

SUMMARY
In neurons, defects in autophagosome clearance have been associated with neurodegenerative disease. Yet, the mechanisms that coordinate trafficking and clearance of synaptic autophagosomes are poorly understood. Here we use genetic screens and in vivo imaging in single neurons of C. elegans to identify mechanisms necessary for clearance of synaptic autophagosomes. We observe that autophagy at the synapse can be modulated in vivo by the state of neuronal activity, that autophagosomes undergo UNC-16/JIP3-mediated retrograde transport, and that autophagosomes containing synaptic material mature in the cell body. Through forward genetic screens we then determine that autophagosome maturation in the cell body depends on the protease ATG-4.2, but not the related ATG-4.1, and that ATG-4.2 can cleave LGG-1/Atg8/GABARAP from membranes. Our studies reveal that ATG-4.2 is specifically necessary for the maturation and clearance of autophagosomes, and that defects in transport and ATG-4.2-mediated maturation genetically interact to enhance abnormal accumulation of autophagosomes in neurons.
eTOC Blurb
Hill et al. use genetic screening and live imaging of autophagosomes in C. elegans to observe activity-dependent synaptic autophagy and discover that autophagosome trafficking and clearance are coordinated in neurons. They discover a unique role for protease isoform ATG-4.2 in cleaving LGG-1/Atg8/GABARAP from autophagosomes to promote their maturation and clearance.
INTRODUCTION
Neurons are highly polarized cells in which autophagy is spatially compartmentalized. Local formation of autophagosomes is observed at the distal end of axons and at synapses (Maday and Holzbaur, 2014; Maday et al., 2012; Soukup et al., 2016; Stavoe et al., 2016). Synaptic autophagy can be enhanced under conditions of prolonged neuronal activity, suggesting a physiological link between neuronal activity and local autophagosome biogenesis (Shehata et al., 2012; Soukup et al., 2016; Wang et al., 2015). Autophagosomes are then transported towards the soma for lysosome fusion and degradation (Hollenbeck, 1993; Kaasinen et al., 2008).
Retrograde transport of autophagosomes is tightly controlled through both the recruitment and regulation of the dynein motor complex (Ikenaka et al., 2013; Katsumata et al., 2010; Maday et al., 2012; Neisch et al., 2017). Dynein is recruited to autophagosomes through fusion of autophagosomes with late endosomes (Cheng et al., 2015), and in vertebrate neurons robust retrograde transport is facilitated by the motor scaffolding protein JIP1 (Fu et al., 2014). During transport, autophagic vacuoles (a general term encompassing autophagic structures of all maturity states (Klionsky et al., 2016)) begin progressive maturation (Maday et al., 2012; Neisch et al., 2017; Vanhauwaert et al., 2017), and once in the cell body, they are targeted for degradation and clearance through mechanisms that are not well understood. The importance of these regulated processes in neuronal autophagy is perhaps best exemplified by the consequences of their disruption, which include autophagosome accumulation at presynaptic terminals, Alzheimer’s disease-like autophagic stress and axonal pathology (Ikenaka et al., 2013; Lee et al., 2011; Nixon et al., 2005; Takats et al., 2013; Tammineni et al., 2017).
In this study we used forward and reverse genetics, combined with in vivo imaging, to identify mechanisms necessary for the clearance of synaptic autophagosomes. We visualized autophagosomes at single-neuron resolution by examining the localization of the autophagosome-associated protein LGG-1/Atg8/GABARAP (Manil-Segalen et al., 2014; Melendez et al., 2003; Stavoe et al., 2016; Zhang et al., 2015). We found that, in vivo, we could predictably vary the number of synaptic autophagosomes by modulating the firing state of the neuron. We also detected synaptic proteins colocalized with autophagic vacuoles in the cell body, suggesting a link between autophagosome biogenesis at the synapse and autophagy-mediated degradation of synaptic proteins in the soma. We identified a role for the motor adaptor protein, UNC-16/JIP3, in facilitating robust retrograde transport of synaptic autophagic vacuoles to the cell body. To then uncover mechanisms that coordinate transport and elimination of autophagosomes from the neurite, we performed an enhancer screen in unc-16/jip3 mutant animals and identified a dominant enhancer mutation in the poorly understood autophagy gene atg-4.2. Mutations in atg-4.2; unc-16 double mutant animals displayed dramatic accumulation of autophagosomes in the neurite, while atg-4.2 single mutant animals accumulated autophagosomes in the cell body, phenotypes not observed for the other atg-4 gene in C. elegans, atg-4.1. We determined through biochemical assays that ATG-4.2 delipidates LGG-1 from membranes, and observed that the accumulated autophagic vacuoles in atg-4.2 mutant animals failed to properly co-localize with lysosomal proteins, mature and degrade. This resulted in a dramatic accumulation of abnormal multilamellar autophagosome-like vacuoles in the cell bodies of atg-4.2 mutant neurons. Our findings support a model in which the cysteine protease atg-4.2 is specialized to remove LGG-1 and LGG-2 (hereafter LGG-1/LGG-2) from autophagosomes, to enable autophagosome fusion with lysosomes and to promote degradation. Our studies uncover mechanisms that regulate transport and clearance of autophagosomes in neurites and provide a framework to understand how disruption of trafficking and degradation genetically interact to result in abnormal accumulation of autophagosomes, as observed in neurodegenerative diseases.
RESULTS
Autophagosomes in C. elegans neurons contain synaptic proteins and form near synapses in response to physiological stimuli
To examine autophagy in neurons of living animals, we imaged LGG-1 and LGG-2 in the AIY interneurons of C. elegans at larval stage 4 (L4) (Figure 1A–1E, Figure S1A-S1C, and as previously described in (Stavoe et al., 2016)). LGG-1 and LGG-2 are C. elegans homologues of Atg8 (in yeast) and GABARAP and LC3 family proteins (in mammals) that associate with immature and mature autophagic structures, and have been used as markers to track autophagosomes in living cells ((Alberti et al., 2010; Manil-Segalen et al., 2014; Melendez et al., 2003), for controls of these markers, please see Figures 1F and S1D-S1F and STAR Methods).
Figure 1. Synaptic autophagosomes occur in response to physiological stimuli and cell body autophagosomes contain synaptic material.

(A) Schematic of the AIY interneuron pair located in the nerve ring within the head of the worm Neurite (red and blue) and cell body (asterisk) are labeled. Dorsal (D), ventral (V), anterior (A), posterior (P), left (L), right (R).
(B) Autophagosomes (visualized with GFP∷LGG-1) in the synaptic regions (visualized with mCh∷RAB-3) of a representative wild-type AIY interneuron. The dashed line traces the path of the AIY neurite (see schematic in 1A). The yellow dotted line highlights the synaptic regions of the neurite, and the white dashed line, the asynaptic region. The synapse-rich portion of the synaptic region (boxed) is enlarged in (B’).
(C) Schematic of AIY with the cell body and the proximal portion of the synaptic region as in (B).
(D) As (B’) but GFP∷LGG-1 only.
(E) Enlargement of the cell body in (B) and showing GFP∷LGG-1 only. Note that we observe autophagic vacuoles in both the cell body and synapse of neurons.
(E) LGG-1(G116A) fails to be lipidated and associate with autophagic vacuoles, and does not form puncta at synapses or the cell body ((Alberti et al., 2010; Manil-Segalen et al., 2014; Stavoe et al., 2016), see also Figure S1 for additional controls).
(G-I) Autophagosomes (visualized with GFP∷LGG-1) in a representative wild-type AIY neuron held at 25°C for 4 hours prior to imaging. In (H-I), the synaptic regions of wild-type AIY neurons are shown in animals held at 20°C (H) or held at 25 °C (I) for 4 hours prior to imaging. The AIY interneurons are primary interneurons in the thermotaxis circuit of C. elegans, and the activity state of AIY is known to increase when animals are held at 25°C for ~4 hours (as compare to animals at 20°C) (Biron et al., 2006; Clark et al., 2006; Hawk et al., 2018). We therefore used the physiological stimuli of temperature in this intact circuit to increase the activity state of the AIY neurons and examine synaptic autophagy in vivo under these conditions. The synaptic enriched-region of (G) (dashed boxed) is enlarged in (I).
(J) Quantification of the average number of autophagosomes in the AIY neurite in wild-type (green line) and exocytosis/unc-13(e450) mutant (purple line) animals. Animals were raised at 20°C and then held at 25°C for variable lengths of time as indicated. Note that after 4 hours at 25°C, higher numbers of synaptic autophagosomes can be observed, and that this increase is suppressed in exocytosis/unc-13(e450) mutant animals (purple line). Our findings suggest that physiological increases in activity state of the neuron result in an increase in the number of synaptic autophagosomes. Our findings also demonstrate activity independent increases in autophagosome number for animals raised at 25°C for 24 hours or longer, likely due to temperature-dependent effects in the cell and consistent with the known detrimental effects of prolonged exposure of C. elegans to 25°C temperature conditions (Byerly et al., 1976; Hosono et al., 1982). For all genotypes and time points, the number of animals examined is n≥45. *p<0.05, **p<0.01, ****p<0.0001, or “ns” not significant by Student’s t test between wild-type and unc-13 mutant animals at each time point (black significance values) or between different wild-type time points (green significance values).
(K) Quantification of the average number of autophagosomes in the AIY neurite in animals expressing the heterologous Drosophila inhibitory chloride channel (HisCl) (Pokala et al., 2014) when exposed to the activating histamine (purple bars) or control (green bars). Error bars show standard error of the mean (SEM). ****p<0.0001 by one-way ANOVA with Tukey’s post hoc analysis between histamine and control conditions. The number of animals examined is shown on each bar.
(L-N) Representative confocal image of presynaptic active zone protein GFP::SYD-1 (L) co-localized with mCh∷LGG-1 (M). Merge images in (N). Insets in (L’-N’) show the enlarged cell body with subcellular localization of these proteins.
(O-Q) As in (L-N), but imaging the subcellular localization of synaptic vesicle protein synaptobrevin (GFP∷SNB-1) and mCh∷LGG-1. Note how in both (L-N) and (O-Q), LGG-1 partially co-localizes in the cell body with synaptic proteins (also seen for active zone protein SYD-2 in Figure S1G-S1I).
Throughout the figures, images depict maximal projections of confocal z stacks, arrowheads refer to LGG-1 puncta in the cell body, arrows point to LGG-1 puncta in the synaptic regions, dashed circles enclose cell bodies. Abbreviation: “hr.” is hours.
Scale bar(s) in (B) for (B), (F), (G) and (L-Q); in (B’) for (B’) and (D); in (E) for (E) and (L’-Q’); in (H) for (H) and (I).
See also Figure S1.
Autophagosomes in AIY were present in the neurite (Figures 1B–1D) and in the cell body (Figure 1E). In the neurite, the majority of autophagosome biogenesis events occur near synaptic regions (Stavoe et al., 2016). To understand how activity of neurons relates to autophagosome biogenesis in vivo, we first examined autophagy in AIY neurons under varying physiological conditions. AIY is an interneuron that predictably changes its firing frequency based on inputs from sensory neurons (Laurent et al., 2015; Luo et al., 2014; Satoh et al., 2014). As part of its role in thermotaxis behavior, the frequency of calcium transients in AIY increases for animals raised at warmer temperatures (Clark et al., 2006; Hawk et al., 2018). We observed that animals raised at warmer temperatures, in which the neuronal activity state of AIY is increased, also had a higher percentage of animals with autophagosomes in AIY, and that this could be physiologically modulated by changing the cultivation temperature of the animals ((Hawk et al., 2018) and Figure 1G–1K). For example, in animals raised at 20°C, a condition known to have lower AIY calcium activity (Hawk et al., 2018), the percentage of animals with one or more autophagosomes in either AIY neurite was 37% (n=73 animals). When animals were cultivated at 25°C for 4 hours, which is known to change their temperature preference through an increased calcium responsiveness of AIY, a concomitant increase in the percentage of animals with one or more autophagosomes in either AIY neurite was observed (81% (n=78 animals) and Figure 1G–1I). We do note that animals raised at warmer temperatures for prolonged periods of time, under conditions associated with noxious stimuli to the animal (and not known to be associated with thermotaxis behavior or increased activity states in AIY), also displayed an activity-independent increase in autophagosomes in the AIY neurite. Yet for the physiological conditions in which AIY is known to be active and required for thermotaxis behavior (exposure to higher temperatures for periods of less than 8 hours), genetically inhibiting synaptic transmission suppressed the observed increase in synaptic autophagosomes (Figure 1J). Moreover, chemo-genetic silencing of AIY through the cell-specific expression of a histamine-activated chloride channel suppressed temperature-related increases in synaptic autophagosomes (Figure 1K). Previous findings using optogenetic approaches established a link between stimulation of the neuron and autophagosome biogenesis in the neurites (Shehata et al., 2012; Soukup et al., 2016; Wang et al., 2015). Our findings are consistent with and extend these studies, demonstrating that increases in autophagosomes near synapses can be induced by activity-dependent mechanisms associated with physiological responses of the neuron, and activity-independent mechanisms associated with noxious stimuli to the system.
Does increased autophagy at active synapses act to recycle synaptic material? To address this we examined if autophagic vacuoles in the cell body, where autophagosomes are transported for degradation, co-localized with synaptic proteins. Simultaneous visualization of active zone protein SYD-1 and autophagy-associated protein LGG-1 revealed partial co-localization in the neuronal cell body (Figure 1L–1N). Similar results were observed for active zone protein SYD-2/Liprin alpha and synaptic vesicle protein synaptobrevin (SNB-1) (Figures 1O–1Q and S1G-S1I). Our findings indicate that autophagic vacuoles in the cell body contain synaptic proteins, and suggest that autophagy might degrade synaptic material such as active zone and synaptic vesicle proteins. Together, our findings are consistent with an association, in vivo, between the state of synaptic activity, presence of autophagosomes at the synapse and degradation of synaptic material in the neuronal cell body.
Dynactin and JIP3/UNC-16 promote autophagosome retrograde transport
To then investigate how autophagosomes at the synapse are transported and degraded in the cell body, we recorded and tracked individual synaptic autophagosomes in wild-type and mutant animals. In C. elegans neurons, as in vertebrate neurons, retrograde transport of autophagosomes depends on the dynein complex (Cheng et al., 2015; Ikenaka et al., 2013; Katsumata et al., 2010; Maday et al., 2012) (Figure 2A–2C). We therefore examined known regulators of transport to investigate their specific requirement in autophagosome trafficking.
Figure 2. Dynactin and UNC-16/JIP3 mediate retrograde transport of autophagosomes from the synaptic regions.

(A-B) Autophagosomes (visualized with GFP∷LGG-1) in a wild-type AIY neuron (A) and in a temperature-sensitive dynactin, dnc-1(or404) mutant animals (B) grown for 48 hours at the restrictive temperature of 25°C.
(C) Quantification of the average number of autophagosomes in the synaptic regions in wild-type and dnc-1(or404) mutant animals kept for 48 hours at the permissive (20°C) or restrictive (25°C) temperature (note an increase in wild-type a nimals at 25°C, as in Figure 1J). Error bars show standard error of the mean (SEM). ****p<0.0001 by one-way ANOVA with Tukey’s post hoc analysis between indicated groups. Numbers on bars represent number of animals examined.
(D) Schematic of the unc-16 genomic region with exons (purple boxes), untranslated regions (grey boxes), introns (black lines) and allelic lesions (early stop codons as red asterisks in ju146 and n730; and deletion as red box in e109). Scale bar shows a 1-kilobase (kb) region.
(E-F) Autophagosomes (visualized with GFP∷LGG-1) in a representative wild-type AIY neuron (E) and in an unc-16(ju146) mutant neuron (F) grown at the standard 20°C.
(G) Quantification of the average number of autophagosomes in the synaptic regions in wild-type, three alleles of unc-16 (ju146, n730, and e109), jip-1(gk466982), unc-116(e2310), and unc-14(e57) mutant animals. Error bars show standard error of the mean (SEM). ****p<0.0001 by one-way ANOVA with Tukey’s post hoc analysis. The number of animals examined is shown on each bar.
