

Abstract
Obtaining atomic level information about the structure and dynamics of biomolecules is critical to understand their function. Nuclear magnetic resonance (NMR) spectroscopy provides unique insights into the dynamic nature of biomolecules and their interactions, capturing transient conformers and their features. However, relaxation-induced line broadening and signal overlap make it challenging to apply NMR to large biological systems. Here, we take advantage of the high sensitivity and the broad chemical-shift range of 19F nuclei, and leverage the remarkable relaxation properties of the aromatic 19F-13C spin pair to disperse 19F resonances in a 2-dimensional transverse relaxation optimized TROSY spectrum. We demonstrate the application of the 19F-13C TROSY to investigate proteins and nucleic acids. This experiment expands the scope of 19F NMR in the study of structure, dynamics and function of large and complex biological systems and provides a powerful background-free NMR probe.
Introduction:
Obtaining structural and dynamics information on biomolecules is critical for understanding their functions and mechanisms. Solution-state nuclear magnetic resonance (NMR) spectroscopy provides such information at atomic resolution and in a quantitative manner. A main advantage of NMR is that it queries individual atoms within a biomolecule to provide information on structure and local and global dynamics. However, extracting such information relies heavily on the ability to resolve individual atomic resonances. Spectral complexity and line broadening severely limit the application of NMR, particularly for larger systems.
One of the most impactful developments in biomolecular NMR has been the implementation of transverse relaxation optimized spectroscopy (TROSY)1, 2. The transverse relaxation rate (R2) determines the width of the NMR signal and it increases with the size of the system. Therefore, signals originating from larger biomolecules are broader. Two major factors that determine R2 at high magnetic field strengths are chemical shift anisotropy (CSA) and the dipole-dipole relaxation (DD). The TROSY experiment selects a component of the NMR signal for which CSA and DD negate each other, resulting in narrower lines. TROSY has enabled studies of large systems such as the 670 kDa 20S proteasome3 and the 900 kDa GroEL-GroES complex4.
Despite these advances, NMR studies of biomolecules larger than 30 kDa remain far from routine and need further methodological developments. Fluorine-19 (19F) NMR is re-emerging as a preferred method to detect weak interactions in biomolecules and to study the structure and dynamics of proteins and nucleic acids. 19F NMR provided early evidence of scalar coupling5 and the nuclear Overhauser effect6 in the 1950s and was introduced into biological NMR in the 1970s7. However, its use for study of biological systems has been limited due to the rapid 19F-CSA-driven relaxation that occurs at high magnetic fields. Nevertheless, the power of 19F NMR has been illustrated by studying GPCR dynamics using site-specific-CF3 labelling strategies8, 9. 19F NMR has also been used to probe DNA duplex formation10, site-specific RNA binding11, RNA invasion12, and telomeric RNA G-quadruplex structures in vitro and in living cells13.
19F is one of the most NMR sensitive nuclei due to its large gyromagnetic ratio (γ = 0.941 relative to 1H), 100% natural abundance, and a large chemical shift range of ~200 ppm in biological systems14. Uniquely, 19F is not naturally present in biomolecules, rendering 19F NMR background-free and thus especially useful for in cell NMR experiments15, 16. The main disadvantage of 19F is its large CSA, which broadens 19F resonances in high molecular weight systems, even at relatively low magnetic field strengths.
To address this problem and enhance the utility of 19F NMR spectroscopy, we developed a 2-dimensional (2D) 19F-13C TROSY experiment. Our theoretical calculations show that the 19F-13C TROSY effect is applicable for ten different 19F incorporations in aromatic amino acids and eight 19F substitutions in nucleobases. Intriguingly, the mutual cancellation of the CSA and DD in aromatic 19F-13C systems yields TROSY resonances for 13C nuclei bonded to 19F (13CF) that are significantly narrower than those of the corresponding 13C resonances bonded to 1H (13CH). We use a 13C-detected experiment with TROSY selection to directly observe these narrow 13CF resonances, which results in excellent spectral resolution. We provide both the theoretical and experimental framework for 19F-13C TROSY experiments and validate the approach on the 7 kDa protein G B1 domain (GB1)17, the 42 kDa maltose binding protein (MBP)18, and the 180-kDa α7 single-ring of the 20S proteasome core particle (CP) from Thermoplasma acidophilum3, 19, as well as a 16-mer, double-stranded DNA at low temperature.
Results:
19F-13C DD-CSA cross-correlation in 3-19F13C Tyr and the TROSY effect
While R2 in spin ½ nuclei like 19F and 13C is dominated by DD and CSA mechanisms, their individual contribution can vary. In the case of aromatic 19F-13C systems, the line width is dominated by the CSA mechanism, especially at high magnetic fields. We calculated chemical shielding tensors for ten 19F-13C spin pairs at various positions in aromatic amino acid side chains using DFT methods (Supplementary Note I; axiality and rhombicity parameters of the CSA tensors are collected in Supplementary Table 1) and then used the Bloch-Redfield-Wangsness relaxation theory module available in the Spinach20 library to compute the R2 relaxation rates for each 13C-19F pair (Supplementary Fig. 1). While there are variations in R2, dependent on the amino acid type and 19F position, the calculations indicate that the R2 of the TROSY component of 13CF is 3 to 18-fold slower than that of the corresponding decoupled resonance. Thus, selective detection of the 13CF TROSY resonance would yield sensitive, high-resolution NMR spectra for 19F-13C pairs in any aromatic amino acid.
CSA contribution to the R2 scales quadratically with the magnetic field, while the DD contribution is much less sensitive to increase in magnetic field strength. This is reflected in the magnetic-field-dependent increase in R2 calculated for decoupled resonances (e.g.: 3-19F Tyr; Fig.1, blue triangles). However, CSA relaxation that is comparable to DD relaxation often generates favourable DD-CSA cross-correlation, which makes one of the components of the 13CF coupled doublet relax slower compared to the other (Fig. 1a, red squares). Which doublet component relaxes slower depends on the angles between the DD and the CSA tensor axes, and on the sign of the J-coupling. The narrower (slower relaxing) line is referred to as the transverse-relaxation-optimized “TROSY” line1, 2. For the 13CF resonances, our estimates indicate that the TROSY component of the 13CF doublet relaxes 8.7 times slower (6.1 s−1) compared to the decoupled signal (53 s−1) for 3-19F Tyr incorporated in a 42 kDa protein at 600 MHz (1H frequency; τc = 25 ns at 25 °C) (Fig. 1a). By comparison, the TROSY components of 13CH resonances relax with a rate of 43 s−1 (Fig. 1b) under identical conditions. On the other hand, the TROSY effect on the 19FC and 1HC resonances is rather small (Fig. 1c and Fig. 1d). Thus, selective detection of TROSY resonances from 13CF would allow detection of the 19F-13C pair with high sensitivity and resolution.
Figure 1: Theoretical transverse relaxation rates (R2) of 13C, 19F, and 1H resonances as a function of magnetic field strength.
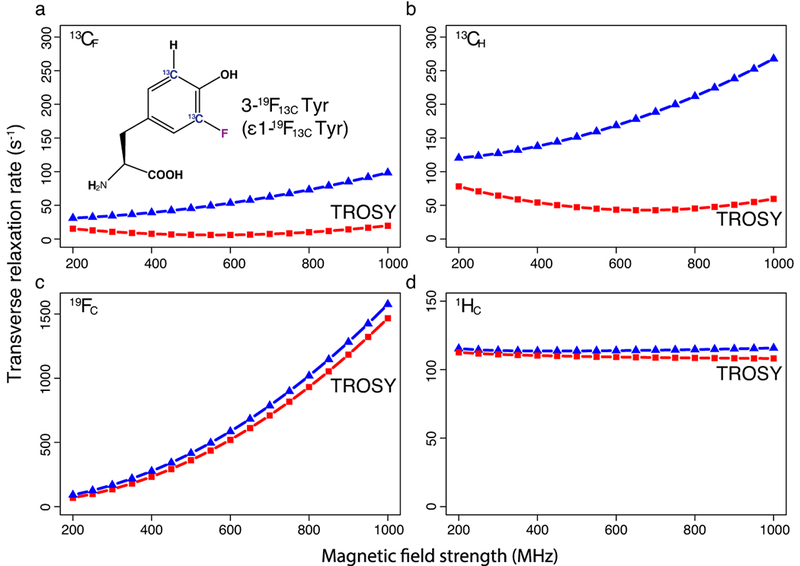
(a) R2 of 13CF in 3-19F Tyr and (b) R2 of 13CH in Tyr. (c) R2 of 19FC in 3-19F Tyr and (d) R2 of 1HC at position 3 in Tyr. Here 3-19F Tyr (panel A inset) and Tyr are part of a 42 kDa protein with a τc of 25 ns. Red squares represent the R2 of the TROSY component and blue triangles represent the R2 of the decoupled resonances. The points are connected by a dashed line to provide visual clarity.
