

Summary
Sound evidence supports a role for interleukin‐17 (IL‐17) ‐producing γδ T cells and IL‐17‐producing helper T (Th17) cells in intestinal homeostasis, especially in intestinal barrier integrity. In the present study, we aimed to evaluate the role of IL‐17 cytokine in the regulation of intestinal immunity and obesity‐induced metabolic syndrome (MetS) in an experimental murine model. C57BL/6 wild‐type (WT) mice and mice lacking the IL‐17 cytokine receptor (IL‐17RA−/−) were fed either a control diet (CD) or a high‐fat diet (HFD) for 9 weeks. Our data demonstrate that IL‐17RA−/− mice are protected against obesity, but develop hyperglycemia, hyperinsulinemia and insulin resistance. In parallel, HFD‐fed IL‐17RA−/− mice display intense inflammation in the ileum compared with WT mice on the HFD. IL‐17RA−/− mice fed the HFD exhibit impaired neutrophil migration to the intestinal mucosa and reduced gene expression of the CXCL‐1 chemokine and CXCR‐2 receptor in the ileum. Interestingly, the populations of neutrophils (CD11b+ Ly6G+) and anti‐inflammatory macrophages (CD11b+ CX3CR1+) are increased in the mesenteric lymph nodes of these mice. IL‐17RA−/− mice on the HFD also display increased commensal bacterial translocation into the bloodstream and elevated lipopolysaccharide (LPS) levels in the visceral adipose tissue (VAT). Metagenomic analysis of bacterial 16S gene revealed increased Proteobacteria and Bacteroidetes phyla, the main representatives of Gram‐negative bacteria, and reduced Akkermansia muciniphila in the fecal samples of IL‐17RA−/− mice fed the HFD. Together, these data indicate that the IL‐17/IL‐17R axis drives intestinal neutrophil migration, limits gut dysbiosis and attenuates LPS translocation to VAT, resulting in protection to MetS.
Keywords: gut microbiota, interleukin‐17‐producing helper T cells, interleukin‐17‐producing γδ T cells, inflammation, metabolic disease, obesity
Abbreviations
- AKT
serine/threonine‐specific protein kinase
- ATP
adenosine triphosphate
- CD
control diet
- CXCL‐1
chemokine (C‐X‐C motif) ligand 1
- CXCL‐2
chemokine (C‐X‐C motif) ligand 2
- CXCR‐2
C‐X‐C chemokine receptor type 2
- ELISA
enzyme‐linked immunosorbent assay
- FACS
flow cytometry
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- Foxp3
forkhead box P3
- HFD
high‐fat diet
- HRP
horseradish peroxidase
- IL
interleukin
- ILP
intestinal lamina propria
- LPS
lipopolysaccharide
- MetS
metabolic syndrome
- MLN
mesenteric lymph nodes
- PBS
phosphate‐buffered saline
- PI3K
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase
- pIgR
polymeric IgA receptor
- PMA
phorbol myristate acetate
- ROR‐γt
Retinoic acid receptor‐related orphan receptor‐γt
- RPMI
Roswell Park Memorial Institute
- TGF‐β
transforming growth factor β
- Th17
IL‐17‐producing helper T cells
- Treg
regulatory T cells
- VAT
visceral adipose tissue
- WT
wild‐type
Introduction
Metabolic syndrome (MetS) is an important and growing public health problem worldwide. Increasing urbanization, excessive consumption of nutrients and energy, and obesity contribute to the substantial increase in the incidence of this disease.1 MetS is characterized by a low‐grade inflammation that, among other factors, leads to increased blood glucose, insulin resistance, visceral adiposity, dyslipidemia and high blood pressure.2 According to a survey by the National Health and Nutrition Examination Survey, the prevalence of this disease is strongly associated with increased body weight.3 Furthermore, MetS increases the risk of developing cardiovascular disease and type 2 diabetes by two and five times, respectively.4
Under homeostatic conditions, innate production of interleukin‐17 (IL‐17) at the intestinal epithelial surface is primarily driven by γδ T cells (γδT17),5 whereas the adaptive production of IL‐17 is mediated mainly by IL‐17‐producing helper T cells (Th17) present in the intestinal lamina propria (ILP).6 The differentiation of Th17 cells in the intestinal mucosa depends on a range of cytokines, such as IL‐6, tumor necrosis factor‐α (TNF‐α) and interleukin‐23, produced by dendritic cells and macrophages present in the ILP.7 Once differentiated, these cells are activated and express the IL‐23 cytokine receptor (IL‐23R) and synthesize molecules such as IL‐17A and IL‐22.8 In particular, IL‐17 cytokine inhibits adipogenesis through the suppression of pro‐adipogenic transcription factors, inhibits adipose tissue accumulation and regulates glucose metabolism in high‐fat diet (HFD) ‐induced obesity models. Additionally, this cytokine can directly prevent glucose uptake by differentiated adipocytes.9, 10 Finally, IL‐17 regulates the levels of adipokines, such as leptin and adiponectin, by the adipose tissue.9
Interleukin‐17A also regulates granulopoiesis and neutrophil recruitment into ILP through chemokine production by epithelial cells and fibroblasts in the intestinal mucosa.11, 12 In addition, IL‐17A plays an important role in progenitor neutrophil expansion in the bone marrow and spleen, as well as in mature neutrophils in the peripheral blood through the induction of other mediators such as IL‐6, granulocyte colony‐stimulating factor, and chemokine (C‐X‐C motif) ligands 1 (CXCL‐1) and 2 (CXCL‐2).13 Also, this cytokine regulates components of the intestinal barrier by reinforcing tight junction proteins like occludins and claudins in epithelial cells14, 15 and induces the expression of mucin‐associated genes and the production of β‐defensins in the colon.16, 17
Mice deficient in the IL‐17 cytokine receptor (IL‐17RA−/−) have lower expression of the polymeric IgA receptor (pIgR) on intestinal epithelial cells, as well as decreased production of IgA in the intestine. Moreover, IL‐17RA−/− mice exhibit gut dysbiosis as a result of reduced α‐defensin expression in the gut and pIgR.18 Recent studies show that mice fed an HFD exhibit an imbalance in the population of commensal bacteria present in the intestine and immune cells present in ILP.19, 20, 21, 22 These events are associated with increased intestinal permeability and translocation of pathogenic bacteria and their products into the bloodstream and tissues such as liver, pancreas and adipose tissue. These changes lead to metabolic inflammation and insulin resistance, which may promote the onset of MetS and type 2 diabetes.23
Despite recent advances in understanding the role of Th17 cells in host immunity and autoimmunity, the expression profile and functions of these cells in the development of obesity and MetS are not well understood. In this report, a strain of C57BL/6 wild‐type (WT) mice and mice lacking the IL‐17 cytokine receptor (IL‐17RA−/−) were fed a control diet (CD) or an HFD, to establish the role of IL‐17 in the development of obesity‐associated MetS. Our findings show that IL‐17RA−/− mice have delayed recruitment of neutrophils into the intestinal mucosa associated with a process of intestinal dysbiosis, increased translocation of commensal bacteria and lipopolysaccharide (LPS) into the visceral adipose tissue (VAT), leading to aggravation of MetS in a murine model of obesity induced by an HFD.
Methods
Animals
Male, 4‐ to 6‐week‐old, C57BL/6 mice deficient in IL‐17RA (IL‐17RA−/−) and their littermate controls were used. Mice were kept in the animal house of the Department of Biochemistry and Immunology, Ribeirao Preto Medical School at the University of Sao Paulo, where they were provided with filtered air and free access to water and food. Mice were reared under specific pathogen‐free conditions. The experiments were carried out in accordance with the National Council for Animal Experimentation Control and were approved by the Ethics Committee on Animal Use of the University of Sao Paulo, Ribeirao Preto, Brazil (protocol number 144/2014).
Obesity and metabolic syndrome (MetS) induction
Obesity and MetS was induced by HFD. After 7 days of adaptation, WT and IL‐17RA−/− mice were divided into four experimental groups. Both WT and IL‐17RA‐deficient mice received a control diet (AIN 93, comprising 9·7% fat, 77·1% carbohydrate and 13·4% protein) or a high‐fat diet (D12492, comprising 60% fat, 20% carbohydrate and 20% protein) for 9 weeks. The diets were purchased by Pragsoluções Biociências company.
Nutritional and metabolic parameters
The nutritional profile was determined by analysis of food intake, caloric intake and body weight. Body weight of animals was measured weekly, using a digital scale. For the glucose tolerance test, mice were submitted to a 12‐hr fasting period. Blood samples were taken at baseline and after intraperitoneal administration of a solution containing 25% glucose (Sigma‐Aldrich, St Louis, MO) equivalent to 2·0 g/kg, being collected at 0, 15, 30, 60 and 120 min. The ACCU‐CHEK®Active equipment was used to read glucose levels. For the insulin tolerance test, mice were submitted to a 6‐hr fasting period. Blood samples were taken from mice at baseline and after intraperitoneal administration of regular insulin equivalent to 1·5 IU/kg, being collected at 0, 5, 10, 15, 20, 15 and 30 min.
Extraction of total lipids in mice feces
For the extraction of lipids, the feces of the different groups of mice were weighed and then macerated in Eppendorf conical tubes, with the aid of a pistil. Subsequently, the macerate was suspended in phosphate buffered saline (PBS) solution, in the proportion of 5 ml of PBS for each 1 g of feces. The solution was homogenized using a vortex and a chloroform–methanol solution (2 : 1) was added to the mixture, with a proportion of 10 ml of chloroform–methanol for each 1 g of feces. The samples were centrifuged at 805 g, 25° for 25 min. At the end, the lower phase was removed with the aid of a syringe and the volume of this phase was measured in the syringe itself.