(H) Schematic of the AIY interneuron colored by region and similar to the schematic in Figure 1A: Zone 1 (asynaptic, green), Zone 2 (enriched synaptic region, brown), and Zone 3 (punctate synaptic region, pink, see (Colon-Ramos et al., 2007)). Modified and reprinted with permission from wormatlas.org.
(I) Quantification of the percentage of autophagosomes in the AIY neurite by location. Bars are colored as zones in (H). Zone-1 localized autophagosomes observed in wild-type animals many times represent synaptic autophagosomes in transit towards the cell soma. Error bars show a 95% confidence interval. ****p<0.0001 by Chi-square test between wild-type animals (n=57 autophagosomes from 36 animals), unc-16(ju146) mutant animals (n=482 autophagosomes from 37 animals) and unc-16(e109) mutant animals (n=145 autophagosomes from 22 animals).
(J-K) Kymographs of autophagosome trafficking in AIY neurites in representative wild-type (J) and unc-16(ju146) mutant (K) animals showing retrograde (right down diagonals), anterograde (left down diagonals), and paused events (vertical lines). Neurite zones are indicated by color above kymographs and as in (H).
(L) Quantification of autophagosome trafficking events in the AIY neurite, showing percentage of autophagosomes that undergo anterograde, retrograde or paused events within the 5-minute imaging window for wild-type and unc-16 mutant animals. ****p<0.0001, **p<0.01 by Fisher’s exact test between wild-type and mutant animals. Error bars show a 95% confidence interval.
Arrows denote autophagosomes in the synaptic region. Dashed circles enclose the cell bodies. Abbreviations: “ns” is not significant; “AV” is autophagic vacuole.
Scale bar(s) in (A) for (A), (B), (E) and (F); in (J) for (J) and (K).
See also Figures S2 and S3.
We observed that while available alleles for jip-1/Mapk8Ip1, unc-116/kinesin-1 and unc-14/RUN mutant animals did not display defects in autophagosome accumulation at synapses (Figure 2G), loss of JIP3/UNC-16/Sunday Driver resulted in a significant accumulation of LGG-1/LGG-2 puncta in the neurite, but not in the cell body (Figures 2E–2G and S2A-S2D). The unc-16/jip3 mutant phenotype was similar to the phenotype seen for dnc-1/p150 dynactin complex subunit mutant animals (which affect the dynein complex) ((Ikenaka et al., 2013) and Figure 2B–2C). JIP3/UNC-16/Sunday Driver is a conserved adaptor protein that binds to kinesin and dynein (Arimoto et al., 2011; Cavalli et al., 2005) to regulate early endosome and lysosome transport (Brown et al., 2009; Byrd et al., 2001; Drerup and Nechiporuk, 2013; Edwards et al., 2015; Edwards et al., 2013; Gowrishankar et al., 2017). Mutant animals containing one of three independent alleles of unc-16 (ju146, e109, or n730) (Figure 2D) displayed an increase in the percentage of animals with LGG-1 puncta in the neurites (from 34% in wild-type animals to 100% in all unc-16 allele mutant animals examined). Moreover, while wild-type animals averaged less than one LGG-1 puncta per neurite, unc-16 mutant animals averaged ~2 LGG-1 puncta in the presynaptic regions (Figure 2G), indicating abnormal accumulation of autophagosomes. Analyses of the subcellular localization of LGG-1-containing structures in unc-16/jip3 mutant animals also revealed a higher probability for autophagosomes to be present in the distal synaptic Zone 3 region (Figures 2H and 2I). The distal accumulation of autophagosomes observed for unc-16 mutant animals was not due to a rearrangement of microtubule polarity, as we observed that the microtubule plus-end-binding protein EBP-2/EB1 (Baas and Lin, 2011; Maniar et al., 2011) was similarly oriented (plus-end-out) in wild-type and unc-16/jip3 mutant animals (Figure S2E-S2G). Our findings indicate that unc-16/jip3 is necessary to prevent the abnormal accumulation of autophagosomes in the neurite, and are consistent with a requirement for unc-16/jip3 in autophagosome retrograde transport.
Previous reports have implicated UNC-16/JIP3/Sunday Driver in the regulation of retrograde transport and retention of lysosomes, early endosomes and Golgi in the cell body ((Brown et al., 2009; Byrd et al., 2001; Edwards et al., 2013) and Figure S3). The role of UNC-16/JIP3 in transport of autophagosomes has not been examined. To directly investigate if unc-16/jip3 is required for retrograde transport of autophagosomes, we performed time-lapse imaging of individual LGG-1 puncta in wild-type neurons (n=71) and for two independent alleles of unc-16 mutant neurons (ju146, n=73; and e109, n=43). We observed that compared to wild-type animals, unc-16/jip3 mutant animals displayed decreased retrograde transport of LGG-1 puncta in neurites and increased anterograde trafficking or paused LGG-1 puncta (defined as puncta that did not traffic within a 5-minute observation window; Figure 2J–2L). In wild-type animals, LGG-1 puncta in the cell body were sometimes transported in an anterograde fashion into the neurite and returned to the cell body via retrograde transport within the 5-minute examination window. In contrast, the percentage of puncta that returned was greatly reduced in unc-16(ju146) mutant neurites (Figure S2H). Our findings are consistent with previous reports that demonstrate that another JIP family member (JIP1) is important for regulating autophagosome retrograde transport in cultured vertebrate neurons (Fu et al., 2014). We now extend those findings and demonstrate an important role for unc-16/jip3 in regulating the retrograde trafficking of autophagosomes in vivo.
To understand how UNC-16-dependent retrograde transport contributes to the degradation of autophagic vacuoles, we then tested if defects in unc-16 mutant animals altered the total number of LGG-1 puncta in neurons. We observed that the total number of LGG-1 puncta in neurons of unc-16/jip3 mutant animals was higher than that seen for wild-type animals (average ±95% confidence interval from 4.1±0.6 in wild-type neurons (n=65) to 8.2±0.8 in unc-16(ju146) mutant neurons (n=64); significant by Student’s t test p<0.0001). Our findings indicate that the increased number of autophagic vacuoles observed in the neurites of unc-16/jip3 mutant animals did not result from a simple redistribution of a fixed number of autophagosomes. Instead, retrograde transport, promoted by unc-16/jip3, appears to be necessary for the effective degradation of autophagic vacuoles, and defects in transport in unc-16/jip3 mutant animals therefore result in an abnormal accumulation of autophagic vacuoles. Our findings underscore the importance of regulated trafficking in the degradation of synaptic autophagic vacuoles.
Mutant allele ola316 enhances the number of autophagic vacuoles in the neurites of unc-16/jip3 mutant animals
Degradation of autophagosomes occurs upon fusion with lysosomes, an important and final step that is tightly regulated but poorly understood (Itakura et al., 2012; Nakamura and Yoshimori, 2017). To better understand the molecular machinery underlying coordinated trafficking and degradation of synaptic autophagic vacuoles, we performed a visual forward genetic screen in the unc-16/jip3 mutant background. We reasoned that molecules important for regulated lysosomal fusion and degradation would result in an enhanced accumulation of autophagosomes, and screened for mutant animals with increased numbers of LGG-1 puncta in the AIY synaptic regions. We uncovered a mutant allele, ola316, in which animals displayed a dramatic accumulation of LGG-1 puncta in the neurites. While unc-16(ju146) mutant animals displayed an average of 2.6 LGG-1 puncta abnormally accumulated in the synaptic regions (n=64 neurites), ola316; unc-16(ju146) double mutant animals enhanced this phenotype and displayed an average of 9.4 LGG-1 puncta in the synaptic regions (n=67 neurites) (Figure 3A–3D). We note that in these double mutant animals with dramatic accumulation of autophagosomes, some LGG-1-positive structures were observed outside the AIY neurite (Figure S4A-S4C). It is possible that these LGG-1 structures might be expelled from the neurite in response to an overburdening of the endo-lysosomal system, a phenomenon that has been observed in the context of neurodegenerative diseases (Borland and Vilhardt, 2017; Miranda et al., 2018; Ojha et al., 2017) (Movie S1).
Figure 3. ATG-4.2 enhances UNC-16/JIP3-mediated accumulation of autophagosomes.
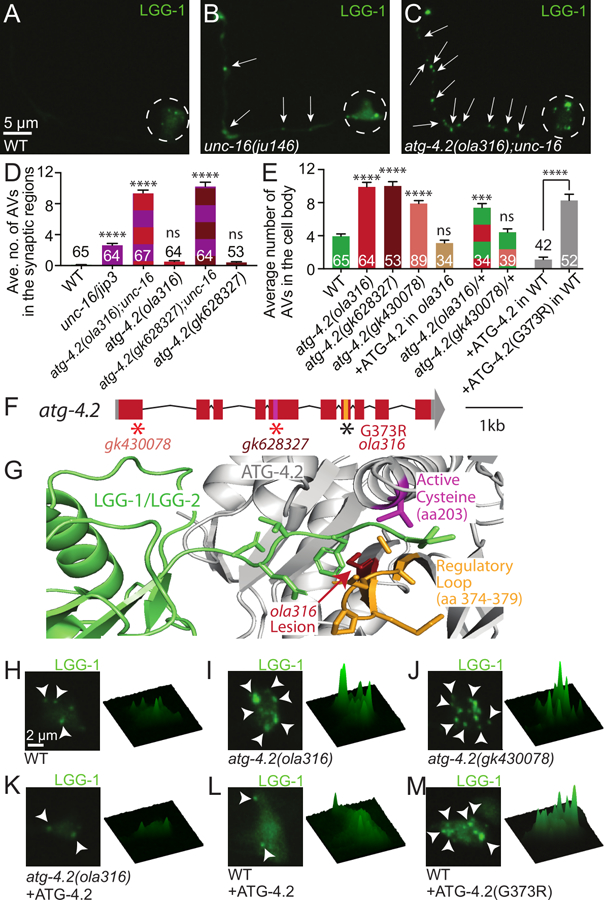
(A-C) Autophagosomes (visualized with GFP∷LGG-1) in representative wild-type (A), unc-16(ju146) mutant (B), and atg-4.2(ola316); unc-16(ju146) double mutant (C) AIY neurons.
(D-E) Quantification of the average number of LGG-1 puncta in the synaptic regions (Zones 2+3) of the AIY neurite (D) or in the cell body of AIY (E). Synaptic quantifications for wild-type and indicated mutant and double animals are shown. Here unc-16 refers to allele ju146 (D). Cell body quantifications are shown for wild-type, indicated mutant animals, cell-specific expression of an ATG-4.2 rescuing array in atg-4.2(ola316) mutant animals, heterozygous animals, and wild-type animals with AIY cell-specific expression of wild-type ATG-4.2 or mutant ATG-4.2 cDNA containing the lesion found in allele atg-4.2(ola316) (corresponding to (G373R)) (E). Error bars show standard error of the mean (SEM). ***p<0.001; ****p<0.0001 by one-way ANOVA with Tukey’s post hoc analysis between wild-type and mutant animals. The numbers on the bars represent number of neurons examined. See also Figure S1.
(F) Schematic of the atg-4.2 genomic region on chromosome IV with exons (red boxes), untranslated regions (grey boxes), and introns (black lines). Alleles affect indicated regions with early stop codons (red asterisks; gk430078 and gk628327) or amino acid change (G373R for ola316). The conserved regulatory loop, important for substrate recognition (orange bar), and cysteine active site (pink bar) are indicated. Scale bar shows a 1-kilobase region.
(G) Model of the crystal structure of the human ATG4B-LC3 complex (Kumanomidou et al., 2006) indicating the location of the G373R lesion from the ola316 allele (red arrow), the conserved regulatory loop (orange) important for LC3 recognition (LGG-1/LGG-2, in green), and the cysteine active site (pink, amino acid 203). Note that the G373R lesion in atg-4.2(ola316) neighbors the regulatory loop, and is also sterically close to the active site in the three dimensional predicted structure of the protein.
(H-M) Autophagic vacuoles in the AIY cell body imaged as maximal projection of confocal z stacks (left) and corresponding surface plots of the LGG-1 signal intensity (right) in wild-type animals (H), atg-4.2(ola316) mutant animals (I), atg-4.2(gk430078) mutant animals (J), atg-4.2(ola316) mutant animals containing an ATG-4.2 rescue array (K), wild-type animals cell-specifically overexpressing either ATG-4.2 (L) or ATG-4.2(G373R) (M).
Arrows denote autophagosomes in the neurite; arrowheads denote autophagic vacuoles in the cell body. Dashed circles enclose cell bodies. Abbreviations: “ns” is not significant; “AV” is autophagic vacuole. Scoring was done by blindly quantifying maximal projections of confocal z-stacks.
Scale bar(s) in (A) for (A-C); in (H) for (H-M).
See also Figure S4.
We then examined if animals containing the ola316 allele displayed a phenotype independent of the unc-16/jip3 mutant lesion. To achieve this, we outcrossed the ola316; unc-16(ju146) animals to remove the unc-16/jip3 lesion and examined LGG-1-containing structures in the ola316 single mutant animals. We observed that the ola316 single mutant animals did not display a significant accumulation of LGG-1 puncta in the synaptic regions when compared with unc-16(ju146) mutant or unc-16(ju146); ola316 double mutant animals (Figure 3D). However, we did observe an increased number of LGG-1 puncta in the cell bodies of the ola316 single mutant animals (Figures 3E, 3H and 3I; we observed similar results for LGG-2, Figures S4D, S4E, S4G and S4H). We also observed an increase in the percentage of neurites with one or more LGG-1 puncta in the synaptic regions (Figure S4I). The gene affected in the ola316 allele is important for most neurons in the nematode’s nervous system, as abnormal panneuronal accumulation of LGG-1 puncta was observed for both the single mutant animals and the ola316; unc-16(ju146) double mutant animals (Figure S4J-S4Q). Together our data indicate that the ola316 allele affects a gene that is important for preventing the abnormal accumulation of autophagosomes both in the neuronal soma and, through cooperation with UNC-16/JIP3, in the neurites.
ola316 is a dominant allele of the autophagy gene atg-4.2
Genetic characterization of animals containing the ola316 allele revealed that the phenotype results from a single genetic dominant lesion (Figure 3E, green and red striped bar). To identify the causative lesion in animals containing ola316, we performed single-nucleotide polymorphism (SNP) mapping, whole genome sequencing, independent allele analyses and rescue experiments. We SNP-mapped ola316 to a 2.2 Megabase (Mb) region between 10.1 and 12.3 Mb on chromosome IV. We then performed whole genome sequencing (Minevich et al., 2012; Sarin et al., 2008) and uncovered a missense lesion in the autophagy gene atg-4.2. Animals containing either of two independent alleles of atg-4.2 (gk430078 or gk628327), which result in early stop codons (Figure 3F), phenocopied the ola316 single mutant phenotype (Figure 3E (maroon and peach bars) and 3J). Moreover unc-16(ju146); atg-4.2(gk628327) double mutant animals also phenocopied the unc-16(ju146); ola316 double mutant animals isolated from the genetic screen (Figure 3D). Consistent with ola316 being an allele of atg-4.2, AIY-specific expression of the wild-type atg-4.2 cDNA in ola316 mutant animals rescued the ola316 mutant phenotype (Figures 3E (brown bar), 3I and 3K). While this rescue experiment might suggest that the dominant allele was acting through happloinsufficiency, we also observed that overexpression of mutant atg-4.2(ola316) cDNA in wild-type animals was sufficient to induce the phenotype (Figures 3E (grey bars), 3L and 3M), indicating that the atg-4.2(ola316) allele acts as a dosage-dependent dominant negative, and that it is required cell autonomously in AIY. Our data indicate that ola316 is an allele of the autophagy gene atg-4.2, and that atg-4.2 acts in neurons to regulate accumulation of autophagic structures.
