

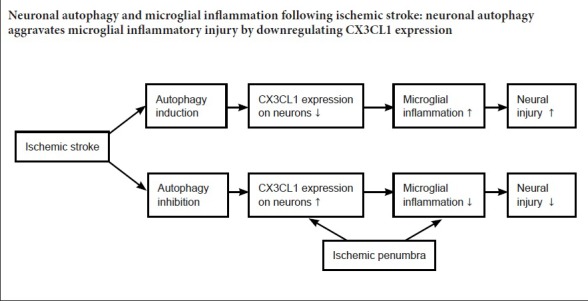
Keywords: nerve regeneration, ischemic stroke, neuronal autophagy, microglial inflammation, CX3CL1, autophagic neurons, autophagy induction, autophagy inhibition, neural regeneration
Abstract
Ischemic stroke often induces excessive neuronal autophagy, resulting in brain damage; meanwhile, inflammatory responses stimulated by ischemia exacerbate neural injury. However, interactions between neuronal autophagy and microglial inflammation following ischemic stroke are poorly understood. CX3CL1/fractalkine, a membrane-bound chemokine expressed on neurons, can suppress microglial inflammation by binding to its receptor CX3CR1 on microglia. In the present study, to investigate whether autophagy could alter CX3CL1 expression on neurons and consequently change microglial inflammatory activity, middle cerebral artery occlusion (MCAO) was established in Sprague-Dawley rats to model ischemic stroke, and tissues from the ischemic penumbra were obtained to evaluate autophagy level and microglial inflammatory activity. MCAO rats were administered 3-methyladenine (autophagy inhibitor) or Tat-Beclin 1 (autophagy inducer). Western blot assays were conducted to quantify expression of Beclin-1, nuclear factor kappa B p65 (NF-κB), light chain 3B (LC3B), and CX3CL1 in ischemic penumbra. Moreover, immunofluorescence staining was performed to quantify numbers of LC3B-, CX3CL1-, and Iba-1-positive cells in ischemic penumbra. In addition, enzyme linked immunosorbent assays were utilized to analyze concentrations of tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), interleukin 1 beta (IL-1β), and prostaglandin E2 (PGE2). A dry/wet weight method was used to detect brain water content, while 2,3,5,-triphenyltetrazolium chloride staining was utilized to measure infarct volume. The results demonstrated that autophagy signaling (Beclin-1 and LC3B expression) in penumbra was prominently activated by MCAO, while CX3CL1 expression on autophagic neurons was significantly reduced and microglial inflammation was markedly activated. However, after inhibition of autophagy signaling with 3-methyladenine, CX3CL1 expression on neurons was obviously increased, whereas Iba-1 and NF-κB expression was downregulated; TNF-α, IL-6, IL-1β, and PGE2 levels were decreased; and cerebral edema was obviously mitigated. In contrast, after treatment with the autophagy inducer Tat-Beclin 1, CX3CL1 expression on neurons was further reduced; Iba-1 and NF-κB expression was increased; TNF-α, IL-6, IL-1β, and PGE2 levels were enhanced; and cerebral edema was aggravated. Our study suggests that ischemia-induced neuronal autophagy facilitates microglial inflammatory injury after ischemic stroke, and the efficacy of this process may be associated with downregulated CX3CL1 expression on autophagic neurons.
Chinese Library Classification No. R459.9; R364; R741
Introduction
Autophagy is a cellular self-recycling process by which damaged organelles, obsolete proteins, and soluble macromolecules are delivered to lysosomes for degradation (Jiang and Mizushima, 2014). Increasing evidence indicates that autophagy extensively participates in immune responses (Levine et al., 2011), protein secretion, and cell metabolism (Kaur and Debnath, 2015), and plays important roles in neurodegenerative diseases, neuroinflammation, and cerebral stroke (Chen et al., 2014). A basal level of autophagy is thought to be required for cell survival by clearing faulty intracellular materials during physiological conditions (Hara et al., 2006). However, excessive autophagy is often induced in pathological situations such as cerebral ischemia, leading to autophagy-mediated cell death (Descloux et al., 2015). However, modest levels of autophagy also provide neuroprotection against ischemic injury (Yang et al., 2017; Wang et al., 2018). Thus, most investigators believe that an appropriate level of autophagy is neuroprotective, while excessive autophagy is harmful. However, it is difficult to define how much autophagy is appropriate or excessive. In fact, complex crosstalk occurs between autophagy signaling and other pathophysiological processes, such as inflammatory responses (Saitoh and Akira, 2010).