Preparation of phospho‐vanillin solution
To prepare the solution, 600 mg of vanillin (Sigma‐Aldrich) was mixed with 100 ml of heated distilled water. After complete dissolution of the vanillin, 400 ml of 85% o‐phosphoric acid was added. The mixture was stirred for 30 min and wrapped in aluminum foil to avoid exposure to light. A glyceryl trioleate standard curve was obtained, using a glyceryl trioleate (Sigma‐Aldrich) stock solution (1 mg/ml in chloroform).
Quantification of total lipids in mice feces
About 5 μl of the lipid‐containing solution, obtained from the mice feces, was evaporated at 90° for 5 min in a heating plate. Then, 0·1 ml of 98% sulfuric acid (Sinth) was added and the mixture was heated at 90° for 10 min. The tubes were cooled on ice for 5 min and 2·4 ml of the phospho‐vanillin reagent was added. The resulting solution was vortexed and kept at room temperature for 10 min. Finally, 200 μl of the solution was aliquoted onto an ELISA plate and absorbance was measured at 530 nm. A solution containing no oils and prepared following the same procedure was considered as blank. For the results, the oil mass detected by the phospho‐vanillin assay was given to the respective feces mass value, expressed in milligrams of fat per gram of feces.
Detection of total cholesterol, triglyceride and LPS levels
Mice were fasted for 12 hr and blood was collected from the tip of the tail vein. Hemolysis‐free serum was collected after centrifugation. Total cholesterol, triglyceride and LPS concentrations were measured using kits from Labtest (St Louis, MO) or Lonza (Basel, Switzerland).
Quantification of serum insulin levels
Insulin concentrations were determined using the Mouse Ultrasensitive Insulin kit (Alpco Diagnostics, Salem, NH) according to the manufacturer's instructions.
Quantification of cytokine and elastase levels
Ileum fragments were removed, weighed and placed in a tube containing 700 μl of Complete Protease Inhibitor Cocktail (Roche Diagnostics, Abbott Park, IL). The tissues were homogenized using a Polytron homogenizer (Thermo Fisher Scientific, Waltham, MA). IL‐6, IL‐17, TNF‐α, IL‐22 and elastase were detected by enzyme‐linked immunosorbent assay (ELISA) using colorimetric kits according to the manufacturer's instructions (R&D Systems, Minneapolis, MN). The results are expressed as the mean ± SEM (nanograms per gram of tissue).
Expression and phosphorylation of AKT and PI3K in skeletal muscle
Insulin administration was carried out intraperitoneally (3·8 IU/kg body weight) and after 5 min the gastrocnemius skeletal muscle was collected. The fragments were homogenized with a polytron in extraction buffer [Tris–HCl 0·05 mol/l, NaCl 0·150 mol/l, ethylenediaminetetraacetic acid (EDTA) 0·001 mol/l, Triton X‐100 1%, deoxycholate 1%, sodium dodecyl sulfate 0·1%, sodium orthovanadate 0·001 mol/l – 1 : 100, sodium fluoride 0·01 mol/l – 1 : 100 and protease inhibitor]. A total of 60 μg of protein were separated by polyacrylamide 10% gel electrophoresis. Then, the proteins were transferred to a nitrocellulose membrane. After blocking the non‐specific sites with 5% milk, the membranes were incubated with primary antibody for 20 hr at 4°. Antibodies were used for tubulin, pSer473‐Akt, pTyr467PI3K, total serine/threonine‐specific protein kinase (AKT) and total phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K) in the 1 : 500 dilution (Cell Signaling, Danvers, MA). Then, the membranes were washed three times for 10 min with Tris‐buffered saline‐Tween [TBS‐T (100 ml TBS 10×; 1 ml Tween‐20 and 1 l distilled water)] and incubated with secondary antibody IgG labeled with peroxidase at a dilution 1 : 1000 (Cell Signaling) for 1 hr with stirring. Finally, the membranes were washed three times for 10 min with TBS‐T and the detection was carried out with a chemiluminescence kit (Pierce Biotechnology, Waltham, MA). The intensity of the bands was evaluated by densitometric analysis using imagej software.
Intestinal permeability by FITC‐Dextran
After 12 hr of fasting, mice received fluorescein isothiocyanate (FITC) ‐dextran by gavage (250 mg/kg) (Sigma‐Aldrich). After 4 hr, blood samples were collected from the tail vein. The blood was centrifuged at 4°, 1000 g for 3 min. The serum was diluted in the same volume of PBS (pH 7·4) for analysis of FITC‐dextran concentrations at excitation wavelength of 485 nm and emission wavelength of 535 nm.
Histopathology and immunohistochemistry analysis
Histopathological evaluations of the pancreas, VAT and liver were performed after staining with haematoxylin & eosin of fixed samples in PBS/10% formaldehyde. The material was sectioned, mounted on glass slides and kept in a dry oven at 60° for 1 hr. Then, the material was hydrated and deparaffinized using xylene, alcohol and water. Immunohistochemistry reactions were performed as previously described.24
Evaluation of bacterial translocation
To evaluate bacterial translocation into the blood and VAT, samples were aseptically collected. Subsequently, 50‐μl blood aliquots were spread with a sterile loop in brain–heart infusion medium‐containing agar plates and placed in an incubator at 37° for 48 hr to count the colony‐forming units.
Analysis of leukocytes by flow cytometry (FACS)
Quantitative analysis of cells taken from mesenteric lymph nodes (MLN) was performed by flow cytometry (FACS). The detection of Th17 lymphocytes (CD3+ CD4+ IL‐17+) and Tγδ (CD3+ CD4− γδ+ IL‐17+) was performed after 4 hr culture of leukocytes in vitro with phorbol myristate acetate (50 ng/ml), ionomycin (500 ng/ml, Sigma‐Aldrich) and Golgi Stop (1000×, BD Pharmingen, San Diego, CA). Additionally, macrophages (CX3CR1+ CD11b+), neutrophils (CD11b+ Ly6G+) and dendritic cells (CD11b+ CD103+) were quantified in these same tissues. Cells were washed and resuspended in Fc block (to block non‐specific binding) for 30 min at 4° and then incubated with 1.5 μg of monoclonal antibodies to specific markers conjugated with FITC, phycoerythrin (PE), peridinin chlorophyll protein (PerCP), allophycocyanin (APC) or allophycocyanin‐Cy7 (APC‐ Cy7) for 30 min at 4°C. After incubation, the samples were washed twice with 1× PBS and centrifuged at 250 g for 10 min. Cells were analyzed using a FACSCanto flow cytometer, and the data were analyzed using the flowjo (Tree Star, Ashland, OR) software.
Cell extraction of the intestinal lamina propria and epithelial cells
The cells of the ILP and epithelial cells were extracted from the digestion of the small intestine. Initially, the intestinal Peyer's patches were removed and cleaned. Subsequently, the intestines were fragmented into 2‐cm segments and incubated under stirring at 37° in 20 ml of RPMI‐1640 medium, 3% fetal bovine serum (FBS), 5 mm EDTA and 0·145 mg/ml dithiothreitol. After incubation, the fragments were stirred in RPMI incomplete medium (2 mm EDTA) and passed in a metal sieve three times. Then, the segments were fragmented and transferred to 3 ml of incomplete RPMI medium containing 0·05% DNAse, 0·2 mg/ml Liberase TL (Roche Diagnostics, Abbott Park, IL) and incubated under stirring at 37° for 30 min. Next, 20 ml of complete RPMI medium with 3% FBS was added to inhibit the activity of digestion enzymes. After centrifugation (604 g, 10 min, 4°), the supernatant was discarded and 10 ml of complete RPMI with 3% FBS was added, and the material was transferred and filtered through a cell strainer and centrifuged. The supernatant was discarded, the cells were resuspended in 1 ml of complete RPMI medium with 5% FBS and counted in a Neubauer chamber. The medium collected for isolation of epithelial cells was centrifuged (604 g, 10 min, 4°) and the cells were resuspended in 1 ml complete RPMI medium with 5% FBS for later counting.
Stimulation of ILP and epithelial cells with recombinant IL‐17
A total of 1 × 106 epithelial cells or 0.5 × 106 ILP cells isolated from mice fed the CD or HFD were plated in 48‐well plates and stimulated with RPMI medium, with 10 ng/ml or 100 ng/ml of recombinant IL‐17. After 24 hr of stimulation, the culture supernatant was collected for subsequent use in ELISAs. The cultured cells were centrifuged (5220 g, 2 min, 4°) and stored at −20° for RNA extraction.
RNA extraction and quantitative real‐time PCR (qPCR)
The extraction of total RNA from ileum and cell culture samples was carried out using Trizol (Life Technologies, Molecular Probes, Carlsbad, CA) following the manufacturer's instructions. cDNA was obtained using a High Capacity reverse transcription kit (Applied Biosystems, Foster City, CA) following DNase treatment (Life Technologies). Quantitative gene expression of IL‐6, IL‐17A, IL‐22, TNF‐α, CXCL‐1, CXCL‐2, IL‐17 receptor gene, occludin, claudin‐2, and mucins 1 and 2 was analyzed by quantitative polymerase chain reaction (PCR) using the SYBR Green PCR Master Mix (Applied Biosystems). Specific mRNA expression levels were normalized relatively to β‐actin mRNA levels using the comparative 2‐ΔΔCt method.