Atg4s are a conserved family of cysteine proteases that bind to and cleave Atg8/GABARAP/LC3/LGG-1/LGG-2 family proteins. Comparison with the human ATG4B-LC3 crystal structure (Kumanomidou et al., 2006) revealed that the atg-4.2(ola316) lesion identified in our screen neighbors a conserved regulatory loop important for LC3 (the vertebrate homologue of LGG-2) recognition by ATG4 (Wu et al., 2012), and is sterically close to the cysteine active site (Figure 3G). Based on its position, and our in vivo genetic data, we hypothesize that the dominant lesion in the atg-4.2(ola316) allele might interfere with ATG-4.2 activity and its interaction with LGG-1/LGG-2.
Atg4s cleave Atg8/GABARAP/LC3/LGG-1/LGG-2 family proteins to achieve two functions: 1) prime LGG-1 (which contains a C terminal tail) for conjugation onto the autophagosomal membrane; and 2) remove LGG-1 and LGG-2 from the autophagosomal membrane (Alberti et al., 2010; Kirisako et al., 2000; Maruyama and Noda, 2017). In yeast one ATG4 gene performs both the priming and delipidation events on Atg8/LGG-1/LGG-2 (Kirisako et al., 2000; Maruyama and Noda, 2017), while in higher metazoans multiple genes encode distinct Atg4 cysteine proteases, some with unknown function, specificity, or redundancy (Kauffman et al., 2018; Li et al., 2011; Marino et al., 2003; Wu et al., 2012; Zhang et al., 2016). Consistent with these, a previous study in C. elegans embryos determined that the two C. elegans atg-4 genes (atg-4.1 and atg-4.2) (Figure 4A) were largely redundant (Wu et al., 2012), with ATG-4.1 displaying enhanced proteolytic activity on a soluble pro-form of LGG-1/Atg8 as compared to ATG-4.2. The specific roles of ATG-4.2 in autophagy, if any, remain unknown.
Figure 4. ATG-4.2, but not ATG-4.1, is necessary for clearance of autophagosomes.

(A) Schematic of the atg-4.1 and atg-4.2 genomic regions on chromosomes I and IV respectively, with exons (blue or red boxes), untranslated regions (grey boxes), and introns (black lines). Alleles affect indicated regions with early stop codons (red asterisks; bp501 and gk127285 in atg-4.1 and gk430078 and gk628327 in atg-4.2) or amino acid changes (G373R for ola316 in atg-4.2). Scale bar shows a 1-kilobase region.
(B-D) Autophagic vacuoles in the AIY cell body imaged as maximal projection of confocal z stacks (top row) and corresponding surface plots of the LGG-1 signal intensity (bottom row) in wild-type animals (B), atg-4.1(gk127286) mutant animals (C) and atg-4.2(ola316) mutant animals (D). Arrowheads denote LGG-1 puncta in the cell body.
(E) Quantification of the average number of LGG-1 puncta in the cell body of AIY in wild-type and indicated mutant and double mutant animals. Error bars show standard error of the mean (SEM). ****p<0.0001 by one-way ANOVA with Tukey’s post hoc analysis between wild-type and mutant animals or as indicated. The numbers on the bars represent number of neurons examined; corresponding to ≥10 animals for each genotype. Abbreviations: “ns” is not significant as compared to wild-type animals; “AV” is autophagic vacuole. Scoring was done by blindly quantifying maximal projections of confocal z-stacks.
(F-H) Representative micrographs obtained by electron microscopy (EM) of neuronal cell bodies in wild-type animals (F), atg-4.1(bp501) mutant animals (G) and atg-4.2(ola316) mutant animals (H), with the cell bodies pseudo-colored in blue, mitochondria in green and multilamellar autophagosome-like vacuoles in orange. See also Figure S5 for non-pseudo-colored images.
(I-J) Enlarged images of boxed areas showing multilamellar autophagosome-like vacuole examples from (H). See also Figure S5 for additional EM images.
(K) Quantification of the percentage of electron microscopy profiles with multilamellar autophagosome-like vacuoles in the cell somas of wild-type and mutant animals. Error bars show a 95% confidence interval. ****p<0.0001 by Fisher’s exact test between wild-type and mutant conditions. The numbers on the bars indicate the number of profiles scored; corresponding to ≥ 5 cell bodies from 2 different animals for each genotype. Abbreviations: “ns” is not significant as compared to wild-type animals. Quantification was done by blindly scoring the electron micrograph somatic profiles.
Scale bar in (B) for (B-D); in (F) for (F-H), in (I) for (I-J).
See also Figure S5.
ATG-4.2, but not ATG-4.1, is required to prevent autophagosome accumulation
The two C. elegans Atg4 homologs display 44% amino acid similarity, including conserved catalytic and regulatory sites (Wu et al., 2012). A previous study investigating the degradation of protein aggregates in embryos found that atg-4.1 is more efficient at the first LGG-1 cleavage event (priming), and more important for removal of protein aggregates than atg-4.2 (Wu et al., 2012). To examine if atg-4.1 also acts as atg-4.2 to prevent autophagosomal accumulation in neurons we examined animals containing a confirmed null allele of atg-4.1(bp501) (Wu et al., 2012) or containing the allele atg-4.1(gk127286) (early stop codon) (Figure 4A). Surprisingly we did not detect LGG-1 or LGG-2 puncta accumulation in the atg-4.1 mutant animals (Figures 4C, 4E, S4F-S4H, and S4R).
We then examined autophagosomes in the neuronal cell bodies of these mutant animals by performing electron microscopy. Consistent with our GFP∷LGG-1 and GFP∷LGG-2 in vivo assays, we observed a dramatic accumulation of abnormal multilamellar autophagosome-like vacuoles in the neuronal somas of atg-4.2(ola316) mutant animals (Figures 4F–4K and S5A-S5P). Our findings indicate that atg-4.1 and atg-4.2 have distinct functions in vivo that result in different phenotypes regarding the accumulation of autophagosomes in neurons. Importantly, our findings indicate that atg-4.2 is specialized for the clearance of autophagic vacuoles.
While we observed distinct phenotypes for atg-4.1 and atg-4.2 in the accumulation of autophagic structures, we also note that both atg-4.1 and atg-4.2 single mutant animals contained LGG-1/LGG-2 puncta in neurons (Figures 4C–4E and S4E-S4H). Comparisons with the corresponding lipidation controls (LGG-1(G116A); explanatory diagrams in Figure S1) suggest that nearly all of the observed LGG-1 puncta in atg-4.1 and atg-4.2 mutant animals were lipidated (Figure S5Q, S5T-S5V). These observations indicate that either protease is able to partially compensate for the absence of the other protease and perform the first priming cleavage necessary to conjugate LGG-1 onto the nascent autophagosome. They also localized similarly throughout the neuronal cytoplasm, consistent with the ability to act either in the synaptic region or the cell body (Figure S6A-S6B). Also consistent with these two proteases performing partially redundant roles, we have previously shown that in AIY, where autophagy is also required for synapse development, the synaptic assembly phenotype was not seen for atg-4.1 or atg-4.2 single mutant animals, but was evident in the atg-4.1; atg-4.2 double mutant animals (Figure S6C-S6G and (Stavoe et al., 2016)), and that the GFP∷LGG-1 phenotype is enhanced in atg-4.1; atg-4.2 double mutant animals (Figures 4E and S6H-S6I). Our results indicate that while atg-4.1 and atg-4.2 can act in a partially redundant fashion, they also display distinct phenotypes in LGG-1/LGG-2 puncta accumulation. Importantly, our findings indicate that atg-4.2 is specifically necessary to prevent abnormal accumulation of autophagosomes in the neuronal soma.
ATG-4.2 promotes autophagosome maturation
Our observations that lesions in atg-4.2 result in animals with an abnormal accumulation of autophagosomes in the neuronal cell body are consistent with a model where atg-4.2 acts late in the autophagy pathway to promote autophagosome maturation and autophagic degradation. Autophagosome maturation into an acidic autolysosome is mediated by fusion between autophagosomes and late endosomes or lysosomes in a process that requires the small GTPase Rab7 (Gutierrez et al., 2004; Hyttinen et al., 2013; Jager et al., 2004). We reasoned that if atg-4.2 mutant animals have a defect at this step, then loss of rab-7 function should phenocopy the atg-4.2 mutant phenotype.
Since null lesions in rab-7 are lethal, we examined animals mutant for the rab-7 activator sand-1/Mon1 (Hegedus et al., 2016; Poteryaev et al., 2007), and the rab-7 effector, epg-5 (Wang et al., 2016). We observed that indeed sand-1 and epg-5 mutant animals displayed increased autophagosome accumulation in the cell body compared to wild type-animals, similar to atg-4.2(ola316) mutant animals (Figure 5A–5C, 5G). The observed phenotypes are specific to late blocks in autophagy, as mutant animals for genes involved in early events prior to autophagosome closure or trafficking, such as atg-9(wy56), atg-2(bp576), or unc-16(ju146), did not display autophagosome accumulation in the cell body (Figure 5G). Also consistent with atg-4.2 acting in a late step of autophagy to promote autophagosome maturation, we found that atg-4.2(gk628327); epg-5(tm3425) double mutant animals phenocopied either single mutant for the cell body accumulation phenotype, suggesting that they act in the same pathway in this process (Figure S6J). We do note however that the atg-4.2(gk628327); epg-5(tm3425) double mutant animals also displayed an enhanced accumulation in the presynaptic region (Figure S6K), suggesting that they might perform distinct roles at presynaptic sites, which we did not further examine in this study. Together our findings indicate that atg-4.2, like sand-1 and epg-5, acts in a late stage of autophagy prior to degradation.
Figure 5. ATG-4.2 promotes autophagosome maturation.
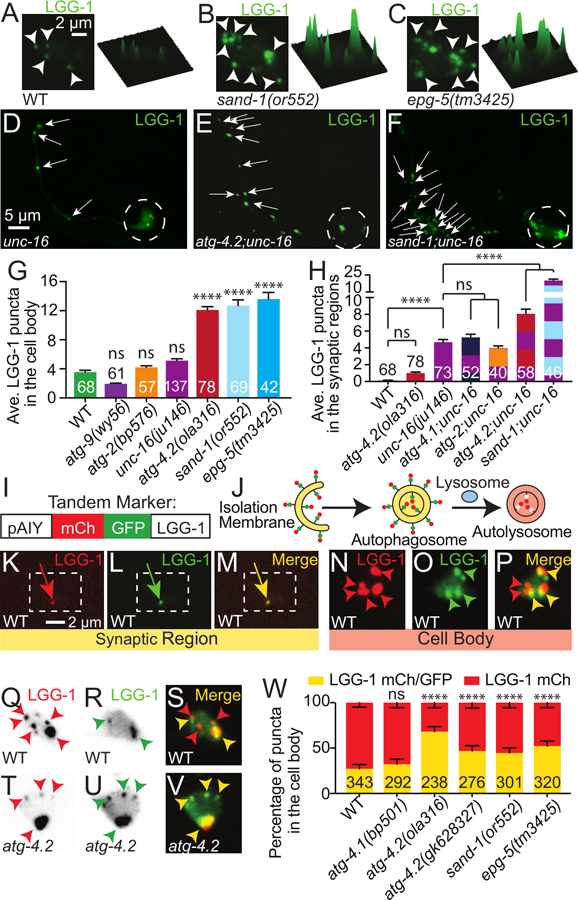
(A-C) Autophagic vacuoles in the AIY cell body imaged as maximal projections of confocal z stacks (left) and corresponding surface plots of the LGG-1 signal intensity (right) in representative wild-type (A), sand-1(or552) mutant (B) and epg-5(tm3425) mutant (C) animals.
(D-F) Autophagic vacuoles (visualized with GFP∷LGG-1) in representative unc-16(ju146) mutant (D), atg-4.2(ola316); unc-16(ju146) double mutant (E) and sand-1(or552); unc-16(ju146) double (F) mutant neurons.
(G-H) Quantification of the average number of autophagic vacuoles in the cell body (G) or synaptic region (H) of AIY in wild-type and mutant or double mutant animals. Alleles examined were unc-16(ju146), atg-4.1(gk127286), atg-2(bp576), atg-4.2(ola316), sand-1(or552) or as indicated. Note that the increased severity of the sand-1 enhancement might suggest a more complete block in autophagosome maturation than that of atg-4.2, which is partially redundant with atg-4.1. Error bars show standard error of the mean (SEM). ***p<0.001; ****p<0.0001 by one-way ANOVA with Tukey’s post hoc analysis between wild-type and mutant animals or as indicated. The numbers on the bars represent number of neurons examined. Abbreviations: “ns” is not significant as compared to wild-type animals; “AV” is autophagic vacuole.
(I-J) Schematic for the tandem LGG-1 marker (mCh::GFP::LGG-1), showing the construct configuration with the AIY promoter (I) and the progression of the tandem LGG-1 marker through the stages of autophagosome biogenesis, fusion with lysosomes and acidification (J and (Chang et al., 2017; Kimura et al., 2007)).
(K-P) Autophagic vacuoles in a representative wild-type AIY neuron, visualized with the tandem marker (mCh∷GFP∷LGG-1) in the synaptic region (K-M) and the cell body (N-P). The mCherry channel (K and N), GFP channel (L and O) and merge (M and P) are shown separately. In (K-M), the dashed box encloses the synapse-rich portion of the synaptic region (similar to Figure 1A–1D).
(Q-V) Autophagic vacuoles in representative wild-type (Q-S) and atg-4.2(ola316) mutant (T-V) cell bodies, visualized with the tandem marker (mCh∷GFP∷LGG-1). The mCherry channel (Q and T), GFP channel (R and U) and merge (S and V) are shown separately. Single channels (Q, R, T and U) are inverted to better show dim red puncta.
(W) Quantification of puncta labeled with the LGG-1 tandem marker in the cell body, showing the percentage of puncta with mCh and GFP colocalized signal (yellow, immature) or mCh only signal (mature). Error bars show a 95% confidence interval. ****p<0.0001 by Fisher’s exact test between wild-type and mutant animals. The numbers on the bars indicate the number of puncta scored; corresponding to ≥27 neurons for each genotype. Abbreviations: “ns” is not significant as compared to wild-type animals.
Arrowheads denote LGG-1 puncta in the cell body. Arrows denote LGG-1 puncta in the synaptic regions. Dashed circles enclose cell bodies. In merge images (M, P, S, V), red arrowheads refer to puncta labeled with only mCh (representing acidified autophagic vacuoles), while yellow arrows and arrowheads refer to structures labeled with both mCh and GFP (representing autophagosomes that have not yet been acidified). Quantifications were done by blindly scoring maximal projections of confocal micrographs.
Scale bar(s) in (A) for (A-C), and (N-V); in (D) for (D-F); in (K) for (K-M).
See also Figure S6.
Atg-4.2 genetically interacts with unc-16 in double mutant animals to enhance the number of autophagosomes observed at the synapse. To test if sand-1 also genetically interacts with unc-16, we created sand-1(or552); unc-16(ju146) double mutant animals. We observed that like atg-4.2; unc-16 double mutant animals, sand-1; unc-16 double mutant animals also displayed dramatically enhanced accumulation of autophagosomes in the synaptic regions (Figure 5D–5F and 5H). Importantly, early autophagy mutants like atg-2 (required for autophagosome closure) (Velikkakath et al., 2012) and atg-4.1 (required for LGG-1 priming) (Wu et al., 2012) failed to enhance the neurite accumulation phenotype observed in unc-16 mutant animals (Figure 5H). Our findings indicate that the observed enhancement of unc-16 with either sand-1 or atg-4.2 is a specific phenotype resulting from a genetic interaction between defects in retrograde transport and defects in autophagosome degradation.