Experimental observation of 19F-13C TROSY effect from 1D 13C spectra
In order to harness the power of aromatic 19F-13C TROSY selection and to avoid splitting of 13C resonances by the one-bond C-C coupling (1JCC ~60Hz), we synthesized a selectively 13C-labeled and fluorinated tyrosine, 3,5-13C2-3-fluoro-L-tyrosine, which has a 19F-13C pair at the 3rd position in the aromatic ring (3-19F13C Tyr), using a modified method described by Kitevski-LeBlanc et al.21. To demonstrate the applicability of the 19F-13C TROSY experiment, we expressed two model systems, a small protein, G B1 domain (GB1), and a larger protein system, maltose binding protein (MBP) in E. coli using 3-19F13C Tyr.
GB1 (τc = 5 ns) which harbours three tyrosine residues, gives three resonance pairs in its 1D 13C spectrum, each of which are separated by ~240 Hz, the expected 19F -13C (1JCF) coupling value (Fig. 2a). The TROSY effect is clearly seen in these 1D spectra acquired at different field strengths. The TROSY resonances which are up-field are intense and narrow while the down-field anti-TROSY signals are weak and broad. 1H decoupling was applied during acquisition to remove remote (>two-bond) 1H couplings. Although our labelling technique removed the strong one-bond 13C-13C coupling (1JCC ~ 60Hz), the 13C-labeling at the 5th position in the aromatic ring, originating from the stable-isotope precursor, caused a two-bond 13C-13C coupling (2JCC ~7 Hz). The narrow TROSY lines make this small coupling clearly visible in the 1D 13C spectra as a testament to the resolution gain (Fig. 2a).
Figure 2: TROSY effect observed in a coupled 1D 13C spectrum of GB1 at multiple magnetic field strengths.
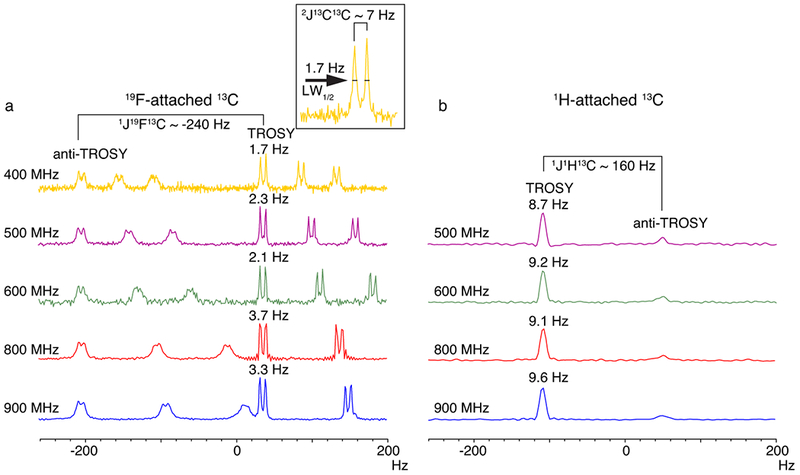
(a) 13CF as compared to (b) 13CH. Linewidths at half height are indicated above the TROSY component of the Tyr 13CF or 13CH resonances, respectively. In (a), the ~7 Hz splitting due to 2JCC coupling is highlighted in the inset panel. Linewidth at half height is abbreviated as LW1/2. Note that there is no 2JCC coupling present in 13CH as it is recorded on natural abundance 13C. Due to signal overlap, only one isolated Tyr 13CH resonance is shown in (b). In the 800 and 900 MHz spectra depicted in (a), one down-field doublet was cut off for clarity.
To compare the effect of TROSY on 13CF and 13CH resonances at equivalent positions, we recorded a 1D 13C spectrum of unlabelled GB1 using natural abundance 13C, to negate the 1JCC coupling. Although the TROSY effect can be seen clearly in the sharper, more intense up-field TROSY component of the 13CH resonances, the linewidths are much broader than those of 13CF (Fig. 2b), highlighting the effect of the significantly slower R2 of 13CF. It should be noted that to see the TROSY effect, 1H decoupling could not be applied during 13CH detection and therefore the remote 1H couplings contribute to the line broadening. Nevertheless, unlike the very narrow 13CF lines, which allowed resolution of the 7Hz 2JCC couplings, the broader lines of the 13CH nuclei obscure the individual detection of these remote couplings. Of note, TROSY resonance of 13CF is the up-field component, whereas the TROSY resonance of 13CH is the down-field component due to the opposite sign of the 19F-13C and 1H-13C 1J-coupling (negative and positive, respectively).
The 2D 19F-13C TROSY experiment
Our 1D-13CF data indicated the need to choose the up-field 13CF component for TROSY selection in a 2D 13C-19F TROSY experiment. Theoretical calculations showed that the 19FC TROSY component relaxed more slowly as compared to the decoupled resonance. To observe the TROSY effect on 19FC and choose the correct TROSY resonance we recorded decoupled and coupled 13C-19F HSQCs of MBP (Supplementary Fig. 2). The coupled spectrum showed that the up-field 19F component was narrower, which, when taken together with the 1D-13C results, indicates that the top-right resonance is the TROSY component.
To selectively choose the TROSY resonance we designed an experiment with an ST2PT pulse scheme for TROSY selection (13C-19F TROSY-SE), similar to that used for the 15N-1H TROSY-HSQC22 (Supplementary Fig. 2). In this sensitivity enhanced pulse-sequence design, the experiment starts with and ends on 19F magnetization, the nucleus with the larger gyromagnetic ratio, with the 13C frequency encoded in the indirect dimension. However, while the experiment worked well for GB1, the experiment was not sufficiently sensitive for the higher molecular weight protein MBP (42 kDa), as we were not able to observe all the fifteen 3-19F13C Tyr resonances at 25 °C. We hypothesized that this was due to faster relaxation of the 19FC nuclei. The CSA of 19FC accelerates its relaxation significantly and is the primary reason for the poor sensitivity observed in the 13C-19F TROSY-SE experiment. Indeed, shortening the experiment by removing the sensitivity enhancement block had no significant effect on signal intensity, as the reduction in time cancels the gain of the sensitivity enhancement block (Supplementary Fig. 3). The contributions of CSA from 19F and 13C nuclei to the R2 of the coherences present in the 13C-19F TROSY pulse sequence are shown in Supplementary Fig. 4. Any coherence with transverse 19F magnetization (19Fx/y,19Fx/y13Cz,19Fx/y13C x/y) experiences ~22-fold faster CSA relaxation as compared to that of 13C (13Cx/y,19Fz13Cx/y), suggesting that it would be beneficial to minimize the time 19F spends in the transverse plane. We estimated that, for a 42 kDa protein, ~30% of the initial magnetization is lost during the first INEPT transfer from 19F to 13C due to relaxation. Consequently, we shortened the pulse sequence by employing an out-and-stay design in which the experiment starts with excitation and indirect encoding of one of the nuclei, 13C or 19F, and ends with the direct detection and encoding of the other. Using this approach, we designed two additional experiments a 13C-19F TROSY, which begins with 13C magnetization and ends on 19F, and a 19F-13C TROSY, which starts with 19F magnetization and ends on 13C, the more slowly relaxing nucleus (Supplementary Fig. 5). These experiments utilize the ST2PT block where the phases of the first 90° pulse in 13C and 19F, respectively, are chosen to observe the upfield 13CF TROSY component (Supplementary Fig. 6).
Although the 13C -19F TROSY was more sensitive than the 19F-13C TROSY, the latter had much better resolution (Fig. 3a–c). Fifteen intense and isolated 19F-13C pairs were detected by the out-and-stay experiments, which correspond to the fifteen Tyr residues in MBP. This resolution demonstrates the advantage of the 2D experiments, as both the 19F and 13C 1D-spectra of MBP cannot resolve individual resonances (Fig. 3d and 3e).
Figure 3: 19F-13C TROSY experiments for the 42 kDa MBP with different excitation and detection schemes.
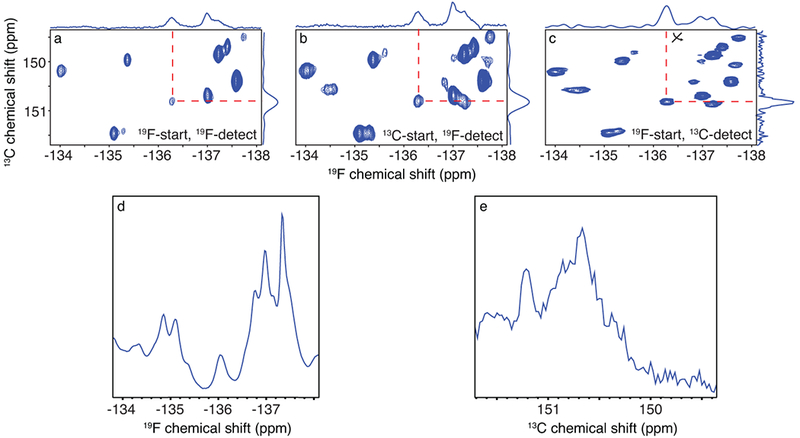
(a) 19F-start, 19F-detected, (b) 13C-start, 19F-detected, and (c) 19F-start, 13C-detected experiments were recorded on a 700 μM 3-19F13C Tyr-labelled MBP sample, at 600 MHz. The axes were transposed in (c) for easier comparison. (d) 1D 19F spectrum of MBP and (e) 1D 13C spectrum of MBP.
