

Abstract
Histone deacetylase (HDAC) inhibition modulates dendritic cells (DCs) functions and regulates experimental graft-versus-host disease (GVHD) and other immune mediated diseases. The mechanisms by which HDAC inhibition modulates immune responses remain largely unknown. Signal transducer and activator of transcription-3 (STAT-3) is a transcription factor shown to negatively regulate DC functions. Herein we report that HDAC inhibition acetylates and activates STAT-3, which regulates DCs by promoting the transcription of indoleamine 2, 3-dioxygenase (IDO). These findings demonstrate (a) novel functional role for post-translational modification of STAT-3 through acetylation and (b) provide mechanistic insights into HDAC inhibition mediated immuno-regulation by induction of IDO.
Keywords: DCs, HDAC inhibitors, cytokines, IDO
Introduction
Histone deacetylase (HDAC) inhibitors potently modulate experimental graft-versus-host disease (GVHD), allograft rejection and autoimmune diseases (1–10), partly through regulation of dendritic cells (DC)(1, 11–13). The molecular mechanisms underpinning their immunosuppressive effects on DCs are not well-understood.
Signal transducer and activator of transcription-3 (STAT-3) negatively regulates DC functions and its activation requires posttranslational phosphorylation (14, 15), (16, 17). However whether other types of posttranslational modifications are relevant for its function in modulating DC responses is not known (18–20). Emerging data suggest that STATs, including STAT-3 can also be acetylated (21–24), but the functional relevance of acetylation in DC responses remains undefined.
We recently demonstrated that HDAC inhibitors partly modulate DC functions through induction of indoleamine 2,3 dioxygenase (IDO) (1). Seminal studies by Puccetti and colleagues demonstrate that STAT-3 activation induces IDO(25). Because HDAC inhibitors can target non-histone proteins such as STAT-3 (24), we hypothesized that STAT-3 acetylation might be critical for induction of IDO and modulation of DCs. Indeed HDAC inhibition acetylated and activated STAT-3 which was critical for induction of IDO and regulation of DCs.
Methods and Materials
Mice:
C57BL/6 (B6) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and were cared for under the regulations of the University Laboratory Animal Medicine (ULAM) guidelines.
Dendritic Cell Isolation and PC3 cell lines:
To obtain DCs, bone marrow cells from B6 mice were cultured with murine recombinant granulocyte macrophage colony-stimulating factor (GM-CSF; 10 ng/mL; PharMingen, San Diego, CA) and IL-4 (10ng/ml, Peprotech) for 7–8 days and harvested as described before (1). DCs were harvested and positively selected by autoMACS™ Pro Separator (Miltenyi Biotec) for CD11c+ cells. Stable PC3 cell lines expressing pcDNA3 empty vector (STAT3-null), wild-type STAT3 (WT), and Stat3k685R(K685R) were obtained as described previously(24).
Reagents, Treatment and Antibodies
SAHA and ITF2357, both hydroxamic acid containing molecules, were obtained and used as before (see Supplemental Methods)(1). DCs were treated for 14–16 hours with SAHA, ITF 2357, LPS and JSI-124 (Cucurtacin I) as in Supplemental Methods. STAT-3, phosphor-STAT-3 (Tyr705), acetylated-lysine(Cell Signaling), polyclonal IDO (Alexis), acetyl-histone H4 antibody (Upstate), p300 and beta Actin antibodies (Upstate) were used.