We then tested the acidification state of autophagic vacuoles by employing a tandem label strategy in which LGG-1 was fused to both mCherry and GFP (Figure 5I; lipidation controls in Figure S7A-S7D and (Chang et al., 2017; Kimura et al., 2007)). Briefly, in this strategy immature autophagosomes are labeled with both GFP and mCherry, but because GFP is preferentially quenched in acidic environments, autophagic structures lose their GFP signal and display solely mCherry signal when mature (schematic, Figure 5J). Comparisons with the tandem marker lipidation control (Figure S7A-S7D), suggest that while GFP/mCh positive structures are lipidated, some mCh positive/GFP negative puncta accumulate in acidified lysosomes (see Figure S7, Figure Legend and STAR Methods). Using this approach in vivo and in single neurons of wild-type animals, we observed that 88% of synaptic LGG-1-labeled structures were positive for mCh and GFP signal (Figures 5K–5M and S7H). In contrast, examination of LGG-1-labeled structures in the cell body region of the neurons demonstrated that over 70% of the structures were positive for mCherry, but not for GFP (Figures 5N–5P). Our findings in wild-type animals are consistent with studies that demonstrate that active lysosomes are preferentially enriched in the neuronal cell soma (Hollenbeck, 1993; Kaasinen et al., 2008), and suggest that most autophagosomes at synapses mature as they are transported to the neuronal cell body.
To better understand the ability of the tandem marker to assess the maturation state of autophagosomes in mutant animals, we first examined it in sand-1/Mon1 (Hegedus et al., 2016; Poteryaev et al., 2007), and epg-5 (Wang et al., 2016) mutant animals, which are known to block autophagosome acidification. We observed that as compared to wild-type animals, the percentage of mature LGG-1 structures (mCh positive, GFP negative) decreases (Figure 5W), as expected given the known mechanism of action of these genes in autophagosome maturation.
We then used the tandem marker to examine the maturation state of autophagosomes in atg-4.2 mutant animals. For two independent alleles of atg-4.2, we observed that a higher percentage of autophagic vacuoles were immature (positive for both GFP and mCh signal) compared to wild-type or atg-4.1 mutant animals (Figure 5Q–5W). The immature state of the autophagic puncta in atg-4.2 mutant animals could be a result of failure to remove LGG-1 from the outside of the autophagosome (where it would not be acidified), failure to undergo internal acidification, or both, possibilities that would need to be further distinguished through biochemical assays (see below). Nevertheless, these findings with the tandem marker indicate that atg-4.2, but not atg-4.1, is required for late stages in the progression of autophagy.
ATG-4.2 delipidates LGG-1 and is necessary for the formation of autolysosomes that degrade synaptic proteins
Why is atg-4.2, but not atg-4.1, required for the late stages of autophagy? Atg4s are cysteine proteases that cleave Atg8/GABARAP/LC3/LGG-1/LGG-2 to: 1) activate LGG-1 for conjugation onto the autophagosomal membrane (early event) or 2) remove LGG-1/LGG-2 from the autophagosomal membrane (late event) ((Kirisako et al., 2000; Maruyama and Noda, 2017) and Figure 6A). We wondered if ATG-4.2 might have distinct biochemical activity towards LGG-1 in these steps.
Figure 6. ATG-4.2 delipidates LGG-1 to promote formation and acidification of autolysosomes, and breakdown of synaptic cargo.

(A-B) Biochemical characterization of ATG-4.2 activity. Cartoon diagram displaying the tested conditions in the reconstituted biochemical system: cleavage of a soluble LGG-1 substrate (LGG1-His) mimicking priming (blue), which precedes lipidation onto autophagosomal membranes, or cleavage of lipidated LGG-1 on liposomes, mimicking release from the autophagic membrane, delipidation (red).
(C-E) Lysosomes (LAAT-1∷GFP, green arrowheads), autophagosomes (mCh∷LGG-1, red arrowheads) and autolysosomes (co-localization of LAAT-1 and LGG-1, yellow arrowheads in merge images) are visualized in the AIY cell body of wild-type (C) and atg-4.2(ola316) mutant animals (D). In (E), quantification of the percentage of puncta in the cell body displaying LAAT-1∷GFP, mCh∷LGG-1 or co-localization of these two markers in the indicated genotypes is shown. Error bars show a 95% confidence interval. ****p<0.0001 by Chi-square test between wild-type and mutant animals. The numbers on the bars indicate the number of puncta scored; corresponding to ≥66 neurons for each genotype.
(F-H) Autophagic vacuoles visualized with the tandem marker (mCh∷GFP∷LGG-1) and lysosomes visualized with LAAT-1:BFP in the AIY cell body of wild-type animals (F) and atg-4.2(ola316) mutant animals (G). In merge images, grey arrowheads refer to immature autolysosomes (AVL) (positive for mCh, GFP, and BFP) while purple arrowheads refer to mature autolysosomes (AL) (or mature lysosomes, represented as L in legend) (positive for mCh and BFP). In (H), quantification of the percentage of puncta in the cell bodies that represent either mature or immature autolysosomes based on the tandem marker analyses. Error bars show a 95% confidence interval. ****p<0.0001 by Fisher’s exact test between wild-type and mutant animals. The numbers on the bars indicate the number of puncta scored; corresponding to ≥39 neurons for each genotype.
(I-K) Quantification of the percentage of autophagic vacuoles (mCh∷LGG-1) displaying co-localization with the presynaptic protein GFP∷SYD-1 in the cell body of AIY interneurons (I). Error bars show a 95% confidence interval. ****p<0.0001 by Fisher’s exact test between wild-type and mutant animals. The numbers on the bars indicate the number of puncta scored; corresponding to ≥50 neurons for each genotype. Abbreviations: “AVs” is autophagic vacuoles. In (J-K), representative confocal micrographs of the colocalization of presynaptic active zone protein GFP∷SYD-1 (first column in J and K), mCh∷LGG-1 (second column in J and K), and merge images (third column in J and K) in wild-type animals (J) and atg-4.2(ola316) mutant animals (K). In merge images, green arrowheads refer to accumulation of SYD-1, red arrowheads to LGG-1 puncta and yellow arrowheads to co-localization of LGG-1 and SYD-1, indicative of autophagic vacuoles with synaptic material.
(L-O) Schematic for the tandem SYD-1 active zone protein marker (mCh∷GFP∷SYD-1), showing the construct configuration with the AIY promoter (L). Quantification of the percentage of SYD-1 puncta displaying mCh only (representing SYD-1 in an acidified compartment), or mCh and GFP (representing SYD-1 in an environment that is not acidified) (M). Error bars show a 95% confidence interval. ****p<0.0001 by Fisher’s exact test between wild-type and mutant animals. The numbers on the bars indicate the number of puncta scored; corresponding to ≥46 neurons for each genotype. In (N and O), representative confocal images of the tandem SYD-1 presynaptic active zone protein marker in wild-type animals (N) and atg-4.2(ola316) mutant animals (O). In merge images, red arrowheads refer to active zone protein SYD-1 puncta labeled with only mCh, while yellow arrowheads refer to structures labeled with both mCh and GFP.
In E, H, I, and M, quantifications were done by blindly scoring maximal projections of confocal micrographs.
Scale bar in (C) for (C), (D), (F), (G), (J), (K), (N) and (O).
See also Figure S7.
We tested ATG-4.2 activity on LGG-1 by generating recombinant C. elegans ATG-4.2 and in a biochemically-reconstituted system examining either: 1) ATG-4.2 priming activity on the C-terminal of LGG-1, using a construct with an extended C-terminus (LGG-1-His); or 2) delipidation activity on LGG-1 conjugated onto liposomes, as described (Kauffman et al., 2018). We observed that after 8 minutes, 89% of liposomal LGG-1 was cleaved, but only 18% of LGG-1-His was cleaved (average of three independent experiments; Figure 6B), indicating that ATG-4.2 preferentially delipidates (or removes) LGG-1 from autophagosomal membranes. Our biochemical results are consistent with our cell biological and genetic data, and together suggest a model whereby delipidation of LGG-1, specifically by ATG-4.2, is a necessary step for correct maturation of autophagosomes.
To gain further insights into the effects of ATG-4.2 in autophagic progression we examined if there were defects in autolysosomes of atg-4.2 mutant animals. Formation of autolysosomes, which occurs through the fusion of autophagosomes and lysosomes, is a necessary step in autophagosome maturation, and can be assayed by probing the co-localization of lysosomal proteins, such as LAAT-1 (Liu et al., 2012) and the autophagy protein LGG-1 (Hyttinen et al., 2013; Maday et al., 2012; Zhang et al., 2015). We observed that in wild-type animals 91% of the LGG-1 puncta in the cell body co-localized with LAAT-1 (Figure S7I-S7P)), consistent with a progression towards autophagosomes-lysosome fusion and acidification as autophagic structures are transported towards the neuronal cell body. In atg-4.2 mutant animals, while there was no change in LGG-1/LAAT-1 colocalization in the synaptic region, nor in the total number of LGG-1 and/or LAAT-1 structures in the cell body (Figure S7I and S7Q), there was a significant reduction in colocalization of LGG-1 and LAAT-1 (Figure 6C–6E). These findings are consistent with a role for atg-4.2 in promoting autolysosome formation in the neuronal cell body.
Next, we examined the maturation state of the autolysosomes by using the LGG-1 tandem marker while simultaneously visualizing LAAT-1:BFP. Consistent with our previous findings, we observed that in wild-type animals 92% of LAAT-1 and LGG-1 colocalized structures were mature (red positive, green negative), while in atg-4.2(ola316) mutant animals only 33% of LAAT-1 and LGG-1 colocalized structures were mature (Figure 6F–6H). This effect was, at least in part, due to a failure to delipidate LGG-1 from the cytoplasmic face of the autolysosomes, but might also reflect a defect in internal acidification. Our observations are consistent with atg-4.2 mutant animals having a defect in the maturation of autophagic structures, and suggest that ATG-4.2 is necessary for the formation of autolysosomes.
To examine how atg-4.2 affects the acidification of autophagic cargo, we visualized the co-localization of synaptic proteins with LGG-1 in the neuronal cell body. Consistent with the role of autophagy in degrading synaptic material, and with the role of ATG-4.2 in the clearance of autophagic structures, we observed that atg-4.2 mutant animals displayed an increased percentage of autophagosomes co-localized with synaptic proteins in the cell soma (Figure 6I–6K and Figure S7R). Moreover generation of a synaptic tandem marker, which enabled visualization of the internal acidification state of compartments containing synaptic material, revealed that in atg-4.2 mutant animals fewer compartments in which synaptic proteins were acidified (Figure 6L–6O). Together, our findings indicate that atg-4.2 mutant animals are defective in acidification, resulting in an accumulation of immature autophagosomes with undigested cargo, including synaptic material.
DISCUSSION
Distinct steps of the autophagy pathway occur in different subcellular compartments. In vivo, autophagosome biogenesis occurs at presynaptic sites (Soukup et al., 2016; Stavoe et al., 2016) and the number of autophagosomes in the neurite can be increased by stimulating the activity of neurons (Figure 1 and (Shehata et al., 2012; Soukup et al., 2016; Wang et al., 2015). Autophagosomes then undergo retrograde trafficking prior to degradation (Maday et al., 2012), indicating a regional decoupling of biogenesis and degradation. In this study we extend these observations by demonstrating that in C. elegans the number of synaptic autophagosomes in the AIY interneuron predictably changed depending on the firing state of the neuron, which we could manipulate by altering physiological stimuli that promote AIY responses, by genetically inhibiting synaptic transmission or by chemo-genetically altering the response state of the neuron. We also observed co-localization of synaptic proteins with autophagosomes in the cell body, further indicating a link between autophagosome biogenesis at the synapse, and autophagy-mediated degradation of synaptic proteins in the soma. Our findings extend our understanding of autophagy regulation in neurons, and uncover mechanisms linking autophagosomes at the synapse with their clearance in the cell soma.
UNC-16/JIP3-dependent retrograde transport plays an important role in transport and clearance of synaptic autophagosomes. In C. elegans, Drosophila and vertebrate neurons, axonal autophagosomes are transported towards the cell body via the dynein complex (Cheng et al., 2015; Ikenaka et al., 2013; Katsumata et al., 2010; Neisch et al., 2017). UNC-16/JIP3/Sunday Driver, a motor adaptor protein that interacts with dynein, plays important and conserved roles in the regulated localization and transport of late endosomes and lysosomes, as well as other organelles (Brown et al., 2009; Byrd et al., 2001; Drerup and Nechiporuk, 2013; Edwards et al., 2015; Edwards et al., 2013; Gowrishankar et al., 2017). Our findings now demonstrate that UNC-16/JIP3 is also required for autophagosome retrograde transport. In vertebrate neurons, autophagosome retrograde transport is promoted by JIP1, a motor adaptor protein that belongs to the same family as UNC-16/JIP3 (Fu and Holzbaur, 2014; Fu et al., 2014). While we did not detect a phenotype for autophagosome retrograde transport in an available C. elegans jip-1 nonsense allele (for which partial function is unknown), we note that JIP1 and JIP3 share sequence and functional similarities, and are also known to cooperate in the transport of cargo (Hammond et al., 2008; Sun et al., 2017). Together, our findings ascribe an important and potentially conserved role for UNC-16/JIP3 in retrograde transport of autophagosomes in C. elegans neurons.
Impaired retrograde transport in unc-16/jip3 mutant animals resulted in accumulation of LGG-1-containing structures in the neurites. In vertebrate neurons, disruption of retrograde transport also causes accumulation of autophagosomes and lysosomes in swollen neuronal processes and synapses. This, in turn, results in autophagic stress and in neurodegeneration phenotypes similar to those seen in the neurons of Alzheimer’s disease patients (Ikenaka et al., 2013; Lee et al., 2011; Nixon et al., 2005; Takats et al., 2013). Specific disruption of JIP3 in vertebrate neurons has also been associated with an increase in soluble Aβ levels, plaque size, plaque abundance, and axonal dystrophy (Gowrishankar et al., 2017). Our findings in C. elegans neurons are consistent with these studies and underscore the importance of retrograde transport in linking the mechanisms of autophagosome biogenesis at the synapse with degradation in the cell body. Furthermore, our observations that combined defects in both retrograde transport and degradation machinery enhanced autophagosome accumulation in the neurites raises the question of whether the accumulated autophagosomes seen in progressive neurodegenerative diseases might result from an accruement of distinct dysfunctions in different stages of autophagy.
ATG-4.2 genetically cooperates with UNC-16/JIP3 in the clearance of autophagic vacuoles from axons (Figure 7A-7D). ATG-4.2 is one of two Atg4 cysteine proteases in C. elegans (Wu et al., 2012). Atg4 cysteine proteases are required during both early and late stages of autophagy to cleave Atg8/LGG-1/LGG-2, but to achieve two distinct functions: the first cleavage (priming) is required for the conjugation of LGG-1 to the autophagosomal membrane to promote autophagosome elongation during biogenesis, while the second cleavage (delipidation) removes LGG-1/LGG-2 from the membrane prior to autophagic degradation (Kirisako et al., 2000; Maruyama and Noda, 2017). Our biochemical and cell biological studies indicate that ATG-4.2 preferentially performs delipidation on LGG-1 to remove it from autophagic membranes, and that this step is necessary for fusion of autophagosomes with lysosomes and degradation.
Figure 7. Synaptic autophagy: a model from birth at the synapse to breakdown in the cell body.