The 13C- and 19F-detected out-and-stay designs had substantially greater sensitivity than the 19F-detected out-and-back style method. In the 19F-13C TROSY pulse program, we take advantage of the large polarization of 19F with its initial magnetization and the faster longitudinal relaxation of 19FC (T1 ~0.5s) to allow faster recycling delays than are possible in a design where we start with 13CF magnetization (T1 ~1.4s) (Supplementary Fig. 7). Furthermore, by starting on 19F, the nuclei with the narrower 13CF TROSY resonances are encoded in the direct dimension, which allows access to high-resolution without sacrificing experimental time.
The choice between the 19F and 13C detected out-and-stay experiments is dependent on the sample concentration, the rotational correlation time of the sample, the sample-dependent spectral width in the individual dimensions, the field strength and the probe design. Although we recorded both 19F- and 13C-detected experiments for comparison and have outlined the advantages of each above, we favour the 19F-13C TROSY pulse program. In this design, the maximum evolution in the indirect 19F dimension is limited to ~20 ms for sensitivity, and the more slowly relaxing 13C is encoded in the direct dimension for >200 ms, giving access to the high resolution unlocked by the 19F-13C TROSY effect.
Comparison of 19F-13C TROSY with 1H-13C TROSY
The TROSY resonances of 13CF in 3-19F Tyr are ~7 times narrower than the corresponding 13CH for systems across a wide range of molecular weights (Supplementary Fig. 8). As an illustration, the linewidth of the 13CF TROSY resonance of a protein with τc = 95 ns is narrower than the 13CH TROSY resonance of a protein with τc = 15 ns. This is due to the more efficient cross-correlation process (or TROSY effect) in the 19F-13C system, where the magnitude of the CSA term in the spin Hamiltonian is comparable to the dipole-dipole interaction. Thus, selecting the TROSY component of 13CF will yield sharper lines than those of 13CH. To experimentally validate this, we took advantage of the presence of two 13C-labelled carbons in 3-19F13C Tyr. While the carbon at position 3 is bonded to a 19F atom, the carbon at position 5 is bonded to 1H. This labelling pattern allows the measurement of 1H-13C and 19F-13C aromatic TROSY on the same sample. We recorded 13C-detected 19F-13C and 1H-13C TROSY spectra of 3-19F13C Tyr-labelled MBP at 25 °C and at 10 °C (Fig. 4). A 42 kDa MBP at 10 °C, behaves with the relaxation properties of a ~65 kDa protein at 25 °C. At 25 °C, in the 19F-13C TROSY we observe 15 sharp and well-resolved cross-peaks as expected for the 15 Tyr. By comparison, only 12 strong and 2 weaker peaks are observed in the 1H-13C TROSY, and the cross-peaks are clearly broader when compared to their 19F-13C TROSY counterparts. At 10 °C, the advantage of the 19F-13C TROSY compared to the 1H-13C TROSY is more pronounced. 19F-13C TROSY lines are moderately broadened at 10 °C, while the linewidths of the 1H-13C TROSY cross-peaks are severely broadened, so that only two cross-peaks can still be recognized.
Figure 4: The 13CF TROSY resolves all expected cross-peaks for 3-19F13C Tyr-labelled MBP at 10 °C.
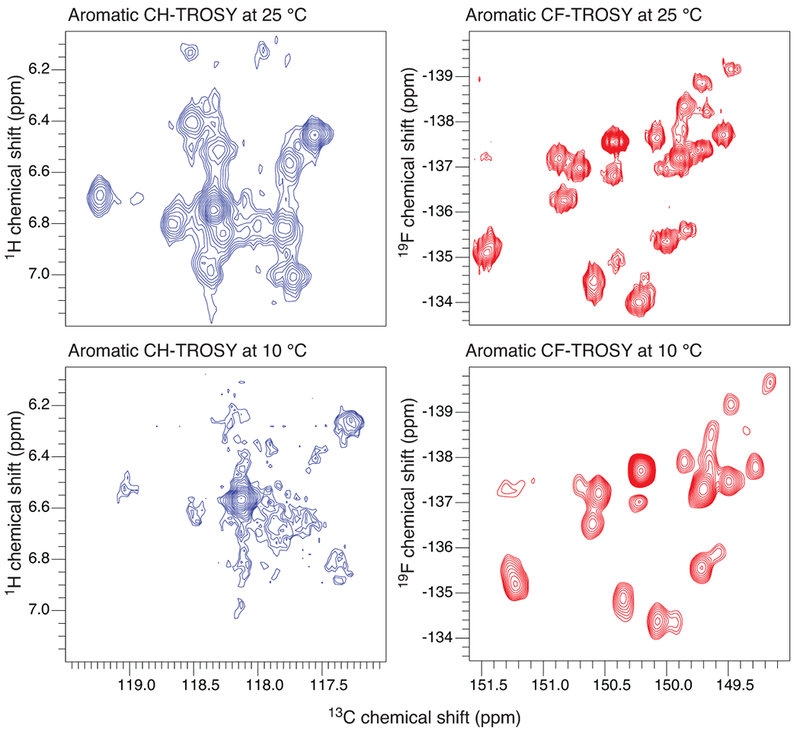
Comparison of the 1H-13C (left) and 19F-13C (right) TROSYs of the 5-1H13C and 3-19F13C Tyr spin-pairs at 25 °C (spectra in the top panel) and 10 °C (spectra in the bottom panel).
The effect of molecular weight on the relaxation of the 19F-13C TROSY component can be further highlighted by comparing the TROSY and the anti-TROSY resonances. (Supplementary Fig. 9). A larger molecular weight system can be mimicked by observation at lower temperatures. The linewidth and intensity of the TROSY resonances were barely affected by lowering the temperature from 25 °C to 10 °C, whereas the anti-TROSY resonances were severely broadened and diminished in intensity.
In addition to the fifteen intense peaks observed for MBP, there are some additional weaker signals observed in the 19F-13C TROSY spectrum (Supplementary Fig. 10). Rotation along the Cγ-Cβ bond is commonly referred to as an aromatic ring flip and has been previously observed for several aromatic amino acids (non-fluorinated). Some of these ring flips have been shown to be in slow exchange on the chemical shift time scale23. In unlabelled Tyr residues this leads to two distinct signals of equal intensity due to the symmetry of the aromatic ring. In contrast, 3-19F13C Tyr is no longer symmetric and the differences between 19F and 1H, e.g. the ability of fluorine to hydrogen bond, could shift the equilibrium towards one conformation, producing the observed additional peaks of lesser intensity
To show the applicability of the 19F-13C TROSY to a more challenging and biologically relevant system, we prepared the 3-19F13C labelled 180-kDa α7 single-ring of the 20S proteasome core particle (CP) from Thermoplasma acidophilum3. This particle lacks residues 97-103 (including one Tyr), to prevent formation of the α7-α7 subunit19, and thus, our final construct contained 10 Tyr residues. 19F-13C TROSY spectra at 45 °C (τc = 66 ns), 50 °C (τc = 60 ns)3, 24 and 60 °C (τc = 50 ns), show 8-10 discrete 3-19F13C Tyr signals (Figure 5).
Figure 5: 13CF TROSY resonances of the 180-kDa proteasome α7 single-ring particle.