ELISAs, RNA Isolation and Reverse Transcriptase (RT)-PCR, Immunoprecipitation (IP), immunoblotting (IB) and Chromatin Immunoprecipitation Assay (ChIP):
PCR of mouse IDO, IP, IB and ChIP were performed as described before (1, 24),(26). Briefly, Concentrations of TNF-α were measured in culture supernatants by ELISA with specific anti-mouse mAb for capture and detection, and the appropriate standards were purchased from Pharmingen ( TNF- α). Total cellular RNA was isolated by using TRIzol reagent (Invitrogen) and then reverse-transcribed (2 μg) using Moloney murine leukemia virus RT (Promega) and random hexamer primers. RT reaction was subjected to PCR analysis using primer pairs specific for mouse IDO ( forward 5’-GAAGGATCCTTGAAGACCAC-3’ and reverse 5’-GAAGCTGCGATTTCCACCAA-3’), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward 5’-AAATCCCATCACCATCTTCC-3’ and reverse 5’-GTCCACCACCCTGTTGCTGC-3’). IP and IB were performed as described before (1, 2). ChIP was performed as described previously, with some modifications(3). Briefly, cells were cross-linked with 1% formaldehyde for 30 min and quenched with glycine. They were then harvested, washed x 2 with cold PBS, resuspended in lysis buffer and sonicated, cooled in ice bath by using a sonic Dimembrator 550 ( Fisher Scientific), followed by centrifugation at 13,000 g for 10 min. One-tenth of the total lysate was used for total genomic DNA as “Input DNA” control. Supernatant was pre-cleared with salmon sperm DNA and BSA coated protein A/G agarose ( Invitrogen) and rabbit IgG ( Santa Cruz Biotechnology) by incubating on a rocker at 4ºC for 1 h. Immunoprecipitation was performed for 15 h at 4ºC with 4 μg each of specific antibodies and control rabbit IgG. Protein A/G were added and incubated at 4ºC for 1 h. Complexes were washed one time in low salt buffer, one time in high salt buffer, and one time in LiCl buffer followed by two washes in TE buffer. Washed complexes were eluted with freshly prepared elution buffer and the Na+ concentration was adjusted to 200 mM by adding NaCl followed by incubation at 55 °C for 3 h and 65°C for 6 h to reverse the formaldehyde cross-linking. DNA fragments were precipitated. For PCR, 2 μl from 30 μl DNA extraction was used. PCR primers correspond to sequences within IDO promoter GAS regions as follows: forward, 5’-CTCCTTTTATGGGTGATTGTTTCC-3’ reverse 5’-GAGAACTCCTAAGTTTATGTCCAC-3’(1).
Generation of reporter constructs, transfection and assessment of promoter activity:
A 1500bp DNA fragment upstream of mouse IDO gene start codon was cloned from mouse small intestine DNA library and the fragment was inserted to luciferase reporter plasmid pGl4.20Luc. Using the Transformer™ site-directed mutagenesis kit various mutants were generated that were transfected into CD11c+ DCs using N-[1-(2,3-Dioleoyloxy)propyl] – N,N,N-trimrtylammonium methysulfate) (see Supplemental Data).
Statistics:
All statistical analyses were performed by using the paired T test.
Results and Discussion
STAT-3 was acetylated and IDO protein was induced in murine BM DCs that were treated with both SAHA and ITF2357 (Figure 1A and 1B). We reasoned that inhibition of HDAC enzymes likely facilitated STAT-3 acetylation by endogenous HAT enzyme, p300 in DCs (24). Accordingly STAT-3 was recovered from the immuno-precipitated p300 prepared from the DCs (Supplemental Figure 1A). Moreover, as shown in Supplemental Figure 1A, STAT-3 formed dimers in DCs treated with HDAC inhibitors but not in the control treated DCs demonstrating that acetylation is associated with dimerization and activation of STAT-3 in DCs (24). These results show that enzymatic activity of p300 in STAT-3-p300 complex was inhibited by HDAC enzymes in untreated DCs and that this inhibition was reversed upon treatment with HDAC inhibitors.
