

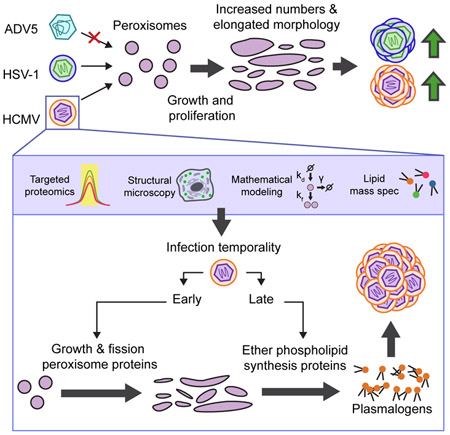
Summary
Viral proteins have evolved to target cellular organelles and usurp their functions for virus replication. Despite the knowledge of these critical functions for several organelles, little is known about peroxisomes during infection. Peroxisomes are primarily metabolic organelles with important functions in lipid metabolism. Here, we discovered that the enveloped viruses human cytomegalovirus (HCMV) and herpes simplex virus (HSV-1) induce the biogenesis of and unique morphological changes to peroxisomes to support their replication. Targeted proteomic quantification revealed a global virus-induced upregulation of peroxisomal proteins. Mathematical modeling and microscopy structural analysis show that infection triggers peroxisome growth and fission, leading to increased peroxisome numbers and irregular disc-like structures. HCMV-induced peroxisome biogenesis increased the phospholipid plasmalogen, thereby enhancing virus production. Peroxisome regulation and dependence were not observed for the non-enveloped adenovirus. Our findings uncover a role of peroxisomes in viral pathogenesis, with likely implications for multiple enveloped viruses.
eTOC
Jean Beltran et al. reveal the virus-induced modulation of peroxisome functions to facilitate viral replication, a mechanism likely relevant across enveloped viruses. Human cytomegalovirus increases peroxisome numbers and proteome abundance, and alters peroxisome shape. This represents a virus strategy to engage peroxisome lipid metabolism for the assembly of infectious particles.
Introduction
Peroxisomes are versatile, multifunctional organelles with essential roles in human health and disease. Their functions include those common across eukaryotes, such as fatty acid oxidation and reactive oxygen species (ROS) detoxification, and those specific to animals, such as ether phospholipid synthesis and innate immune response (Di Cara et al., 2017; Dixit et al., 2010; Wanders and Waterham, 2006). In humans, patients with peroxisome biogenesis disorders (PBD) develop severe or fatal metabolic alterations and developmental defects (Steinberg et al., 2006).
Peroxisomes are highly dynamic as external stimuli or mutations alter their numbers, morphology, composition, and function. A set of cellular proteins, known as peroxins (PEXs), exert control over peroxisome dynamics (as reviewed by (Smith and Aitchison, 2013)). Peroxisome proliferation can occur via de novo biogenesis from ER membranes or growth/fission of pre-existing peroxisomes. While the relative contribution of these processes has been long debated, both processes require growth of membranes and targeting of peroxisome membrane proteins, and are facilitated by PEX3, PEX16, and PEX19. Peroxisome matrix proteins bind to PEX5 and PEX7 in the cytosol, facilitating their peroxisome docking and import through channels formed by PEX13 and PEX14. Additionally, peroxisome fission is initiated by a complex formed by PEX11 (isoforms α/β/γ), MFF, FIS1, and DRP1. The balance of these processes is crucial for correct peroxisome function and maintenance, and for cellular response to environmental stress (Schrader and Fahimi, 2006).
Although peroxisome biology is understudied in the context of infection, accumulating evidences reveal an important interplay between peroxisomes and diverse viral families. So far, peroxisomes have been primarily associated with host antiviral innate immunity through the peroxisome-localized MAVS in reovirus, vesicular stomatitis virus (VSV), and influenza virus infections (Dixit et al., 2010). Several viruses are known to actively inhibit peroxisome antiviral functions. Two flaviviruses, West Nile and dengue, decrease peroxisome numbers (You et al., 2015), while human cytomegalovirus (HCMV) inhibits the peroxisomal MAVS (Magalhães et al., 2016). However, given the critical metabolic roles of peroxisomes, it is likely that the contribution to innate immunity is only one facet of its functions during infection. Whether viruses target peroxisome metabolic functions to favor viral replication remains largely unexplored, but several findings support this possibility. For example, HCMV triggers an increase in the abundance of peroxisome proteins (Jean Beltran et al., 2016). Similarly, Kaposi’s Sarcoma associated herpesvirus increases peroxisome abundance and regulates peroxisome lipid metabolism during latency (Sychev et al., 2017), while influenza virus increases ether phospholipid production via peroxisomes (Tanner et al., 2014). The metabolic and signaling roles of peroxisomes, combined with their capability for morphological and functional adaptation, led us to predict that peroxisomes are important mediators of infection and can be coopted by viruses for viral replication and spread.
HCMV infection provides an important model system to study peroxisome functions, as it causes a global rewiring of organelle composition and structure (Jean Beltran et al., 2016). HCMV is a world-wide spread virus, with 50% to >90% seroprevalence, and a significant cause of birth defects and disease in immunocompromised patients (Manicklal et al., 2013). HCMV infection modulates cellular metabolic and stress processes that are associated with peroxisomes. For instance, HCMV alters lipid metabolism and incorporates ether phospholipids (e.g., plasmalogens) in virions (Liu et al., 2011). Peroxisomes catalyze the first two committed steps for the synthesis of plasmalogens (Smith and Aitchison, 2013). HCMV also generates an anti-ROS environment to prevent ROS toxicity from the upregulated cellular metabolism (Tilton et al., 2011). Peroxisomes contain important ROS detoxifying enzymes, such as catalase, PMP20, and GSTK1 (Schrader and Fahimi, 2006). Furthermore, we observed increased abundance of peroxisome proteins (Jean Beltran et al., 2016). From this evidence, an attractive hypothesis is that HCMV modulates peroxisome metabolic functions to support production of virus components (e.g, lipids) or an environment amenable for virus replication (e.g., ROS detoxification).