A recent study revealed intimate participation of inflammatory mechanisms in all pathological stages of cerebral ischemia (Iadecola and Anrather, 2011). Within minutes after a cerebral stroke, ischemia elicits severe oxidative stress that rapidly activates inflammatory signaling (Eltzschig and Carmeliet, 2011) and, in turn, leads to reactive oxygen species production, microglial activation, and blood-brain barrier damage. This inflammation eventually becomes self-protective to permit structural and functional reorganization of injured tissues during the late stage of cerebral ischemia (Spite and Serhan, 2010). Microglia, which are the resident inflammatory cells of the brain, play pivotal roles in inflammatory responses in response to ischemia. During physiological conditions, microglia exert basic functions, such as maintenance of synaptic structure, remodeling of presynaptic environments, and release of neurotrophic factors for brain tissues (Tremblay et al., 2011; Martinez and Peplow, 2017). However, during pathological conditions, microglia become activated and act as key mediators of both neuropathology and neuroprotection (Kohman and Rhodes, 2013). Activated microglia even can determine the fate of neurons by releasing multiple cytokines and modulatory molecules to alter microenvironmental homeostasis (Walker et al., 2014; Hass and Barnstable, 2016; Plaza-Zabala et al., 2017). Both autophagic mechanisms and microglial inflammatory responses are simultaneously involved in the pathological process of cerebral stroke. Indeed, studies have shown that autophagy is able to control the state of microglial activation to balance the detrimental and beneficial efficacy of inflammation in response to cerebral ischemia (Puyal et al., 2013; Su et al., 2016). However, precise interactions between autophagy and microglial inflammatory signaling are complex and widely unknown.
CX3CL1 (fractalkine), a membrane-bound chemokine expressed on neurons, can suppress microglial activation by binding to its receptor CX3CR1 on microglia in brain tissue (Cardona et al., 2006). Under normal conditions, microglia are kept quiescent by contact with neurons through conjugation of CX3CL1-CX3CR1 (Iadecola and Anrather, 2011). In this manner, microglial inflammatory activity can be appropriately repressed through CX3CL1-CX3CR1 cross-communication between neurons and microglia. However, expression of CX3CL1 on neurons undergoing autophagy may be downregulated in pathological conditions (Cardona et al., 2006), such as cerebral ischemia. Thus, the efficacy of neuronal inhibition of microglial inflammatory activity is weakened as a result of reduced CX3CL1 expression on autophagic neurons. Subsequently, microglia become activated to amplify ischemia-stimulated inflammatory responses, which likely result in excessive inflammation and damage.
Based on the interaction between neurons and microglia through conjugation of CX3CL1-CX3CR1, we hypothesized that CX3CL1 expression on autophagic neurons may be reduced, consequently activating microglia to aggravate ischemia-induced inflammatory injury. To test this hypothesis, we first verified whether cerebral ischemia could induce detrimental autophagy in a rat model of ischemic stroke, and then evaluated CX3CL1 expression levels and the state of microglial inflammatory activation. Furthermore, to investigate whether CX3CL1 expression on neurons can be specifically altered by autophagy, an autophagy inducer and inhibitor were individually administered in rats who had undergone middle cerebral artery occlusion (MCAO). Finally, the effects of altered CX3CL1 expression on autophagic neurons upon microglial inflammatory responses were observed. The results of this study identify targets against inflammatory injury that may be useful for promoting novel stroke treatments.
Materials and Methods
Animals
A total of 96 male specific-pathogen-free Sprague-Dawley rats (aged 9–10 weeks and weighing 250–280 g) were provided by the Experimental Animal Center at Kunming University of Science and Technology, China (Animal license number: SYXK (Dian) K2013-0003). Rats were fed according to animal welfare practices. All experiments were approved by the Animal Experiment Committee of Kunming University of Science and Technology.
Rats were randomly divided into four groups: sham (sham operation), MCAO, MCAO + 3-methyladenine (3-MA; 3-MA treatment), and MCAO + Tat-Beclin 1 (Tat; Tat treatment).
Preparation of cerebral stroke model and treatment with 3-MA or Tat-Beclin 1
To prepare cerebral stroke model rats, animals were first anesthetized with chloral hydrate (10%, 360 mg/kg) by intraperitoneal injection. Thereafter, the common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA) on the left were separated away from adjacent nerves and muscles. A 4-0 nylon monofilament coated with a round poly-lysine tip (tip diameter of 0.36 mm was similar to the diameter of the middle cerebral artery; Beijing Cinontech, Beijing, China) was inserted from a mini-incision on the ECA and went back into the CCA, and then gently into the middle cerebral artery (MCA) from the ICA. The nylon monofilament was withdrawn to allow reperfusion after 90 minutes of MCAO. A laser Doppler flowmeter (PeriFlux 5000, Perimed, Stockholm, Sweden) was used to detect regional blood flow to guarantee the robustness of ischemic stroke. This method of preparing the MCAO rat model was based on our previous study (Hongyun et al., 2017).
To observe the association between neuronal autophagy and microglial inflammatory responses following cerebral stroke, the autophagy inhibitor 3-MA (0.5 ng in 10 μL of 0.1% dimethyl sulfoxide; Sigma, Milwaukee, WI, USA) and autophagy inducer Tat (0.5 mg in 10 μL 0.1% dimethyl sulfoxide; Millipore, Billerica, MA, USA) were separately injected intracerebroventricularly 40 minutes before the onset of reperfusion. For stereotaxic injection, bregma was viewed as an original point, and then the injection point was fitted: 1.1-mm posterior, 1.5-mm lateral and 4.5-mm depth.