Figure 1:
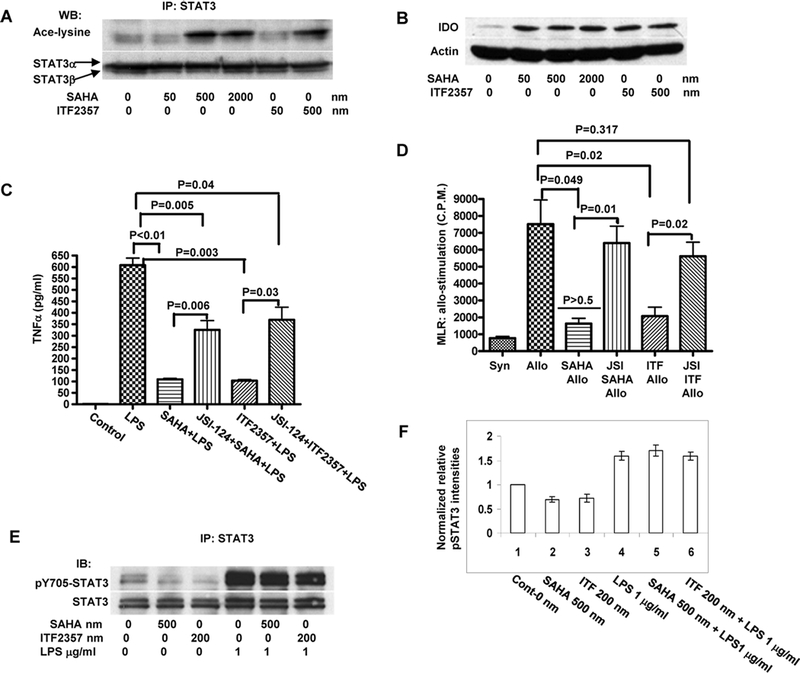
(A) HDAC inhibition acetylates STAT-3: BM DCs were harvested and treated with HDAC inhibitors SAHA and ITF 2357. Whole cell lysates were prepared and immunoprecipitated with anti-Stat3 and then analyzed with antibodies to acetylated lysine. Data shown are from one of 3 similar experiments. (B) HDAC inhibition induces IDO protein: BM DCs were harvested and treated with HDAC inhibitors SAHA and ITF 2357. Cell lysates were analyzed for IDO by Western Blot. Data are from one of two similar experiments. (C) Blockade of STAT-3 signaling with JSI-124 reverses HDAC inhibitor mediated suppression TNFα secretion from BM DCs: BM DCs were pretreated with 5 μM of JSI-124 or diluent for 30 min and then treated with 500 nM of SAHA or 200 nM ITF2357 or H20 for another 12–16 hours. They were then stimulated with 1μg/ml of LPS for 8 hours after which supernatants were harvested and measured for TNFα by ELISA. Results are representative of 3 experiments. (D) JSI-124 reverses the suppression of allogeneic T cell proliferation by the HDAC inhibitor treated BM DCs: BM DCs were pretreated with 5 μM of JSI-124 or diluent and then treated with 500 nM of SAHA or 200 nM ITF2357 or H20 for another 12–16 hours. They were then washed and used as stimulators in allogeneic MLR. T cell proliferation was determined after 72 h of culture. Results are representative of 2 experiments. (E) HDAC inhibitors acetylate but do not enhance phosphorylation of STAT-3. BM DCs were treated with SAHA, ITF 2357, LPS or the diluent control. Whole cell lysates were processed for immunoprecipitation with STAT-3 antibody and then blotted with phosphorylated 705-tyrosine STAT-3 antibody. The data are representative of 2 similar experiments. (F): Quantification of normalized pSTAT-3 levels combined from the above experiments is shown.
We next explored the functional relevance of STAT-3 acetylation in DCs. BM DCs were pretreated with either diluent control or JSI-124, a drug that specifically disrupts STAT-3 DNA complex formation (27). DCs were then conditioned with HDAC inhibitors and thereafter stimulated with LPS for additional 6 hours. SAHA and ITF 2357 markedly reduced LPS induced secretion of TNFα from the DCs (Figure 1C). By contrast, pretreatment with STAT-3 inhibitor JSI-124 abrogated the suppressive effect of HDAC inhibitors on LPS induced TNFα secretion (1). JSI-124 pre-treatment also abrogated HDAC inhibition mediated reduction of allogeneic T cell proliferation by the DCs (Figure 1D).
DCs were next pretreated with JSI-124 or vehicle and then conditioned with HDAC inhibitors alone or with LPS (a potent inducer of STAT-3 phosphorylation). As expected, LPS induced STAT-3 phosphorylation, while treatment with HDAC inhibitors alone did not enhance phosphorylation of STAT-3 (Figure 1E and 1F). Collectively these data show that HDAC inhibitors acetylate and activate STAT-3 without altering its phosphorylation status and that disruption of STAT-3 activity with JSI-124 reverses HDAC inhibition mediated suppression of DC functions.