Here, we discover that HCMV increases peroxisome biogenesis and hijacks peroxisome metabolic functions to facilitate its replication and spread. The broader impact of peroxisome functions on enveloped viruses is highlighted by our finding that peroxisomes are similarly modulated and needed for another herpesvirus, herpes simplex virus type 1 (HSV-1), but not for the non-enveloped virus, adenovirus type 5 (ADV5). Using primary human fibroblasts and clinical PBD patient cells, we demonstrate that viral-induced peroxisome biogenesis and morphological changes are associated with increased plasmalogen levels required for efficient HCMV virion assembly. Therefore, our study establishes peroxisomes as key targets of viral manipulation and as proviral organelles during herpesvirus infection. Our results have implications for long-standing questions in peroxisome biology, such as the mechanism of peroxisome biogenesis and the relation between peroxisome morphology and function.
Results
HCMV infection temporally upregulates proteins that function in peroxisome biogenesis and lipid metabolism
We previously observed an increase in the average abundance of peroxisome proteins during HCMV infection (Fig S1A) (Jean Beltran et al., 2016). Since peroxisomes have been understudied during infection, we aimed to monitor different peroxisome functions and their temporality during the progression of HCMV infection. We designed a targeted mass spectrometry (MS) assay to obtain a comprehensive quantification of peroxisome proteins and gain insight into specific pathways regulated (Fig 1A). From public databases, literature mining, and recent proteomic datasets for peroxisome localization, we confirmed experimental evidence for 83 proteins being peroxisomal in humans. To quantify these proteins in primary human fibroblasts (HFs), a model system for cellular HCMV replication, we constructed a spectral library of peroxisome peptides for parallel reaction monitoring (PRM) MS analysis (see STAR Methods). A total of 60 proteins were found suitable for MS-based quantification in HFs, while the remaining 23 included tissue-specific proteins (e.g., PEX11A, PEX11G), low abundance proteins, or proteins that did not generate detectable signature peptides. Importantly, our assay monitors proteins from all primary peroxisome functions (Fig S1B). In addition, we observed high correlation between peptides within proteins (average Pearson’s coefficient of 0.85) and reproducibility across three biological replicates (average coefficient of variation of 0.15) (Fig S1C).
Figure 1. HCMV infection induces a temporal upregulation of the peroxisome proteome.

A) Workflow for targeted MS-based quantification of peroxisome proteins during infection.
B) Heatmap of peroxisome protein abundances throughout HCMV infection. Functional groups (left) and log2 abundances (right) for columns. Rows sorted by hierarchical clustering. See also Fig S1 and Table S1.
C) Hierarchical clustering of peroxisome proteins show 4 different expression kinetics.
D) Schematic of temporal profiles for proteins associated with peroxisome biogenesis and metabolism processes. Line-graphs show protein abundance over time for each functional group (hpi; hours post infection). See also Fig S2.
We used the developed PRM assay to quantify peroxisome proteins throughout the time course of HCMV infection. HCMV replication progresses through a temporal cascade of viral gene expression defined by the immediate early (IE), delayed early (DE), and late (L) gene classes. These stages of infection are represented in our experiment by the time points 6 (IE), 24 (DE), and 48 to 120 (L) hours post infection (hpi). The temporality of infection was confirmed by monitoring the viral proteins IE1 (IE), pUL26 (DE), and pp65 (L) (Fig S1D). PRM quantification of peroxisome proteins showed an overall increase in peroxisome protein abundance (Fig 1B), supporting our previous observation. Most of the peroxisome proteins (52 out of 60) showed a statistically significant association between abundance and infection (Table S1). Since the increase was observed starting at 24hpi (Fig 1B, S1E), this data suggests that DE viral genes are responsible for the regulation of peroxisome proteins. To better distinguish whether DE or L viral genes modulate peroxisome proteins, we tested infection with UV-irradiated virus, unable to express viral genes (Fig S2A), and infection in the presence of phosphonoformate (PFA), an inhibitor of viral DNA replication. Infected cells treated with PFA severely reduce L viral gene expression (Fig S2A). PRM analysis of these samples showed that increased peroxisome protein abundance requires viral gene expression, but does not require L gene expression (Fig S2B). Therefore, the expression of IE or DE HCMV genes triggers a robust increase in the abundance of the peroxisome proteome, starting at early stages of infection (24 hpi) and continuing to increase towards late stages of infection (72 and 120 hpi).
We next used hierarchical clustering to uncover distinct temporal profiles of peroxisome protein regulation (Fig 1C). This temporality was related to the various functions of peroxisomes (Fig 1D, S2C). We found that proteins involved in early steps of peroxisome biogenesis (i.e., membrane assembly and cargo docking) are already 2-fold upregulated by 48 hpi, while proteins with functions in lipid metabolism are upregulated 2-fold at 72 hpi. Among those showing the largest fold-changes were peroxisome biogenesis proteins involved in peroxisome membrane assembly (PEX3 and PEX16) and those involved in import to the peroxisome matrix (PEX13 and PEX14). Furthermore, of particular interest was the upregulation of ether lipid and plasmalogen synthesis proteins at late stages of infection; ether lipid synthesis is a unique pathway of peroxisomes, and plasmalogens are enriched in viral particles (Liu et al., 2011). Therefore, these proteomic results led us to hypothesize that upon HCMV infection, peroxisomes first undergo a stage of increased biogenesis, followed by altered peroxisome lipid metabolism to support either host defense or the formation of new viral particles.