Western blot assay
Rats (n = 6 in each group) were sacrificed without pain 12 hours after MCAO/reperfusion. (In our previous study, we found that at 12 hours after MCAO/reperfusion, autophagic activity in the penumbra was obviously reduced, but CX3CL1 expression was contrarily increased; meanwhile, nuclear factor kappa B p65 (NF-κB) level was decreased, suggesting a potential correlation between them. Therefore, we chose this time point to investigate the relationship between autophagy and microglial inflammatory injury following MCAO.) Tissues in the penumbric area were isolated. After homogenizing using an abrasive approach, samples were treated with radioimmune precipitation assay buffer (containing 50 mM Tris pH 7.4, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 150 mM NaCl, and 1% sodium deoxycholate; Beijing BLKW Biotechnology, Beijing, China) for 40 minutes. After centrifugation at 13,000 × g and 4°C for 15 minutes, the supernatant was obtained. To perform western blot assays, sodium dodecyl sulfate polyacrylamide gel electrophoresis was used to separate proteins from supernatants, and separated proteins were then transferred onto a polyvinylidene fluoride membrane (Millipore). Next, 10% nonfat milk was added to cover membranes for 2 hours while shaking to block non-specific antigen binding. After washing with PBS containing 0.1% polysorbate 20 (PBST), proteins on membranes were separately labeled using primary rabbit antibodies against rat Beclin-1, NF-κB p65 (1:1000; Cell Signaling Technology, Danvers, MA, USA), LC3B (1:1000; Cell Signaling Technology), CX3CL1 (1:1000; Abcam, Cambridge, UK), and β-actin (1:10,000; Sigma, St. Louis, MO, USA) for 12 hours at 4°C. Membranes were then washed with PBST and incubated with fluorescence-conjugated second goat antibody (1:5000; Cell Signaling Technology) for 1 hour at room temperature. After washing for 2 hours with shaking, the reaction band was amplified through electrochemiluminescence. Fluorescence densitometry was analyzed by a system (Bio-Rad Image Lab 4.1, Hercules, CA, USA). Protein signals were normalized against the fluorescence densitometry of β-actin.
Immunofluorescence
Animals (n = 6 in each group) were sacrificed without pain 12 hours after MCAO/reperfusion. Brains were quickly removed onto ice and placed in sucrose solution for dehydration (30%; Invitrogen, Shanghai, China). Brains were then sliced into 20-μm-thick sections with a cryostat (SLEE, Mainz, Germany). After washing with PBS, sections were treated with Triton X-100 for 15 minutes for permeabilization. After a wash step, 10% normal goat serum was used to block non-specific antigen binding for 40 minutes. Sections were then separately labeled with primary rabbit antibodies against rat LC3B and CX3CL1 (1:400; Cell Signaling Technology), mouse antibodies against Iba-1 and NeuN (1:500; Abcam), or PBS (negative control) for 4 hours at room temperature. After washing with PBS, sections were incubated with DyLight 488-conjugated anti-rabbit IgG (1:800; Invitrogen) and Alexa Fluor® 594-conjugated anti-mouse IgG (1:800; Invitrogen) for 2 hours in the dark. Brain sections were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen) in PBS (1:1000) for 5 minutes. After a wash, the immune reaction was measured with a fluorescent microscope (Nikon Instruments, Tokyo, Japan). Outcomes were indicated as percentages of positive cells. Under high magnification (400×), numbers of total cells and positive cells were counted in 10 randomly selected fields from each section, and 10 detected sections from each animal were counted.
Enzyme-linked immunosorbent assay (ELISA)
Penumbral tissues (n = 6 in each group) were obtained 12 hours after the insult. A 0.5-g sample of brain tissue from each rat was isolated and homogenized by abrasiveness. Brain tissue was then diluted in 4.5 mL of 0.86% physiological saline and ultrasonicated with an ultrasonic generator (Ningbo Scientz Biotechnology, Ningbo, China). Supernatants of brain homogenates were obtained after centrifugation at 3000 × g and 4°C for 15 minutes. Concentrations of tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), interleukin 1 beta (IL-1β), and prostaglandin E2 (PGE2) in supernatant were measured by ELISA kits (Invitrogen) according to the manufacturer’s protocols.
Detection of brain water content
Animals (n = 6 in each group) were painlessly sacrificed after deep anesthesia. Brains were quickly removed onto ice and immediately weighed for wet weight. Next, brains were dried overnight at 105°C for dry weight measurements. Water content in brain tissue was evaluated by an Electronic Microbalance (METTLER TOLEDO, Zurich, Switzerland) to assess percentage of water content (%) = (wet weight − dry weight) / wet weight × 100%.