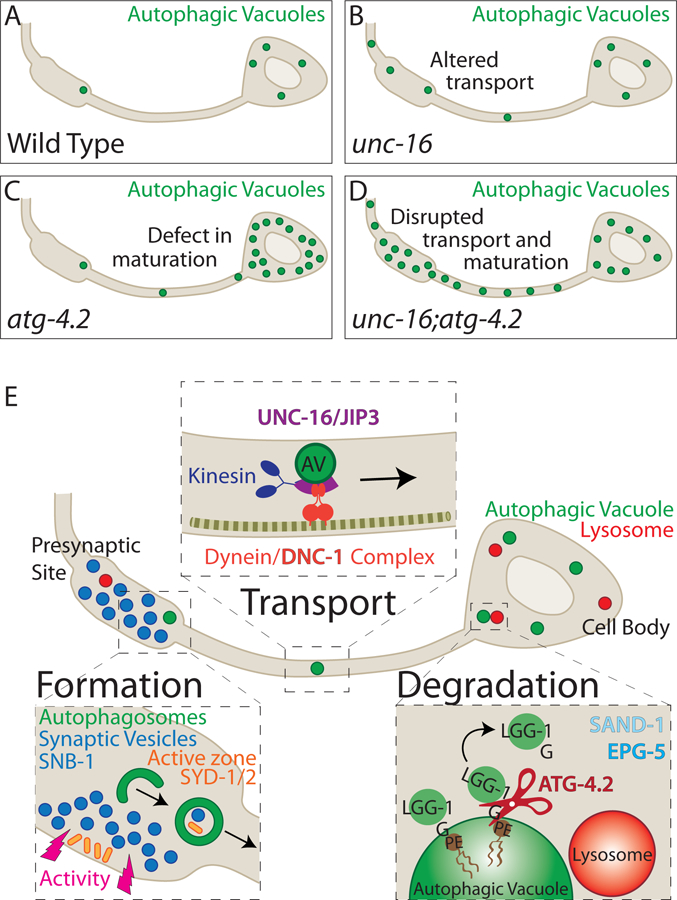
(A-D) Schematics of autophagic vacuole localization in wild-type (A), unc-16 mutant (B), atg-4.2 mutant (C), and unc-16; atg-4.2 double mutant (D) animals. Disruption of retrograde transport (unc-16 mutant) or maturation (atg-4.2 mutant) leads to autophagosome accumulation in the neurite or the cell body respectively, and that these pathways cooperate to clear autophagosomes from the synapse (D).
(E) Model schematic illustrating the steps of synaptic autophagy in the neuron, and key molecules involved. Formation: Autophagosomes form at presynaptic sites, influenced by the state of synaptic activity of the neuron (Figure 1 and (Shehata et al., 2012; Soukup et al., 2016; Wang et al., 2015)) through ordered transport and assembly of autophagy molecules (Maday and Holzbaur, 2014), including ATG-9 (Stavoe et al., 2016). Synaptic autophagosomes contain post-synaptic proteins, synaptic vesicles (Nikoletopoulou et al., 2017; Okerlund et al., 2017; Rowland et al., 2006) and active zone proteins.
Transport: Autophagosomes undergo retrograde transport towards the cell body, a process dependent on the dynein activator dynactin/DNC-1, and the motor adaptor UNC-16/JIP3.
Degradation: Once in the cell body, the autophagy protease ATG-4.2 cleaves LGG-1 and LGG-2 off the autophagic vacuole membrane to facilitate maturation and degradation in a process that also requires the RAB-7 activator SAND-1/Mon1 and effector EPG-5.
Our findings, together with previous in vitro studies showing that ATG-4.1 is the dominant priming protease (Wu et al., 2012), are consistent with a model whereby distinct cleavage roles in LGG-1/Atg8 priming and delipidation might be regulated in vivo by distinct ATG4 proteases. In support of this idea, a recent biochemical study examining mammalian ATG4 proteins revealed that different ATG4s possess different activities for cleaving soluble or membrane-bound GABARAP/Atg8/LGG-1 (Kauffman et al., 2018). Distinct ATG4 proteases could therefore modulate the association of LGG-1/LGG-2 with autophagic membranes either during priming or delipidation, and these regulated steps could “bookend” the progression of autophagy from biogenesis to degradation (model presented in Figure 7E). Disruption of retrograde transport of synaptic autophagosomes, or defective delipidation of LGG-1/LGG-2 by ATG-4.2, would enhance abnormal accumulation of autophagosomes in neurons and result in cell biological phenotypes reminiscent to those seen in neurodegenerative diseases.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel A. Colón-Ramos (daniel.colon-ramos@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans strains and genetics
All C. elegans strains were raised on NGM plates with OP50 Escherichia coli as a food source, kept at 20°C, unless otherwise noted, and analyzed as larval 4 (L4) stage animals. We used N2 Bristol as the wild-type reference strain. We used the balancer nT1[qIs51] isolated from VC3476 to assess escapers from olaIs35; atg-4.1; atg-4.2/nT1, which is sterile in its unbalanced state. For a full list of strains used in this study, please see the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Levamisol hydrochloride | Sigma-Aldrich | 31742 |
| Ethyl methanesulfonate (EMS) | Sigma-Aldrich | M0880 |
| IPTG | AmericanBio | AB00841 |
| Binding buffer (25mM Tris, pH7.6, 150mM NaCl, 10mM Imidazole, 1mM BME) | Modified from Wu et al., 2012 | N/A |
| cOmplete™, EDTA-free Protease inhibitor cocktail tablet | Roche | 11873580001 |
| Nickel beads | Qiagen | 1018244 |
| Washing buffer (25mM Tris, pH 7.6, 150mM NaCl, 60mM Imidazole, 1mM BME) | Modified from Wu et al., 2012 | N/A |
| Elution buffer (25mM Tris, pH 7.6, 150mM NaCl, 250mM Imidazole, 1mM BME) | Modified from Wu et al, 2012 | N/A |
| Thrombin buffer (20 mM Tris, pH 7.6, 100 mM NaCl, 5 mM MgCl2, 2 mM CaCl2, 1 mM DTT) | Choy et al., 2012 | N/A |
| Glutathione beads | Sigma Aldrich | G4501 |
| Thrombin | Sigma Aldrich | T6884 |
| SN buffer (20mM Tris, pH 7.6, 100mM NaCl, 5mM MgCl2) containing 1 mM DTT | Choy et al, 2012 | N/A |
| LDS Loading Buffer | ThermoFisher Scientific | NP007 |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) | Avanti Polar Lipids | 850752 |
| 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) | Avanti Polar Lipids | 857457 |
| L-α-phosphatidylinositol (liver, bovine) (PI) | Avanti Polar Lipids | 840042 |
| Nycodenz | Accurate Chemical and Scientific Corp. | 1002424 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: olaIs35 X [Pttx-3::egfp::lgg-1; Pttx-3::mCh]) | Stavoe et al., 2016 | DCR4750 |
| C. elegans: unc-13(e450); olaIs35 | This paper | DCR5581 |
| C. elegans: olaEx3465 [Pmod-1::HisCl (30ng/ul)]; olaIs35 | This paper | DCR5896 |
| C. elegans: dnc-1(or404) IV | Caenorhabditis Genetics Center | EU1006 |
| C. elegans: dnc-1(or404); olaIs35 | This paper | DCR5549 |
| C. elegans: unc-16(ju146) III | Caenorhabditis Genetics Center | CZ3011 |
| C. elegans: unc-16(ju146); olaIs35 | This paper | DCR5353 |
| C. elegans: unc-16(e109) III | Caenorhabditis Genetics Center | CB109 |
| C. elegans: unc-16(e109); olaIs35 | This paper | DCR5579 |
| C. elegans: unc-16(n730) III | Caenorhabditis Genetics Center | MT1542 |
| C. elegans: unc-16(n730);olaIs35 | This paper | DCR5597 |
| C. elegans: jip-1(gk466982) II | Caenorhabditis Genetics Center | VC40110 |
| C. elegans: jip-1(gk466982); olaIs35 | This paper | DCR5650 |
| C. elegans: unc-116(e2310) III | Caenorhabditis Genetics Center | FF41 |
| C. elegans: unc-116(e2310); olaIs35 | This paper | DCR5440 |
| C. elegans: unc-14(e57) I | Caenorhabditis Genetics Center | CB57 |
| C. elegans: unc-14(e57); olaIs35 | This paper | DCR5454 |
| C. elegans: atg-4.1(bp501) I | Caenorhabditis Genetics Center | HZ1685 |
| C. elegans: atg-4.1(bp501); olaIs35 | This paper | DCR6360 |
| C. elegans: atg-4.1(gk127286) I | Caenorhabditis Genetics Center | VC30126 |
| C. elegans: atg-4.1(gk127286); olaIs35 | This paper | DCR5752 |
| C. elegans: atg-4.1(gk127286); unc-16(ju146); olaIs35 | This paper | DCR6134 |
| C. elegans: atg-4.2(gk430078) IV | Caenorhabditis Genetics Center | VC30172 |
| C. elegans: atg-4.2(gk430078); olaIs35 | This paper | DCR5649 |
| C. elegans: atg-4.2(gk628327) IV | Caenorhabditis Genetics Center | VC40419 |
| C. elegans: atg-4.2(gk628327); olaIs35 | This paper | DCR6874 |
| C. elegans: unc-16(ju146); atg-4.2(gk628327); olaIs35 | This paper | DCR6876 |
| C. elegans: atg-4.2(ola316) IV | This paper | DCR5765 |
| C. elegans: atg-4.2(ola316); olaIs35 | This paper | DCR5551 |
| C. elegans: unc-16(ju146); atg-4.2(ola316); olaIs35 | This paper | DCR5652 |
| C. elegans: atg-4.2(ola316)/nT1[qIs51]; atg-4.1(bp501); olaIs35 | This paper | DCR6390 |
| C. elegans: atg-4.2(gk430078)/nT1[qIs51]; atg-4.1(bp501); olaIs35 | This paper | DCR6391 |
| C. elegans: sand-1(or552) | Caenorhabditis Genetics Center | FA85 |
| C. elegans: sand-1(or552); olaIs35 | This paper | DCR6672 |
| C. elegans: unc-16(ju146); sand-1(or552); olaIs35 | This paper | DCR6736 |
| C. elegans: epg-5(tm3425);olaIs35 | Stavoe et al., 2016 | DCR4859 |
| C. elegans: atg-4.2(gk628327); epg-5(tm3425); olaIs35 | This paper | DCR7240 |
| C. elegans: atg-2(bp576); olaIs35 | Stavoe et al., 2016 | DCR4906 |
| C. elegans: unc-16(ju146); atg-2(bp576); olaIs35 | This paper | DCR5770 |
| C. elegans: atg-9(wy56);olaIs35 | Stavoe et al., 2016 | DCR4851 |
| C. elegans: atg-4.2(gk628327); atg-9(wy56); olaIs35 | This paper | DCR7365 |
| C. elegans: olaEx3430 [Pttx-3::atg-4.2 (30ng/ul)]; olaIs35 | This paper | DCR5843 |
| C. elegans: olaEx3438 [Pttx-3::atg-4.2 (30ng/ul)]; atg-4.2(ola316); olaIs35 | This paper | DCR5854 |
| C. elegans: olaEx3426 [Pttx-3::atg-4.2(G1117A) (30ng/ul)]; olaIs35 | This paper | DCR5833 |
| C. elegans: olaEx3293 [Pelt-7::gfp (10ng/ul)]; olaIs35 | This paper | DCR5559 |
| C. elegans: olaEx3986 [Pttx-3::mCh::rab-3 (30ng/ul)]; olaIs44 [Pttx-3::egfp::lgg-1 (15ng/ul)] | This paper | DCR6670 |
| C. elegans: olaEx4440 [Pmod-1::gfp::lgg-1(G116A) (5ng/ul), Pttx-3::mCh (30ng/ul)] | This paper | DCR7354 |
| C. elegans: olaEx4440; atg-4.2(ola316) | This paper | DCR7393 |
| C. elegans: olaEx4440; atg-4.2(gk628327) | This paper | DCR7394 |
| C. elegans: olaEx4440; epg-5(tm3425) | This paper | DCR7395 |
| C. elegans: olaEx4440; sand-1(or552) | This paper | DCR7396 |
| C. elegans: olaEx4440; unc-16(ju146) | This paper | DCR7397 |
| C. elegans: olaEx4440; unc-16(n730) | This paper | DCR7398 |
| C. elegans: olaEx4440; atg-4.1(bp501) | This paper | DCR7411 |
| C. elegans: olaEx4493 [Pttx-3::gfp::syd-1A (15 ng/ul), Pttx-3::mCh::lgg-1 (20ng/ul)] | This paper | DCR7457 |
| C. elegans: olaEx4493; atg-4.2(ola316) | This paper | DCR7470 |
| C. elegans: olaEx4472 [Pttx-3::gfp::snb-1 (3 ng/ul), Pttx-3::mCh::lgg-1 (20ng/ul)] | This paper | DCR7415 |
| C. elegans: olaEx4472; atg-4.2(ola316) | This paper | DCR7462 |
| C. elegans: olaEx3013 [Pttx-3::mCh::egfp::lgg-1) (1ng/ul)] | This paper | DCR5074 |
| C. elegans: olaEx3013; atg-4.2(ola316) | This paper | DCR6384 |
| C. elegans: olaEx3013; atg-4.1(bp501) | This paper | DCR7125 |
| C. elegans: olaEx3013; atg-4.2(gk628327) | This paper | DCR7362 |
| C. elegans: olaEx3013; sand-1(or552) | This paper | DCR7065 |
| C. elegans: olaEx3013; epg-5(tm3425) | This paper | DCR7064 |
| C. elegans: olaEx3015 [Pttx-3::mCh::egfp::lgg-1(G116A)) (1ng/ul)] | This paper | DCR5077 |
| C. elegans: olaEx4354 [Pttx-3::laat-1::gfp (1ng/ul), Pttx-3::mCh::lgg-1 (20ng/ul)] | This paper | DCR7225 |
| C. elegans: olaEx4354; atg-4.2(gk628327) | This paper | DCR7419 |
| C. elegans: olaEx4354;atg-4.2(ola316) | This paper | DCR7421 |
| C. elegans: olaEx4431 [Pttx-3::mCh::egfp::lgg-1 (1ng/ul), Pttx-3::laat-1::tagbfp (2ng/ul)] | This paper | DCR7345 |
| C. elegans: olaEx4431; atg-4.2(ola316) | This paper | DCR7404 |
| C. elegans: olaEx4374 [Pttx-3::mCh::egfp::lgg-1(G116A) (1ng/ul), Pttx-3::laat-1::tagbfp (1ng/ul)] | This paper | DCR7252 |
| C. elegans: olaEx4496 [Pttx-3::mCh::egfp::syd-1A (10ng/ul)] | This paper | DCR7460 |
| C. elegans: olaEx4496; atg-4.2(ola316) | This paper | DCR7468 |
| C. elegans: olaEx1802 [Pttx-3::gfp::lgg-2 (30ng/ul), Pttx-3::mCh (30ng/ul)] | This paper | DCR3114 |
| C. elegans: olaEx1802; unc-16(ju146) | This paper | DCR7215 |
| C. elegans: olaEx1802; unc-16(n730) | This paper | DCR7265 |
| C. elegans: olaEx1802; atg-4.1(bp501) | This paper | DCR7213 |
| C. elegans: olaEx1802; atg-4.2(ola316) | This paper | DCR7214 |
| C. elegans: olaEx1802; atg-4.2(gk628327) | This paper | DCR7256 |
| C. elegans: olaEx4438 [Pttx-3::gfp::lgg-2(G130A) (15ng/ul), Pttx-3::mCh (30ng/ul)] | This paper | DCR7352 |
| C. elegans: olaEx4456 [Pttx-3::gfp::syd-2 (15 ng/ul), Pttx-3::mCh::lgg-1 (20ng/ul)] | This paper | DCR7378 |
| C. elegans: olaEx4456; atg-4.2(ola316) | This paper | DCR7455 |
| C. elegans: olaEx3358 [Pttx-3::ebp-2::gfp (5ng/ul), pttx-3::mCh (30ng/ul)] | This paper | DCR5675 |
| C. elegans: olaEx3358; unc-16(ju146) | This paper | DCR5757 |
| C. elegans: olaEx3331 [Pttx-3::laat-1::gfp (1ng/ul), Pttx-3::mCh (30ng/ul)] | This paper | DCR5616 |
| C. elegans: olaEx3331; unc-16(ju146) | This paper | DCR5651 |
| C. elegans: olaEx1706 [Pttx-3::SP12::gfp (5ng/ul)] | This paper | DCR2843 |
| C. elegans: olaex1706; unc-16(ju146) | This paper | DCR5972 |
| C. elegans: olaEx1700 [Pttx-3:aman-2:gfp (0.5 ng/ul)] | This paper | DCR2837 |
| C. elegans: olaex1700; unc-16(ju146) | This paper | DCR5968 |
| C. elegans: olaEx1951 [Pttx-3::tom-20::gfp (30 ng/ul), Pttx-3:mCh:rab-3 (30 ng/uL)] | This paper | DCR3373 |
| C. elegans: olaex1951; unc-16(ju146) | This paper | DCR5974 |
| C. elegans: olaIs58 [Paex-3::egfp::lgg-1 (15ng/ul)] | This paper | DCR5495 |
| C. elegans: olaIs58; unc-16(ju146) | This paper | DCR5582 |
| C. elegans: olaIs58; atg-4.2(ola316) | This paper | DCR5892 |
| C. elegans: olaIs58; atg-4.2(ola316); unc-16(ju146) | This paper | DCR6169 |
| C. elegans: olaEx3644 [Pttx-3::atg-4.1B::gfp (20ng/ul)] | This paper | DCR6199 |
| C. elegans: olaEx3648 [Pttx-3::atg-4.2::gfp (20ng/ul)] | This paper | DCR6203 |
| C. elegans: wyIs45 [Pttx-3:gfp:rab-3] | Colon-Ramos et al., 2007 | TV392 |
| C. elegans: wyIs45; atg-4.2(gk430078) | Stavoe et al., 2016 | DCR2323 |
| C. elegans: wyIs45; atg-4.2(ola316) | This paper | DCR5858 |
| C. elegans: wyIs45; atg-4.1(gk127286) | Stavoe et al., 2016 | DCR2322 |
| C. elegans: wyIs45; atg-9(wy56) | Stavoe et al., 2016 | DCR1468 |
| C. elegans: wyIs45; atg-4.1(gk127286); atg-4.2(gk430078) | Stavoe et al., 2016 | DCR2832 |
| C. elegans: wyIs45; atg-4.1(bp501) | This paper | DCR6347 |
| C. elegans: wyIs45; sand-1(or552) | This paper | DCR1510 |
| Oligonucleotides | ||