Peroxisome numbers are increased late in infection, and this is a common feature of infections with enveloped viruses HCMV and HSV-1
As peroxisome biogenesis proteins were among the most upregulated, we asked whether HCMV infection elevates peroxisome numbers. We measured peroxisome numbers by immunofluorescence (IF) microscopy and 3D reconstruction of peroxisome structures (Fig 2A, Movie S1). Supporting our proteomic results, peroxisome numbers per cell increased by 3.6-fold at 96 hpi (Fig 2B, 2C). Since infected cells increase in size, we also calculated the peroxisome concentration as numbers per area, observing a 2.6-fold increase at 96hpi (Fig S3A). Other studies have shown that a 30% decrease in peroxisome numbers is associated with significant neuronal cell death in mice, and a 25% increase is associated with altered peroxisome metabolism during osteoblast differentiation (Ahlemeyer et al., 2012; Qian et al., 2015). Therefore, the large increase caused by HCMV infection is likely to have a significant impact on cell function and metabolism. In agreement with our PRM analyses (Fig S2B), the increase in peroxisome numbers was still evident following PFA treatment, but not when using an UV-irradiated virus (Fig 2D and S3B). The combined microscopy and proteomic results show an upregulation of peroxisome biogenesis caused by HCMV gene expression, likely from the DE class.
Figure 2. HCMV and HSV-1 increase peroxisome numbers late in infection.

A) Workflow for the analysis of peroxisome numbers and structure during viral infection.
B) IF analysis of peroxisomes (PEX14) in HFs infected by HCMV AD169-GFP (96 hpi). GFP and pUL99 used as markers for infection. Individual cells delineated in yellow. Uninfected (right) and infected (left, pUL99/GFP positive) cells are shown.
C) Peroxisome numbers in HCMV-infected HFs. N > 22 cells.
D) Peroxisome numbers in PFA-treated HCMV-infected HFs and HFs infected with UV-treated HCMV. N > 19 cells. See also Fig S3.
E) IF analysis of peroxisomes in HSV1-infected HFs at 16 hpi. Uninfected cell (left) and infected cell (right, ICP0 positive marker of infection) can be observed.
F) Number of peroxisomes in HSV1-infected HFs. N > 17 cells.
G) IF analysis of peroxisomes in HFs infected with ADV5-GFP at 20 hpi. Two uninfected cells (left) and one infected cell (right, GFP positive) can be observed.
H) Number of peroxisomes in ADV5-infected HFs. N > 18 cells.
Scale bars = 10μm. Significance determined by one-way ANOVA with a Dunnett’s post-test (C, D, F, H), marked with (*) for P<0.05 and (**) for P<0.01.
Given the critical functions of peroxisomes in lipid metabolism, regulation of peroxisome biogenesis may be a common feature of several viruses, particularly enveloped viruses that rely on lipids to form viral envelopes. Therefore, we investigated peroxisomes in HFs infected with herpes simplex virus 1 (HSV-1), another enveloped virus, or adenovirus type 5 (ADV5), a non-enveloped virus. An increase in peroxisome numbers was observed late in HSV-1 infection (16 hpi), with a magnitude strikingly similar to HCMV infection (Fig 2E, F). In contrast, peroxisome numbers showed a modest increase early in ADV5 infection (4 and 12 hpi) and no significant increase late in infection (20 hpi) (Fig 2G, H). Our results demonstrate a common upregulation of peroxisome numbers by HCMV and HSV-1, suggesting a role for peroxisomes during infection with enveloped herpesviruses.
Peroxisomes are required for efficient HCMV and HSV-1 replication
Increased peroxisome biogenesis can have cellular effects that either benefit host defense (i.e., restricting metabolism or inducing immune response) or promote viral replication (i.e., producing lipids or protecting against oxidative species). To test the role of peroxisomes in the outcome of infection, we induced peroxisome biogenesis in HFs using 4-phenylbutyrate (4-PBA) (Kemp et al., 1998) and infected them with HCMV, HSV-1, or ADV5 (Fig 3A, B). The increased peroxisome numbers enhanced the replication and production of HCMV and HSV-1 infectious particles, but not for ADV5. HCMV displayed a striking 7-fold increase in virus produced, prompting us to ask which steps of peroxisome biogenesis are important for virus replication. We generated a panel of knockout (KO) cell lines using CRISPR/Cas9 in HFs, targeting genes involved in peroxisome membrane assembly (PEX3 and PEX19), matrix import (PEX7), and fission (PEX11B) (Fig 3C). PEX3 and PEX19 KO cells are devoid of peroxisomes because peroxisome membrane assembly is stopped, while PEX11B KO produces enlarged and elongated peroxisomes because of hindered fission but not growth (Fig 3D). These phenotypes agree with previous reports in mice and humans (Li et al., 2002; Muntau et al., 2000). Furthermore, knockout efficiency was validated by PRM (Fig 3E), and these cells showed good viability (Fig S4A, B). We found that PEX3 and PEX19 KO cells, devoid of peroxisomes, were defective in replicating HCMV (Fig 3F). PEX7 KOs also showed reduced viral output, indicating that functional peroxisome matrix is required for HCMV replication. Surprisingly, PEX11B KOs with enlarged peroxisomes did not inhibit HCMV, and even showed an increase in virus produced.
Figure 3. Peroxisomes are required for efficient HCMV replication.

A) Peroxisome counts in control and 4-PBA treated cells. N = 30 cells.
B) HCMV, HSV-1, and ADV5 infectious units produced from infected cells following 4-PBA treatment to induce peroxisome biogenesis. N = 4 biological replicates.
C) Diagram of proteins involved in different processes of the peroxisome biogenesis pathway.
D) Representative images of peroxisome morphology (anti-PEX14) in PEX3, PEX19, and PEX11B knockout (KO) cells and negative control CRISPR cells. Note the absence of peroxisomes in PEX3 KO and PEX19 KO cells, and weak PEX14 signal observed at endomembranes.
E) Confirmation of CRISPR-mediated KO of PEX3, PEX19, PEX11B in HFs. Protein abundance measured by PRM relative to negative control cells.