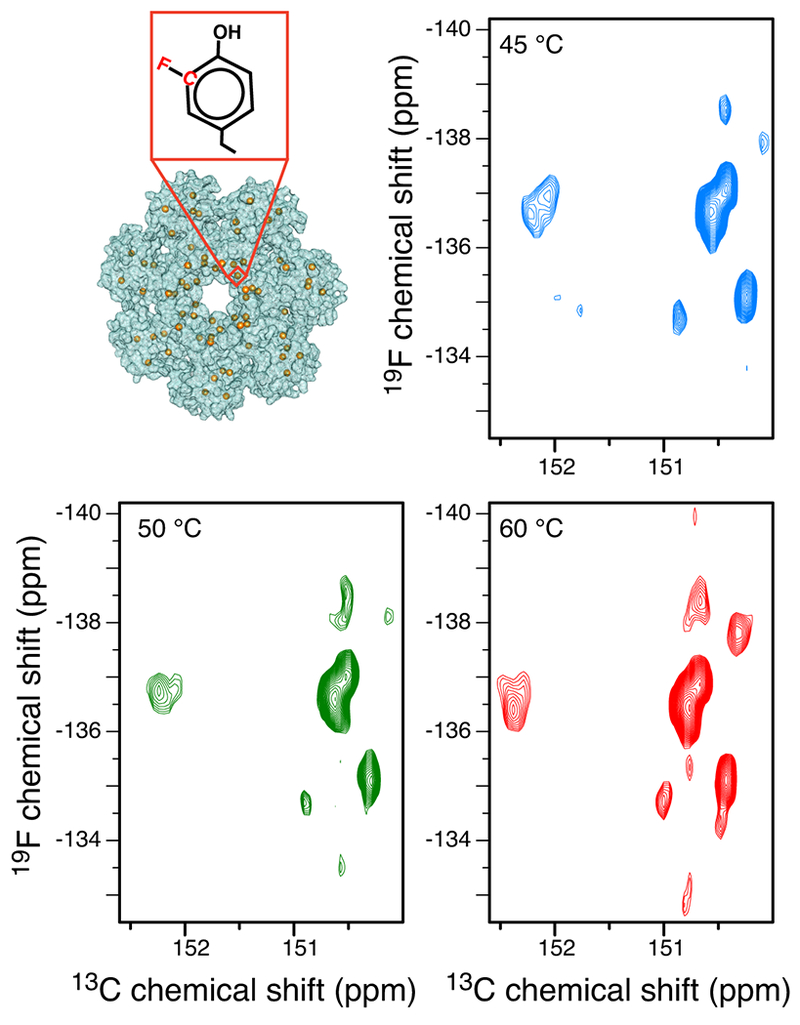
19F-13C TROSY spectra of the 180-kDa proteasome α7 single-ring particle recorded at 45 °C (τc = 66 ns, blue), 50 °C (τc = 60 ns, green) and 60 °C (τc = 50 ns, red), respectively.
19F-13C TROSY effect in ribonucleic acids
The aromatic 1H-13C TROSY effect has been previously harnessed to obtain narrow lines in nucleic acid NMR25, and independently, 19F-substituted nucleobases have been used in the study of nucleic acids, including in cell NMR applications13, 26. Therefore, we posited that the aromatic 19F-13C TROSY described here could also be applied to nucleic acids. We calculated R2 to determine the TROSY effect for guanine, uracil, thymine, cytosine and adenine nucleobases, with 19F substitutions in eight different positions (Supplementary Fig. 11). For the following discussion, we will consider a fluorinated nucleotide and its non-fluorinated counterpart, to be part of a nucleic acid with a τc of 25 ns measured at 600 MHz.
For a nucleobase harbouring a 19F-13C pair, our calculations predicted that the TROSY component of 13CF would, on average, relax 4 times slower than the equivalent decoupled resonance. Similar to the aromatic amino acids, the cross-correlation of 13C-19F pairs is more effective than that of 13C-1H pairs in nucleic acids (e.g. 5-19F Uracil, Supplementary Fig. 12). This makes the R2 of the TROSY component on average 3 times slower for 13CF than that of 13CH at the same position. 2-fluoroadenine represents a special case, where the TROSY component of 13CF relaxes 58 times slower as compared to the decoupled resonance, yielding theoretically predicted linewidths of < 1Hz. This 13CF TROSY relaxation rate is approximately 29 times slower than the TROSY component of the corresponding 13CH. Intriguingly, the 19FC R2 rates of several nucleotide configurations are considerably slower than those of aromatic amino acids. For example, the 19FC relaxation rate of the TROSY component in 5-fluorocytosine is only 181 Hz in comparison to the 520 Hz of 3-19F Tyr.
To experimentally observe the 13C-19F TROSY effect in nucleic acids, we synthesized a double stranded 16-mer 5-fluorouracil-substituted DNA. We used natural abundance 13C to record 19F-13C correlations at 5 °C (Fig. 6). An approximate rotational correlation time of 11.6 ns was calculated using HYDRONMR27. The coupled 19F-13C HSQC spectrum shows four resonances with a sharp and intense top right component. Similar to what was previously observed for 3-19F13C Tyr, the upfield shifted carbon components are clearly narrower than the downfield counterparts. Figure 6b and c, show selection for the TROSY and anti-TROSY components in the 5-fluoruracil 16-mer at 5 °C, respectively. The TROSY effect can also be observed for the upfield 19FC component, confirming the prediction of an effective 19FC TROSY for 5-fluorouracil.
Figure 6: TROSY selection of the narrowest component in 19F-13C correlation spectra of a 16-mer 5-fluorouracil-substituted DNA, at 5 °C.
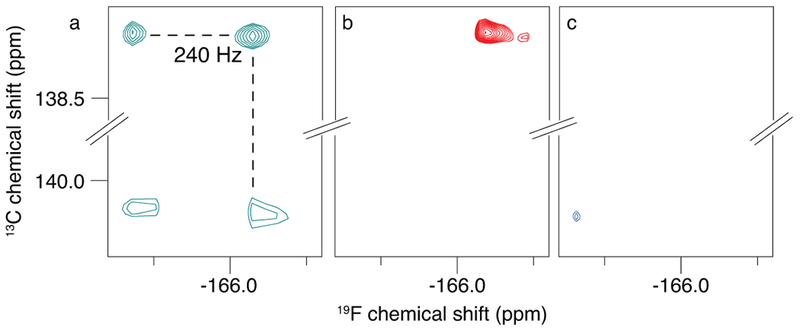
a) Coupled HSQC. In b) the top right “TROSY” component, and in c) the bottom left “anti-TROSY” component is selected. The spectra were recorded at 600 MHz, b and c are time-equivalent spectra with equivalent contour levels.
Discussion:
Fluorinated amino acids and nucleobases are easily incorporated into proteins and nucleic acids, respectively, as demonstrated here and reviewed elsewhere28, 29. Indeed, 19F is often considered an isostere of 1H, due to their similar van der Waals radii30. Unlike other chemical modifications, fluorine incorporation into the aromatic rings of amino acids seems to have minimal effect on the structure and function of the resulting protein31, although this might be protein-specific. If incorporation of a given 19F-labelled amino acid is found to be detrimental, fractional labelling can be used to dial down the percentage of incorporation. This strategy was recently applied to improve the stability and spectral quality of 3-fluoro-phenylalanine labelled calmodulin32.
In general, incorporation of fluorinated aromatic amino acids is usually not a technical obstacle for those proteins that express in high yields in E. coli. In our hands, expression of MBP and GB1 yielded 100 mg of purified protein per litre of E. coli culture. Other fluorinated aromatic amino acids have been expressed with similarly excellent yields, using auxotrophic strains or by incorporation of 5-fluoroindole as a precursor for 5-fluorotryptophan33, 34. However, incorporation of fluorinated aromatic amino acids using yeast or other eukaryotic expression systems will require further optimization. For example, P. pastoris strain X33 Δaro1 has been previously used in this context35, although in this strain incorporation of the fluorinated amino acids was stochastic, yielding mixtures of differentially fluorinated proteins36. Additionally, while fluorinated amino acids can be recognized as substrates by aminoacyl-tRNA synthetases (AARSs), their efficiency of recognition is generally worse than that of their natural counterparts. In light of this, efficient incorporation of 19F-labeled aromatic amino acids in a eukaryotic expression system may require directed evolution of AARSs, or co-expression of AARSs specific for the given non-canonical amino acid. Thus, some additional development of eukaryotic overexpression systems will be needed to further broaden the range of proteins that can be subjected to 19F-13C TROSY.
Site specific 19F resonance assignment can be accomplished through the use of site-directed mutagenesis of the relevant residue or nearby residues31, in conjunction with an HCCF-COSY experiment21, or with heteronuclear NOESY (HOESY) between 19F and 1H37. Alternatively, one can exploit the scalar coupling between the fluorine nucleus and adjacent aromatic protons38, or use solvent-induced isotope shift experiments to evaluate solvent accessibility23, 39. The latter would greatly benefit from the 19F-13C TROSY effect. The pulse programs described here can be incorporated into the aforementioned NMR experiments, and introduce an additional dimension using the 13C-19F correlation, thus facilitating resonance assignment. The unique chemical shift of the fluorinated 13C resonances at ~150 ppm, separated from the aromatic 13CH resonances (at ~120 ppm), will further aid the assignment of 19F resonances. With the appropriate NMR probe design, experiments that encode both 1H-13C and 19F-13C correlations can be recorded using parallel receivers40.
The specific NMR probe design we used here does not allow 1H decoupling during the detection period, resulting in lines broadened by not only 2JCC couplings (~7 Hz) but 2JCH and 3JCH couplings as well, as seen in the 19F-13C TROSY spectra of MBP (Fig. 4). Thus, a probe design that allows 1H decoupling during the detection period would result in further improvement of linewidths and signal height. We demonstrate the effect of additional 1H decoupling using a room temperature probe that allows pulsing on 1H, in addition to pulses applied on the 13C and 19F channels (Supplementary Fig. 13). Since the interference of 19F-13C dipole-dipole coupling and CSA is most effective on 13CF nuclei, and only marginally affects 19FC relaxation rates, the 13C spin could be decoupled during 19F evolution in a 13C-19F TROSY experiment to enhance sensitivity, as done in aromatic 13C-1H TROSY experiments.