2,3,5,-Triphenyltetrazolium chloride (TTC) staining
Animals (n = 6 in each group) were sacrificed under deep anesthesia 12 hours after ischemia/reperfusion. Brains were quickly removed onto ice and immediately frozen at −20°C for 20 minutes. Brain samples were then sliced into 2-mm-thick coronal sections and immediately put into a 0.5% TTC solution (Beijing Leagene Biotechnology, Beijing, China) at 37°C. After staining for 30 minutes, brain sections were fixed in 4% paraformaldehyde buffer (Invitrogen) for 12 hours at room temperature. With regard to TTC staining, infarct tissues were pale, while normal tissues were red. Afterwards, Adobe Photoshop 7.0 imaging software (Adobe Systems, Dublin, Ireland) was used to calculate infarct volume. Results are represented as infarction ratio (%) = A°/A’ × 100%, whereby A’ is the volume of the homolateral hemisphere, and A° represents the infarct volume.
Statistical analysis
All data are presented as the mean ± SEM. Statistical differences were evaluated by one-way analysis of variance followed by Dunett’s test using SPSS 11.0 software (SPSS, Chicago, IL, USA). A value of P < 0.05 was considered statistically significant.
Results
Autophagy level negatively correlates with CX3CL1 expression, but positively associates with inflammatory signaling after cerebral stroke
Tissues from the penumbra region were obtained to perform western blot assays at 1, 2, 3, 4, 5, 6, 12, and 24 hours after MCAO/reperfusion. The results illustrated that autophagy signaling was remarkably activated and maintained at a high level within 6 hours after MCAO/reperfusion (Figure 1A, B); however, two markers of autophagy, Beclin-1 and light chain 3B (LC3B) (Klionsky et al., 2012), were markedly decreased 12 hours after the insult. At this time point, NF-κB expression was also decreased (Figure 1D), but expression of CX3CL1/fractalkine [a ligand for microglial activation (Sheridan and Murphy, 2013)] was contrarily enhanced (Figure 1C).
Figure 1.

Correlation between autophagy and CX3CL1/fractalkine expression or NF-κB p65 activation after ischemic stroke (western blot assay).
Autophagic signaling in the penumbra was remarkably activated by ischemia within 6 hours(h) after cerebral ischemia (A, B); however, the autophagy level was obviously declined at 12 h after insult. At this time point, the NF-κB p65 expression level (D) was also decreased, but CX3CL1 expression (C) was contrarily increased. There is a trend that autophagy level was negatively correlated with CX3CL1 expression, but positively correlated with activation of inflammatory signaling (as shown in the red squares). Relative protein expression was expressed as the optical density ratio to β-actin. Data are presented as the mean ± SEM (n = 6; one-way analysis of variance followed by Dunett’s test). *P < 0.01. NF-κB: Nuclear factor kappa B; LC3B: light chain 3B.
Autophagy induction reduces CX3CL1/fractalkine expression and enhances microglial activation
To test the effect of autophagy on stroke-induced inflammatory responses, MCAO rats were additionally administered with 3-MA and Tat-Beclin 1, separately. Brain tissues from the penumbra were obtained to perform western blot assays 12 hours after MCAO/reperfusion. The results showed that ischemia-induced autophagy was further increased after treatment with an autophagy inducer (Figure 2A), while CX3CL1 expression levels were markedly reduced (Figure 2B), and both Iba-1 (Figure 2C) and NF-κB expression levels (Figure 2D) were markedly increased. In contrast, after treatment with the autophagy inhibitor 3-MA, the MCAO-induced reduction of CX3CL1 expression was effectively suppressed, and both Iba-1 and NF-κB expression were downregulated compared with the MCAO group.
Figure 2.

Effect of altered autophagy on CX3CL1 expression and inflammatory responses after ischemic stroke (western blot assay).
Autophagy inducer Tat-Beclin 1 and autophagy inhibitor 3-MA were additionally administered in MCAO rats, separately. CX3CL1, Iba-1, and NF-κB p65 expression were detected 12 hours after insult. The results indicated that autophagy inhibition (A) dramatically enhanced CX3CL1 expression (B); meanwhile, both Iba-1 (C) and NF-κB p65 (D) expression levels were markedly declined. Compared with the MCAO group, CX3CL1 expression was further reduced after treatment with an autophagy inducer in the MCAO + Tat group; whereas, Iba-1 and NF-κB p65 were increasingly expressed. Relative protein expression was expressed as the optical density ratio to β-actin. Data are presented as the mean ± SEM (n = 6; one-way analysis of variance followed by Dunett’s test). *P < 0.01. MCAO: Middle cerebral artery occlusion; 3-MA: 3-methyladenine; Tat: Tat-Beclin 1; NF-κB: nuclear factor kappa B.