16S rRNA gene sequencing
16S rRNA gene sequencing was performed as previously described by Palm et al.25 The Ribosomal Database Project classifier (RDP) and the May 2013 GreenGenes taxonomy were used to assign taxonomy to representative operational taxonomic units26, 27, 28 and the Linear Discriminant Analysis Effect size Galaxy module was used for additional statistical analyses.29
Analysis of bacterial 16S genes by qPCR
Fecal DNA was isolated using the QIAamp Fast DNA Stool Mini Kit (Qiagen) following the manufacturer's instructions. qPCR was performed using universal 16S rRNA gene primers or specific for Akkermansia muciniphila. Differences (ΔCT) between cycle threshold (CT) values of 16S and specific bacterial group were used to obtain normalized levels (2 ‐ΔCT). The relative levels of the bacterial group were obtained after normalization with that of control group.
Statistical analysis
The data are expressed as mean ± SEM. The differences observed among the several experimental groups were analyzed by one‐way analysis of variance followed by the parametric Tukey test for comparing multiple groups. All analyses were performed using PRISM 50 software (GraphPad software, San Diego, CA). Statistical significance was set at P < 0·05.
Results
Increased expression of Th17 pattern‐related cytokines in the ileum after HFD
Initially, we evaluated gene and protein expression of IL‐6, TNF‐α, IL‐17A and IL‐22 in the ileum of WT mice fed a CD or an HFD for 9 weeks. At this time, there was a significant increase in gene and protein expression of these cytokines in the ileum of HFD‐fed mice, compared with mice on the CD (Fig. 1a–d). Considering that mice fed the HFD had increased Th17 profile cytokine expression in the ileum, our next step was to identify the IL‐17‐producing cell subtypes in the MLN of WT mice fed the CD or HFD. For this reason, the populations of Th17 and IL‐17‐producing γδ cells were analyzed. Although not significantly different, there was a trend to increased percentage of Th17 cells in the MLN (Fig. 1e,f). In addition, the percentage of IL‐17‐producing γδ cells was significantly increased in the MLN of HFD‐fed mice when compared with mice on CD (Fig. 1g,h). These findings demonstrated that both Th17 and IL‐17‐producing γδ cells are up‐regulated during obesity induced by HFD.
Figure 1.
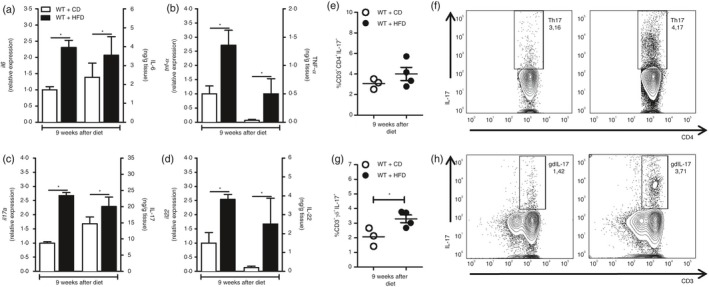
Gene and protein expression of the T helper type 17 (Th17) pattern‐related cytokines in the ileum and Th17 or interleukin‐17 (IL‐17) ‐producing γδ cell numbers in the mesenteric lymph nodes (MLN) of wild‐type (WT) mice at 9 weeks of control diet (CD) or high‐fat diet (HFD). Gene and protein expression of IL‐6 (a), tumor necrosis factor‐α (TNF‐α) (b) IL‐17A (c) and IL‐22 (d) cytokines in the ileum, by RT‐PCR and ELISA, respectively. Percentage of Th17 (CD4+ CD3+ IL‐17+) and IL‐17‐producing γδ cells (CD3+ γδ + IL‐17+) in the MLN by FACS (e, g). Percentage of Th17 and IL‐17‐producing γδ cells is shown in representative dot plots (f, h). Values are expressed as mean ± SEM. *P < 0·05 was considered statistically significant when compared with WT mice on CD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test.
IL‐17RA effect in the development of obesity after HFD
Subsequently, we determined whether IL‐17RA deficiency affects the development of obesity and the metabolic alterations observed in mice with MetS, as a relationship between IL‐17 and adipocyte differentiation has recently been described.9, 10 Therefore, nutritional parameters such as body weight, weight gain, food and caloric intake were evaluated. As expected, WT mice fed an HFD for 9 weeks had an increase in body weight. On the other hand, mice deficient in the IL‐17RA fed the HDF for 9 weeks had a reduced body weight compared with WT mice (Fig. 2a,b). Of importance, IL‐17RA−/− mice showed weight loss, starting on week 7, after receiving either a CD or HFD (Fig. 2a). In addition, WT mice on the HFD had a lower food intake when compared with the same mice after CD (Fig. 2c). No differences in caloric intake were observed between the different groups (data not shown). To investigate why IL‐17RA−/− mice fed the HFD had reduced body weight when compared with WT mice on the same diet, the total lipid content in the feces was determined. IL‐17RA−/− mice fed the HFD diet had statistically higher total fecal lipid levels when compared with the other experimental groups (Fig. 2d). These data indicate that IL‐17RA deficiency confers protection against obesity induced by HFD, possibly through an inability to adequately absorb lipids present in the diet.
Figure 2.
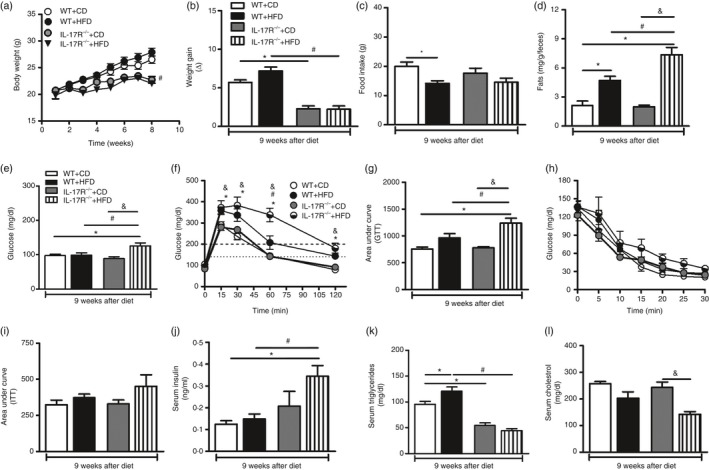
Nutritional and metabolic parameters for wild‐type (WT) mice and mice lacking the interleukin‐17 (IL‐17) cytokine receptor (IL‐17RA−/−) at 9 weeks of control diet (CD) or high‐fat diet (HFD). Time course of body weight (a), body weight gain (b), food intake (c) and fat content in feces (d) of WT or IL‐17RA−/− mice fed either a CD or an HFD. Blood glucose levels after fasting (e) and after the glucose tolerance test (GTT) (f); area under the curve for the GTT (g), insulin tolerance test (ITT) (h) and area under the curve for the ITT (i) in WT and IL‐17RA−/− mice fed a CD or an HFD. Concentrations of insulin (j), triglycerides (k) and cholesterol (l) were determined in the serum of these mice. Values are expressed as mean ± SEM. *P < 0·05 was considered statistically significant when compared with WT mice after CD or # P < 0·05 compared with WT with HFD or & P < 0·05 compared with IL‐17RA−/− mice after CD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test.
IL‐17RA effect in the control of metabolic syndrome after HFD
In parallel, we evaluated parameters related to glucose and lipid metabolism after 9 weeks of HFD or CD. For this purpose, blood samples were collected for monitoring fasting blood glucose, serum insulin, triglycerides and cholesterol levels. Glucose tolerance and insulin tolerance tests were also performed. Interestingly, we observed that HFD‐fed mice deficient in IL‐17RA had increased fasting glycemia compared with CD‐fed IL‐17RA−/− mice and WT mice on HFD and CD (Fig. 2e). In accordance, these mice displayed greater glucose intolerance after glucose tolerance test (Fig. 2f,g). A slow increase in insulin resistance was also observed in HFD‐fed IL‐17RA−/− mice, compared with HFD‐fed WT mice, on the insulin tolerance test (Fig. 2h,i). In addition, HFD‐fed IL‐17RA−/− mice exhibited higher serum insulin levels compared with WT on HFD (Fig. 2j). However, HFD‐fed IL‐17RA−/− mice showed a significant reduction of serum triglyceride and cholesterol levels compared with WT mice fed the HFD or IL‐17RA−/− mice on the CD, respectively (Fig. 2k,l). These data indicate that IL‐17RA deficiency aggravates metabolic alterations, including glucose intolerance and insulin resistance, associated with the MetS.
IL‐17RA effect on the improvement of insulin resistance after HFD
Insulin is the main hormone associated with the regulation of glucose levels in the blood, and defects in insulin signaling result in chronic hyperglycemia.30 In an attempt to elucidate the intracellular mechanisms responsible for increased levels of glucose observed in IL‐17RA−/− mice fed the HFD, we assessed expression of the phosphorylated and total forms of PI3K and AKT kinases in the gastrocnemius skeletal muscle by Western blotting. Although not significant, a relative decrease in phosphorylated PI3K, but not in total PI3K, was observed in HFD‐fed IL‐17RA−/− mice when compared with WT mice fed the same diet (Fig. 3a–c). As expected, the phosphorylation form of AKT was increased in HFD‐fed WT mice compared with WT mice on CD. However, phosphorylation of AKT was significantly reduced in IL‐17RA−/− mice compared with WT mice on the HFD (Fig. 3d,e). However, no alterations on total AKT expression were observed in skeletal muscle from the several groups, with the exception of IL‐17RA−/− mice, which showed increased total AKT expression compared with WT on CD (Fig. 3f). These results indicate that IL‐17RA deficiency causes a defect in the phosphorylation and activation of kinases associated with insulin signaling, resulting in insulin resistance and MetS onset.