The fact that we were able to observe the ~7Hz splitting of the 13CF TROSY resonance (Fig. 2) is a powerful illustration of the linewidth improvements that can be obtained using 19F-13C TROSY. However, the 2JCC coupling does introduce additional level of complexity and could be removed by selective 13C labelling or band selective 13C decoupling in a 13C-19F TROSY experiment. Furthermore, it should be noted that 4-19F-13C Phe can be synthesized from commercially available 4-13C Tyr41. This amino acid would have a single 13C connected to 19F, thereby obviating the need for 13C decoupling. Fluorinated analogues of tryptophan, 4-, 5-, 6- and 7-fluorotryptophan have been used quite extensively as fluorescent and NMR probes42, 43. However, synthesizing the corresponding isotopomers containing a 13C-19F bond is challenging because the 13C-fluoroindole moiety has to be chemically or enzymatically assembled from simpler 13C-labeled precursors44. Treatment of tryptophan with an electrophilic fluorinating agent under the conditions reported here for tyrosine yields 3-fluorooxindole instead of fluorotryptophan45.
The ability to disperse the 19FC peaks with the narrow 13CF resonances provides a new strategy for investigating dynamic processes, including weak binding, enzyme kinetics, conformational exchange, and protein folding, as well as the physical and thermodynamic properties of proteins and nucleic acids29. 19F-13C TROSY combined with 19F relaxation dispersion measurements would enable us to monitor a wide range of dynamics at different timescales. The large chemical shift dispersion and the sensitivity of the 19F chemical shift, coupled with the dispersion of the 19F chemical shift by the narrow 13C resonances, enables the resolution of minor states and the detection of exchange processes (in the range of 100-500 μs) that cannot be explored with traditional 13C- or 15N-relaxation-dispersion experiments46, 47.
Reduced spectral crowding and the strong 1JCF coupling make the 13C-19F spin-pair an excellent tool to measure residual dipolar couplings (RDCs). RDCs provide distance independent information about structure and dynamics48. Since the TROSY effect is largely experienced by the 13C nuclei, measuring both resonances corresponding to the 13CF TROSY component, will yield sharp and intense peaks, which could be used to extract residual dipolar couplings (RDC).
The 2D 19F-13C correlation spectrum can also be used in conjunction with protein-observed 19F-fragment screening28 to elucidate information about the binding sites of fragments on the surface of the protein. The method could further be extended to study the interactions of fluorine-bearing small molecules with bio-molecules, particularly if the cross correlation between the 19F-13C DD and CSA relaxation is favourable.
Fluorinated nucleotides are frequently employed as cancer drug antimetabolites. For example, 5-fluorouracil and its prodrug capecitabine are used for the treatment of cancers, including colorectal, breast, and head and neck malignancies49. Fludarabine, a prodrug of 2-fluoroadenosine, has been approved for clinical use for the treatment of leukaemia and has shown strong potential for prostate cancer50. However, the mechanisms of action for these drugs are incompletely understood, and their use is associated with serious side effects50. With the 19F-13C -TROSY, we could leverage the slow 13CF relaxation predicted for 2-fluoroadenosine and 5-fluorouracil, in combination with in cell NMR, to investigate binding, number of targets and metabolism of these drugs.
In summary, we have demonstrated that the 19F-13C TROSY experiment with 13C direct detection can generate spectra with sensitive, high-resolution signals from a high molecular weight system up to 180 kDa. Theoretical calculations of transverse relaxation rates for the 13CF, 13CH and the 15N-1H (15NH) TROSY components reveal that the narrow 13CF TROSY resonances are sharper than any other resonance in biological NMR, including the extremely narrow 15NH TROSY resonances (Supplementary Fig. 14). It should also be noted that detection of 13CF TROSY does not need deuteration, which is required for the methyl TROSY. In addition, observing one resonance per aromatic amino acid of choice vastly reduces the spectral complexity as compared to detecting resonances of methyl bearing amino acids.
Given that aromatic residues are critical in providing the structural framework and play an important role in the function of biomolecules, the 19F-13C TROSY we describe here will expand the utility of the existing arsenal of NMR methods and allow the detection of these residues in both high molecular weight systems as well as in biomolecular systems with complex conformational behaviours where standard NMR strategies are impeded.
Online Methods:
Synthesis of 3,5-13C2-3-fluoro-L-tyrosine (3-F Tyr)
Selectfluor® was purchased from Fluorochem (Hadfield, UK). HPLC-grade acetonitrile and methanol as well as all other reagents were purchased from Labimex Ltd (Sofia, Bulgaria) and used without further purification. 3,5-13C2-L-tyrosine was purchased from Cambridge Isotope Laboratories (Tewksbury, MA). Preparative HPLC was performed on a Waters® 600 Delta system equipped with a Phenomenex® Luna C-18 column (250 × 21 mm, 5μ) and electrospray ionization mass spectrometry (ESI-MS) detection in the positive mode using a Waters® Micromass ZQ 2000. NMR spectra were recorded on a Bruker 500 MHz spectrometer equipped with an AvanceIII console.
The synthesis follows a modified procedure of Kitevski-LeBlanc et al.21 (Supplementary Note II). L-tyrosine (phenol-3,5-13C2, 95-99%) (100 mg, 0.55 mmol) was suspended in 20 mL of acetonitrile:water (4:1) in a screw cap vial and heated to 80°C for 30 minutes in a polyethylene glycol 400 bath. The vial was removed from the bath briefly, Selectfluor® (N-Fluoro-N’-chloromethyltriethylenediaminebis(tetrafluoro-borate)) (586 mg, 2.4 equivalents) was added, and the vial was returned to the heating bath. The suspension cleared within minutes and stirring was continued for 1h at 80°C. The course of the reaction was monitored by mass spectrometry. At the end of the 1 h incubation, the ratio of tyrosine to 3-fluorotyrosine and 3,5-difluorotyrosine was 30:60:10. Longer reaction times or excess of fluorinating agent resulted in larger fractions of difluorinated product, which could not be recycled, unlike unreacted tyrosine. The reaction solution was diluted with 20 mL of 0.1 M HCl and the acetonitrile was evaporated fully under reduced pressure. The remaining aqueous solution was filtered through a 0.45 μm PTFE syringe filter and purified in 5 mL volumes by preparative HPLC with ESI-MS detection. Gradient elution over a 15-min period at a flow rate of 13 mL/min with a starting solvent ratio of 1.25:1.25:97.5 (methanol:acetonitrile:0.1% HCOOH) and an ending solvent ratio of 10:10:80 (methanol:acetonitrile:0.1% HCOOH) was used. Retention times of tyrosine, 3-fluorotyrosine and 3,5-difluorotyrosine were 9.4 min, 10.3 min and 11.8 min, respectively. 3-fluorotyrosine-containing fractions were evaporated under reduced pressure to a film, which was then triturated with diethyl ether to yield 40 mg of tan powder (36% from tyrosine). In addition, another 15 mg of unreacted tyrosine was recovered.
1H NMR (500 MHz CD3OD:D2O (1:1)) δ 3.10 (dd, J = 14.7, 7.5 Hz, 1H), δ 3.23 (dd, J = 14.7, 5.5 Hz, 1H), δ 4.08 (dd, J = 7.5, 5.5 Hz, 1H), δ 6.97 (d, J = 8.3, 1H), δ 6.98 (dq, J = 161.3, 8.7 Hz, 1H), δ 7.08 (ddd, J= 11.9, 5.6, 1.63 Hz). 13C NMR (126 MHz, CD3OD:D2O (1:1)) δ 118.20 (d, J = 7.2 Hz), δ 151.28 (dd, J = 245.3, 7.2 Hz). 19F NMR (471 MHz, CD3OD:D2O (1:1)) δ −136.5 (d, J = 245.3 Hz). LRMS calculated for C713C2H11FNO3+ [M+H]⁺ 202.08, found 202.12.
Expression and purification of 3-F Tyr GB1, MBP and the single-ring a7 particle of the 20S proteasome core particle:
BL21 (DE3) E. coli were transformed with plasmids encoding the protein G B1 domain (GB1; pET9d), maltose binding protein (MBP; pMALC4X), or the single-ring α7 particle (α7, modified pET3). Cultures were grown at 37 °C in a shaker-incubator in M9 minimal medium supplemented with 2 g/L 13C-glucose and 1 g/L 15NH4Cl. To achieve uniform incorporation of 3-19F13C tyrosine, cells were first grown to an optical density of 0.5 measured at a wavelength of 600 nm (OD600). Then 1 g/L glyphosate was added, along with 50 mg/L 3-19F13C l-tyrosine, 50 mg/L l-phenylalanine, and 50 mg/L l-tryptophan for GB1 and MBP expression. α7 was expressed with 80% 3-19F13C l-tyrosine labelling. To this end 70 mg/L l-phenylalanine, 70 mg/L l-tryptophan, 5 mg/L l-tyrosine and 30 mg/L 3-19F13C l-tyrosine were added alongside 1 g/L glyphosate. Cultures were grown to an OD600 of 0.7-0.8 at which point protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). The cultures were incubated for an additional 16 h at 28 °C (MBP and GB1) or 30 °C (α7) to allow for protein expression. Cells were pelleted by centrifugation for 20 min at 4 °C at 3,500 × g.