Neuronal autophagy enhances microglial inflammatory responses but downregulates CX3CL1/fractalkine expression
To further investigate the association between CX3CL1 expression and activation of microglial inflammatory responses, immunofluorescence was performed using antibodies against NeuN [a neuron marker (Gusel’nikova and Korzhevskiy, 2015)], CX3CL1, NF-κB, and Iba-1 [a microglia marker (Korzhevskiy and Kirik, 2015)] at 12 hours after the insult. The results illustrated that induction of autophagy remarkably increased the percentage of autophagic neurons (Figure 3A, E), but obviously decreased the ratio of CX3CL1-positive neurons in the MCAO + Tat group (Figure 3B, F). However, the percentage of Iba-1- (Figure 3C, G) and NF-κB-positive microglia (Figure 3D, H) was contrarily increased after treatment with the autophagy inducer. Comparatively, after treatment with the autophagy inhibitor 3-MA, CX3CL1 expression on neurons was markedly increased, whereas Iba-1 and NF-κB expression were reduced.
Figure 3.

Neuronal autophagy reduces CX3CL1 expression on neurons and activates microglial inflammatory responses after cerebral stroke.
Immunofluorescence was performed to determine cellular localization of CX3CL1 and its effect on microglial inflammatory responses. The results showed that enhanced neuronal autophagy [A, E (MCAO + Tat group)] reduced CX3CL1 expression on neurons (B, F), and further boosted microglial activity (C, G). In contrast, inhibition of neuron autophagy improved the percentage of CX3CL1-positive neurons, and reduced the ratio of NF-κB p65-positive microglia (D, H). In A and B, green staining (DyLight 488) indicates LC3B and CX3CL1-positive cells, while red staining (Alexa Fluor® 594) indicates NeuN-positive cells and yellow (merged) indicated the LC3B and CX3CL1-positive neurons. In C, red indicates Iba-1-positive cells; blue indicates total cells; and the merged picture shows the percentage of activated microglia. In D, green staining (DyLight 488) indicates NF-κB p65-positive cells, while red staining (Alexa Fluor® 594) indicates Iba-1-positive cells and yellow (merged) indicates the ratio of NF-κB p65-positive microglia. Arrows indicate positive neurons or microglia. Scale bars: 10 μm. Data are presented as the mean ± SEM (n = 6; one-way analysis of variance followed by Dunett’s test). *P < 0.01. MCAO: Middle cerebral artery occlusion; 3-MA: 3-methyladenine; Tat: Tat-Beclin 1; NF-κB: nuclear factor kappa B.
Autophagy induction boosts production of inflammatory markers after cerebral stroke
Inflammatory markers indirectly reflect the activation of microglial inflammatory responses. Thus, we measured production of TNF-α, IL-6, IL-1β, and PGE2 in the penumbra 12 hours after MCAO/reperfusion. The results illustrated that inhibition of autophagy (MCAO + 3-MA group) could reduce the production of inflammatory molecules, but induction of autophagy (MCAO + Tat group) further promoted their production after stimulation by ischemia (MCAO group). Moreover, concentrations of inflammatory molecules in the sham group exhibited lower levels (Figure 4).
Figure 4.

Effect of neuronal autophagy on production of inflammatory molecules after ischemic stroke (enzyme-linked immunosorbent assay).
Tissues in the penumbra area were obtained 12 hours after insult to detect the production of inflammatory markers. (A–D) IL-1β, TNF-α, IL-6, and PGE2 production, respectively. Concentrations of inflammatory molecules in brain tissues from the MCAO group were markedly higher than those from the sham or MCAO + 3-MA groups, but notably lower than those from the MCAO + Tat group. Data are presented as the mean ± SEM (n = 6; one-way analysis of variance followed by Dunett’s test). *P < 0.01. IL: Interleukin; TNF-α: tumor necrosis factor alpha; PGE2: prostaglandin E2; MCAO: middle cerebral artery occlusion; 3-MA: 3-methyladenine; Tat: Tat-Beclin 1.
Autophagy induction aggravates ischemia-induced brain edema after cerebral stroke
To investigate the correlation between autophagy and inflammatory edema, brain water content was evaluated 12 hours after the insult. After MCAO, ischemia/reperfusion induced prominent brain edema in brains. Moreover, brain water content increased in the MCAO + Tat group compared with the MCAO group. In contrast, MCAO-induced brain edema was obviously attenuated in the MCAO + 3-MA group after treatment with the autophagy inhibitor 3-MA (Figure 5).
Figure 5.
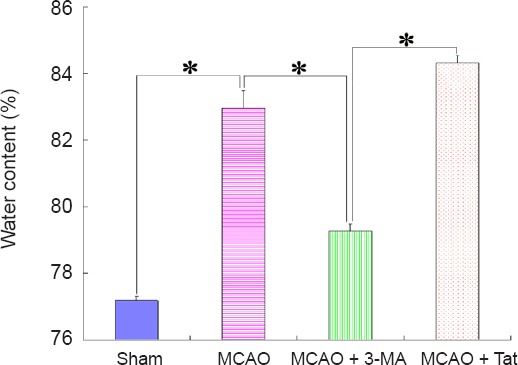
Effect of neuronal autophagy on brain edema in ischemic stroke rats.