We had previously demonstrated that HDAC inhibition modulates DC functions, in vivo and in vitro, partly through induction of IDO (1). Because blockade of STAT-3 with JSI-124 mitigated the suppressive effects of HDAC inhibition, we determined whether HDAC inhibition mediated induction of IDO is dependent on STAT-3. Although we have treated the DCs with non-cytotoxic doses of SAHA, it is possible that JSI-124 might be directly cytotoxic either alone or after subsequent treatment with SAHA. To rule out any confounding effects of cytotoxicity, we first evaluated the viability of DCs with annexin staining after treatment with JSI-124 alone or followed by treatment with SAHA. As shown in supplemental Figure 1B, JSI-124 and SAHA treatment did not significantly alter DC viability at the doses that were used. Induction of IDO mRNA by HDAC inhibition was abrogated upon pretreatment with JSI-124 suggesting that STAT-3 is necessary for transcription of IDO (Figure 2A). To confirm a direct role for STAT-3 we utilized TESS promoter analysis software and searched the IDO promoter for the potential STAT binding consensus sequence TTCN3GAA, designated as gamma activated sequences (GAS) (28). We found two such sites upstream of start codon designated as GAS-1 and GAS-2 (Fig. 2B). ChIP assay demonstrated that SAHA induced STAT-3 binding to IDO gene promoter (Fig. 2C). As expected, SAHA also acetylated of histone 4 (H4) at the IDO promoter but STAT-3 and acetylated H4 were not bound to each other (Supplemental Fig. 1C).
Figure 2:
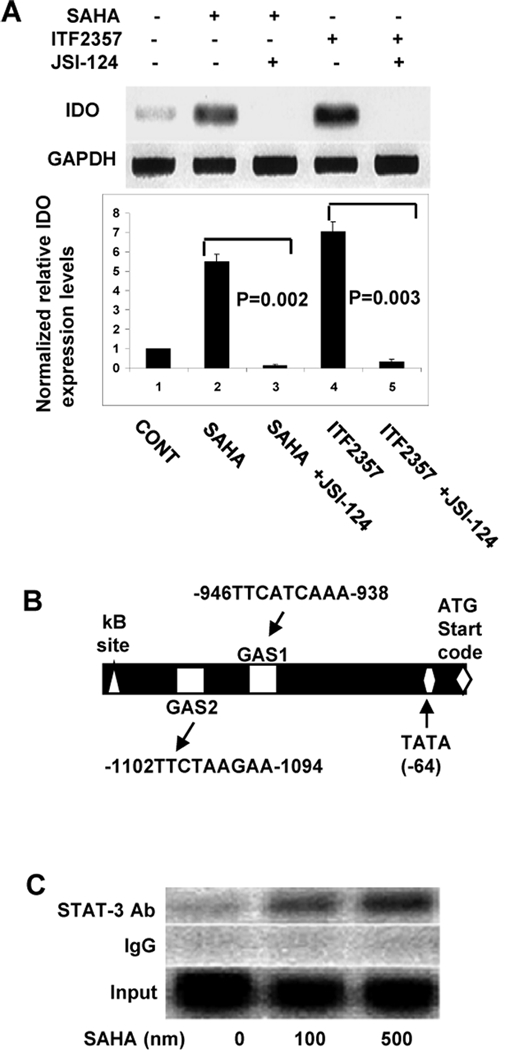
(A) Inhibition of HDAC inhibitor mediated induction of IDO by JSI-124: BM DCs were pretreated with 5 μM of JSI-124 or diluent for 30 min and then treated with 500 nM of SAHA or 200 nM ITF2357 or H20 for another 14–16 hours. Expression of IDO was evaluated with RT-PCR for IDO mRNA. Results combined from three similar representative similar experiments are shown in bar graph. P < 0.05, solid bars vs. open bars (B) IDO promoter analysis: Nucleotide sequence of the 5’ region of the mouse IDO gene (−1500 bp upstream of IDO gene start codon) was obtained from GenBank database and analyzed with TESS promoter analysis software (http://www.cbil.uppen.edu/tess/). STAT binding sites (GAS) and NF-κB binding site (κB site) are shown. (C) STAT-3 binds to IDO promoter: BM DCs were treated with SAHA or diluent for 14–18 hours and assessed for the occupancy of STAT-3 to the IDO GAS regions. DCs were harvested and ChIP assay was performed as described in Materials and Methods. Chromatin complexes were immunoprecipitated with antibodies to STAT-3 or with control rabbit IgG. One-tenth of the total lysates were used as for total genomic DNA as input DNA control.