Figure 3.
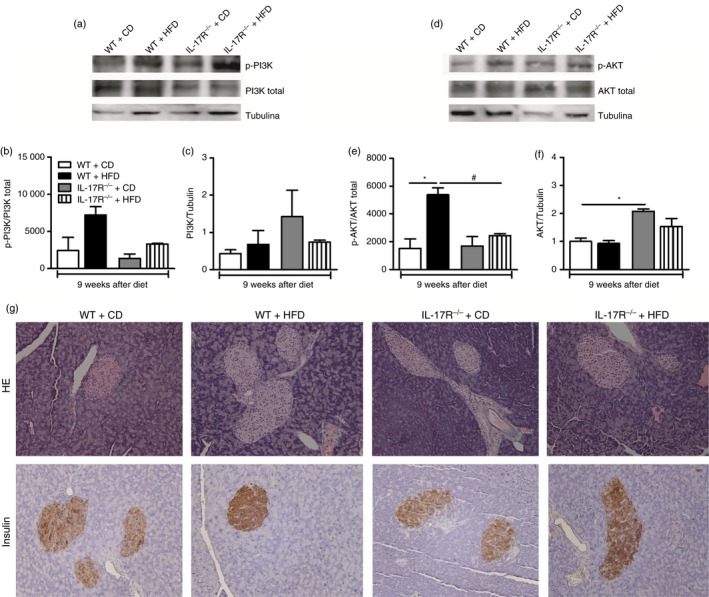
Expression of total and phosphorylated forms of phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K) and serine/threonine‐specific protein kinase (AKT) in skeletal muscle and histopathological analysis of pancreatic tissues of wild‐type (WT) mice and mice lacking the interleukin‐17 (IL‐17) cytokine receptor (IL‐17RA−/−) fed a control diet (CD) or a high‐fat diet (HFD) for 9 weeks. Representative images of total and phosphorylated PI3K and AKT expression (a, d). Quantification of phosphorylated and total PI3K (b, c) and AKT (e, f) by densitometry. The skeletal muscle was collected 5 min after administration of insulin in a concentration of 3·8 IU/kg body weight. Pancreatic histological sections were fixed and stained with hematoxylin & eosin (g, top panels) or immunostained for insulin by immunohistochemistry (g, bottom panels). Values are expressed as mean ± SEM. *P < 0·05 was considered statistically significant when compared with WT mice on CD or # P < 0·05 compared with WT on HFD or & P < 0·05 compared with IL‐17RA−/− mice on CD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. Images were acquired in increased 200× and are representative of one animal per group.
IL‐17RA effect on insulitis and insulin‐producing β‐cell damage after HFD
Later, we examined possible alterations in the morphology of pancreatic islets, which are present in the insulin‐producing β cells. In addition, immunohistochemistry was performed to assess insulin expression in these cells. There are reports in the literature showing that in the initial phase of MetS a process of proliferation and/or regeneration of insulin‐producing β cells occurs as a compensatory mechanism to normalize high blood glucose levels.31 Indeed, we noted a hyperplasia of pancreatic islets in both WT and IL‐17RA−/− mice fed the HFD compared with mice on the CD (Fig. 3g, top panels). In addition, increased expression of insulin in pancreatic islets of IL‐17RA−/− mice on the HFD (Fig. 3g, bottom panels). These data suggest that IL‐17RA deficiency promotes pancreatic hyperplasia to compensate the insulin resistance during MetS induced by HFD.
IL‐17RA effect on tight junction expression and intestinal inflammation after HFD
As we detected a reduction of triglyceride and cholesterol levels in the blood of HFD‐fed IL‐17RA−/− mice (Fig. 2k,l), we evaluated lipid accumulation in the liver of these mice. In parallel, we analyzed the presence of inflammatory infiltrate in the intestine (ileum), as IL‐17 directly affects the physical components that maintain the epithelial barrier, such as the expression of intercellular junctions like claudin 1 and 2, occludin and junctional adhesion molecule.32 Surprisingly, we did not detect hepatic fat deposition in the different groups (Fig. 4a, top panels). However, an intense inflammatory infiltrate was observed in the ILP, which was associated with destruction of the intestinal microvilli in the ileum of HFD‐fed IL‐17RA−/− mice compared with the WT mice on HFD (Fig. 4a, bottom panels). Whereas expression of claudin‐2 is positively associated with intestinal permeability, expression of occludin is inversely correlated with intestinal permeability. In fact, there was increased claudin‐2 expression and reduced expression of occludin in the ileum of WT mice on HFD compared with WT mice on CD, indicating elevated intestinal permeability in these mice. However, a significant decrease in claudin‐2 and increased occludin expression was observed in the ileum of IL‐17RA−/− mice compared with WT mice fed the HFD (Fig. 4b,c). In addition, a significant increase in gene expression of Muc‐1, but not Muc‐2, was observed in the ileum of IL‐17RA−/− mice compared with WT mice on HFD (Fig. 4d,e). Together, these data show that IL‐17RA deficiency induces a profound intestinal inflammation during MetS development induced by HFD.
Figure 4.
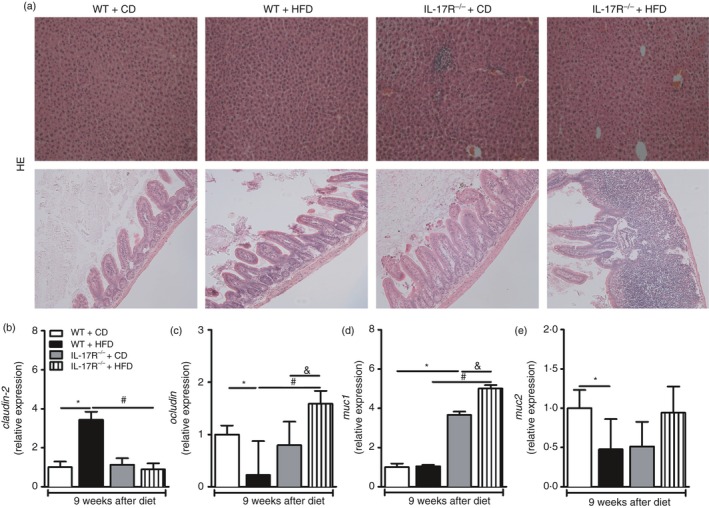
Histopathological analysis of the liver and small intestine (ileum) and gene expression of claudin‐2, occludin, mucin‐1 and mucin‐2 in the ileum of the wild‐type (WT) and mice lacking the interleukin‐17 (IL‐17) cytokine receptor (IL‐17RA−/−) fed a control diet (CD) or a high‐fat diet (HFD) for 9 weeks. Histological sections of the livers (a, top panels) and the ileum (a, bottom panels) were fixed and stained with hematoxylin & eosin. Gene expression of claudin‐2 (b), occludin (c), muc‐1 (d) and muc‐2 (e) in the ileum by RT‐PCR. Values are expressed as mean ± SEM. *P < 0·05 was considered statistically significant when compared with WT mice on CD or # P < 0·05 compared with WT fed the HFD or & P < 0·05 compared with IL‐17RA−/− mice on CD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. Images were acquired in increased 200X and are representative of one animal per group.
IL‐17RA effect on neutrophil, macrophage and dendritic cell homing to MLN after HFD
Subsequently, we evaluated the percentage and absolute numbers of dendritic cells (CD11b+ CD103+), intestinal macrophages (CD11b+ CX3CR1+) and neutrophils (CD11b+ Ly6G+) in the MLN, because these cell populations are important for gut homeostasis.33, 34, 35 Increased percentage and absolute numbers of neutrophils were detected in MLN of IL‐17RA−/− mice fed the HFD compared with IL‐17RA−/− mice on CD (Fig. 5a,d,g, top panels). However, no alterations either in the percentage or in absolute numbers of dendritic cells were observed among the several groups (Fig. 5b,e,g, middle panels). Finally, an increased percentage of intestinal macrophages was observed in MLN of IL‐17RA−/− mice compared with WT on HFD (Fig. 5c,g, bottom panels). Although not significant, increased absolute numbers of macrophages were observed in IL‐17RA−/− mice compared with WT on HFD (Fig. 5f). These results suggest that IL‐17RA deficiency promotes the retention of intestinal macrophages and neutrophils in MLN during MetS development induced by HFD.
Figure 5.
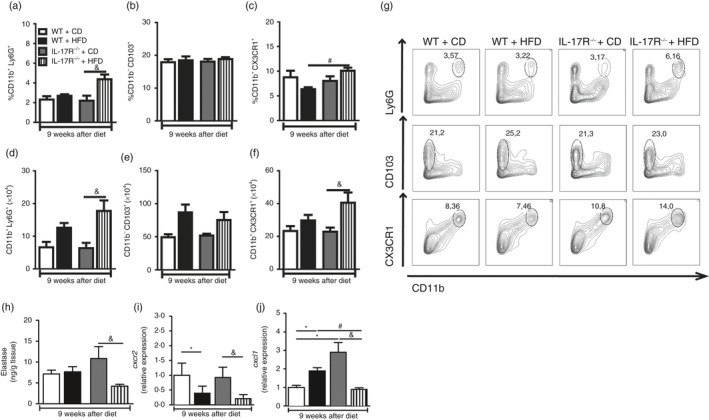
Percentage and absolute number of cells in mesenteric lymph nodes (MLN) and elastase protein levels, CXCR‐2 and CXCL‐1 gene expression in the ileum of mice lacking the interleukin‐17 (IL‐17) cytokine receptor (IL‐17RA−/−) fed a control diet (CD) or a high‐fat diet (HFD) for 9 weeks. Percentage and absolute number of neutrophils (CD11b+ Ly6G+) (a, d), dendritic cells (CD11b+ CD103+) (b, e) and macrophages (CD11b+ CX3CR1+) (c, f) in MLN, determined by FACS. Percentages of neutrophils, dendritic cells or macrophages are shown in representative dot plots (g). Elastase concentration (h), CXCR‐2 (i) and CXCL‐1 (j) gene expression were quantified in the ileum by ELISA and RT‐PCR, respectively. Values are expressed as mean ± SEM. *P < 0·05 was considered statistically significant when compared with WT mice on CD or # P < 0·05 compared with WT fed the HFD or & P < 0·05 compared with IL‐17RA−/− mice on CD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. The results are representative of a single experiment repeated three times.