To observe the effect of autophagy on ischemia-induced inflammatory edema, brain water content was assessed. The ratio of water content in brain tissues from MCAO + 3-MA group was obviously lower than that from MCAO + Tat or MCAO groups, and was similar to that of the sham group. Data are presented as the mean ± SEM (n = 6; one-way analysis of variance followed by Dunett’s test). *P < 0.01. MCAO: Middle cerebral artery occlusion; 3-MA: 3-methyladenine; Tat: Tat-Beclin 1.
Autophagy induction enlarges brain infarct volume after cerebral stroke
The results described above confirmed that an autophagy inducer could aggravate microglial inflammatory responses. We next investigated the effect of facilitated inflammatory responses on infarct volume by TTC staining (Figure 6A). As shown in Figure 6B, infarct volume was markedly enlarged in the MCAO + Tat group compared with the MCAO group. Comparatively, the infarct volume was reduced after treatment with an autophagy inhibitor in the MCAO + 3-MA group.
Figure 6.
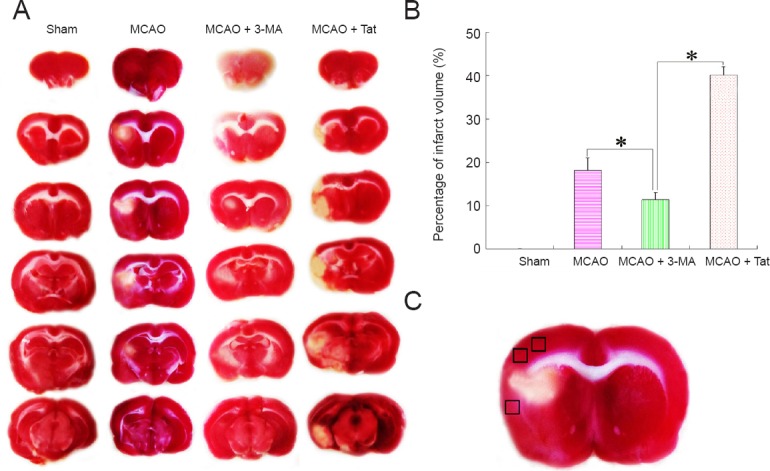
Effect of neuronal autophagy on cerebral infarct volume in rats with ischemic stroke.
(A) To observe the effect of microglial inflammation on cerebral infarct volume after ischemic stroke, TTC staining was performed to assess infarct size. (B) Cerebral infarct volume was obviously reduced after treatment with the autophagy inhibitor of 3-MA compared with MCAO rats, and was similar to that observed in sham animals. In contrast, infarct volume was further expanded after administration with the autophagy inducer Tat. Data are presented as the mean ± SEM (n = 6; one-way analysis of variance followed by Dunett’s test). *P < 0.01. (C) Black squares indicate the penumbral area (derived from the third graph in the MCAO group from A). MCAO: Middle cerebral artery occlusion; 3-MA: 3-methyladenine; Tat: Tat-Beclin 1; TTC: 2,3,5-triphenyltetrazolium chloride.
Discussion
Tissue plasminogen activator is an efficacious drug for ischemic cerebral stroke; however, less than 5% of stroke patients can benefit from this treatment, as a result of safety concerns and a narrow window for delivery within 4.5 hours (Fonarow et al., 2011). Therefore, more practical treatments for ischemic stroke are urgently needed. Identifying pathological mechanisms of cerebral ischemia may provide better insights into stroke treatment (Rana et al., 2018). Most clinical stroke patients exhibit an infraction in the middle cerebral artery (Moskowitz et al., 2010); thus, the MCAO rat model was prepared in the present study. After stroke, cells in the ischemic core rapidly die within several minutes, owing to complete interruption of blood supply. However, in areas localized around the ischemic core (also known as the ischemic penumbra), cells can be partly fueled by the adjacent vascular supply; thus, cell death in the penumbra spreads slowly (Moskowitz et al., 2010) and a large number of cells in the penumbral area undergo autophagy. Because autophagic injury is reversible (Lo, 2008), we investigated the rescue of autophagic cells in ischemic penumbra.
During the acute stage of ischemic stroke, dead and injured neurons are the key stimulus to activate inflammatory responses, as they release so-called “danger signals”, such as the nucleotides UTP and ATP (Kono and Rock, 2008; Gao et al., 2017; Liu et al., 2017). These danger signals activate microglial inflammatory responses through recognizable receptors that are widely expressed on microglia. Once activated, microglial inflammatory responses are amplified to reinforce the expression of adhesion molecules, chemokines, and cytokines that drive more neuronal death (Iadecola and Anrather, 2011). CX3CL1, a chemokine expressed on neurons, can suppress microglial activation by binding with its receptor CX3CR1 on microglia (Cardona et al., 2006). When CX3CL1 expression in autophagic neurons is reduced, subsequent depression of the CX3CL1-CX3CR1 interactions between neurons and microglia consequently weakens the efficacy of neurons to inhibit microglial inflammatory activity. Ultimately, microglial inflammation is excessively activated and may worsen brain damage (Cardona et al., 2006). Our previous study (Deng et al., 2016) and another relevant report (Button et al., 2015) revealed that cerebral stroke often induces excessive neuronal autophagy, resulting in brain injury. Therefore, we proposed that microglial inflammatory responses might be excessively amplified by neuronal autophagy to aggravate brain damage after cerebral stroke. To verify our hypothesis, we focused on the efficacy of neuronal autophagy on microglial inflammation following MCAO in the present study.