| Genotyping primers | This paper, See Table S1 | N/A |
| Recombinant DNA | ||
| Plasmid: Pttx-3:eGFP:lgg-1 | Stavoe et al., 2016 | DACR1321 |
| Plasmid: Pmod-1::HisCl | This paper | DACR1963 |
| Plasmid: Pttx-3::atg-4.2 | This paper | DACR1426 |
| Plasmid: Pttx-3::atg-4.2(G1117A) | This paper | DACR2458 |
| Plasmid: Pmod-1::gfp::lgg-1(G116A) | Stavoe et al., 2016 | DACR2196 |
| Plasmid: Pttx-3::GFP::syd-1A | This paper | DACR3153 |
| Plasmid: Pttx-3::mCh::lgg-1 | This paper | DACR2099 |
| Plasmid: Pttx-3::gfp::snb-1 | This paper | DACR2736 |
| Plasmid: Pttx-3::mCh::egfp::lgg-1 | This paper | DACR2199 |
| Plasmid: Pttx-3::mCh::egfp::lgg-1(G116A) | This paper | DACR2204 |
| Plasmid: Pttx-3::laat-1::gfp | This paper | DACR1669 |
| Plasmid: Pttx-3::laat-1::tagbfp | This paper | DACR2899 |
| Plasmid: Pttx-3::mCh::egfp::syd-1A | This paper | DACR3154 |
| Plasmid: Pttx-3::gfp::lgg-2 | This paper | DACR1288 |
| Plasmid: Pttx-3::gfp::lgg-2(G130A) | This paper | DACR3113 |
| Plasmid: Pttx-3::ebp-2::gfp | This paper | DACR2441 |
| Plasmid: Paex-3::egfp::lgg-1 | This paper | DACR2239 |
| Plasmid: Pttx-3::atg-4.1B::gfp | This paper | DACR2564 |
| Plasmid: Pttx-3::atg-4.2::gfp | This paper | DACR1492 |
| Software and Algorithms | ||
| Volocity | Improvision by Perkin Elmer | N/A |
| Adobe Photoshop | Adobe | CS4 and CS6 |
| (Fiji is Just) ImageJ (FIJI) | Schindelin, J.et al. (2012) | https://fiji.sc/ |
| PyMOL Molecular Graphics System Version 2.1.1 | Schrodinger LLC | https://pymol.org |
| PRISM 7 | Graphpad Software Inc | https://www.graphpad.com/ |
| Other | ||
| UltraView VoX spinning disc confocal microscope with a 60x CFI Plan Apo VC, NA 1.4, oil objective on a NikonTi-E stand | PerkinElmer | N/A |
| Hammamatsu C9100–50 camera | Hammamatsu | N/A |
| Electron microscope: JEOL JEM-1220 TEM | JEOL USA, MA | N/A |
| Ultramicrotome: Leica EM UC6 | Leica Microsystems, IL | N/A |
| Knife: Diatome Ultra 45 Diamond Knife | Diatome, PA | N/A |
| Grids: FCF2010-CU-50 FORMVAR/CARBON 2X1MM CU 50/BX | Electron Microscopy Sciences, PA | N/A |
| Substitution: Leica AFS2 | Leica Microsystems, IL | N/A |
Molecular biology and transgenic lines
The plasmids used in this study for the creation of transgenic lines are derived from the pSM vector (Shen and Bargmann, 2003). We created transgenic strains using standard injection techniques. We used the following plasmids as co-injection markers: Punc-122∷gfp (10–15ng/ul) and Punc-122∷dsRed (30ng/ul). We also generated olaEx3293 [Pelt-7∷gfp (10ng/ul)] to distinguish heterozygous cross offspring from self progeny for the dominant/recessive tests. For a full list of transgenic strains used in this study, please see the Key Resources Table.
For the cDNA constructs generated for use in this study (ATG-4.2, ATG-4.1B, LAAT-1 and EBP-2), we PCR amplified cDNA from a mixed stage population of C. elegans. A Q5 mutagenesis kit (NEB) was then used to introduce the DNA mutation G1117A (which corresponds to the allelic lesion in atg-4.2(ola316)) into the ATG-4.2 cDNA. The introduction of this lesion should result in expression of the mutant protein ATG-4.2(G373R). See the Key Resources Table for a plasmid list. Detailed sub-cloning information is available upon request.
METHOD DETAILS
Fluorescence microscopy and confocal imaging
We used an UltraView VoX spinning disc confocal microscope with a 60x CFI Plan Apo VC, NA 1.4, oil objective on a NikonTi-E stand (PerkinElmer) with a Hammamatsu C9100–50 camera. We imaged the following fluorescently tagged fusion proteins, eGFP, GFP, RFP, tagBFP and mCherry at 405, 488 or 561 nm excitation wavelength. For three-channel imaging, in addition to eGFP and mCh, we expressed a worm-optimized blue fluorophore, tagBFP ((Chai et al., 2012), kindly provided by the Shen lab at Stanford University). For experiments involving colocalization on punctate structures, we acquired channels first then z slices. We anesthetized C. elegans at room temperature in 10mM levamisole (Sigma) or as indicated.
Images were obtained using Volocity software (Improvision by Perkin Elmer) and processed using Adobe Photoshop CS4 and (Fiji is Just) ImageJ (FIJI) software. Image processing included maximal projection, rotation, cropping, brightness/contrast, pseudo coloring, and making surface plots. Kymographs were made using the “reslice” function in FIJI. Between genotypes for compared groups the confocal laser and camera settings and the FIJI brightness/contrast settings were kept identical. All quantifications from confocal images were conducted on maximal projections of the raw data. All images are oriented anterior to the left and dorsal up.
The crystal structure of human ATG4B-LC3(1–120) (Kumanomidou et al., 2006) was visualized using PyMOL Molecular Graphics System Version 2.1.1 (Schrodinger LLC), using PDB file 2Z0D.
SNP Mapping and Whole-Genome Sequencing
We obtained mutant allele atg-4.2(ola316) from a visual forward genetic enhancer screen in the unc-16(ju146); olaIs35 mutant background (integrated line olaIs35 contains Pttx-3∷egfp∷lgg-1; Pttx-3∷mCh). Ethyl methanesulfonate (EMS) mutagenesis was performed and animals were screened for enhanced number of autophagosomes (visualized with GFP∷LGG-1) in the AIY neurite. F2 progeny were viewed on a Leica DM 5000 B compound microscope with an HCX PL APO 63x/1.40–0.60 oil objective.
The novel lesion ola316 was out-crossed from the unc-16 lesion and found to have an independent dominant cell body accumulation of autophagic vacuoles. We then used single-nucleotide polymorphism (SNP) mapping as described (Davis and Hammarlund, 2006; Davis et al., 2005) to map the ola316 lesion. Briefly, this involves crossing mutant animals to a divergent strain, Hawaiian CB4865, and detecting sites of recombination via comparison of dissimilar SNPs. To avoid selecting heterozygous recombinants of the dominant lesion, we mapped loss of the ola316 locus by selecting animals with the wild-type phenotype. SNP mapping was then repeated for verification by selecting and confirming homozygous animals with the unc-16; ola316 enhanced phenotype.
We then performed whole-genome sequencing on unc-16; ola316; olaIs35 animals and for comparison unc-16; olaIs35 animals at the Yale Center for Genome Analysis (YCGA), as previous (Sarin et al., 2008). We analyzed the results with www.usegalaxy.org, the “Cloudmap Unmapped Mutant workflow (w/ subtraction of other strains),” (Minevich et al., 2012) and verified lesions by Sanger sequencing.
Electron Microscopy
Strains were prepared using high-pressure freeze (HPF) fixation as previously described (Weimer, 2006). Briefly, twenty to thirty young adult hermaphrodites were placed in specimen chambers filled with Escherichia coli and frozen at −180°C under high pressure (Leica SPF HPM 100). Samples then underwent freeze substitution (Reichert AFS, Leica, Oberkochen, Germany) using the following program: Samples were held at −90°C for 107 hours with 0.1% tannic acid and 2% OsO4 in anhydrous acetone, then incrementally warmed at a rate of 5°C/hour to −20°C, and kept at −20°C for 14 hours, before increasing temperature by 10°C/hour to 20°C. After fixation, samples were infiltrated w ith 50% Epon/acetone for 4 hours, 90% Epon/acetone for 18 hours, and 100% Epon for 5 hours. Finally, samples were embedded in Epon and incubated for 48 hours at 65°C. Specimens were blinded for genotype prior to sectioning. Thin (70 nm) serial sections were cut using an Ultracut 6 (Leica) and collected on formvar-covered, carbon-coated copper grids (EMS, FCF2010-Cu). Post-staining was performed using 2.5% aqueous uranyl acetate for 4 minutes, followed by Reynolds lead citrate for 2 minutes. Images were obtained on a Jeol JEM-1220 (Tokyo, Japan) transmission electron microscope operating at 80 kV. Micrographs were collected using a Gatan digital camera (Pleasanton, CA) at a magnification of 40 k.
Images were taken from 2 animals for each strain (olaIs35 (wild-type control), atg-4.2(ola316); olaIs35, and atg-4.1(bp501); olaIs35), using the isthmus of the pharynx as a marker to locate neuronal cell bodies within the nerve ring (White et al., 1986). Image collection and scoring of multi-lamellar (autophagosome) structures was performed blinded to the genotype.
Purification of C. elegans proteins
Mouse Atg3 and Atg7 proteins were purified as previously described (Horenkamp et al., 2015; Nath et al., 2014). In brief, mouse ATG3 was expressed with a GST tag in E. coli, and then affinity purified on a glutathione-bead column, eluted by cleavage of the GST, and mixed with 10% glycerol for storage at −80°C. Mouse ATG7 was expressed with a his-tag in a baculoviral system and then affinity purified on nickel resin, eluted by thrombin protease cleavage of the his-tag and mixed with 10% glycerol for storage at −80°C. The C. elegans His-ATG-4.2 was cloned into the pET28A vector for bacterial expression. C. elegans LGG-1(G116) and LGG-1-His were cloned into the pGEX-2T vector to include a thrombin-cleavable N-terminal GST to be used for affinity purification.
His-ATG-4.2 was then expressed as previously described for mammalian ATG4 proteins (Kauffman et al., 2018). Plasmids were transformed into BL21-Gold(DE3) E. coli (Agilent Technologies, 230132). Cells were grown to an OD of 0.6–0.8 at 37°C before induction with 0.2 mM IPTG (AmericanBio, AB00841) at 18°C overnigh t. Cells were then collected by centrifugation and resuspended in binding buffer (25mM Tris, pH7.6, 150mM NaCl, 10mM Imidazole, 1mM BME) also supplemented with a cOmplete™, EDTA-free Protease inhibitor cocktail tablet. Cells were lysed with a cell disruptor and cleared by centrifugation. Supernatants were incubated with nickel beads for 4h at 4°C. Bea ds were collected and washed three times with washing buffer (25mM Tris, pH 7.6, 150mM NaCl, 60mM Imidazole, 1mM BME) before elution with elution buffer (25mM Tris, pH 7.6, 150mM NaCl, 250mM Imidazole, 1mM BME). His-ATG4.2 was then stored at −80°C in 20% glycerol
LGG-1(G116) and LGG-1-His were expressed as previously described for mammalian Atg8 homologues (Kauffman et al., 2018). Briefly, cells were resuspended in thrombin buffer (20 mM Tris, pH 7.6, 100 mM NaCl, 5 mM MgCl2, 2 mM CaCl2, 1 mM DTT), which was supplemented with a cOmplete™, EDTA-free Protease inhibitor cocktail tablet (Roche, 11873580001). Cells were lysed using a cell disruptor and cleared by centrifugation. The supernatant was incubated with glutathione beads (Sigma Aldrich, G4501) for 4 hours at 4°C. Beads were collected and washed three times with thrombin buffer before the addition of thrombin (Sigma Aldrich, T6884). Thrombin was allowed to cut at 4°C overnight. Fract ions of LGG-1(G116) or LGG-1-His were then collected and stored at −80°C in 20% glycerol.
Proteoliposome preparation
Liposomes were prepared as previously described and all lipids were purchased from Avanti (Kauffman et al., 2018). Briefly, lipids were mixed to obtain a composition of 55 mol percent 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 35 mol percent 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), and 10 mol percent L-α-phosphatidylinositol (liver, bovine) (PI) and dried to a thin film under nitrogen. The lipid film was dried for an hour under vacuum before resuspension in SN buffer (20mM Tris, pH 7.6, 100mM NaCl, 5mM MgCl2) and subjected to seven freeze-thaw cycles. Liposomes were extruded to 400nm and further sonicated to ≤ 50nm immediately prior to the lipidation reaction. LGG-1(G116) (15µM), mAtg3 (2μM), mAtg7 (2 μM), and sonicated liposomes (3 mM) were mixed in SN buffer containing 1 mM DTT. Lipidation was initiated by adding 1 mM ATP and reactions were incubated at room temperature for 2 hours. Protein-conjugated liposomes were then purified using a Nycodenz Density gradient (Kauffman et al., 2018). The bottom layer of the gradient was composed of the lipidation reaction in 40% Nycodenz, the middle layer was 30% Nycodenz, and the top layer was SN buffer. Following ultracentrifugation, LGG-1-conjugated liposomes were collected from the 30% Nycodenz/buffer interface. LGG-1-conjugated liposomes were stored on ice at 4°C.
In vitro proteolysis assays
LGG-1-conjugated liposomes or 7 μM LGG-1-His were incubated with 250nM His-Atg-4.2 in SN buffer at room temperature. Samples were removed at the indicated times and mixed with LDS Loading Buffer (ThermoFisher Scientific, NP007) and boiled to stop the reaction. Samples were then visualized using SDS-PAGE and Coomassie Brilliant Blue staining and were analyzed by densitometry using ImageJ. Graph displays the average of three independent assays with standard deviation.