Next, to directly test whether binding of STAT-3 to IDO promoter is absolutely necessary for HDAC inhibition mediated transcription of IDO we next performed mutagenesis studies. We cloned the IDO promoter and designed site-directed GAS deletion mutants (Supplemental Fig. 1D) that were then inserted it to a pGL4 luciferase reporter vector. As shown in Figure 3A, IDO promoter driven luciferase induction was enhanced several fold upon treatment with SAHA. Although SAHA treatment also enhanced the induction of the reporter genes containing deletions of either GAS-1 (MUT-1) or GAS-2 (MUT-2) sites, it was significantly less than the expression driven by the wild type promoter (Figure 3A). By contrast, constructs with deleted GAS-1 and GAS-2 (MUT-3) were unable to respond to SAHA treatment. Furthermore, induction of luciferase by SAHA was completely blocked after treatment with JSI-124 in the wild type promoter controls (Figure 3A). A similar pattern was observed in cells that were treated with ITF2357 (data not shown) demonstrating a direct requirement for IDO induction by the HDAC inhibitors induced STAT-3 activation and binding to the GAS regions of IDO promoter.
Figure 3.
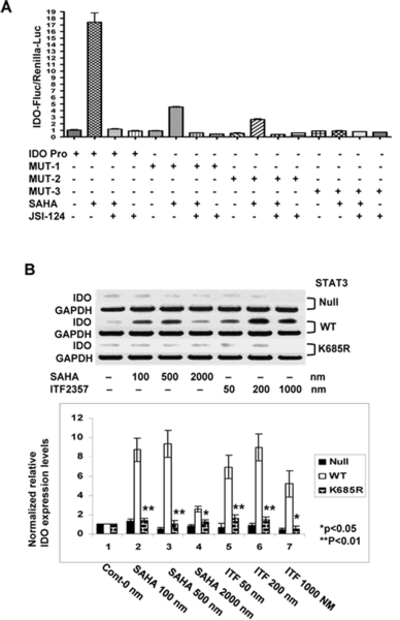
(A): STAT-3 binding and IDO promoter driven luciferase: BM DCs were transfected with different IDO promoter driven luciferase constructs as in Materials and Methods. DCs were treated with JSI-124 and then with SAHA as above. The expression of luciferase was analyzed as in Materials and Methods. The error bars and statistics are representative of the technical replicates from one of 3 similar experiments. (B): STAT-3 acetylation is required for induction of IDO: PC3 cell lines expressing pcDNA3 empty vector (STAT-3 null), wild type STAT-3 (WT) and STAT-3 mutant K685R (K685R) were treated with either SAHA, ITF 2357 or diluent for 12–14 hours. They were then harvested and analyzed for IDO by RT-PCR. Data are from 1 of 2 experiments with similar results. (C): Quantification of normalized IDO levels from the above experiments is shown.solid bars vs. striped bars, P < 0.05.
Functional relevance of STAT-3 activation in DCs has been attributed to its posttranslational phosphorylation and previous reports have suggested that JSI-124 regulates STAT-3 activity by inhibiting phosphorylation (27, 29). By contrast, our data show that the HDAC inhibitors acetylate STAT-3 but do not alter its phosphorylation status (Fig 1E and 1F), induce IDO and modulate the function of DCs, which was blocked by JSI-124, a small molecule that disrupts STAT-3 binding to DNA.
These observations show that HDAC inhibition increased acetylation with no alteration of phosphorylation of STAT-3; but do not show a critical requirement for acetylation of STAT-3 in the induction of IDO. To this end we tested the effects of SAHA and ITF 2357 on the induction of IDO in cell lines expressing pcDNA3 empty vector (STAT-3 null), wild type STAT-3 (WT) and STAT-3 mutant K685R that contains Lys685-to-Arg substitution and therefore cannot be acetylated (K685R)(24). HDAC inhibition enhanced IDO expression in the WT STAT-3 transfected cells but not in the null controls (Figure 3B and 3C). HDAC inhibition also failed to increase IDO expression in the cells transfected with acetylation resistant STAT-3 mutantK685R. These data thus demonstrate a critical role for STAT-3 acetylation in the induction of IDO.