IL‐17RA effect on chemokine expression and intestinal neutrophil migration after HFD
Our next step was to investigate whether the deficiency of IL‐17RA impairs intestinal neutrophil migration, because several studies have shown that the IL‐17/IL‐17RA axis modulates expression of the cytokines and chemokines involved in the recruitment and activation of neutrophils, such as IL‐6, IL‐8, IL‐1β, CXCL‐1 and CXCL‐2.36, 37 In fact, a significant reduction in expression of elastase, a specific protease in neutrophils, was observed in the ileum of IL‐17RA−/− mice on HFD compared with IL‐17RA−/− mice on CD (Fig. 5h). We also found a significant reduction in CXCR‐2 gene expression in the ileum of WT and IL‐17RA−/− mice fed the HFD when compared with mice of the respective groups fed the CD (Fig. 5i). Interestingly, we observed a significant decrease in CXCL‐1 gene expression in the ileum of IL‐17RA−/− mice compared with IL‐17RA−/− mice on CD or WT mice on HFD (Fig. 5j). These results demonstrate that IL‐17RA deficiency compromises chemokine expression and neutrophil migration in the intestinal mucosa during MetS development induced by HFD.
IL‐17RA effect on diversity and abundance of gut microbiota after HFD
Our next step was to evaluate the composition of the gut microbiota by metagenomic analysis of the 16S bacterial gene. It is already well described and characterized that, in humans and mice, the development of obesity correlates with a change of the dominant bacteria in the gut phyla: Firmicutes and Bacteroidetes.20, 21, 22 The metagenomic analyses revealed an increase of two phyla of bacterial Proteobacteria and Bacteroidetes in relation to the phyla Firmicutes and Actinobacteria in the feces of the lL‐17RA−/− mice fed the HFD (Fig. 6a). In addition, depletion of the Verrucomicrobia phylum in the feces of lL‐17RA−/− mice has been shown (Fig. 6a). Interestingly, analysis of the bacterial genera identified an increase of Bacteroides and Sporosarcina and a decrease of Bifidobacterium in IL‐17RA−/− mice compared with WT on HFD (Fig. 6b). Overall, these data indicate that IL‐17/IL‐17RA deficiency worsens gut dysbiosis and leads to MetS progression induced by HFD.
Figure 6.
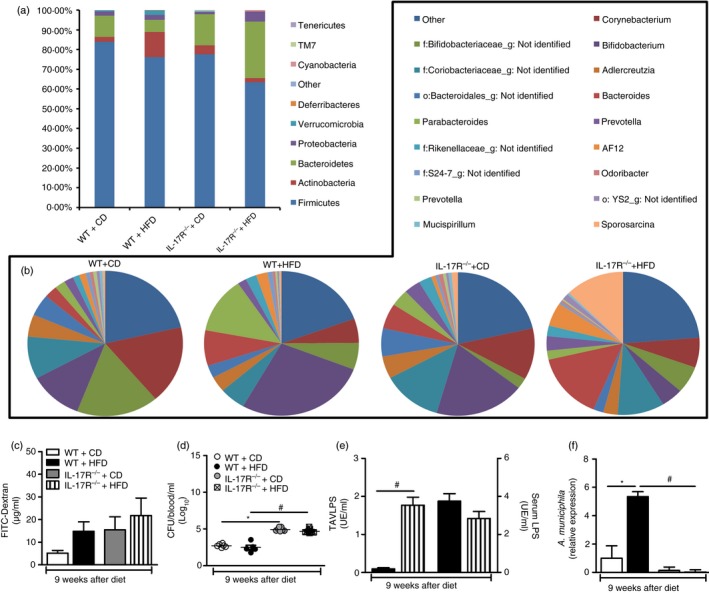
Evaluation of intestinal dysbiosis, permeability, bacterial translocation and lipopolysaccharide (LPS) levels in blood and visceral adipose tissue (VAT) of wild‐type (WT) mice and mice lacking the interleukin‐17 (IL‐17) cytokine receptor (IL‐17RA−/−) fed a control diet (CD) or a high‐fat diet (HFD) for 9 weeks. Relative abundance of fecal bacterial phylum (a) and genera (b) was evaluated by 16S rRNA gene sequencing. FITC‐Dextran concentration (c), number of colony forming units (CFU) (d), LPS levels in the serum or visceral adipose tissue (VAT) (e) A. muciniphila relative levels in the feces were determined by qPCR (f) were determined. Values are expressed as mean ± SEM. *P < 0·05 was considered statistically significant when compared with WT mice on CD or # P < 0·05 compared with WT fed the HFD or & P < 0·05 compared with IL‐17RA−/− mice on CD. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test. The results are representative of a single experiment repeated three times.
IL‐17RA effect on intestinal permeability and bacterial translocation after HFD
The association between obesity and increased intestinal permeability has been recently suggested.32 It is proposed that obesity and MetS in patients lead to a change in composition of the intestinal microbiota due to increased proliferation of pathogenic bacteria over beneficial bacteria, called intestinal dysbiosis, which can influence the integrity of the intestinal epithelium and elevate LPS levels in the blood.19, 38 Additionally, HFD induces high levels of circulating LPS, promoting metabolic inflammation and insulin resistance.39, 40, 41 Therefore, we evaluated whether IL‐17RA‐deficient mice exhibit alterations in intestinal barrier integrity, bacterial translocation and increased LPS levels. For analysis of the intestinal permeability, we used FITC‐Dextran assays. FITC is a non‐metabolized molecule whose concentration in the blood, after administration by gavage, is proportional to the degree of intestinal permeability. Notably, HFD‐fed IL‐17RA−/− mice exhibited increased intestinal permeability compared with WT mice on CD, although no significant statistical differences were observed between IL‐17RA−/− mice on CD and HFD (Fig. 6c). Accordingly, IL‐17RA−/− mice, both on the CD and HFD, exhibited higher blood bacteremia compared with WT mice on the respective diets (Fig. 6d). Interestingly, a significant increase in LPS levels was observed in the VAT of IL‐17RA−/− mice compared with WT mice on HFD, although the circulating LPS levels were equivalent between the groups (Fig. 6e). Recently Akkermansia muciniphila has been described as a beneficial bacterium for the maintenance of intestinal barrier. Akkermansia muciniphila represents up to 3–5% of the microbial community in healthy individuals and its abundance has been inversely correlated with body weight and MetS.42 Considering the increased LPS levels in the VAT of IL‐17RA−/− mice on HFD and the worsening of the metabolic parameters, a PCR was developed to detect A. muciniphila bacteria in the feces of the different experimental groups. Our results show that HFD‐fed WT mice exhibited a significant increase in A. muciniphila abundance in the intestine relative to WT mice on CD. However, HFD‐fed IL‐17RA‐deficient mice had a depletion of this bacterial group relative to WT mice on HFD (Fig. 6f). These findings imply that the IL‐17/IL‐17RA axis favors the abundance of A. muciniphila bacteria and improves the intestinal barrier function, inhibiting LPS translocation to the VAT, which results in protection against MetS induced by HFD.
IL‐17 effect on CXCL‐1 and CXCL‐2 expression in epithelial and ILP cells
Although not statistically different, a trend to increased expression of IL‐17RA in epithelial cells, but not in ILP cells, was observed in HFD‐fed mice compared with mice on the CD (Fig. 7a,d). We evaluated IL‐17‐induced chemokine production by epithelial and ILP cells in mice fed HFD and CD. The cells were isolated and stimulated with increasing concentrations of recombinant IL‐17 (rIL‐17). After 24 hr, the cultured cell supernatant was collected for measurement of CXCL‐1 and CXCL‐2 chemokines. Similar to in vivo results, epithelial cells from HFD‐fed WT mice exhibited increased CXCL‐1, but not CXCL‐2 concentrations after stimulation with 100 ng/ml rIL‐17 (Fig. 7b,c). However, no changes in CXCL‐1 or CXCL‐2 expression in response to rIL‐17 were observed in ILP cells in the same conditions (Fig. 7e,f). These data indicate that IL‐17/IL‐17R axis signaling promotes CXCL‐1 chemokine expression by epithelial cells involved in the effective neutrophil migration during HFD‐induced MetS.
Figure 7.