F) HCMV virus produced in PEX3, PEX19, PEX11B, and PEX7 KO HFs relative to control (120 hpi). N> 3 biological replicates.
G) HCMV virus titers from supernatant or cell-associated (120 hpi) cell cultures of normal human donor cells (NHF) or fibroblasts from patients with peroxisome biogenesis disorders (GM16513, GM07371, and GM16866).
Scale bars = 10μm. Significance determined by Mann-Whitney test (A, B) and one-way ANOVA with a Dunnett’s post-test relative to control cells (E, F),marked with (*) for P<0.05, (**) for P<0.01, and (***) for P<0.001.
To demonstrate the relevance of peroxisome functions in a clinical model, we tested viral replication in primary skin fibroblasts from PBD patients. The cell lines originate from patients with mutations in PEX1 (GM16513) (Collins and Gould, 1999) and PEX26 (GM16866 and GM07371) (Matsumoto et al., 2003). GM16513 and GM07371 cells showed a 2-3 fold reduction in HCMV virus production relative to normal donor cells (NHF), while GM16866 showed a severe defect (Fig 3G). By measuring cell-associated virus, we observed that this decrease is not due to a defect in virus release, but at the intracellular stages (Fig 3G). While some PEX proteins, such as PEX3, can participate in functions outside peroxisomes (Lee et al., 2017), the use of a panel of CRISPR KOs and samples from human PBD patients having mutations in different PEX genes show that peroxisome deficiency leads to reduced HCMV replication. Altogether, these results show that efficient HCMV replication requires functional peroxisomes and that viral output is a function of peroxisome numbers.
HCMV induces peroxisome biogenesis through growth and fission of pre-existing peroxisomes
We next sought to determine how peroxisome proliferation occurs during HCMV infection. Peroxisome numbers are controlled by three processes and their associated rates, de novo biogenesis from ER membranes (kd), growth followed by fission of pre-existing peroxisomes (kf), and pexophagy (γ) (Fig 4A). Whether kd or kf dominates during proliferation is a long-standing debate (Hua and Kim, 2016). Experimentally, it is difficult to distinguish these two processes, as proteins involved in early de novo biogenesis (e.g., PEX3) are also part of the machinery needed for membrane growth prior to fission. Moreover, knockout of fission factors (e.g., PEX11B), exclusively involved in fission, does not stop pre-existing peroxisomes from growing (Fig 3D). Therefore, we developed a biochemical stochastic model and inference procedure to estimate peroxisome biogenesis rates without perturbing the peroxisome biogenesis system (Galitzine et al., 2018). Our method uses time-lapse peroxisome count data obtained by live microscopy and a Bayesian inference approach to estimate kd, kf, and γ (Fig 4B). We now expanded our method to monitor peroxisome time evolution in primary fibroblasts (Fig 4C, see STAR Methods). Our parameter estimates show that kf dominates over kd as each growth/fission event is ~10- to 20-fold more frequent than formation of a peroxisome de novo (Fig 4D and Table S2). These inference results suggest that peroxisome abundance in uninfected HFs is primarily controlled by growth and fission of pre-existing peroxisomes, and it is likely the process used for proliferation during HCMV infection.
Figure 4. Mathematical modeling and localization of MFF and FIS1 show peroxisome proliferation primarily occurs via growth/fission of pre-existing peroxisomes.

A) Schematics of processes regulating peroxisome numbers.
B) Experimental workflow for peroxisome counting (n) over time (t) used in the inference of peroxisome biogenesis rates. See also START Methods.
C) Traces of peroxisome counts obtained by long-term live microscopy of EGFP-PTS1. N = 20 cells for replicate 1, 22 for replicate 2, and 13 for replicate 3.
D) Posterior distributions of inferred rates kd, kf, and γ. Peroxisome rates kf and γ at the cell level depend on peroxisome numbers (N) and are shown as products to mean N for comparison to kd. See also Table S2.
E and F) Mock (top) and infected (120 hpi, lower) HFs labelled with EGFP-PTS1 (peroxisomes), mito-BFP (mitochondria), and anti-MFF (E) or anti-FIS1 (F). Left, Maximum projections of the whole cell (top) and region of interest (ROI, lower white box). Right, single-channel images from the ROI depicting peroxisomes, mitochondria, and MFF (E) or FIS1 (F). Arrowheads show peroxisomes colocalized with MFF or FIS1. Scale bars = 10μm.
G) Average Pearson’s correlation coefficients for peroxisomes and mitochondria with MFF (top) and FIS1 (lower) throughout HCMV infection. For MFF, N = 41 cells in mock, 11 in 24 hpi, 9 in 72 hpi, and 9 in 120 hpi. For FIS1, N = 35 cells in mock, 31 in 24 hpi, 9 in 72 hpi, and 11 in 120 hpi. Significance determined by Student’s t-test, marked with (*) for P<0.05, (**) for P<0.01, and (***) for P<0.001.
To further support our inference results, we investigated peroxisome fission components during infection. The fission proteins MFF and FIS1 are shared between peroxisomes and mitochondria and are primarily localized to mitochondria (Gandre-Babbe and Bliek, 2008). Indeed, in uninfected HFs, MFF and FIS1 primarily marked mitochondrial ends and constrictions, while rarely localizing to peroxisomes (Fig 4E-G; Mock). Therefore, an increase in their abundance does not necessarily indicate a specific increase in peroxisome fission. If peroxisome growth and fission is upregulated upon infection, we would expect enhanced association of fission factors with peroxisomes. We observed a striking increase in co-localization of MFF and FIS1 with peroxisomes at 120 hpi, while co-localization with mitochondria did not change (Fig 4E-G). Although the inference and microscopy results support the use of growth/fission, they do not rule out that decreased pexophagy is leading to an increase in peroxisome numbers upon infection. Therefore, we measured localization of the peroxisome-specific autophagy factor NBR1, and observed no significant change in NBR1-positive peroxisomes upon infection (Fig S5). Altogether, our results demonstrate that HCMV infection causes an increased association of the fission machinery with peroxisomes, and further supports a model in which HCMV-induced peroxisome proliferation occurs through increased growth and fission of pre-existing peroxisomes.