Heretofore the critical pathways responsible for induction of IDO and the relevant post-translational requirements for the function of STAT-3 remained unknown. Herein we demonstrate a novel role for STAT-3 acetylation in the induction of IDO with HDAC inhibition. This, to our knowledge is also the first direct demonstration of a role for acetylation of non-histone proteins in direct regulation of DCs. It is however possible that acetylated STAT-3 might target other genes in addition to IDO, which might contribute either directly or indirectly to the regulation of DC function (17, 30). Furthermore, our data also do not rule out the requirement for phosphorylation of STAT-3 in mediating its other downstream effects in DCs. Future studies will determine the specific gene targets that are dependent on the acetylation or phosphorylation or both modifications of STAT-3 protein regulation DC functions. Although IDO is partly critical for the HDAC inhibition mediated regulation of DCs (1), it remains to be determined whether this alone or other distinct pathways of suppression of co-stimulatory molecules and proinflammatory cytokines (1, 11–13) might act in concert to cause the overall DC suppressive effects of HDAC inhibitors. Further studies will need to be performed to carefully determine the effects of these pathways in the absence of IDO secretion. Nonetheless, together our data demonstrate that post-translational modification of STAT-3 through acetylation is responsible, in part, for the induction of IDO and the immuno-regulatory effects of HDAC inhibitors on DCs.
Supplementary Material
Acknowledgments
Supported by NIH AI-075284 (P.R.), HL-090775 (P.R.), AI-15614 (C.A.D.) and HL-68743 (C.A.D.). P.R. is a recipient of the Alaina J. Enlow Scholar Award and the Doris Duke Clinical Scientist Development Award.
Abbreviations used:
- HDAC
histone deacetylase
- DC
dendritic cells
- BMT
bone marrow transplantation
- STAT
signal transducers and activators of transcription
- IDO
indoleamine 2,3dioxygenase
Footnotes
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party.
The final, citable version of record can be found at www.jimmunol.org
Contributor Information
Yaping Sun, Email: yapings@umich.edu.
Y. Eugene Chin, Email: y_eugene_chin@brown.edu.
Elizabeth Weisiger, Email: eweisige@umich.edu.
Chelsea Malter, Email: cmalter@med.umich.edu.
Isao Tawara, Email: itawara@umich.edu.
Tomomi Toubai, Email: tomomit@umich.edu.
Erin Gatza, Email: egatza@umich.edu.
Paolo Mascagni, Email: P.MASCAGNI@italfarmaco.com.
Charles A. Dinarello, Email: CDinare333@aol.com.
Pavan Reddy, Email: reddypr@umich.edu.
References:
- 1.Reddy P, Sun Y, Toubai T, Duran-Struuck R, Clouthier SG, Weisiger E, Maeda Y, Tawara I, Krijanovski O, Gatza E, Liu C, Malter C, Mascagni P, Dinarello CA, and Ferrara JL. 2008. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest 118:2562–2573. PMC2430497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA, and Ferrara JL. 2004. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci U S A 101:3921–3926. PMC374345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leng C, Gries M, Ziegler J, Lokshin A, Mascagni P, Lentzsch S, and Mapara MY. 2006. Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp Hematol 34:776–787. [DOI] [PubMed] [Google Scholar]
- 4.Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, Wells AD, and Hancock WW. 2007. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 13:1299–1307. [DOI] [PubMed] [Google Scholar]
- 5.Mishra N, Reilly CM, Brown DR, Ruiz P, and Gilkeson GS. 2003. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest 111:539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skov S, Rieneck K, Bovin LF, Skak K, Tomra S, Michelsen BK, and Odum N. 2003. Histone deacetylase inhibitors: a new class of immunosuppressors targeting a novel signal pathway essential for CD154 expression. Blood 101:1430–1438. [DOI] [PubMed] [Google Scholar]