GB1-expressing cell pellets were resuspended in 40 mL of GB1 lysis buffer (50 mM Tris-HCl (pH 8.0), 350 mM NaCl, 10 mM imidazole, 5 mM β-mercaptoethanol (β-ME)) per liter of original bacterial culture and lysed by sonication. Cell debris was removed from the crude lysate by centrifugation at 4 °C for 40 min at 33,000 × g. GB1 was purified from the resulting supernatant by gravity-flow affinity chromatography using 5 mL (10-mL of a 50% slurry) of Ni-NTA resin (Qiagen). After washing the resin with 40 mL of 50 mM Tris-HCl (pH 8.0), 350 mM NaCl, 40 mM imidazole, and 5 mM β-ME, the protein was eluted in an identical buffer containing 350 mM imidazole. GB1 was then further purified using size exclusion chromatography (GE Healthcare Life Sciences, Superdex 75 10/300 GL pre-packed size exclusion chromatography column) into NMR buffer (10 mM Na2HPO4 (pH 6.5), 50 mM NaCl, 1mM EDTA).
MBP-expressing cell pellets were resuspended in 40 mL MBP lysis buffer (25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA) per liter of original bacterial culture, lysed by sonication, and clarified by centrifugation for 40 min at 4 °C at 33,000 × g. The lysate was loaded onto 5 mL of amylose resin. After washing the resin with resuspension buffer, MBP was eluted with the addition of 40 mM d(+)-maltose. MBP was further purified using size exclusion chromatography (GE Healthcare Life Sciences, Superdex 75 10/300 GL pre-packed size exclusion chromatography column) into NMR buffer (10 mM HEPES (pH 6.5), 1mM EDTA, 1mM β-cyclodextrin).
α7 cell pellets were disrupted by sonication, and the insoluble fraction was removed by centrifugation for 40 min at 16,000 rpm. The protein was initially purified by gravity-flow affinity chromatography using 5 ml (10-ml of a 50% slurry) of Ni-NTA resin (Qiagen). After washing the resin with 40 ml of 50 mM Tris-HCl (pH 8.0), 350 mM NaCl, 20 mM imidazole, and 5 mM β-ME, the protein was eluted in an identical buffer containing 350 mM imidazole. The elution fraction was dialyzed against a buffer containing 50 mM Tris-HCl (pH 8.0), NaCl (100 mM), and DTT (1 mM), and the His6-tag was removed using TEV protease. The digested proteasome was further purified using size exclusion chromatography (GE Healthcare Life Sciences “Superdex 200 10/300 GL”) and eluted in 20 mM sodium phosphate buffer (pH: 6.6) with 50 mM NaCl and 0.5 mM EDTA. The α7 single-ring particle was concentrated to 143 μM, which corresponds to a monomer concentration of 1 mM.
Typically, a 1 L expression culture produced approximately 100 mg GB1 or MBP and 50 mg α7.
5-fluorouracil-substituted DNA:
The 16-mer, double-stranded DNA sample containing a single 5-fluorodeoxyuridine nucleotide was made from two individual strands obtained from IDT (Coralville, IA) as polyacrylamide-purified samples. The 5-fluorodeoxyuridine nucleotide is known to form a base pair with an adenosine nucleotide.
The DNA sequences of the individual strands are shown below.
5’ – GCT AGG /5F-dU/ CA ATA CTC G - 3’
5’ - CGA GTA TTG ACC TAG C – 3’
where /5F-dU/ is the 5-fluorodeoxyuridine nucleotide (refer to Supplementary Note III for the structure).
The samples were annealed in TE buffer (5 mM Tris-HCl (pH 8.0), 1 mM EDTA) by heating to 80 °C and cooling to 20 °C with a ramp of 1 °C/ 5 sec. The concentration of the double stranded DNA 16-mer was determined using an extinction coefficient of 265,113 L·mol−1·cm-1. The final concentration was 1.5 mM.
NMR spectroscopy
Experiments were performed on 1 mM GB1, 700 μM MBP, 1 mM α7 single-ring proteasome particle, or 1.5 mM 5-fluorouracil-substituted DNA. All NMR spectra were collected in the indicated NMR buffer at 298 K, unless otherwise noted. All the 2D NMR spectra were processed with NMRPipe51 and analysed with CcpNmr52. The 1D spectra were processed with Bruker TopSpin® (version 3.2).
1D 13C spectra on 3-19F13C Tyr GB1 and unlabelled GB1
The 1D 13C spectra of 3-19F13C Tyr GB1 (1 mM) were collected on 400, 500, 600, 800, and 900 MHz (1H frequency) spectrometers. The 400 MHz spectrometer is a Varian spectrometer equipped with an INOVA console and a room temperature probe. The 500, 600, 800, and 900 MHz (1H frequency) spectrometers are Bruker spectrometers, equipped with cryogenically cooled probes. 1H decoupling was applied during the acquisition and recovery time periods. The 1D 13C spectra of unlabelled GB1 (1 mM) were collected on 500, 600, 800, and 900 MHz (1H frequency) Bruker spectrometers, equipped with cryogenically cooled probes. 1H decoupling was not applied on the unlabelled sample to allow for observation of the TROSY component of the natural abundant 13CH resonance at the 3rd position of Tyr.
2D 13C-19F correlation spectra on MBP.
The spectra of MBP were recorded on a 600 MHz (1H frequency) Bruker spectrometer equipped with a 5mm triple resonance inverse CryoProbe (TCI, 1H tunable to 19F) with 1H/19F, 13C, and 15N frequencies. The 1H channel was tuned to 19F. This configuration does not allow for 1H decoupling.
For the 19F-detected out-and-back HSQC, the spectral width in the indirect 13C dimension was set to 5 ppm and the spectral width in the direct 19F dimension was set to 40 ppm in both decoupled and coupled experiments. 256 complex points (real + imaginary) were collected in the indirect 13C dimension (acquisition time = 170 ms) and 4096 complex points were collected in the direct 19F dimension (acquisition time = 180 ms). 200 and 512 scans were collected for the coupled and decoupled HSQC, respectively, with a recycling delay of 1 second. The carrier frequencies were centred at 151.5 and −134 ppm in the 13C and 19F dimensions, respectively.
For the out-and-back style TROSY experiments (including the sensitivity enhanced version) the spectral width in the indirect 13C dimension was set to 5 ppm and the spectral width in the direct dimension was set to 40 ppm. 128 complex points were collected in the indirect 13C dimension (acquisition time = 170 ms) and 4096 complex points were collected in the direct 19F dimension (acquisition time = 180 ms). The carrier frequencies were centred at 151.5 and −134.0 ppm in the 13C and 19F dimensions, respectively. 200 scans were collected with a recycling delay of 1 seconds. The regular ST2PT and the sensitivity enhanced versions of the experiments were recorded in a time-equivalent fashion with the same acquisition parameters.
Two different types of out-and-stay experiments were recorded with the following parameters:
1) 19F-detected, 13C-19F out-and-stay TROSY: Here the spectral width in the indirect 13C dimension was set to 20 ppm and the spectral width in the direct dimension was set to 40 ppm. 128 complex points were collected in the indirect 13C dimension (acquisition time = 21 ms) and 4096 complex points were collected in the direct 19F dimension (acquisition time = 90 ms). 80 scans were collected with a recycling delay of 2 seconds. The carrier frequency was centred at 151.5 ppm in the 13C dimension and −135.0 ppm in the 19F dimension.
2) 13C-detected, 19F-13C out-and-stay TROSY: Here the spectral width in the indirect 19F dimension was set to 10.6 ppm and the spectral width in the direct 13C dimension was set to 39 ppm. 116 complex points were collected in the indirect 19F dimension (acquisition time = 10 ms) and 4096 complex points were collected in the direct 13C dimension (acquisition time = 348 ms). 80 scans were collected with a recycling delay of 2 seconds. The carrier frequency was centred at 151.5 ppm in the 13C dimension and −135 ppm in the 19F dimension.
To mimic a higher molecular weight system, we recoded the 19F-13C out-and-stay TROSY experiment on MBP at 10 °C. Here the spectral width in the indirect 19F dimension was set to 6.9 ppm and the spectral width in the direct 13C dimension was set to 39 ppm. 32 complex points were collected in the indirect 19F dimension (acquisition time = 4 ms) and 1024 complex points were collected in the direct 13C dimension (acquisition time = 87 ms). 400 scans were collected with a recycling delay of 1 seconds. The carrier frequency was centred at 151.5 ppm in the 13C dimension and −136.25 ppm in the 19F dimension. Spectra generated at 25 °C were truncated when compared to spectra recorded at 10 °C.