First, we observed the dynamic variations of autophagy activity, CX3CL1 expression level, and state of microglial activation in the ischemic penumbra after MCAO/reperfusion. Western blot assays illustrated that ischemia-induced autophagy is negatively correlated with CX3CL1 expression, but positively associated with activation of microglial inflammation. According to the characteristic variations of autophagy following cerebral ischemia, we chose the time point of 12 hours after MCAO/reperfusion to investigate a potential correlation between autophagy and microglial inflammation. CX3CL1 expression levels on neurons in penumbral tissues were obviously decreased after treatment with an autophagy inducer in the MCAO + Tat group compared with the MCAO group. In contrast, Iba-1 and NF-κB expression levels, brain water content, and production of inflammatory molecules were increased, indicating that autophagy induction facilitated MCAO-induced microglial inflammation. Moreover, these results suggested this efficacy might be correlated with a reduction of CX3CL1 expression on autophagic neurons. Furthermore, after administration of an autophagy inducer, the infarct volume was markedly expanded, suggesting that autophagy augmentation led to serious inflammatory injury. In contrast, after treatment with the autophagy inhibitor 3-MA, levels of autophagy were expectantly reduced, whereas CX3CL1 expression on neurons was obviously increased in the MCAO + 3-MA group; moreover, brain injury and microglial inflammatory responses were also alleviated. These results indicated that inhibition of autophagy could attenuate MCAO-stimulated microglial inflammatory injury by promoting CX3CL1 expression.
The main objective of our study was to observe the association between neuronal autophagy and microglial inflammation after cerebral stroke. We found that autophagy induction with Tat-Beclin 1 led to downregulation of CX3CL1 expression on neurons after cerebral ischemia; meanwhile, serious microglial inflammation and brain injury were observed compared with the MCAO group. In contrast, after treatment with the autophagy inhibitor 3-MA, ischemia-induced autophagy activity, microglial inflammation, and brain injury were obviously alleviated; however, the CX3CL1 expression level was contrarily increased. Therefore, we concluded that ischemia-induced neuronal autophagy facilitates microglial inflammatory injury in cerebral stroke, and this efficacy might be associated with downregulation of CX3CL1 expression on autophagic neurons.
Our study preliminarily explores the association between neuronal autophagy and microglial inflammation after cerebral ischemia. However, several issues should be resolved in future research. First, why is CX3CL1 down-regulated on autophagic neurons? Second, as CX3CL1 is partly expressed by astrocytes, are there associations between autophagic astrocytes and microglial inflammation after ischemic stroke? Third, does neuronal autophagy exacerbate microglial inflammation in a rat model of transient cerebral ischemia and does it play a similar role in permanent ischemic stroke? Determining the answers to these queries may identify novel therapeutic targets aimed at autophagy signaling or microglial inflammatory mechanisms that can improve stroke treatment.
Footnotes
Conflicts of interest: There was no any conflict of interest among authors.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 81660383 (to YHD), 81860411 (to HYH); a grant from the Applied Basic Research Projects of Yunnan Province of China, No. 2017FB113 (to YHD); and the Scientific Research Fund of Yunnan Provincial Department of Education of China, No. 2018JS016 (to HYH). The conception, design, execution, and analysis of experiments, as well as the preparation of and decision to publish this manuscript, were made independent of any funding organization.
Institutional review board statement: The study was approved by the Animal Experiment Committee in Kunming University of Science and Technology, China.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Deusen AV, Norman C, Qiu Y, Song LP; T-Editor: Liu XL
Funding: This study was supported by the National Natural Science Foundation of China, No. 81660383 (to YHD), 81860411 (to HYH); a grant from the Applied Basic Research Projects of Yunnan Province of China, No. 2017FB113 (to YHD); and the Scientific Research Fund of Yunnan Provincial Department of Education of China, No. 2018JS016 (to HYH).