- 7.Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, Dona G, Fossati G, Sozzani S, Azam T, Bufler P, Fantuzzi G, Goncharov I, Kim SH, Pomerantz BJ, Reznikov LL, Siegmund B, Dinarello CA, and Mascagni P. 2002. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A 99:2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, Modena D, Moras ML, Pozzi P, Reznikov LL, Siegmund B, Fantuzzi G, Dinarello CA, and Mascagni P. 2005. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med 11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glauben R, Batra A, Fedke I, Zeitz M, Lehr HA, Leoni F, Mascagni P, Fantuzzi G, Dinarello CA, and Siegmund B. 2006. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol 176:5015–5022. [DOI] [PubMed] [Google Scholar]
- 10.Glauben R, Batra A, Stroh T, Erben U, Fedke I, Lehr HA, Leoni F, Mascagni P, Dinarello CA, Zeitz M, and Siegmund B. 2008. Histone deacetylases: Novel targets for prevention of colitis-associated cancer in mice. Gut 57:613–622. [DOI] [PubMed] [Google Scholar]
- 11.Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D, and Huang Q. 2007. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood 109:1123–1130. [DOI] [PubMed] [Google Scholar]
- 12.Nencioni A, Beck J, Werth D, Grunebach F, Patrone F, Ballestrero A, and Brossart P. 2007. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin Cancer Res 13:3933–3941. [DOI] [PubMed] [Google Scholar]
- 13.Bode KA, Schroder K, Hume DA, Ravasi T, Heeg K, Sweet MJ, and Dalpke AH. 2007. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology 122:596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reich NC 2007. STAT dynamics. Cytokine Growth Factor Rev 18:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray PJ 2007. The JAK-STAT signaling pathway: Input and output integration. J Immunol 178:2623–2629. [DOI] [PubMed] [Google Scholar]
- 16.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, and Yu H. 2004. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 10:48–54. [DOI] [PubMed] [Google Scholar]
- 17.Barton BE 2006. STAT3: A potential therapeutic target in dendritic cells for the induction of transplant tolerance. Expert Opin Ther Targets 10:459–470. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, and Stark GR. 2005. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 65:939–947. [PubMed] [Google Scholar]
- 19.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, and Stark GR. 2007. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev 21:1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, and Stark GR. 2008. Roles of unphosphorylated STATs in signaling. Cell Res 18:443–451. [DOI] [PubMed] [Google Scholar]
- 21.Tang X, Gao JS, Guan YJ, McLane KE, Yuan ZL, Ramratnam B, and Chin YE. 2007. Acetylation-dependent signal transduction for type I interferon receptor. Cell 131:93–105. [DOI] [PubMed] [Google Scholar]
- 22.Nadiminty N, Lou W, Lee SO, Lin X, Trump DL, and Gao AC. 2006. Stat3 activation of NF-{kappa}B p100 processing involves CBP/p300-mediated acetylation. Proc Natl Acad Sci U S A 103:7264–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang R, Cherukuri P, and Luo J. 2005. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem 280:11528–11534. [DOI] [PubMed] [Google Scholar]
- 24.Yuan ZL, Guan YJ, Chatterjee D, and Chin YE. 2005. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307:269–273. [DOI] [PubMed] [Google Scholar]
- 25.Orabona C, Belladonna ML, Vacca C, Bianchi R, Fallarino F, Volpi C, Gizzi S, Fioretti MC, Grohmann U, and Puccetti P. 2005. Cutting edge: silencing suppressor of cytokine signaling 3 expression in dendritic cells turns CD28-Ig from immune adjuvant to suppressant. The journal of immunology 174:6582–6586. [DOI] [PubMed] [Google Scholar]
- 26.Boyd KE, and Farnham PJ. 1999. Coexamination of site-specific transcription factor binding and promoter activity in living cells. Mol Cell Biol 19:8393–8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, and Sebti SM. 2003. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res 63:1270–1279. [PubMed] [Google Scholar]
- 28.Brierley MM, and Fish EN. 2005. Functional relevance of the conserved DNA-binding domain of STAT2. J Biol Chem 280:13029–13036. [DOI] [PubMed] [Google Scholar]
- 29.Brierley MM, and Fish EN. 2005. Stats: Multifaceted regulators of transcription. J Interferon Cytokine Res 25:733–744. [DOI] [PubMed] [Google Scholar]
- 30.Stepkowski SM, Chen W, Ross JA, Nagy ZS, and Kirken RA. 2008. STAT3: An important regulator of multiple cytokine functions. Transplantation 85:1372–1377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.