QUANTIFICATION and STATISTICAL ANALYSIS
Quantification of autophagosomes in AIY
Quantifications were performed taking into consideration best practices as suggested in (Landis et al., 2012), including randomization, blinding, and data handling procedures. Particulars for each assay as follows:
Activity-dependent autophagy:
To assess synaptic autophagy, animals containing olaIs35 in a wild-type, or unc-13(e450) mutant background were grown at 20°C and then shifted to 25°C for variable lengths of time (as indicated) and assessed for number of LGG-1 puncta in the AIY neurite on a Leica DM 5000 B compound microscope. For the HisCl experiments, the animals additionally contained the extrachromosomal array olaEx3465 (Pmod-1∷HisCl) and were transferred to 10mM histamine-containing or control NGM plates at 20°C or 25°C fo r 4 hours prior to assessment (Pokala et al., 2014). Animals were then mounted in either 5 mM levamisole in M9 buffer (compared to 10 mM used in all other experiments) or in M9 buffer containing 5 mM levamisole and 10 mM histamine and scored blindly. Results are reported in Figure 1J and 1K.
AV accumulation in neurites and cell bodies:
To investigate the accumulation of autophagosomes in the AIY neuron, we tested mutant alleles in the olaIs35 (Pttx-3∷egfp∷lgg-1; Pttx-3∷mCh) background. For the temperature sensitive dnc-1(or404) allele (Koushika et al., 2004) and a wild-type control, animals were held at either 20°C (permissive) or 25°C (restrictive) for 48 hours prior to examination and analyzed at larval stage 4 (L4). Other candidate transport mutant animals (unc-16, jip-1, unc-116, unc-14 and controls) were kept at 20°C. Autophagosome accumulation in the neurite was then scored on a Leica DM 5000 B compound microscope. For each animal, the sum of autophagosomes in both AIY neurons was counted, then divided by two and reported as an average per neuron in Figure 2C and 2G. Additional quantifications (presented in Figures 3D, 3E, 4E, 5G, 5H, S4I, S4R, S6J, S6K) using the olaIs35 background to detect presence of autophagosomes in the cell bodies and neurites of mutant animals (unc-16, atg-4.2, atg-4.1, epg-5, sand-1 etc.), double mutant animals, and animals overexpressing ATG-4.2 arrays, were scored as follows: raw z-stacks were divided to separate individual AIY neurons, maximal projections were created, and images were randomized and scored blindly. The same method was used to assess the LGG-1(G116A) and LGG-2(G130A) lipidation controls, in olaEx4440 [Pmod-1∷gfp∷lgg-1(G116A) (5ng/ul), Pttx-3∷mCh (30ng/ul)] and olaEx4438 [Pttx-3∷gfp∷lgg-2(G130A) (15ng/ul), Pttx-3∷mCh (30ng/ul)], results described in the Figure S1 and S5 figure legends. For LGG-2-labeled autophagosomes in the olaEx1802 [Pttx-3∷gfp::lgg-2 (30ng/ul), Pttx-3::mCh (30ng/ul)] background (reported in Figures S2C, S2D, S4G and S4H), however, due to the variability in expression for the olaEx1802 array in both wild-type and mutant animals, neurons were only assessed for puncta if the GFP::LGG-2 cytoplasmic signal was within the dynamic range for a chosen set of confocal laser and camera settings that were kept constant between all genotypes examined.
Autophagosome dynamics:
To assess autophagosome dynamics over time, we used the integrated line olaIs35 (Pttx-3∷egfp∷lgg-1; Pttx-3∷mCh). We imaged at maximal speed for 5 minutes in wild-type (n=71 neurons), unc-16(ju146) mutant (n=73 neurons), and unc-16(e109) mutant (n=43 neurons) animals. These videos were scored for percentage of autophagosomes present in each of the three sub-neurite zones (Figure 2I) (Colon-Ramos et al., 2007), for anterograde and retrograde directionality of autophagosome trafficking in the neurite (Figure 2L), and for percentage of autophagosomes leaving the cell body towards the neurite, and then returning to the cell body within the imaging window of 5 minutes (Figure S2H).
Tandem markers and colocalization assays:
For quantifications, images were acquired channels first then z slice (unless otherwise stated) and image settings were kept identical between compared groups. Raw z-stacks were then divided to separate individual AIY neurons and images were randomized and scored blindly. Puncta counts across neurons were then pooled and reported as percentages.
The tandem marker used in this study is based on similar probes used in mammalian neurons (Kimura et al., 2007) and in C. elegans tissues (Chang et al., 2017). Autophagosome maturation was assessed using extrachromosomal lines olaEx3013 and olaEx4431. For olaEx3013, (Pttx-3∷mCh∷egfp∷lgg-1) puncta were scored as “mCh/GFP” (immature) if green and red colocalized puncta could be detected and as “LGG-1 mCh” (mature) if the puncta displayed primarily red signal (reported in Figures 5W and S7H). For olaEx4431 [Pttx-3∷mCh∷egfp∷lgg-1 (1ng/ul), Pttx-3∷laat-1∷tagbfp (2ng/ul)], puncta were scored as “Immature AVL” if positive for tagBFP, mCherry and GFP, and as “Mature AL or L” if positive for tagBFP and mCherry, but negative for GFP (reported in Figure 6H).
To examine the tandem-labeled LGG-1 probe for lipidation onto membranes and to control for the possibility of marker aggregation, we also examined mutant versions of the tandem probe (olaEx3015 (Pttx-3∷mCh∷egfp∷lgg-1(G116A)) and olaEx4374 [Pttx-3∷mCh∷egfp∷lgg-1(G116A) (1ng/ul), Pttx-3∷laat-1∷tagbfp (1ng/ul)]), which prevents LGG-1 from associating with the autophagosomal membrane (Schematic in Figure S1D; (Mizushima et al., 2010; Zhang et al., 2015)). We observed that in the LGG-1(G116A) mutant probe, GFP was diffuse throughout the neuron (Figure S7A), indicating that lipidation is necessary for formation of LGG-1 puncta, and suggesting that most mCh∷GFP∷LGG-1 puncta correspond to autophagic vacuoles, as expected (Chang et al., 2017; Kimura et al., 2007).
We also note that for the tandem-labeled LGG-1 probe, we observed mCherry puncta in the cell body. We observed mCherry puncta even when the marker contained the LGG-1(G116A) point mutation that prevents LGG-1 lipidation and association with autophagosomes (Figure S7B). These mCherry puncta did not co-localize with GFP, indicating they do not simply correspond to protein aggregates of the tandem-labeled marker. Red fluorophores like mCherry are stable at low pH (Shaner et al., 2004) and accumulate in lysosomal compartments (Han et al., 2014; Katayama et al., 2008). Consistent with this, we observed that most mCh puncta from the LGG-1(G116A) tandem probe colocalized with lysosomes in the cell body (Figure S7A-S7D). Indeed, even cytoplasmic expression of mCherry resulted in accumulation of mCherry into puncta that co-localized with lysosomes (Figure S7E-S7G). Our findings indicate that mCherry accumulates in lysosomal compartments, and suggest that most of the observed non-lipidated mCh∷GFP∷LGG-1(G116A) puncta are inside acidic lysosomes.
To assess colocalization between LGG-1 and other proteins of interest, including lysosomal protein LAAT-1 (Liu et al., 2012) and synaptic proteins SYD-1, SYD-2, and SNB-1 (Dittman and Kaplan, 2006), we examined animals containing olaEx4354 [Pttx-3∷laat-1∷gfp (1ng/ul), Pttx-3∷mCh∷lgg-1 (20ng/ul)] (data reported in Figures 6E and S7I), olaEx4493 [Pttx-3∷gfp∷syd-1A (15 ng/ul), Pttx-3∷mCh∷lgg-1 (20ng/ul)] (Figure 6I), olaEx4472 [Pttx-3∷gfp∷snb-1 (3 ng/ul), Pttx-3∷mCh∷lgg-1 (20ng/ul)] (Figure S7R), and olaEx4456 [Pttx-3∷gfp∷syd-2 (15 ng/ul), Pttx-3∷mCh∷lgg-1 (20ng/ul)] (Figure S7R). We also examined the acidification state of synaptic compartments by examining a tandem SYD-1 marker in olaEx4496 [Pttx-3∷mCh∷egfp∷syd-1A (10ng/ul)]. Puncta were scored as “SYD-1 mCh only” if positive for mCh and negative for GFP (acidic compartments) and “SYD-1 mCh/GFP” if positive for both GFP and mCh (non-acidified compartments) (reported in Figure 6M). To assess the percentage of mCh puncta inside lysosomes, we examined animals containing olaEx3331 [Pttx-3∷laat-1∷gfp (1ng/ul), Pttx-3∷mCh (30ng/ul)] (Figure S7E-S7G). Images were acquired z first, then channels, and puncta were only quantified if there was no detectable motion artifact, and if puncta were clearly distinguished from the cytosolic pool of mCherry signal. As a result, it is likely our results (81% of cytosolic mCh puncta colocalize with LAAT-1∷GFP; n= 27 puncta from 9 neurons) under-sample mCherry puncta and might underestimate the extent to which mCh and LAAT-1 colocalize.
Other Quantifications in AIY
Microtubule EBP-2 quantification:
To examine microtubule polarity, we analyzed the direction of movement for EBP-2 comets (Baas and Lin, 2011; Maniar et al., 2011), which track with the growing ends of microtubules, along the AIY neurite using the extrachromosomal line olaEx3358 (Pttx-3∷ebp-2∷gfp, Pttx-3∷mCh). Confocal z-stacks were acquired at maximal speed for one minute and comets were assessed for movement towards the cell body (minus end out microtubule) or movement away from the cell body (plus end out microtubule) in wild-type (n=18 animals) and unc-16 mutant animals (n=16 animals), and reported in Figure S2G.
Lysosomes:
To quantify lysosome number in AIY, we observed the extrachromosomal transgenic line olaEx3331 (Pttx-3∷laat-1∷gfp, pttx-3∷mCh) in wild-type or mutant backgrounds on a Leica DM 5000 B compound microscope. The number of GFP puncta in the two AIY neurons in the animal were counted by location, divided by two and then reported as an average per neuron (Figure S3C).
Endoplasmic reticulum, Golgi, Mitochondria:
For examination of ER, Golgi, and mitochondria the following extrachromosomal lines were used, respectively: olaEx1706 (Pttx-3∷SP12∷gfp), olaEx1700 (Pttx-3:aman-2:gfp), and olaEx1951 (Pttx-3∷tom-20∷gfp, pttx-3:mCh:rab-3) (Abe et al., 2000; Qi et al., 2012; Rolls et al., 2002) in wild-type and unc-16(ju146) mutant backgrounds. Quantifications were performed on maximal projections of confocal micrographs. Fluorescence intensity was measured by tracing the neurite and background in FIJI (Schindelin et al., 2012) and reported as an average of background-subtracted fluorescence intensity (Figure S3F and data not shown). The percent of the neurite occupied by mitochondria was calculated as a ratio of length of the neurite with background-subtracted fluorescence signal greater than zero divided by the total length of the neurite (Figure S3I).
Presynaptic enrichment:
To quantify presynaptic enrichment in AIY, we used the integrated transgenic line wyIs45, expressing pttx-3∷gfp∷rab-3 in wild-type and mutant backgrounds, as described (Colon-Ramos et al., 2007; Stavoe et al., 2016). The AIY neurite is divided into three zones as follows, consistent with the C. elegans EM reconstruction (White et al., 1986): Zone 1 is proximal to the cell body and asynaptic; Zone 2 is defined morphologically as a ~5-µm synapse-rich region at the dorsal turn of the neuron into the nerve ring; Zone 3 is the distal part of the neurite that traverses the nerve ring and contains punctate synaptic sites. We calculated the GFP::RAB-3 enrichment using maximal projection confocal micrographs and measured fluorescence intensity across presynaptic Zones 2 and 3 with the line scan function in FIJI (Schindelin et al., 2012). Fluorescence intensity was background subtracted. All settings, for the confocal microscope and camera, were kept identical between genotypes, and neurons were only quantified when all neurite fluorescence signal was within the measurable range. Synaptic enrichment was reported as signal in Zone 2 (the first 20% of the synaptic region) divided over Zones 2 and 3 (as described in (Stavoe and Colon-Ramos, 2012)) and reported as a percentage and shown in Figure S6G.
Statistical Analyses
Statistical analyses were conducted with PRISM 7 software. For each case, the chosen statistical test is described in the figure legend and “n” values are reported. Briefly, for continuous data and for cases of counting structures, comparisons between two groups were determined by the Student’s t test, while groups of three or more were analyzed with a one-way ANOVA with post hoc analysis by Turkey’s multiple comparison test. Error bars were reported as standard errors of the mean (SEM). For categorical data, groups were compared with Fisher’s exact test, or for samples corresponding to large datasets, the chi-square test was used. Error bars show a 95% confidence interval. Significance was defined as a p value where p>0.05. The p values for significant differences are reported in the figure legends.
Supplementary Material
Time-lapse video of GFP::LGG-1 in an atg-4.2(ola316); unc-16(ju146) double mutant AIY neuron showing accumulated structures trafficking inside and outside of the AIY neuronal process.
HIGHLIGHTS.
Retrograde transport of autophagic vacuoles requires the motor adaptor UNC-16/JIP3
Protease ATG-4.2, but not related ATG-4.1, is required for autophagosome maturation
ATG-4.2 cleaves LGG-1/Atg8 to remove it from membranes and enable lysosomal fusion
Transport and maturation mechanisms interact to clear autophagosomes from synapses
ACKNOWLEDGEMENTS
We thank members of the Colón-Ramos lab, Melia lab, Shawn Ferguson (Yale University) and Andrea Stavoe (University of Pennsylvania) for their thoughtful comments. We thank Enrique Cruz-Reyes (Universidad de Puerto Rico, Cayey), Alec Rodriguez, Sisi Yang and Joon Lee (Colón-Ramos lab) for experimental assistance. We thank the Caenorhabditis Genetics Center (supported by NIH P40 OD010440), the Mitani laboratory ( Tokyo Women’s Medical University) and the Hong Zhang laboratory (Institute of Biophysics, Chinese Academy of Sciences) for strains. We thank Z. Altun (www.wormatlas.org) for diagrams used in figures. We thank the Research Center for Minority Institutions program, the Marine Biological Laboratories (MBL) and the Instituto de Neurobiología de la Universidad de Puerto Rico for providing a meeting and brainstorming platforms. This work made use of the BioCryo facility of Northwestern University’s NUANCE Center (supported by NSF ECCS-1542205; NSF DMR-1720139; and the International Institute for Nanotechnology by the State of Illinois) and the CryoCluster equipment (funded by NSF DMR-1229693). Images were acquired using instruments in the Electron Microscopy Core of UIC’s Research Resources Center. Support for TJM was provided by NIH grant (GM1000930), and support for KJK and SEH was provided by T32-GM007223. SEH was also supported by the NSF GRF DGE-1122492. Research in the DACR lab was supported by by NSF IOS 1353845, by NIH R01NS076558 and by HHMI Scholar Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing financial interests.