References
- 1.Button RW, Luo S, Rubinsztein DC. Autophagic activity in neuronal cell death. Neurosci Bull. 2015;31:382–394. doi: 10.1007/s12264-015-1528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, Sun Y, Liu K, Sun X. Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res. 2014;9:1210–1216. doi: 10.4103/1673-5374.135329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng YH, He HY, Yang LQ, Zhang PY. Dynamic changes in neuronal autophagy and apoptosis in the ischemic penumbra following permanent ischemic stroke. Neural Regen Res. 2016;11:1108–1114. doi: 10.4103/1673-5374.187045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Descloux C, Ginet V, Clarke PG, Puyal J, Truttmann AC. Neuronal death after perinatal cerebral hypoxia-ischemia: Focus on autophagy-mediated cell death. Int J Dev Neurosci. 2015;45:75–85. doi: 10.1016/j.ijdevneu.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 6.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fonarow GC, Smith EE, Saver JL, Reeves MJ, Bhatt DL, Grau-Sepulveda MV, Olson DM, Hernandez AF, Peterson ED, Schwamm LH. Timeliness of tissue-type plasminogen activator therapy in acute ischemic stroke: patient characteristics, hospital factors, and outcomes associated with door-to-needle times within 60 minutes. Circulation. 2011;123:750–758. doi: 10.1161/CIRCULATIONAHA.110.974675. [DOI] [PubMed] [Google Scholar]
- 8.Gao FJ, Gao SE, Chen X, Sun J, Wang JY. Anti-inflammatory effect of stem cells in the treatment of ischemic stroke. Zhongguo Zuzhi Gongcheng Yanjiu. 2017;21:4088–4093. [Google Scholar]
- 9.Gusel’nikova VV, Korzhevskiy DE. NeuN as a neuronal nuclear antigen and neuron differentiation marker. Acta Naturae. 2015;7:42–47. [PMC free article] [PubMed] [Google Scholar]
- 10.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 11.Hass DT, Barnstable CJ. Uncoupling protein 2 in the glial response to stress: implications for neuroprotection. Neural Regen Res. 2016;11:1197–1200. doi: 10.4103/1673-5374.189159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hongyun H, Tao G, Pengyue Z, Liqiang Y, Yihao D. Puerarin provides a neuroprotection against transient cerebral ischemia by attenuating autophagy at the ischemic penumbra in neurons but not in astrocytes. Neurosci Lett. 2017;643:45–51. doi: 10.1016/j.neulet.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. doi: 10.1038/cr.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 16.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohman RA, Rhodes JS. Neurogenesis, inflammation and behavior. Brain Behav Immun. 2013;27:22–32. doi: 10.1016/j.bbi.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korzhevskiy DE, Kirik OV. Cerebral microglia and microglial markers. Morfologiia. 2015;147:37–44. [PubMed] [Google Scholar]
- 20.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu YM, Zhao YY, Chen HB. Neural stem cell transplantation effects on neuronal apoptosis, differentiation and neurobehavior changes in rats with cerebral ischemia. Zhongguo Zuzhi Gongcheng Yanjiu. 2017;21:2029–2035. [Google Scholar]
- 22.Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- 23.Martinez B, Peplow PV. Immunomodulators and microRNAs as neurorestorative therapy for ischemic stroke. Neural Regen Res. 2017;12:865–874. doi: 10.4103/1673-5374.208540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci. 2017;18:E598. doi: 10.3390/ijms18030598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puyal J, Ginet V, Clarke PG. Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol. 2013;105:24–48. doi: 10.1016/j.pneurobio.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Rana DS, Anand I, Batra A, Sethi PK, Bhargava S. Serum levels of high-sensitivity C-reactive protein in acute ischemic stroke and its subtypes: a prospective case-control study. Asia Pac J Clin Trials Nerv Syst Dis. 2018;3:128–135. [Google Scholar]
- 28.Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. 2010;189:925–935. doi: 10.1083/jcb.201002021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheridan GK, Murphy KJ. Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol. 2013;3:130181. doi: 10.1098/rsob.130181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res. 2010;107:1170–1184. doi: 10.1161/CIRCRESAHA.110.223883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su P, Zhang J, Wang D, Zhao F, Cao Z, Aschner M, Luo W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience. 2016;319:155–167. doi: 10.1016/j.neuroscience.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 32.Tremblay MÈ, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker FR, Beynon SB, Jones KA, Zhao Z, Kongsui R, Cairns M, Nilsson M. Dynamic structural remodelling of microglia in health and disease: a review of the models, the signals and the mechanisms. Brain Behav Immun. 2014;37:1–14. doi: 10.1016/j.bbi.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 34.Wang JF, Mei ZG, Fu Y, Yang SB, Zhang SZ, Huang WF, Xiong L, Zhou HJ, Tao W, Feng ZT. Puerarin protects rat brain against schemia/reperfusion injury by suppressing autophagy via the AMPK-mTOR-ULK1 signaling pathway. Neural Regen Res. 2018;13:989–998. doi: 10.4103/1673-5374.233441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Y, Chen S, Zhang Y, Lin X, Song Y, Xue Z, Qian H, Wang S, Wan G, Zheng X, Zhang L. Induction of autophagy by spermidine is neuroprotective via inhibition of caspase 3-mediated Beclin 1 cleavage. Cell Death Dis. 2017;8:e2738. doi: 10.1038/cddis.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]