in fungal selenoprotein sequences. We used RNALfold26 v2.4.6 with default settings to predict locally stable structures. This resulted in tens of candidate structures per gene (Supplementary Figure 13), without clear criteria to distinguish the functional ones. We thus turned to a comparative approach. The program RNAz25 v2.1 predicts thermodynamically stable conserved structures in multiple sequence alignments. Since requiring closely-related orthologous sequences, we ran RNAz only on SelenoH and SPS (i.e., the selenoproteins in Harpellales). We aligned gene sequences with both mafft37 v7.123b and Clustal Omega45 v1.2.1, and removed nearly identical sequences (>95% identity). We ran RNAz on the resulting alignments in sliding windows of 80, 100 and 120 nucleotides, using a step size of 10, on the forward strand only, with options no-reference and opt-id=50. We then filtered out RNAz predictions supported by less than three sequences, or with probability <90%. Next, we merged overlapping candidates (predicted using different parameters or alignment methods): for each group of structures with any partial overlap, we kept the one with lowest energy (and highest probability, in case of ties). This resulted in two candidate structures, both within coding sequences: one for SelenoH and one for SPS. We thus used RNAalifold26 v.2.4.6 to fold, plot and predict the free energy of Harpellales aligned sequences. We also employed alignment viewers Jalview46 v2.10.4b1 and Emacs RALEE47 v0.8 to inspect nucleotide and structural conservation across all fungi. All these analyses are presented in Supplementary Figure 14 and 15 for SelenoH and SPS, respectively. All searches described in this section were performed on the coding sequences of fungal selenoproteins extended by 600 nucleotides on each side. SECIS elements were also searched further downstream but that did not add other suitable candidates.
Phylogenetic profiling
We designed a custom procedure of phylogenetic profiling27 to identify fungal proteins involved in the Sec pathway. Aiming to detect proteins present in Sec-utilizing genomes but absent in selenoproteinless species, we searched with tblastn the NCBI proteome of Sec-utilizing Smittium culicis in all non-dikarya, non-microsporidia fungal genomes, and analysed the e-values of the best matches in each genome. For each query protein, we computed a gene presence threshold based on e-values in Sec-utilizing species, and required e-values worse than such threshold in >90% of non-Sec species. The threshold was computed as the second worst e-value in Sec-utilizing species (thus allowing one Sec-utilizing species to lack candidate proteins), and was further adjusted 20% on log scale (e.g. 1e-10 becomes 1e-8) for higher stringency. We then analysed the resulting candidates to assess the possibility that some constituted undiscovered selenoproteins. We built alignments for each homologous group, and searched them in Sec-utilizing fungal genomes using Selenoprofiles. Other than known selenoproteins in our list, the search did not report any additional in-frame UGA-containing genes. However, selenoprotein genes are typically mis-annotated by standard pipelines, with the Sec codon located either downstream of the annotated gene structure, or downstream of it, or within an artifactually predicted intron17. By relying on annotated proteins, our profile alignments may have included their deficiencies. We thus considered the possibility that potential Sec residues resided in the immediate proximity of our gene predictions. Separately for upstream, downstream and intron sequences, we considered those regions containing in-frame UGA, but not any other stop codon. Introns were further filtered to require a length multiple of 3. Finally, we translated the regions passing these filters to amino acids, and aligned and manually inspected sequences across species in order to assess their conservation. Based on lack of conservation around all possible Sec-UGAs, all candidates were dismissed as unlikely to encode for undiscovered selenoproteins.
Genome completeness
We used BUSCO48 v3.0.2 to estimate the completeness of fungal genome assemblies. This program is based on pre-defined sets of genes that are expected universally in single copy in genomes. These genes are searched in the genome assemblies under examination, and the number of absent / fragmented genes is taken as indicator of poor genome quality. We ran BUSCO with default parameters, using the fungal-specific gene set “Fungi odb9”. Results are shown in Supplementary Figure 16. We also used BUSCO predictions to obtain profiles of stop codon usage across species (Supplementary Figure 17).