HCMV infection alters peroxisome morphology to increase the peroxisome membrane-to-lumen ratio
Our findings that elongated peroxisomes (caused by PEX11B KO) seem to benefit HCMV replication (Fig 3) and that growth/fission from pre-existing peroxisomes is a predominant process in HFs (Fig 4) led us to investigate the morphology of peroxisomes during infection. In uninfected HFs, peroxisomes appear spherical and consistent in size and shape throughout the cell (Fig 5A, S6A, Movie S2). However, at 120 hpi, we observed both highly enlarged and slightly smaller peroxisomes (Fig 5A, S6B, Movie S3). Structural analysis of individual peroxisomes revealed that the peroxisome size distribution, measured in both surface area and volume, becomes polarized late in infection (Fig 5B). However, cell-averaged peroxisome length, surface area, and volume increased significantly at 120 hpi (Fig 5C). Therefore, in addition to the increase in peroxisome numbers, the average size of peroxisomes increases.
Figure 5. HCMV infection alters peroxisome morphology to increase the membrane-to-lumen ratio.

A) Mock (top) and HCMV-infected (120 hpi, lower) wild type (WT) cells labelled with anti-PEX14. ROI squares (left) show z-stack maximum projection, a single slice, and masks of peroxisomes from object analysis. Small peroxisomes (arrows) and enlarged peroxisomes (arrowheads) indicated. See also Fig S6.
B) Analysis of peroxisome size distribution in mock and HCMV-infected (120 hpi) cells. Peroxisome surface area (SA) and volume (VOL) were quantified and binned into small (SA: <0.9 μm2; VOL: <0.06 μm3), medium (SA: 1.1-3.3 μm2; VOL: 0.07-0.34 μm3), and large (SA: >3.3 μm2; >0.34 μm3) categories. N = 23868 peroxisomes in mock; N = 33958 in HCMV.
C) Average peroxisome morphology per cell in mock and HCMV-infected (120 hpi) WT and PEX11B KO conditions. N = 70 cells in WT Mock; N = 25 in WT 120 hpi; N = 18 in PEX11B KO Mock; N = 19 in Pex11B KO Mock; N = 21 in Pex11B KO 120 hpi.
D) Peroxisome morphology changes during HCMV infection. Examples of peroxisome masks from 3D-analysis are shown, with SA and VOL values indicated.
E) Theoretical diagram of WT peroxisome shape based on SA:V. Average SA:V of peroxisomes in each WT condition is specified.
F) Surface area to volume ratio (SA:V) regression curves from data in C. Plotted in the physiologically relevant range of observed mock peroxisome volumes (1-5μm3). The 95% confidence interval is shown (shaded region). See also Fig S6.
G) PEX11B KO cells in mock and infected conditions, as in A.
H) Analysis of peroxisome size distribution in mock and HCMV-infected PEX11B KO cells, as in B. N = 7391 in PEX11B KO Mock; N = 16708 in PEX11B KO 120 hpi.
Scale bars = 10μm. Significance of P<0.001 determined by Chi Squared test for B, H. Significance by Student’s t-test for C, marked with (*) for P<0.05, (**) for P<0.01, and (***) for P<0.001.
We also noted that peroxisomes at 120 hpi exhibited an irregular, flattened morphology, which was especially apparent on enlarged peroxisomes (Fig 5A, 5D, Movie S3). Peroxisomes, like cells and other organelles, can adapt their shapes to increase surface area, enhancing the efficiency of processes associated with their membranes. To investigate if these morphological alterations complement the expansion of peroxisome membranes, we calculated the surface area-to-volume ratio (SA:V) of each peroxisome. The SA:V of a geometric object with a set shape is inversely proportional to its size, while irregular objects (e.g., tubules) have higher SA:V relative to regular objects of similar volumes (e.g., spheres). Peroxisomes with irregular morphology would have a greater SA:V than those with spherical structure. HCMV infection caused a significant increase in the average peroxisome SA:V (Fig 5E). We next fitted a quadratic curve to this data, showing that SA:V at 120 hpi is higher at larger volumes (Fig 5F, S7). Therefore, HCMV-infected cells generate enlarged peroxisomes with a higher membrane-to-lumen ratio through induction of an irregular, flattened morphology.
As the irregular peroxisome population at 120 hpi (Fig 5A) was reminiscent of the elongated peroxisomes in PEX11B KO cells (Fig 3D), we asked if the enlarged peroxisome phenotype was due to perturbations of peroxisome fission. Peroxisomes from uninfected PEX11B KO cells exhibited an elongated morphology that varied in size but remained tubular in shape (Fig 5G, upper). In contrast, peroxisomes from 120 hpi PEX11B KO cells were asymmetrical and flattened, with both small and large peroxisomes present (Fig 5G, lower). These morphologies are similar to peroxisomes in wild type (WT) HFs at 120 hpi (Fig 5A). Accordingly, PEX11B KO peroxisomes exhibited a polarized size distribution to larger and smaller peroxisomes at 120 hpi compared to uninfected (Fig 5H), similar to 120 hpi WT cells (Fig 5B), and the average peroxisome dimensions were significantly increased compared to uninfected WT cells (Fig 5C). Moreover, the SA:V curve of peroxisomes in 120 hpi PEX11B KO cells was more similar to 120 hpi WT than to uninfected PEX11B KO cells (Fig 5F). Therefore, while knockout of PEX11B is sufficient to induce an elongated, cylindrical phenotype, HCMV infection triggers additional peroxisome changes leading to an irregular, disc-like morphology. Interestingly, PEX11B KO slightly promoted HCMV replication (Fig 3F), which may show that the elongated peroxisomes in PEX11B KOs are already primed for the structural changes induced by HCMV infection. Taken together, these results show that HCMV causes an altered peroxisome morphology, independent of peroxisome fission, to enhance the membrane-to-lumen ratio, which in turn may enhance membrane-related functions that facilitate viral replication.