2D 13C-detected- 1H-13C TROSY on MBP.
Spectra were recorded on 600 MHz (1H frequency) Bruker spectrometers equipped with 5mm CPTCI and CPPTCI Prodigy Z-GRD Cryoprobes at 25 °C and 10 °C, respectively.
At 25 °C the spectral width in the indirect 1H dimension was set to 2 ppm and the spectral width in the direct 13C dimension was set to 52 ppm. 72 complex points were collected in the indirect 1H dimension (acquisition time = 30 ms) and 2048 complex points were collected in the direct 13C dimension (acquisition time = 131 ms). 160 scans were collected with a recycling delay of 1 second. The carrier frequency was centred at 125 ppm in the 13C dimension and 7 ppm in the 1H dimension.
At 10 °C the spectral width in the indirect 1H dimension was set to 2 ppm and the spectral width in the direct 13C dimension was set to 52 ppm. 128 complex points were collected in the indirect 1H dimension (acquisition time = 53 ms) and 2048 complex points were collected in the direct 13C dimension (acquisition time = 131 ms). 400 scans were collected with a recycling delay of 1 second. The carrier frequency was centred at 125 ppm in the 13C dimension and 7 ppm in the 1H dimension.
2D 19F-13C TROSY on the proteasome single-ring α7 particle.
Spectra were recorded on a 600 MHz (1H frequency) Bruker spectrometer equipped with a 5mm inverse triple resonance CryoProbe (TCI, 1H tunable to 19F) with 1H/19F, 13C and 15N frequencies. The 1H channel was tuned to 19F.
All experiments recorded were 13C-detected, 19F-13C out-and-stay TROSYs. The spectral width in the indirect 19F dimension was set to 8 ppm and the spectral width in the direct 13C dimension was set to 97 ppm. 28 complex points were collected in the indirect 19F dimension (acquisition time = 3 ms) and 2048 complex points were collected in the direct 13C dimension (acquisition time = 70 ms). 1024 scans were collected with a recycling delay of 1 second. The carrier frequency was centred at 151.5 ppm in the 13C dimension and −136.5 ppm in the 19F dimension. The total experiment time was ~8 hours.
2D 13C-19F correlation spectra on GB1 with 1H decoupling.
The GB1 spectra were recorded on a 500 MHz (1H frequency) Bruker spectrometer equipped with an Avance III HD console and 5mm triple resonance observe probe (TXO) tuned to 19F, 13C and 1H frequencies. The spectral width in the indirect 13C dimension was set to 5 ppm and the spectral width in the direct dimension was set to 20 ppm. 150 points were collected in the indirect 13C dimension (acquisition time = 238 ms) and 2048 points were collected in the direct 19F dimension (acquisition time = 218 ms). 48 scans were collected with a recycling delay of 1 second. The carrier frequency was centred at 151.5 ppm in the 13C dimension and −134.0 ppm in the 19F dimension. The carrier frequency in the 1H channel was set to 8.0 ppm. During 19F acquisition in the direct dimension, 1H decoupling was applied using a WALTZ-16 scheme53.
2D 13C-19F correlation spectra on 5-fluorouracil-substituted DNA.
The spectra of the DNA 16-mer containing a 5-fluorodeoxyuridine nucleotide were recorded on a 600 MHz (1H frequency) Bruker spectrometer equipped with a 5 mm inverse triple resonance CryoProbe (TCI, H tunable to F) with 1H/19F, 13C and 15N frequencies. The spectra were recorded at 5 °C to mimic a larger molecular weight system. Here we recorded a 19F-detected, out-and-back style, coupled HSQC and a 19F-detected, out-and-stay 13C-19F TROSY spectrum. The spectral width in the indirect 13C dimension was set to 3 ppm and the spectral width in the direct 19F dimension was set to 40 ppm. 55 points were collected in the indirect 13C dimension (acquisition time = 121 ms) and 1024 points were collected in the direct 19F dimension (acquisition time = 45 ms). 256 scans were collected with a recycling delay of 2 seconds. The carrier frequency was centred at 139.0 ppm in the 13C dimension and −165.5 ppm in the 19F dimension.
Calculating the correlation time of 16-mer DNA
The crystal structure of a 16-mer DNA duplex (pdb id: 420D) was used as a proxy for a 16-mer oligo nucleotide to estimate rotational correlation times for 5-fluorouracil-substituted DNA at 5 °C. All water molecules were stripped from the 16-mer DNA duplex and hydrodynamic calculations were performed with HYDRONMR27. The recommended value of 3.2 Å was chosen for the atomic element radius. The parameter NSIG (number of values of the radius of the minibead) was set to 6 and SIGMIN and SIGMAX were defined as 1.0 and 2.0, respectively. The temperature was set to 278.15 K and solvent viscosity was set to 1.5215 mPa·s.
T1 Measurements on 13CF and 19FC
The longitudinal relaxation times (T1) for 13CF and 19FC were estimated using a 1D inversion recovery NMR experiment. The experiments were recorded using GB1 to observe the isolated 19FC and 13CF resonances. The T1 for 19FC was measured on a Bruker Avance HD NMR spectrometer operating at 500 MHz (1H frequency) and equipped with a room temperature probe. 8 experiments were recorded with variable inversion recovery delays of 1, 50, 100, 500, 800, 1000, 1500, and 2000 ms. 32 scans were averaged with a recycle delay of 10 s. The carrier frequency was set to −135 ppm.
The T1 for 13CF was measured on a Bruker Avance NMR spectrometer operating at 500 MHz (1H frequency) and equipped with a cryogenically cooled probe. 24 experiments were recorded with variable inversion recovery delays of 1, 40, 100, 200, 200, 500, 800, 1000, 1250, 1500, 1750, 2000, 2250, 2500, 2750, 3000, 3250, 3500, 3750, 4000, 4250, 4500, 4750, and 5000 ms. 104 scans were collected with a recycle delay of 10 s. The carrier frequency was set to 150 ppm.
The signal intensity was measured using Bruker TopSpin® (version 3.2). The T1 value was estimated by fitting an exponential curve described by
where M0 corresponds to the intrinsic intensity of the 19FC and 13CF resonances without relaxation.
Simulation of the free induction decay
We simulated free induction decays (FID) for the TROSY components of aromatic 13CF and 13CH spin-pairs, based on transverse relaxation rates calculated for 3-19F13C Tyr and 3-1H13C Tyr. To compare these to 1H-15N spin pairs, we also calculated the FID for the TROSY component of an average 1H-15N spin pair. The FIDs were simulated using the following expression:
Where I0 is the initial magnetization, t is the evolution time, R2 is the relaxation rate and Ω is the chemical shift.
The parameters I0, and Ω were set to 0.005 and 200 Hz respectively. The signal was evolved for 1 s and sampled at 2048 points, captured by variable t. The time domain signal was Fourier transformed and the real part is displayed. The TROSY component linewidths for a macromolecule with τc = 25 ns are 1.9, 2.3, and 13.7 Hz, for 13CF, 15NH, and 13CH, respectively. The TROSY component linewidths for a macromolecule with τc = 65 ns are 5.0, 6.1, and 35.7 Hz, for 13CF, 15NH, and 13CH, respectively.
Supplementary Material
Acknowledgments:
We thank Dr. Kendra E. Leigh and Dr. Milka Kostic for help in proofreading of the manuscript presented here and the discussions regarding the work, and Dr. Donna Baldisseri for her help with the NMR experiments. We are especially grateful to Dr. Lewis E. Kay (Departments of Molecular Genetics, Biochemistry and Chemistry, University of Toronto, Toronto, Ontario, Canada) for sharing the plasmid carrying the single-ring α7 proteasome particle with us. This research was supported by NIH grants GM047467 and AI03758 to G.W. H.A. acknowledges funding from the Claudia Adams Barr Program for Innovative Cancer Research. A.B. thanks the Austrian Science Fund FWF for the Schrödinger Fellowship J3872-B21. S.C. acknowledges National Health and Medical Research Council Australia for the C. J. Martin Fellowship APP1090444. Maintenance of the NMR instruments used for this research was supported by NIH grant EB002026.
Footnotes
Competing Financial Interests
The author Vladimir Gelev is the founder of FB Reagents Ltd., a company that provides isotopically enriched NMR reagents and declares a competing interest. Clemens Anklin and Helena Kovacs work for Bruker Biospin Corporation, which is manufacturer of the equipment used in this work.
All other authors declare no competing financial interests.
Data availability:
Pulse sequences, parameter sets are available as Supplementary Software and at the lab website.
https://artlab.dana-farber.org/19f_13c_aromatictrosy.html
A life sciences reporting summary is available.