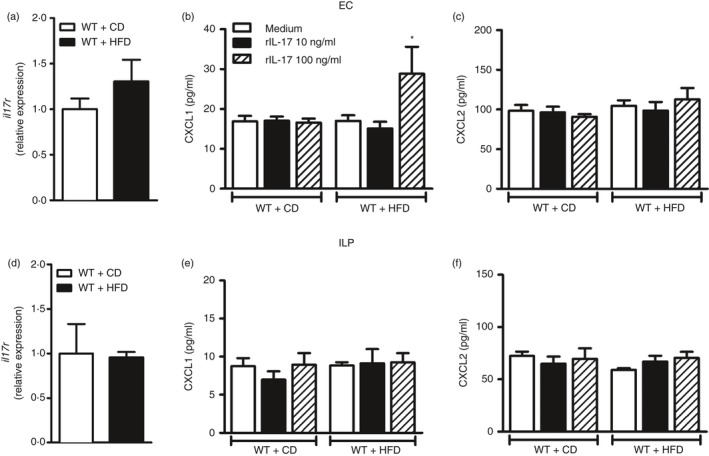
Gene expression of the interleukin‐17 receptor (IL‐17R) and CXCL‐1 or CXCL‐2 protein expression in epithelial cells (EC) and in intestinal lamina propria (ILP) cells stimulated with recombinant IL‐17 (rIL‐17). Cells were isolated from wild‐type (WT) mice fed the control diet (CD) or high‐fat diet (HFD). Relative expression of IL‐17RA in EC or ILP (a, d). After stimulation of cells for 24 hr with culture medium or recombinant IL‐17 (rIL‐17) at concentrations of 10 or 100 ng/ml, supernatants were collected from EC (b, c) or from ILP cells (e, f). Quantification of CXCL‐1 and CXCL‐2 chemokines was performed by ELISA. Values are expressed as mean ± SEM. *P < 0·05 was considered statistically significant when compared with EC from WT mice on HFD stimulated with medium. Significant differences between the groups were compared by one‐way analysis of variance followed by Tukey's multiple‐comparison test.
Discussion
Our study shows the importance of the IL‐17/IL‐17R axis in metabolic and immunological alterations associated with the development of obesity and MetS. An up‐regulation of Th17‐pattern‐related cytokines such as IL‐6, TNF‐α, IL‐17 and IL‐22 in the intestine and increased Th17 cell and IL‐17‐producing γδ T‐cell populations in the MLN were observed in WT mice fed an HFD. In agreement, a recent study reported an increase in Th17 and γδ T cells in the inguinal lymph node and epididymal adipose tissues in WT mice fed an HFD.9
IL‐17RA−/− mice fed an HFD were protected from weight gain and obesity, which was associated with higher total lipid content in feces. A compromised digestion and/or intestinal absorption of lipids from the diet may account for the reduced body weight in IL‐17RA−/− mice. An intense inflammatory infiltrate in the intestinal mucosa associated with the destruction of intestinal microvilli was observed in IL‐17RA−/− mice on HFD. These results allow us to propose that IL‐17RA deficiency causes intestinal inflammation and damage, preventing the absorption and subsequent deposition of lipids in adipose tissue. In agreement with our results, ROR‐γt‐deficient mice fed an HFD rapidly lose weight and die within 1 month after starting the HFD.43
Although IL‐17RA−/− mice did not develop obesity, they exhibited elevated fasting blood glucose levels and glucose intolerance when compared with WT mice on the HFD. In parallel, increased serum insulin levels, indicating a hyperinsulinemic condition, insulin resistance and reduced phosphorylation of key kinases involved in the insulin signaling pathway, such as PI3K and AKT, were observed in HFD‐fed IL‐17RA−/− mice. In obesity, metabolic inflammation is triggered by the activation of resident cells of the adipose tissue, such as adipocytes and immune cells, which secrete cytokines in response to products released by metabolism such as fatty acids or microbial components from the gut microbiota. In this context, TNF‐α reduces the phosphorylation of serine residues of the insulin receptor substrate‐1, impairing the coupling of the substrate to the insulin receptor and inhibiting its function. Another component associated with metabolic inflammation is LPS, which interacts with Toll‐like receptor 4/CD14 receptors present on the surface of immune cells such as macrophages, leading to activation of nuclear transcription factors that directly or indirectly induce insulin resistance.44, 45, 46, 47
Insulin resistance is a multifactorial condition that results in glucose and lipid metabolism abnormalities.48 The concept of lipotoxicity is associated with increased free fatty acids in the circulation and the accumulation of lipids in skeletal muscle and liver, which can lead to insulin resistance.49 Although IL‐17RA−/− mice exhibited insulin resistance, cholesterol and triglyceride levels were unaltered and no fat accumulation was detected in hepatocytes. Mice and diabetic patients progressively produce larger amounts of insulin as a result of increased size of pancreatic islets, characterized as hyperplasia.50 Of interest, histological analysis of the pancreatic tissue sections suggests a hyperplasia process in pancreatic islets of IL‐17RA−/− mice fed the HFD. In addition, increased expression of insulin‐producing β cells in the pancreatic islets, confirming the increased β‐cell mass, which correlates with elevated insulin levels in the serum, was observed in IL‐17RA−/− mice.
To clarify the exact role of IL‐17 in intestinal immunity during the MetS, we evaluated CXCR‐2 receptor expression and its chemokine ligand CXCL‐1, which are involved in neutrophil recruitment. Our results show decreased CXCR‐2 receptor expression in the intestine of WT and IL‐17RA−/− mice fed the HFD. Interleukin‐17 is an important cytokine in granulopoiesis by inducing expression of granulocyte colony‐stimulating factor and chemokines such as CXCL‐1, CXCL‐2 and CXCL‐5 by gut epithelial cells, macrophages and fibroblasts.51, 52 In fact, IL‐17RA−/− mice had decreased CXCL‐1 expression and elastase levels in the intestine, suggesting reduced recruitment of neutrophils into the ILP in these mice. Supporting our data, a major cause of bacterial infections in patients with metabolic syndrome and diabetes is a failure in migration and microbicidal activity of neutrophils.53
In order to understand the influence of the IL‐17/IL‐17R axis in populations of immune cells in the intestinal mucosa during MetS, we evaluated the percentage and absolute numbers of neutrophils, intestinal macrophages and dendritic cells. Interestingly, accumulation of neutrophils was observed in the MLN of IL‐17RA−/− mice fed the HFD compared with IL‐17RA−/− mice on the CD. Neutrophils are the first cells of the immune system to increase in the MLN and are recruited to the adipose tissue and ILP by an inflammatory process after HFD. The presence of neutrophils in adipose tissue occurs after 3 days of HFD, indicating that this recruitment occurs rapidly and transiently54 and contributes to the maintenance of a homeostatic microbiota in the gut, as recently reported.55
We also observed a trend towards an increased absolute number of dendritic cells in WT and IL‐17RA−/− mice fed the HFD in comparison with CD‐fed mice, suggesting increased migration of CD11b+ CD103+ dendritic cells to MLN. It is possible that the HFD‐induced intestinal dysbiosis promotes differentiation/expansion of these cells in order to control replication and colonization by commensal bacteria, so contributing to the integrity of the intestinal barrier. Dendritic cells express Toll‐like receptor 5 receptors, which mediate the differentiation of intestinal Th17 cells after flagellin stimulation of the gut microbiota. In addition, these cells are capable of producing IL‐23, which acts on the innate lymphoid cells inducing IL‐22 production by epithelial cells and synthesis of antimicrobial peptides as regenerating islet‐derived protein 3γ.56
Intestinal macrophages CD11b+ CX3CR1+ have also been described in the control of gut homeostasis as these cells are responsible for the induction of intestinal Treg cells, through IL‐10 secretion, which promotes Foxp3 expression and suppressive activity of these cells.56, 57 Another interesting fact is that adenosine triphosphate (ATP) generated by some commensal bacteria induces the expansion of a population of intestinal CX3CR1+ cells that induce the differentiation of Th17 cells. It is not clear whether the CX3CR1+ cells are resident macrophages. A recent study demonstrated that CX3CR1‐deficient mice exhibit reduced numbers of macrophages in the ILP, higher translocation of bacteria to MLN and greater severity of colitis.35 We also noted that IL‐17RA−/− mice have higher percentage and absolute numbers of intestinal macrophages in MLN, suggesting a compensatory mechanism to counter‐regulate the exacerbated intestinal inflammation observed in these mice.
Given that obese and diabetic patients have an impairment of the intestinal barrier and increased permeability,38, 44 we also investigated whether IL‐17RA deficiency interferes with the maintenance of gut integrity, and if this process is associated with increased bacterial translocation. As expected, HFD caused increased intestinal permeability, and IL‐17RA deficiency worsened the intestinal alteration induced by HFD. In accordance, obese mice exhibited alterations in intestinal permeability after 16 weeks of HFD consumption in association with lower expression of tight junction proteins, showing that the disruption in the intestinal barrier seems to be a later event.58 However, another study found no differences in intestinal permeability between obese and lean individuals, despite the presence of changes in commensal bacteria populations in the gut.59 Lipopolysaccharide is a molecule that crosses the epithelial cells via transcellular transport through the intestinal monolayer of epithelial cells. Patients with diabetes exhibit increased transcellular transport across intestinal M cells.60, 61, 62 The consumption of an HFD increases formation of chylomicrons, which exhibit high affinity for an insoluble fraction (lipid A) of LPS structure, allowing the transcellular transport of this molecule from the intestine to the circulation and to other tissues such as liver, skeletal muscle and adipose tissue.23, 63 Although LPS transport by chylomicrons may represent a physiological mechanism that favors the hepatic removal of LPS, high consumption of HFD leads to an excessive accumulation of chylomicrons, increasing the possibility of extra‐hepatic LPS leakage.23, 63 Although no changes were observed in circulating LPS levels in IL‐17RA‐deficient mice on HFD, LPS levels were increased in VAT of IL‐17RA−/− mice compared with WT mice, indicating that LPS may negatively affect insulin signaling and aggravate insulin resistance in these mice.