Analysis of the G. prolifera transcriptome
We searched public databases for transcriptomic data of any Sec utilizing fungi. The only expression data we could find was at the JGI49 for G. prolifera (Ganpr1_EST_20111215_cluster_consensi). This came in the form of assembled transcriptome (“EST clusters”) with expression levels annotated for each transcript in RPKM (Reads Per Kilobase of transcript, per Million mapped reads). We could map all genomic predictions of Sec machinery and selenoproteins to the transcriptome, except for tRNASec. Its absence may be due to the fact that tRNAs are short and lack a poly-A tail, and thus evade detection by sequencing protocols used for mRNA quantification. Indeed, while 115 tRNAs were predicted in the genome of G. prolifera by Aragorn50 v.1.2.36, only 5 tRNAs were predicted in its transcriptome. We compared the expression levels of selenoproteins and Sec machinery to that of all other transcripts (Supplementary Figure 11, orange line). Since low-quality or non-coding transcripts might skew this analysis to overestimate expression ranks, we also considered an alternative background distribution of bona-fide genes: universal single copy orthologs, predicted by BUSCO in the transcriptome. This gene set is enriched in essential house keeping genes, expected to have expression higher than average. Thus, expression ranks (Supplementary Figure 11, blue line) may actually be underestimated when comparing to this set.
DATA AVAILABILITY
The list of fungal species and corresponding genomes (NCBI assembly accession IDs) used in this study is provided in Supplementary Table 1. Supplementary Data 1 contains the sequences of all genes and RNA elements mentioned in this work, as well as their genomic coordinates to derive these sequences from genomes. For each species, coordinates are mapped to Genbank nucleotide entries (contigs or scaffolds) found within their corresponding genome. Our re-annotated ORFs are in process of being assigned Genbank IDs.
CODE AVAILABILITY
The latest version of the selenoprotein gene finder software Selenoprofiles is available at https://github.com/marco-mariotti/selenoprofiles. The script ncbi_assembly, used to download NCBI assemblies in batch, is available at https://github.com/marco-mariotti/ncbi_db.
Supplementary Material
ACKNOWLEDGEMENTS
Not applicable.
FUNDING
This work was supported by the National Institutes of Health grants DK117149, AG021518 and CA080946.
Funding for open access charge: National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
- 1.Labunskyy VM, Hatfield DL & Gladyshev VN Selenoproteins: molecular pathways and physiological roles. Physiol. Rev. 94, 739–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu XM et al. Biosynthesis of selenocysteine on its tRNA in eukaryotes. PLoS Biol 5, e4 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Romero H, Salinas G & Gladyshev V Dynamic evolution of selenocysteine utilization in bacteria: a balance between selenoprotein loss and evolution of selenocysteine from redox active cysteine residues. Genome Biol. 7, R94 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mariotti M et al. Evolution of selenophosphate synthetases: emergence and relocation of function through independent duplications and recurrent subfunctionalization. Genome Res. 25, 1256–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lobanov AV, Hatfield DL & Gladyshev VN Eukaryotic selenoproteins and selenoproteomes. Biochim. Biophys. Acta 1790, 1424–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapple CE & Guigó R Relaxation of selective constraints causes independent selenoprotein extinction in insect genomes. PLoS One 3, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otero L et al. Adjustments, extinction, and remains of selenocysteine incorporation machinery in the nematode lineage. RNA 20, 1023–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang L et al. Evolution of selenoproteins in the metazoan. BMC Genomics 13, 446 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krol A Evolutionarily different RNA motifs and RNA-protein complexes to achieve selenoprotein synthesis. Biochimie 84, 765–774 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Gupta N, DeMong LW, Banda S & Copeland PR Reconstitution of selenocysteine incorporation reveals intrinsic regulation by SECIS elements. J. Mol. Biol. 425, 2415–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castellano S et al. Low exchangeability of selenocysteine, the 21st amino acid, in vertebrate proteins. Mol. Biol. Evol. 26, 2031 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reich HJ & Hondal RJ Why Nature Chose Selenium. ACS Chem. Biol. 11, 821–841 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Lin J et al. Comparative Genomics Reveals New Candidate Genes Involved in Selenium Metabolism in Prokaryotes. Genome Biol. Evol. 7, 664–676 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mariotti M et al. Lokiarchaeota Marks the Transition between the Archaeal and Eukaryotic Selenocysteine Encoding Systems. Mol. Biol. Evol. 33, 2441–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mariotti M et al. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS One 7, e33066 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lobanov AV et al. Evolutionary dynamics of eukaryotic selenoproteomes: large selenoproteomes may associate with aquatic life and small with terrestrial life. Genome Biol. 8, R198 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mariotti M & Guigó R Selenoprofiles: profile-based scanning of eukaryotic genome sequences for selenoprotein genes. Bioinformatics 26, 2656–63 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santesmasses D, Mariotti M & Guigó R Computational identification of the selenocysteine tRNA (tRNASec) in genomes. PLOS Comput. Biol. 13, e1005383 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox AG et al. Selenoprotein H is an essential regulator of redox homeostasis that cooperates with p53 in development and tumorigenesis. Proc. Natl. Acad. Sci. 113, E5562–E5571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castellano S et al. Reconsidering the evolution of eukaryotic selenoproteins: a novel nonmammalian family with scattered phylogenetic distribution. EMBO Rep. 5, 71–7 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mariotti M, Lobanov AV, Guigo R & Gladyshev VN SECISearch3 and Seblastian: new tools for prediction of SECIS elements and selenoproteins. Nucleic Acids Res. 41, e149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee BC, Dikiy A, Kim H-Y & Gladyshev VN Functions and evolution of selenoprotein methionine sulfoxide reductases. Biochim Biophys Acta 1790, 1471–1477 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darras VM & Van Herck SLJ Iodothyronine deiodinase structure and function: from ascidians to humans. J. Endocrinol. 215, 189–206 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Arnér ES & Holmgren A Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267, 6102–9 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Gruber AR, Findeiß S, Washietl S, Hofacker IL & Stadler PF RNAz 2.0: improved noncoding RNA detection. Pac. Symp. Biocomput. 69–79 (2010). [PubMed] [Google Scholar]
- 26.Lorenz R et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D & Yeates TO Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. U. S. A. 96, 4285–8 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spatafora JW et al. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108, 1028–1046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howard MT et al. Recoding elements located adjacent to a subset of eukaryal selenocysteine-specifying UGA codons. EMBO J. 24, 1596–607 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Labunskyy VM et al. The Insertion Green Monster (iGM) Method for Expression of Multiple Exogenous Genes in Yeast. G3: Genes|Genomes|Genetics 4, 1183–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slater GSC & Birney E Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birney E, Clamp M & Durbin R GeneWise and Genomewise. Genome Res. 14, 988–995 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gladyshev VN in Selenium 127–139 (Springer International Publishing, 2016). doi: 10.1007/978-3-319-41283-2_11 [DOI] [Google Scholar]
- 34.Zhang Y in Selenium 141–150 (Springer International Publishing, 2016). doi: 10.1007/978-3-319-41283-2_12 [DOI] [Google Scholar]
- 35.Camacho C et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.NCBI Resource Coordinators. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 45, D12–D17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K & Standley DM MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Capella-Gutiérrez S, Silla-Martínez JM & Gabaldón T trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huerta-Cepas J, Serra F & Bork P ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 33, 1635–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huerta-Cepas J, Capella-Gutiérrez S, Pryszcz LP, Marcet-Houben M & Gabaldón T PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 42, D897–D902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallace IM, O’Sullivan O, Higgins DG & Notredame C M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 34, 1692–1699 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guindon S et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–21 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Wang Y et al. Comparative Genomics Reveals the Core Gene Toolbox for the Fungus-Insect Symbiosis. MBio 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nawrocki EP, Kolbe DL & Eddy SR Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sievers F et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waterhouse AM, Procter JB, Martin DMA, Clamp M & Barton GJ Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–91 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griffiths-Jones S RALEE--RNA ALignment editor in Emacs. Bioinformatics 21, 257–9 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Waterhouse RM et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nordberg H et al. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 42, D26–D31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laslett D & Canback B ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–6 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The list of fungal species and corresponding genomes (NCBI assembly accession IDs) used in this study is provided in Supplementary Table 1. Supplementary Data 1 contains the sequences of all genes and RNA elements mentioned in this work, as well as their genomic coordinates to derive these sequences from genomes. For each species, coordinates are mapped to Genbank nucleotide entries (contigs or scaffolds) found within their corresponding genome. Our re-annotated ORFs are in process of being assigned Genbank IDs.