REFERENCES
- Abe Y, Shodai T, Muto T, Mihara K, Torii H, Nishikawa S, Endo T, and Kohda D (2000). Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell 100, 551–560. [DOI] [PubMed] [Google Scholar]
- Alberti A, Michelet X, Djeddi A, and Legouis R (2010). The autophagosomal protein LGG-2 acts synergistically with LGG-1 in dauer formation and longevity in C. elegans. Autophagy 6, 622–633. [DOI] [PubMed] [Google Scholar]
- Arimoto M, Koushika SP, Choudhary BC, Li C, Matsumoto K, and Hisamoto N (2011). The Caenorhabditis elegans JIP3 protein UNC-16 functions as an adaptor to link kinesin-1 with cytoplasmic dynein. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 2216–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, and Lin S (2011). Hooks and comets: The story of microtubule polarity orientation in the neuron. Dev Neurobiol 71, 403–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron D, Shibuya M, Gabel C, Wasserman SM, Clark DA, Brown A, Sengupta P, and Samuel AD (2006). A diacylglycerol kinase modulates long-term thermotactic behavioral plasticity in C. elegans. Nature neuroscience 9, 1499–1505. [DOI] [PubMed] [Google Scholar]
- Borland H, and Vilhardt F (2017). Prelysosomal Compartments in the Unconventional Secretion of Amyloidogenic Seeds. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HM, Van Epps HA, Goncharov A, Grant BD, and Jin Y (2009). The JIP3 scaffold protein UNC-16 regulates RAB-5 dependent membrane trafficking at C. elegans synapses. Dev Neurobiol 69, 174–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byerly L, Cassada RC, and Russell RL (1976). The life cycle of the nematode Caenorhabditis elegans. I. Wild-type growth and reproduction. Developmental biology 51, 23–33. [DOI] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, and Jin Y (2001). UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron 32, 787–800. [DOI] [PubMed] [Google Scholar]
- Cavalli V, Kujala P, Klumperman J, and Goldstein LS (2005). Sunday Driver links axonal transport to damage signaling. The Journal of cell biology 168, 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Li W, Feng G, Yang Y, Wang X, and Ou G (2012). Live imaging of cellular dynamics during Caenorhabditis elegans postembryonic development. Nature protocols 7, 2090–2102. [DOI] [PubMed] [Google Scholar]
- Chang JT, Kumsta C, Hellman AB, Adams LM, and Hansen M (2017). Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XT, Zhou B, Lin MY, Cai Q, and Sheng ZH (2015). Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. The Journal of cell biology 209, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DA, Biron D, Sengupta P, and Samuel AD (2006). The AFD sensory neurons encode multiple functions underlying thermotactic behavior in Caenorhabditis elegans. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 7444–7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colon-Ramos DA, Margeta MA, and Shen K (2007). Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science 318, 103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MW, and Hammarlund M (2006). Single-nucleotide polymorphism mapping. Methods in molecular biology 351, 75–92. [DOI] [PubMed] [Google Scholar]
- Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, and Jorgensen EM (2005). Rapid single nucleotide polymorphism mapping in C. elegans. BMC genomics 6, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, and Kaplan JM (2006). Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proceedings of the National Academy of Sciences of the United States of America 103, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drerup CM, and Nechiporuk AV (2013). JNK-interacting protein 3 mediates the retrograde transport of activated c-Jun N-terminal kinase and lysosomes. PLoS genetics 9, e1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Morrison LM, Yorks RM, Hoover CM, Boominathan S, and Miller KG (2015). UNC-16 (JIP3) Acts Through Synapse-Assembly Proteins to Inhibit the Active Transport of Cell Soma Organelles to Caenorhabditis elegans Motor Neuron Axons. Genetics 201, 117–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Yu SC, Hoover CM, Phillips BC, Richmond JE, and Miller KG (2013). An organelle gatekeeper function for Caenorhabditis elegans UNC-16 (JIP3) at the axon initial segment. Genetics 194, 143–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu MM, and Holzbaur EL (2014). Integrated regulation of motor-driven organelle transport by scaffolding proteins. Trends in cell biology 24, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu MM, Nirschl JJ, and Holzbaur ELF (2014). LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Developmental cell 29, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar S, Wu Y, and Ferguson SM (2017). Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. The Journal of cell biology 216, 3291–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MG, Munafo DB, Beron W, and Colombo MI (2004). Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. Journal of cell science 117, 2687–2697. [DOI] [PubMed] [Google Scholar]
- Hammond JW, Griffin K, Jih GT, Stuckey J, and Verhey KJ (2008). Co-operative versus independent transport of different cargoes by Kinesin-1. Traffic 9, 725–741. [DOI] [PubMed] [Google Scholar]
- Han L, Zhao Y, Zhang X, Peng J, Xu P, Huan S, and Zhang M (2014). RFP tags for labeling secretory pathway proteins. Biochem Biophys Res Commun 447, 508–512. [DOI] [PubMed] [Google Scholar]
- Hawk JD, Calvo AC, Liu P, Almoril-Porras A, Aljobeh A, Torruella-Suarez ML, Ren I, Cook N, Greenwood J, Luo L, et al. (2018). Integration of Plasticity Mechanisms within a Single Sensory Neuron of C. elegans Actuates a Memory. Neuron 97, 356–367 e354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus K, Takats S, Boda A, Jipa A, Nagy P, Varga K, Kovacs AL, and Juhasz G (2016). The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Molecular biology of the cell 27, 3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ (1993). Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. The Journal of cell biology 121, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horenkamp FA, Kauffman KJ, Kohler LJ, Sherwood RK, Krueger KP, Shteyn V, Roy CR, Melia TJ, and Reinisch KM (2015). The Legionella Anti-autophagy Effector RavZ Targets the Autophagosome via PI3P- and Curvature-Sensing Motifs. Developmental cell 34, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono R, Mitsui Y, Sato Y, Aizawa S, and Miwa J (1982). Life span of the wild and mutant nematode Caenorhabditis elegans. Effects of sex, sterilization, and temperature. Exp Gerontol 17, 163–172. [DOI] [PubMed] [Google Scholar]
- Hyttinen JM, Niittykoski M, Salminen A, and Kaarniranta K (2013). Maturation of autophagosomes and endosomes: a key role for Rab7. Biochimica et biophysica acta 1833, 503–510. [DOI] [PubMed] [Google Scholar]
- Ikenaka K, Kawai K, Katsuno M, Huang Z, Jiang YM, Iguchi Y, Kobayashi K, Kimata T, Waza M, Tanaka F, et al. (2013). dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PloS one 8, e54511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, and Mizushima N (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269. [DOI] [PubMed] [Google Scholar]
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, and Eskelinen EL (2004). Role for Rab7 in maturation of late autophagic vacuoles. Journal of cell science 117, 4837–4848. [DOI] [PubMed] [Google Scholar]
- Kaasinen SK, Harvey L, Reynolds AJ, and Hendry IA (2008). Autophagy generates retrogradely transported organelles: a hypothesis. Int J Dev Neurosci 26, 625–634. [DOI] [PubMed] [Google Scholar]
- Katayama H, Yamamoto A, Mizushima N, Yoshimori T, and Miyawaki A (2008). GFP-like proteins stably accumulate in lysosomes. Cell structure and function 33, 1–12. [DOI] [PubMed] [Google Scholar]
- Katsumata K, Nishiyama J, Inoue T, Mizushima N, Takeda J, and Yuzaki M (2010). Dynein- and activity-dependent retrograde transport of autophagosomes in neuronal axons. Autophagy 6, 378–385. [DOI] [PubMed] [Google Scholar]
- Kauffman KJ, Yu S, Jin J, Mugo B, Nguyen N, O’Brien A, Nag S, Lystad AH, and Melia TJ (2018). Delipidation of mammalian Atg8-family proteins by each of the four ATG4 proteases. Autophagy, 1–56. [DOI] [PMC free article] [PubMed]
- Kimura S, Noda T, and Yoshimori T (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460. [DOI] [PubMed] [Google Scholar]
- Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, and Ohsumi Y (2000). The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. The Journal of cell biology 151, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushika SP, Schaefer AM, Vincent R, Willis JH, Bowerman B, and Nonet ML (2004). Mutations in Caenorhabditis elegans cytoplasmic dynein components reveal specificity of neuronal retrograde cargo. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 3907–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumanomidou T, Mizushima T, Komatsu M, Suzuki A, Tanida I, Sou YS, Ueno T, Kominami E, Tanaka K, and Yamane T (2006). The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J Mol Biol 355, 612–618. [DOI] [PubMed] [Google Scholar]
- Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, Crystal RG, Darnell RB, Ferrante RJ, Fillit H, et al. (2012). A call for transparent reporting to optimize the predictive value of preclinical research. Nature 490, 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent P, Soltesz Z, Nelson GM, Chen C, Arellano-Carbajal F, Levy E, and de Bono M (2015). Decoding a neural circuit controlling global animal state in C. elegans. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sato Y, and Nixon RA (2011). Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hou Y, Wang J, Chen X, Shao ZM, and Yin XM (2011). Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. The Journal of biological chemistry 286, 7327–7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Du H, Rutkowski R, Gartner A, and Wang X (2012). LAAT-1 is the lysosomal lysine/arginine transporter that maintains amino acid homeostasis. Science 337, 351–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Cook N, Venkatachalam V, Martinez-Velazquez LA, Zhang X, Calvo AC, Hawk J, MacInnis BL, Frank M, Ng JH, et al. (2014). Bidirectional thermotaxis in Caenorhabditis elegans is mediated by distinct sensorimotor strategies driven by the AFD thermosensory neurons. Proceedings of the National Academy of Sciences of the United States of America 111, 2776–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, and Holzbaur EL (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Developmental cell 30, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Wallace KE, and Holzbaur EL (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. The Journal of cell biology 196, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniar TA, Kaplan M, Wang GJ, Shen K, Wei L, Shaw JE, Koushika SP, and Bargmann CI (2011). UNC-33 (CRMP) and ankyrin organize microtubules and localize kinesin to polarize axon-dendrite sorting. Nature neuroscience 15, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manil-Segalen M, Lefebvre C, Jenzer C, Trichet M, Boulogne C, Satiat-Jeunemaitre B, and Legouis, (2014). The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Developmental cell 28, 43–55. [DOI] [PubMed] [Google Scholar]
- Marino G, Uria JA, Puente XS, Quesada V, Bordallo J, and Lopez-Otin C (2003). Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. The Journal of biological chemistry 278, 3671–3678. [DOI] [PubMed] [Google Scholar]
- Maruyama T, and Noda NN (2017). Autophagy-regulating protease Atg4: structure, function, regulation and inhibition. J Antibiot (Tokyo) [DOI] [PMC free article] [PubMed]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, and Levine B (2003). Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391. [DOI] [PubMed] [Google Scholar]
- Minevich G, Park DS, Blankenberg D, Poole RJ, and Hobert O (2012). CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda AM, Lasiecka ZM, Xu Y, Neufeld J, Shahriar S, Simoes S, Chan RB, Oliveira TG, Small SA, and Di Paolo G (2018). Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat Commun 9, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, and Levine B (2010). Methods in mammalian autophagy research. Cell 140, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, and Yoshimori T (2017). New insights into autophagosome-lysosome fusion. Journal of cell science 130, 1209–1216. [DOI] [PubMed] [Google Scholar]
- Nath S, Dancourt J, Shteyn V, Puente G, Fong WM, Nag S, Bewersdorf J, Yamamoto A, Antonny B, and Melia TJ (2014). Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nature cell biology 16, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisch AL, Neufeld TP, and Hays TS (2017). A STRIPAK complex mediates axonal transport of autophagosomes and dense core vesicles through PP2A regulation. The Journal of cell biology 216, 441–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoletopoulou V, Sidiropoulou K, Kallergi E, Dalezios Y, and Tavernarakis N (2017). Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab 26, 230–242 e235. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, and Cuervo AM (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. Journal of neuropathology and experimental neurology 64, 113–122. [DOI] [PubMed] [Google Scholar]
- Ojha CR, Lapierre J, Rodriguez M, Dever SM, Zadeh MA, DeMarino C, Pleet ML, Kashanchi F, and El-Hage N (2017). Interplay between Autophagy, Exosomes and HIV-1 Associated Neurological Disorders: New Insights for Diagnosis and Therapeutic Applications. Viruses 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okerlund ND, Schneider K, Leal-Ortiz S, Montenegro-Venegas C, Kim SA, Garner LC, Waites CL, Gundelfinger ED, Reimer RJ, and Garner CC (2017). Bassoon Controls Presynaptic Autophagy through Atg5. Neuron 93, 897–913 e897. [DOI] [PubMed] [Google Scholar]
- Pokala N, Liu Q, Gordus A, and Bargmann CI (2014). Inducible and titratable silencing of Caenorhabditis elegans neurons in vivo with histamine-gated chloride channels. Proceedings of the National Academy of Sciences of the United States of America 111, 2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteryaev D, Fares H, Bowerman B, and Spang A (2007). Caenorhabditis elegans SAND-1 is essential for RAB-7 function in endosomal traffic. EMBO J 26, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi YB, Garren EJ, Shu X, Tsien RY, and Jin Y (2012). Photo-inducible cell ablation in Caenorhabditis elegans using the genetically encoded singlet oxygen generating protein miniSOG. Proceedings of the National Academy of Sciences of the United States of America 109, 7499–7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls MM, Hall DH, Victor M, Stelzer EH, and Rapoport TA (2002). Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neurons. Molecular biology of the cell 13, 1778–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AM, Richmond JE, Olsen JG, Hall DH, and Bamber BA (2006). Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S, Prabhu S, O’Meara MM, Pe’er I, and Hobert O (2008). Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nature methods 5, 865–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Sato H, Kunitomo H, Fei X, Hashimoto K, and Iino Y (2014). Regulation of experience-dependent bidirectional chemotaxis by a neural circuit switch in Caenorhabditis elegans. The Journal of neuroscience : the official journal of the Society for Neuroscience 34, 15631–15637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, and Tsien RY (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22, 1567–1572. [DOI] [PubMed] [Google Scholar]
- Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, and Inokuchi K (2012). Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 10413–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, and Bargmann CI (2003). The immunoglobulin superfamily protein SYG-1 determines the location of specific synapses in C. elegans. Cell 112, 619–630. [DOI] [PubMed] [Google Scholar]
- Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernandez-Diaz S, Swerts J, Schoovaerts N, Vilain S, Gounko NV, Vints K, et al. (2016). A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 92, 829–844. [DOI] [PubMed] [Google Scholar]
- Stavoe AK, Hill SE, Hall DH, and Colon-Ramos DA (2016). KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Developmental cell 38, 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavoe AKH, and Colon-Ramos DA (2012). Netrin instructs synaptic vesicle clustering through Rac GTPase, MIG-10, and the actin cytoskeleton. Journal of Cell Biology 197, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Li Y, Li T, Ma H, Guo Y, Jiang X, Hou M, Huang S, and Chen Z (2017). JIP1 and JIP3 cooperate to mediate TrkB anterograde axonal transport by activating kinesin-1. Cell Mol Life Sci 74, 4027–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takats S, Nagy P, Varga A, Pircs K, Karpati M, Varga K, Kovacs AL, Hegedus K, and Juhasz G (2013). Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. The Journal of cell biology 201, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammineni P, Ye X, Feng T, Aikal D, and Cai Q (2017). Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhauwaert R, Kuenen S, Masius R, Bademosi A, Manetsberger J, Schoovaerts N, Bounti L, Gontcharenko S, Swerts J, Vilain S, et al. (2017). The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J 36, 1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velikkakath AK, Nishimura T, Oita E, Ishihara N, and Mizushima N (2012). Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Molecular biology of the cell 23, 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Martin S, Papadopulos A, Harper CB, Mavlyutov TA, Niranjan D, Glass NR, Cooper-White JJ, Sibarita JB, Choquet D, et al. (2015). Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type a. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 6179–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J, et al. (2016). The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol Cell 63, 781–795. [DOI] [PubMed] [Google Scholar]
- Weimer RM (2006). Preservation of C. elegans tissue via high-pressure freezing and freeze-substitution for ultrastructural analysis and immunocytochemistry. Methods in molecular biology 351, 203–221. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, and Brenner S (1986). The structure of the nervous system of the nematode Caenorhanditis elegans. Philosophical Transactions of the Royal Society of London 314, 1–340. [DOI] [PubMed] [Google Scholar]
- Wu F, Li Y, Wang F, Noda NN, and Zhang H (2012). Differential function of the two Atg4 homologues in the aggrephagy pathway in Caenorhabditis elegans. The Journal of biological chemistry 287, 29457–29467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chang JT, Guo B, Hansen M, Jia K, Kovacs AL, Kumsta C, Lapierre LR, Legouis R, Lin L, et al. (2015). Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy 11, 9–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li J, Ouyang L, Liu B, and Cheng Y (2016). Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett 373, 19–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse video of GFP::LGG-1 in an atg-4.2(ola316); unc-16(ju146) double mutant AIY neuron showing accumulated structures trafficking inside and outside of the AIY neuronal process.