Gene expression of claudin‐2 and occluding proteins and expression of molecules that form the intestinal epithelial mucus layer, as mucins 1 and 2, were detected in this study. Claudin‐2 is a pore‐forming molecule in the membrane of enterocytes, and increased expression of claudin‐2 is directly related to increased intestinal permeability. However, increased expression of occludin is indicative of increased intestinal integrity.64 Unexpectedly, decreased expression of claudin‐2 and increased expression of occludin were observed in the ileum of IL‐17RA−/− mice fed the HFD. The severity of MetS in IL‐17RA−/− mice was associated with increased intestinal permeability, confirmed by FITC‐Dextran, so it is possible that increased occludin expression is a consequence of the intense inflammatory process observed in these mice. Furthermore, we also detected increased mucin‐1 gene expression in the ileum of IL‐17RA−/− mice compared with WT mice on HFD.
Our results also revealed greater prevalence of Proteobacteria and Bacteroidetes in relation to the Firmicutes and Actinobacteria phyla in feces of IL‐17RA−/− mice in comparison to WT mice fed the HFD. Furthermore, the Verrucomicrobia phylum was depleted in IL‐17RA−/− mice on HFD. A correlation between increased phyla Firmicutes/Bacteroidetes ratio and higher body mass index has been reported in obese individuals and mice.65, 66 However, decreased Firmicutes/Bacteroidetes ratio in obese individuals compared to lean controls has also been reported.67 Other studies could not establish a direct correlation between the abundance of these bacterial phyla with the development of obesity and MetS.68 In agreement with our findings, a significant prevalence of Bacteroidetes and Proteobacteria phyla was observed in the feces of diabetic patients compared with controls.69 Of importance, Gram‐negative bacteria represented by Proteobacteria and Bacteroidetes have a more pathogenic and inflammatory potential, which may be the source of increased LPS levels in the VAT of IL‐17RA‐deficient mice on HFD.
Interestingly, we identified an increase of Bacteroides and Sporosarcina and a decrease of Bifidobacterium in IL‐17RA−/− mice compared with WT mice on HFD. The Bifidobacterium bacteria have been reported to reduce LPS levels in rodents and to improve features of the intestinal barrier, so influencing glucose homeostasis control and insulin sensitivity. The increase in these bacteria has been shown to be associated with reduced macrophage infiltrate with a pro‐inflammatory profile in the visceral adipose tissue of mice.32, 40 In contrast, an increase in the Bacteroides genus in the intestinal tract of obese patients was associated with higher carbohydrate digestion, increased production of acetate and thereafter increased energy absorption compared with lean individuals.70 On the other hand, the genus Sporosarcina is associated with carnitine metabolism in humans,71 but there are no reports in the literature about its role in MetS. Together, these data demonstrate a role of IL‐17/IL‐17RA axis in the maintenance of the homeostatic gut microbiota, impairment of LPS translocation, and insulin resistance in MetS.
An interesting aspect is that WT mice on HFD display increased abundance of A. muciniphila in the feces, which could be an attempt to protect these mice from the deleterious effects of the HFD. On the other hand, there was a significant decrease in A. muciniphila bacterial abundance in the feces of the IL‐17RA−/− mice, relative to WT mice on HFD. IL‐17 signaling through the IL‐17RA receptor mediates IL‐6 production, which, in turn, induces the activation of tyrosine kinases such as JAK/STAT and MAPK, resulting in increased gene expression that encode for mucins by goblet cells.16, 72 Mucin is a substrate for A. muciniphila bacteria, which promotes degradation of these components, favoring their growth and proliferation in the intestine. Elevated abundance of A. muciniphila bacteria in the intestine is associated with intestinal integrity and reduced blood glucose levels in mice and humans.73, 74, 75 In addition, A. muciniphila is related to increased intestinal levels of endocannabinoids, which, in turn, regulate the secretion of glucagon‐like peptide (GLP‐1) by specific cells within the small intestine and colon. GLP‐1 is an incretin hormone that increases insulin sensitivity, protects insulin‐producing β cells from the pancreas against injury, improves adipocyte metabolism and serves as an anti‐inflammatory mediator.76
Our ultimate goal was to analyze the chemokine gene expression profile on epithelial cells and ILP cells isolated from mice fed the HFD or CD. Although no statistically significant differences were found, there was a trend to increased expression of IL‐17RA in EC from mice on HFD compared with mice on CD. We also detected an increased expression of CXCL‐1 chemokine in supernatants of cultured EC isolated from mice fed an HFD, after stimulation with recombinant IL‐17. Overall, these data show that HFD promotes IL‐17R expression on intestinal epithelial cells, making these cells prone to CXCL‐1 production induced by IL‐17. In general, our results revealed that IL‐17 in vitro induces CXCL‐1 expression by epithelial cells, which promotes neutrophil recruitment and confers protective immunity from pathobiont bacteria, thereby preventing the MetS onset.
In summary, our study demonstrates that deficiency of the IL‐17/IL‐17RA axis impairs the migration of intestinal neutrophils, is associated with gut dysbiosis, favors prevalence of Gram‐negative bacteria and increases LPS translocation into the VAT in mice fed an HFD, resulting in aggravation of MetS. Therefore, IL‐17 appears to induce neutrophil migration in the intestinal mucosa through the expression of CXCL‐1 chemokine by epithelial cells. Hence, IL‐17 administration may be used as an immunomodulatory intervention to prevent or treat MetS.
Authors contributions
M.M.P. performed experiments and analyzed the results; L.M.S.M., C.A.P., J.A.L., M.S.D, E.C., S.G., P.Z.A., E.N.F., and G.G.O., contributed to the analysis and helped with in vivo experiments; S.G.R. supported us with histology and imaging data. M.Z. helped with the metagenomic analyses. B.R. genotyped the IL‐17RA‐deficient mice and provided scientific assistance. R.C.T and J.S.S. edited the manuscript, provided scientific assistance and revised it critically. D.C. provided intellectual support in addition to directing and supervising the study.
Disclosures
The authors declare no competing financial interests.
Acknowledgements
This study was supported by grants from the São Paulo Research Foundation (FAPESP) under grant agreement nos. 2014/21020‐2, 2012/10395‐0 (Project Young Researcher) and 2013/08216‐2 (Center for Research in Inflammatory Disease) and from the University of São Paulo (Núcleo de Pesquisa em Doenças Inflamatórias) under grant agreement no. 11.1.21625.01.0. We are grateful to Elaine Medeiros Floriano from the Laboratory of Pathology of the Ribeirão Preto Medical School, University of São Paulo, for helping with the histological analysis. We are grateful to Noah W. Palm and Richard A. Flavell from the School of Medicine, Yale University, New Haven (USA) for support with the metagenomic analysis.
Contributor Information
João S. Silva, Email: danicar@usp.br
Daniela Carlos, Email: danicar@usp.br.