References:
- 1.Pervushin K, Riek R, Wider G & Wuthrich K Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A 94, 12366–12371 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pervushin K, Riek R, Wider G & Wuthrich K Transverse relaxation-optimized spectroscopy (TROSY) for NMR studies of aromatic spin systems in 13C-labeled proteins. J Am Chem Soc 120, 6394–6400 (1998). [Google Scholar]
- 3.Sprangers R & Kay LE Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature 445, 618–622 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Fiaux J, Bertelsen EB, Horwich AL & Wuthrich K NMR analysis of a 900K GroEL GroES complex. Nature 418, 207–211 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Meyer LH & Gutowsky HS Electron Distribution in Molecules. II. Proton and Fluorine Magnetic Resonance Shifts in the Halomethanes. The Journal of Physical Chemistry 57, 481–486 (1953). [Google Scholar]
- 6.Solomon I Relaxation Processes in a System of Two Spins. Physical Review 99, 559–565 (1955). [Google Scholar]
- 7.Sykes BD & Hull WE in Methods in Enzymology, Vol. 49 270–295 (Academic Press, 1978). [DOI] [PubMed] [Google Scholar]
- 8.Liu JJ, Horst R, Katritch V, Stevens RC & Wuthrich K Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim TH et al. The Role of Ligands on the Equilibria Between Functional States of a G Protein-Coupled Receptor. J Am Chem Soc 135, 9465–9474 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barhate NB, Barhate RN, Cekan P, Drobny G & Sigurdsson ST A nonafluoro nucleoside as a sensitive 19f NMR probe of nucleic acid conformation. Org Lett 10, 2745–2747 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kreutz C, Kahlig H, Konrat R & Micura R A general approach for the identification of site-specific RNA binders by 19F NMR spectroscopy: proof of concept. Angewandte Chemie 45, 3450–3453 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Kiviniemi A & Virta P Characterization of RNA invasion by (19)F NMR spectroscopy. J Am Chem Soc 132, 8560–8562 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Bao HL et al. Characterization of human telomere RNA G-quadruplex structures in vitro and in living cells using 19F NMR spectroscopy. Nucleic Acids Res 45, 5501–5511 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerig JT Fluorine NMR of proteins. Progress in Nuclear Magnetic Resonance Spectroscopy 26, 293–370 (1994). [Google Scholar]
- 15.Brindle K, Williams S-P & Boulton M 19F NMR detection of a fluorine-labelled enzyme in vivo. FEBS Letters 255, 121–124 (1989). [Google Scholar]
- 16.Li C et al. Protein 19F NMR in Escherichia coli. J Am Chem Soc 132, 321–327 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gronenborn AM et al. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science 253, 657–661 (1991). [DOI] [PubMed] [Google Scholar]
- 18.Gardner KH, Zhang X, Gehring K & Kay LE Solution NMR Studies of a 42 KDa Escherichia Coli Maltose Binding Protein/β-Cyclodextrin Complex: Chemical Shift Assignments and Analysis. J Am Chem Soc 120, 11738–11748 (1998). [Google Scholar]
- 19.Sprangers R et al. TROSY-based NMR evidence for a novel class of 20S proteasome inhibitors. Biochemistry 47, 6727–6734 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Hogben H, Krzystyniak M, Charnock G, Hore P & Kuprov I Spinach–a software library for simulation of spin dynamics in large spin systems. J Magn Reson 208, 179–194 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Kitevski-LeBlanc JL, Al-Abdul-Wahid MS & Prosser RS A mutagenesis-free approach to assignment of (19)F NMR resonances in biosynthetically labeled proteins. J Am Chem Soc 131, 2054–2055 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Nietlispach D Suppression of anti-TROSY lines in a sensitivity enhanced gradient selection TROSY scheme. J Biomol NMR 31, 161–166 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Khan F, Kuprov I, Craggs TD, Hore PJ & Jackson SE 19F NMR studies of the native and denatured states of green fluorescent protein. J Am Chem Soc 128, 10729–10737 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Religa TL, Sprangers R & Kay LE Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science 328, 98–102 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Riek R, Pervushin K, Fernandez C, Kainosho M & Wuthrich K [(13)C,(13)C]- and [(13)C,(1)H]-TROSY in a triple resonance experiment for ribose-base and intrabase correlations in nucleic acids. J Am Chem Soc 123, 658–664 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Seela F & Xu K DNA with stable fluorinated dA and dG substitutes: syntheses, base pairing and 19F-NMR spectra of 7-fluoro-7-deaza-2’-deoxyadenosine and 7-fluoro-7-deaza-2’-deoxyguanosine. Org Biomol Chem 6, 3552–3560 (2008). [DOI] [PubMed] [Google Scholar]
- 27.García de la Torre J, Huertas ML & Carrasco B HYDRONMR: Prediction of NMR Relaxation of Globular Proteins from Atomic-Level Structures and Hydrodynamic Calculations. Journal of Magnetic Resonance 147, 138–146 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Arntson KE & Pomerantz WC Protein-Observed Fluorine NMR: A Bioorthogonal Approach for Small Molecule Discovery. J Med Chem 59, 5158–5171 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Kitevski-LeBlanc JL & Prosser RS Current applications of 19F NMR to studies of protein structure and dynamics. Progress in Nuclear Magnetic Resonance Spectroscopy 62, 1–33 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Marsh EN & Suzuki Y Using (19)F NMR to probe biological interactions of proteins and peptides. ACS Chem Biol 9, 1242–1250 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Danielson MA & Falke JJ Use of 19F NMR to probe protein structure and conformational changes. Annu Rev Biophys Biomol Struct 25, 163–195 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitevski-LeBlanc JL, Evanics F & Scott Prosser R Optimizing (1)(9)F NMR protein spectroscopy by fractional biosynthetic labeling. J Biomol NMR 48, 113–121 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Gee CT et al. Protein-observed (19)F-NMR for fragment screening, affinity quantification and druggability assessment. Nat Protoc 11, 1414–1427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang M et al. Fast Magic Angle Spinning (1)(9)F NMR of HIV-1 Capsid Protein Assemblies. Angewandte Chemie (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whittaker MM & Whittaker JW Construction and characterization of Pichia pastoris strains for labeling aromatic amino acids in recombinant proteins. Protein Expr Purif 41, 266–274 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Budisa N, Wenger W & Wiltschi B Residue-specific global fluorination of Candida antarctica lipase B in Pichia pastoris. Mol Biosyst 6, 1630–1639 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Watts JK et al. Differential stability of 2’F-ANA*RNA and ANA*RNA hybrid duplexes: roles of structure, pseudohydrogen bonding, hydration, ion uptake and flexibility. Nucleic Acids Res 38, 2498–2511 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitevski-LeBlanc JL, Evanics F & Scott Prosser R Approaches to the assignment of 19F resonances from 3-fluorophenylalanine labeled calmodulin using solution state NMR. J Biomol NMR 47, 113–123 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Kitevski-LeBlanc JL, Evanics F & Prosser RS Approaches for the measurement of solvent exposure in proteins by 19F NMR. J Biomol NMR 45, 255–264 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Kovacs H & Kupce E Parallel NMR spectroscopy with simultaneous detection of (1) H and (19) F nuclei. Magn Reson Chem 54, 544–560 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Tang P, Furuya T & Ritter T Silver-catalyzed late-stage fluorination. J Am Chem Soc 132, 12150–12154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frieden C, Hoeltzli SD & Bann JG The preparation of 19F-labeled proteins for NMR studies. Methods in enzymology 380, 400–415 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Salwiczek M, Nyakatura EK, Gerling UI, Ye S & Koksch B Fluorinated amino acids: compatibility with native protein structures and effects on protein-protein interactions. Chem Soc Rev 41, 2135–2171 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Konas DW, Seci D & Tamimi S Synthesis of (L)-4-Fluorotryptophan. Synthetic Communications 42, 144–152 (2012). [Google Scholar]
- 45.Takeuchi Y, Tarui T & Shibata N A novel and efficient synthesis of 3-fluorooxindoles from indoles mediated by Selectfluor. Org Lett 2, 639–642 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Kim TH et al. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science 355 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Hoang J & Prosser RS Conformational selection and functional dynamics of calmodulin: a (19)F nuclear magnetic resonance study. Biochemistry 53, 5727–5736 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Schwieters CD et al. Solution structure of the 128 kDa enzyme I dimer from Escherichia coli and its 146 kDa complex with HPr using residual dipolar couplings and small- and wide-angle X-ray scattering. J Am Chem Soc 132, 13026–13045 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Depetris I et al. Fluoropyrimidine-induced cardiotoxicity. Crit Rev Oncol Hematol 124, 1–10 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Voeks D et al. Gene therapy for prostate cancer delivered by ovine adenovirus and mediated by purine nucleoside phosphorylase and fludarabine in mouse models. Gene Ther 9, 759–768 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Delaglio F et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of biomolecular NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 52.Fogh R et al. The CCPN project: an interim report on a data model for the NMR community. Nat Struct Biol 9, 416–418 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Shaka AJ, Keeler J, Frenkiel T & Freeman R An improved sequence for broadband decoupling: WALTZ-16. Journal of Magnetic Resonance (1969) 52, 335–338 (1983). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.