Alterations in peroxisome morphology are similar for HCMV and HSV-1 infection, but not for ADV5 infection
Since peroxisome numbers increase upon, and are necessary for, both HCMV and HSV-1 infections, but not in ADV5, we investigated whether peroxisome structures also differ in HSV-1 and ADV5 infections (Fig 6, S7). Late in HSV-1 infection (16 hpi), we observed both highly enlarged and slightly smaller peroxisomes (Fig 6A, S7), like those in HCMV infection. Also, the peroxisome size distribution was polarized (Fig 6B), and the average peroxisome length and surface area significantly increased (Fig 6C). In agreement to an irregular elongated morphology, the peroxisome SA:V was enhanced at 16hpi compared to mock, indicating increased membrane-to-lumen ratio (Fig 6D). In contrast, peroxisomes at both early and late stages of ADV5 infection did not exhibit significant changes in morphology (Fig 6C, E). Our findings reveal commonalities in the regulation of peroxisome morphology between the enveloped viruses, HCMV and HSV-1, but not by the non-enveloped virus, ADV5 (Fig 6F). The higher membrane-to-lumen ratio may thus influence peroxisome functions that benefit both HCMV and HSV-1 replications.
Figure 6. Peroxisome morphology is altered by HSV-1, but not ADV5 infection.

A) Mock (top) and late HSV-1 infected (16 hpi, lower) cells as described in Fig 5A. Small peroxisomes (arrows) and enlarged peroxisomes (arrowheads) are indicated. See also Fig S7.
B) Analysis of peroxisome size distribution in mock and late HSV-1 infection (16 hpi), as in Fig 5B. Mean peroxisome length (L) is included, with small <0.24μm, medium = 0.25-0.5μm, and large >0.5μm. N = 23868 peroxisomes in mock; N = 24417 in HSV-1 16 hpi. Significance was *** by the Chi Squared test.
C) Average peroxisome morphology per cell in mock, HSV-1 (16 hpi), and ADV5 (4 hpi, 20 hpi) infected conditions. N = 70 cells in Mock; N = 28 in HSV-1 16 hpi; N = 19 in ADV5 4 hpi; N = 10 in ADV5 20 hpi. Significance determined by Student’s t-test.
D) Surface area to volume ratio (SA:V) regression curves from data in C of peroxisomes in mock and late HSV-1 infection, as in Fig 5F.
E) Cells at early (4 hpi, top), and late (20 hpi, lower) stages of ADV5 infection, imaged at 60X. Infection was confirmed by infection with GFP-ADV5.
F) The enveloped viruses HCMV and HSV-1 cause an enlarged, flattened peroxisome morphology during infection, while the non-enveloped virus ADV5 does not alter peroxisome structure. Scale bars = 10μm. Statistical significance (*) for P<0.05, (**) for P<0.01, and (***) for P<0.001.
Increased peroxisome biogenesis enhances plasmalogen synthesis to facilitate virus replication
We next investigated how increased peroxisome biogenesis and altered morphology support virus production. Higher peroxisome numbers have been linked to increased plasmalogen abundance (Braverman and Moser, 2012). Plasmalogens are a select class of phospholipids containing a vinyl-ether bond at the sn-1 position. The peroxisome enzymes, GNPAT and AGPS, catalyze the first two committed steps for plasmalogen synthesis producing 1-0-alkyl-DHAP, the plasmalogen precursor containing an ether bonded alkyl group at the sn-1 position (Braverman and Moser, 2012) (Fig 7A). This precursor is transported to the ER where it undergoes addition of the sn-2 acyl group, head group, and other modifications. Plasmalogens provide unique physical and chemical properties for cell membrane functions. These include decreased membrane fluidity and formation of non-bilayer phases at lower temperatures, producing more rigid cell membranes and facilitating membrane fission and fusion events (Lohner et al., 1991; Phuyal et al., 2015). Plasmalogens are enriched in HCMV viral membranes (Liu et al., 2011), but their regulation and function remain unknown.
Figure 7. Plasmalogen abundance increases upon infection, and plasmalogen synthesis is required for HCMV replication.

A) Schematic of plasmalogen synthesis in peroxisomes and ER. The carbon chain at the sn-1 position (R1, red) is added in peroxisomes, while the sn-2 chain (R2, blue) is added in the ER.
B) LC-MS quantification of phosphatidylethanolamine (PE) plasmalogens. Abundances normalized to PE 17:0/17:0 internal standards are shown as HCMV 120 hpi relative to mock. Significance between infected and mock assessed by Student’s t-test followed by Bonferroni correction. N = 3 biological replicates. See also Table S3.
C) LC-MS quantification of PE plasmalogens in PEX3 KO, PEX11B KO, or control HF cells during mock (upper) or HCMV infection (lower). Plasmalogen abundance normalized to internal standards. Plasmalogens with differential abundances >5-fold in (A) are displayed. N = 3 biological replicates. See also Table S3.
D) Confirmation of RNAi-mediated knockdown of GNPAT by WB.
E) Representative WB showing viral protein abundances from DE (pUL26) or L (pp65 and pp28) genes at 120 hpi during GNPAT siRNA knockdown or a non-targeting control. Loading control was α-tubulin.
F) Supernatant and cell-associated virus produced at 120 hpi by cells during GNPAT siRNA knockdown cells. Significance determined by one-way ANOVA with a Dunnett’s post-test.