References
- 1. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract 2014; 2014:943162. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2. Goldberg RB, Mather K. Targeting the consequences of the metabolic syndrome in the Diabetes Prevention Program. Arterioscler Thromb Vasc Biol 2012; 32:2077–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Park YW, Zhu S, Palaniappan L, Heshka S, Carnethon MR, Heymsfield SB. The metabolic syndrome: prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988–1994. Arch Intern Med 2003; 163:427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA et al Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009; 120:1640–5. [DOI] [PubMed] [Google Scholar]
- 5. Cua DJ, Tato CM. Innate IL‐17‐producing cells: the sentinels of the immune system. Nat Rev Immunol 2010; 10:479–89. [DOI] [PubMed] [Google Scholar]
- 6. Shaw MH, Kamada N, Kim YG, Nunez G. Microbiota‐induced IL‐1β, but not IL‐6, is critical for the development of steady‐state TH17 cells in the intestine. J Exp Med 2012; 209:251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL‐17 and Th17 cells. Annu Rev Immunol 2009; 27:485–517. [DOI] [PubMed] [Google Scholar]
- 8. Atarashi K, Tanoue T, Honda K. Induction of lamina propria Th17 cells by intestinal commensal bacteria. Vaccine 2010; 28:8036–8. [DOI] [PubMed] [Google Scholar]
- 9. Zuniga LA, Shen WJ, Joyce‐Shaikh B, Pyatnova EA, Richards AG, Thom C et al IL‐17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol 2010; 185:6947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahmed M, Gaffen SL. IL‐17 in obesity and adipogenesis. Cytokine Growth Factor Rev 2010; 21:449–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL‐17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 2007; 25:821–52. [DOI] [PubMed] [Google Scholar]
- 12. Aujla SJ, Dubin PJ, Kolls JK. Th17 cells and mucosal host defense. Semin Immunol 2007; 19:377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flannigan KL, Ngo VL, Geem D, Harusato A, Hirota SA, Parkos CA et al IL‐17A‐mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol 2017; 10:673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee JS, Tato CM, Joyce‐Shaikh B, Gulen MF, Cayatte C, Chen Y et al Interleukin‐23‐independent IL‐17 production regulates intestinal epithelial permeability. Immunity 2015; 43:727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kinugasa T, Sakaguchi T, Gu X, Reinecker HC. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology 2000; 118:1001–11. [DOI] [PubMed] [Google Scholar]
- 16. Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)‐17 through IL‐6 paracrine/autocrine loop. J Biol Chem 2003; 278:17036–43. [DOI] [PubMed] [Google Scholar]
- 17. Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y et al Differential roles of interleukin‐17A and ‐17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009; 30:108–19. [DOI] [PubMed] [Google Scholar]
- 18. Cao AT, Yao S, Gong B, Elson CO, Cong Y. Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J Immunol 2012; 189:4666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tagliabue A, Elli M. The role of gut microbiota in human obesity: recent findings and future perspectives. Nutr Metab Cardiovasc Dis 2013; 23:160–8. [DOI] [PubMed] [Google Scholar]
- 20. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006; 444:1022–3. [DOI] [PubMed] [Google Scholar]
- 21. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 2009; 1:6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sommer F, Backhed F. The gut microbiota – masters of host development and physiology. Nat Rev Microbiol 2013; 11:227–38. [DOI] [PubMed] [Google Scholar]
- 23. Moreira AP, Texeira TF, Ferreira AB, Peluzio Mdo C, Alfenas Rde C. Influence of a high‐fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr 2012; 108:801–9. [DOI] [PubMed] [Google Scholar]
- 24. Carlos D, Yaochite JN, Rocha FA, Toso VD, Malmegrim KC, Ramos SG et al Mast cells control insulitis and increase Treg cells to confer protection against STZ‐induced type 1 diabetes in mice. Eur J Immunol 2015; 45:2873–85. [DOI] [PubMed] [Google Scholar]
- 25. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L et al Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014; 158:1000–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 2005; 71:8228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007; 73:5261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 2010; 7:335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS et al Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005; 365:1333–46. [DOI] [PubMed] [Google Scholar]
- 31. Masjkur J, Poser SW, Nikolakopoulou P, Chrousos G, McKay RD, Bornstein SR et al Endocrine pancreas development and regeneration: noncanonical ideas from neural stem cell biology. Diabetes 2016; 65:314–30. [DOI] [PubMed] [Google Scholar]
- 32. Frazier TH, DiBaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity‐induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr 2011; 35:14S–20S. [DOI] [PubMed] [Google Scholar]
- 33. Cildir G, Akincilar SC, Tergaonkar V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol Med 2013; 19:487–500. [DOI] [PubMed] [Google Scholar]
- 34. Johansson‐Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Forster R et al Functional specialization of gut CD103+ dendritic cells in the regulation of tissue‐selective T cell homing. J Exp Med 2005; 202:1063–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Medina‐Contreras O, Geem D, Laur O, Williams IR, Lira SA, Nusrat A et al CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest 2011; 121:4787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Onishi RM, Gaffen SL. Interleukin‐17 and its target genes: mechanisms of interleukin‐17 function in disease. Immunology 2010; 129:311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Flannigan KL, Ngo VL, Geem D, Harusato A, Hirota SA, Parkos CA et al IL‐17A‐mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol 2016; 10:673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cani PD, Delzenne NM. Interplay between obesity and associated metabolic disorders: new insights into the gut microbiota. Curr Opin Pharmacol 2009; 9:737–43. [DOI] [PubMed] [Google Scholar]
- 39. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D et al Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007; 56:1761–72. [DOI] [PubMed] [Google Scholar]
- 40. Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM et al Selective increases of bifidobacteria in gut microflora improve high‐fat‐diet‐induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007; 50:2374–83. [DOI] [PubMed] [Google Scholar]
- 41. Saito T, Hayashida H, Furugen R. Comment on: Cani et al. (2007) Metabolic endotoxemia initiates obesity and insulin resistance: Diabetes 56:1761‐1772.. Diabetes 2007; 56:e20. author reply e1. [DOI] [PubMed] [Google Scholar]
- 42. Everard A, Cani PD. Diabetes, obesity and gut microbiota. Best Pract Res Clin Gastroenterol 2013; 27:73–83. [DOI] [PubMed] [Google Scholar]
- 43. Garidou L, Pomie C, Klopp P, Waget A, Charpentier J, Aloulou M et al The gut microbiota regulates intestinal CD4 T cells expressing RORγt and controls metabolic disease. Cell Metab 2015; 22:100–12. [DOI] [PubMed] [Google Scholar]
- 44. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM et al Changes in gut microbiota control metabolic endotoxemia‐induced inflammation in high‐fat diet‐induced obesity and diabetes in mice. Diabetes 2008; 57:1470–81. [DOI] [PubMed] [Google Scholar]
- 45. Cani PD, Delzenne NM, Amar J, Burcelin R. Role of gut microflora in the development of obesity and insulin resistance following high‐fat diet feeding. Pathol Biol (Paris) 2008; 56:305–9. [DOI] [PubMed] [Google Scholar]
- 46. Gual P, Le Marchand‐Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS‐1 phosphorylation. Biochimie 2005; 87:99–109. [DOI] [PubMed] [Google Scholar]
- 47. He C, Shan Y, Song W. Targeting gut microbiota as a possible therapy for diabetes. Nutr Res 2015; 35:361–7. [DOI] [PubMed] [Google Scholar]
- 48. Henriksen EJ, Diamond‐Stanic MK, Marchionne EM. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med 2011; 51:993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 2012; 148:852–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tourrel C, Bailbe D, Lacorne M, Meile MJ, Kergoat M, Portha B. Persistent improvement of type 2 diabetes in the Goto‐Kakizaki rat model by expansion of the β‐cell mass during the prediabetic period with glucagon‐like peptide‐1 or exendin‐4. Diabetes 2002; 51:1443–52. [DOI] [PubMed] [Google Scholar]
- 51. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P et al Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony‐stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 2001; 194:519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol 2012; 5:354–66. [DOI] [PubMed] [Google Scholar]
- 53. Hatanaka E, Monteagudo PT, Marrocos MS, Campa A. Neutrophils and monocytes as potentially important sources of proinflammatory cytokines in diabetes. Clin Exp Immunol 2006; 146:443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Elgazar‐Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra‐abdominal fat early in the course of high‐fat feeding. J Lipid Res 2008; 49:1894–903. [DOI] [PubMed] [Google Scholar]
- 55. Mori K, Suzuki T, Minamishima S, Igarashi T, Inoue K, Nishimura D et al Neutrophil gelatinase‐associated lipocalin regulates gut microbiota of mice. J Gastroenterol Hepatol 2016; 31:145–54. [DOI] [PubMed] [Google Scholar]
- 56. Farache J, Zigmond E, Shakhar G, Jung S. Contributions of dendritic cells and macrophages to intestinal homeostasis and immune defense. Immunol Cell Biol 2013; 91:232–9. [DOI] [PubMed] [Google Scholar]
- 57. Schulz O, Jaensson E, Persson EK, Liu X, Worbs T, Agace WW et al Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med 2009; 206:3101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci 2013; 70:631–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brignardello J, Morales P, Diaz E, Romero J, Brunser O, Gotteland M. Pilot study: alterations of intestinal microbiota in obese humans are not associated with colonic inflammation or disturbances of barrier function. Aliment Pharmacol Ther 2010; 32:1307–14. [DOI] [PubMed] [Google Scholar]
- 60. Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 2010; 72:463–93. [DOI] [PubMed] [Google Scholar]
- 61. Harris K, Kassis A, Major G, Chou CJ. Is the gut microbiota a new factor contributing to obesity and its metabolic disorders? J Obes 2012; 2012:879151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Caricilli AM, Saad MJ. The role of gut microbiota on insulin resistance. Nutrients 2013; 5:829–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res 2009; 50:90–7. [DOI] [PubMed] [Google Scholar]
- 64. Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M et al Intestinal permeability – a new target for disease prevention and therapy. BMC Gastroenterol 2014; 14:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cani PD, Delzenne NM. The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Des 2009; 15:1546–58. [DOI] [PubMed] [Google Scholar]
- 66. Wu X, Ma C, Han L, Nawaz M, Gao F, Zhang X et al Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr Microbiol 2010; 61:69–78. [DOI] [PubMed] [Google Scholar]
- 67. Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C et al Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 2010; 18:190–5. [DOI] [PubMed] [Google Scholar]
- 68. Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P et al Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) 2008; 32:1720–4. [DOI] [PubMed] [Google Scholar]
- 69. Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK et al Gut microbiota in human adults with type 2 diabetes differs from non‐diabetic adults. PLoS ONE 2010; 5:e9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Abdallah Ismail N, Ragab SH, Abd Elbaky A, Shoeib AR, Alhosary Y, Fekry D. Frequency of Firmicutes and Bacteroidetes in gut microbiota in obese and normal weight Egyptian children and adults. Arch Med Sci 2011; 7:501–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhu Y, Jameson E, Crosatti M, Schafer H, Rajakumar K, Bugg TD et al Carnitine metabolism to trimethylamine by an unusual Rieske‐type oxygenase from human microbiota. Proc Natl Acad Sci U S A 2014; 111:4268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gu C, Wu L, Li X. IL‐17 family: cytokines, receptors and signaling. Cytokine 2013; 64:477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dao MC, Everard A, Aron‐Wisnewsky J, Sokolovska N, Prifti E, Verger EO et al Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 2016; 65:426–36. [DOI] [PubMed] [Google Scholar]
- 74. Naito Y, Uchiyama K, Takagi T. A next‐generation beneficial microbe: Akkermansia muciniphila . J Clin Biochem Nutr 2018; 63:33–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chelakkot C, Choi Y, Kim DK, Park HT, Ghim J, Kwon Y et al Akkermansia muciniphila‐derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp Mol Med 2018; 50:e450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bland J. Intestinal microbiome, Akkermansia muciniphila, and medical nutrition therapy. Integr Med (Encinitas) 2016; 15:14–6. [PMC free article] [PubMed] [Google Scholar]