G) Model for HCMV regulation of peroxisomes and resulting effects on viral replication. Infection upregulates PEX proteins required for peroxisome membrane assembly, matrix protein import, and fission. Increased peroxisome biogenesis occurs through growth of pre-existing peroxisomes rather than by de novo. The involvement of proteins with asterisks was shown by CRISPR-mediated KO (bold) or microscopy (italic), rather than PRM. Increased plasmalogen production is required for the cytoplasmic stage of viral assembly and maturation. Statistical significance (*) for P<0.05 and (**) for P<0.01.
To determine whether infection increases plasmalogen production, we measured plasmalogen levels in HCMV-infected cells at 120 hpi by LC-MS (Fig 7B, Table S3). We detected a total of 15 ethanolamine plasmalogen species across the three common sn-1 alkyl groups, saturated C16 (16:0), saturated C18 (18:0), and mono-unsaturated C18 (18:1). Eight plasmalogen species showed >5-fold increase in abundance. Although no apparent bias was observed for sn-1 alkyl groups, only sn-2 acyl groups with less than 4 double bonds showed an increase, in agreement with previous reports of increased saturated fatty acids during HCMV infection (Koyuncu et al., 2013). This suggests that the ER metabolism regulates specific plasmalogen species through modification of the sn-2 alkyl group, while the viral-induced peroxisome biogenesis triggers an overall increase in plasmalogen abundance through production of the plasmalogen precursor, 1-0-alkyl-DHAP.
To show that the plasmalogen increase is specific to peroxisome functions, we measured plasmalogens in PEX3 KO and PEX11B KO cells (Fig 7C). PEX3 mutations lead to severe defects in plasmalogen production, while PEX11B mutations mildly reduce plasmalogen levels as peroxisomes are still present and the ability to produce plasmalogens is retained (Fig 3D) (Li et al., 2002; Muntau et al., 2000). Our PEX3 and PEX11B KO cells had decreased plasmalogen abundance relative to control in uninfected cells (Fig 7C). However, plasmalogen abundance in infected PEX11B KO cells was restored to similar levels as infected controls, while the abundance in infected PEX3 KO cells remained lower than controls. Therefore, PEX11B KO cells retain their ability to increase plasmalogen abundance upon infection. However, PEX3 KO cells are devoid of functional peroxisomes and unable to increase plasmalogen levels upon infection, demonstrating that peroxisomes are directly involved in plasmalogen upregulation during infection.
Lastly, we assessed the functional role of plasmalogens during HCMV infection. Since plasmalogens facilitate membrane fusion/fission, their depletion could block steps in the viral life cycle in which membrane fusion/fission occurs. Namely, plasmalogens could be involved in virus secondary envelopment during assembly, virus exocytosis, and infection of new cells. Since GNPAT deficiencies cause severely reduced plasmalogen levels (Itzkovitz et al., 2012), we probed viral replication upon RNAi-mediated GNPAT knockdown (KD) (Fig 7D). GNPAT KD did not affect the expression of DE and L viral genes (Fig 7E). However, GNPAT KD decreased virus produced in both supernatant and cell-associated preparations (Fig 7F). These findings indicate that plasmalogen synthesis is not required for virus exocytosis or virus gene expression, but rather for secondary envelopment during virus assembly or virion fusion upon the next round of infection. To distinguish between the last two alternatives, we measured cell-associated and released viral genomes during GNPAT KD (Fig 7G). If plasmalogen depletion decreases the ability of the virions to infect new cells, this would translate into a loss of infectivity measured as decreased infectious units-to-viral genome ratio in both cell-associated and released virus. Instead, GNPAT KD caused a decrease in virus genomes released and an increase in cell-associated virus genomes (Fig 7G). Therefore, plasmalogens are necessary for efficient secondary envelopment, and their depletion leads to accumulation of viral genomes and a reduction of infectious virus particles within the cell.
Discussion
This study reports that virus-mediated upregulation of peroxisome biogenesis is a mechanism that benefits virus replication. By performing an integrative analysis of peroxisome biogenesis, morphology, and temporal protein composition during HCMV and HSV-1 infections, we show that fundamental peroxisome functions are regulated to enhance replication of these enveloped viruses. In conjunction with lipid analysis during HCMV infection, we propose a model in which peroxisome biogenesis and morphology are altered upon infection to promote plasmalogen metabolism and facilitate virus production (Fig 7H). This finding opens a new avenue for understanding the roles of peroxisomes during herpesvirus infection, placing these organelles in a similar vein with ER, Golgi, and mitochondria, whose regulation and diverse functions are well-recognized during infection. While peroxisomes were previously thought to act only in host antiviral response (Magalhães et al., 2016), we discover their finely-tuned manipulation for the benefit of HCMV replication. Therefore, HCMV has evolved a dual strategy to manipulate peroxisomes, blocking peroxisome antiviral signaling using its immediate early protein, vMIA (Magalhães et al., 2016), and promoting peroxisome biogenesis and metabolism to benefit virus replication. This provides another example of the dynamic interplay between viruses and hosts and highlights the importance of the multifunctional nature of peroxisomes during infection.
The investigation of peroxisomes in the context of virus infection gives insight into two outstanding questions in peroxisome biology: (1) What process drives peroxisome proliferation in mammalian cells? (2) Whether changes in morphology dictate peroxisome functions?
How peroxisome numbers are regulated in mammalian cells has been long debated and challenging to study (Hua and Kim, 2016). Our integration of mathematical modeling and microscopy provides evidence that peroxisome numbers are primarily regulated through growth/fission of pre-existing peroxisomes in human fibroblasts. This discovery supports previous experimental and mathematical models in yeast, which demonstrated that growth/fission is a preferred mechanism for cells with a large number of peroxisomes, or those that demand quick changes to population (Mukherji and O’Shea, 2014). Therefore, we now expand this understanding to human cells and show that herpesvirus infection is a biological context that requires the rapid increase in peroxisome numbers.
In addition to peroxisome abundance, peroxisome morphology has been proposed to influence cellular homeostasis and metabolism. Peroxisome elongation was reported during proliferation and external stress, and is thought to enhance processes associated with peroxisome membranes (Qian et al., 2015; Schrader and Fahimi, 2006). Here, we provide evidence for a link between peroxisome morphology and metabolic capacity in the context of viral infection. We find that irregular peroxisome morphology is induced by the enveloped viruses, HCMV and HSV-1, increasing the peroxisome membrane-to-lumen ratio. These altered structures resemble those observed upon PEX11B KO cells, which show both enhanced plasmalogen production during infection (Fig 7C) and a slight increase in HCMV replication (Fig 3F). Since plasmalogen synthesis requires a concerted mechanism between the peroxisome lumen, membrane, and peroxisome-ER interface, it is likely that the increased peroxisome membrane-to-lumen ratio we describe drives the increased plasmalogen synthesis observed during late HCMV infection. In addition, our proteomic analysis showed that components of other peroxisome membrane-associated pathways, such as ROS processing (e.g., GSTK1) and fatty acid oxidation, are also increased during infection. As peroxisomes are known to couple metabolic pathways (Lodhi and Semenkovich, 2014), it is tempting to speculate that the morphological shift we identified here may be of general benefit to many of these pathways.
The elevation in peroxisome-mediated plasmalogen production may represent a mechanism broadly relevant to infections with enveloped viruses. Virions of enveloped viruses, including HCMV, influenza virus, human immunodeficiency virus (HIV), and West Nile Virus (WNV), are enriched in plasmalogens (Liu et al., 2011; Lorizate et al., 2013; Martin-Acebes et al., 2014; Tanner et al., 2014). Our study shows that plasmalogens are required during intracellular HCMV virus assembly, supporting a role in vesicle fission during envelopment (Fig 7). Indeed, plasmalogens exhibit biophysical properties known to facilitate membrane fission and fusion events (Lohner et al., 1991; Phuyal et al., 2015). Given the preponderance of such events during the replication cycles of enveloped viruses, the enhancement of plasmalogen synthesis is likely a common strategy to aid viral fitness. This is supported by our study of peroxisomes in infections with enveloped HCMV and HSV-1 and unenveloped ADV5, showing that only HCMV and HSV-1 alter peroxisome morphology, and benefit from increased peroxisome numbers to replicate.
Taken together, this study advances the knowledge of fundamental peroxisome biology in both health and disease states. We defined mechanisms involved in the homeostatic regulation of peroxisome proliferation and discovered the requirement of peroxisome biogenesis and functions for herpesvirus production. We anticipate this work will fuel studies aimed at characterizing the impact of peroxisome functions on a range of virus infections. Furthermore, by defining the relationship between peroxisome morphology and metabolism, our study has broad implications for understanding the roles of peroxisomes in biological contexts and diseased states beyond viral infection.
STAR Methods
1. Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ileana M. Cristea (icristea@princeton.edu).
2. Experimental model and subject details
2.1. Cell lines and primary cultures
MRC5 primary human fibroblasts (HFs) (ATCC CCL-171, passage number 18 - 28) were cultured under standard conditions (37°C and 5% CO2) in complete growth medium (DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin antibiotics). The original MRC5 cell line, as well as the CRISPR KO cell lines produced, were authenticated by morphology check by microscope and growth curve analysis, and subject to mycoplasma detection tests. Peroxisome biogenesis disorder (PBD) dermal fibroblasts GM16513, GM16866, and GM07371 were obtained from the Coriell Institute and used within 5 passages.
2.2. Virus strains and viral infection
Viral stocks of wild-type HCMV strain AD169 and AD169-GFP, a modified strain encoding green fluorescent protein (GFP), were produced from bacteria artificial chromosomes in HFs (Wang et al., 2004; Yu et al., 2002). Adenovirus type 5 (ADV5) encoding a GFP cassette (ADV5-GFP) was a gift from Jane Flint (Chahal and Flint, 2012). Wild-type HSV-1 (strain 17+) was propagated on Vero cells. Virus stocks were kept at −80C for up to 6 months, and virus titers were determined using tissue culture infectious dose (TCID50) for HCMV and plaque assays for HSV-1 and ADV5. To determine relative infectious units (IUs) produced under experimental conditions, the supernatant was used to infect a reporter plate and asses the number of infected cells with anti-IEl (HCMV), anti-ICPO (HSV-1), or anti-ICP4 (HSV-1). Cell-associated virus was collected by washing cells with PBS in the dish and scrapping in complete growth medium. The cells were subject to a single freeze/thaw cycle and sonication to release intracellular virus. To inhibit late gene expression, HCMV infections were performed with complete media supplemented with 300 μg/ml phosphonoformate (PFA). For infection with UV-irradiated virus, virus stock was irradiated using a Stratalink UV crosslinker in auto setting for five cycles. This setting was the minimum to achieve less than 99.9% infected cells as measured by anti-IE1 immunofluorescence. Infections were performed at a multiplicity of infection of 3 for HCMV, 10 for HSV-1, and 30 for ADV5.
3. Method details
3.1. 4-Phenylbutyric acid treatment
To induce peroxisome biogenesis, cells were treated with 5mM 4-Phenylbutyric acid (4PBA, Sigma-Aldrich) for 10 days with media changes every third day, as previously described (Kemp et al., 1998). A parallel cell culture of untreated cells was used as a control.
3.2. Cell death assays
Cell death was monitored with trypan blue or by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and was observed to remain at a rate below 5%. TUNEL assay was performed using the in situ Cell Death Detection Kit (Roche # 11684795910) as per manufacturer recommendations. Cells were imaged by fluorescence microscopy, and number of apoptotic cells were determined by fluorescein-dUTP signal in the nucleus.
3.3. HCMV genomes quantification
Cell-associated virus or virus from supernatant were treated with DNAseI to remove capsid-free DNA followed by DNAseI inactivation and Proteinase K digestion in 0.5% SDS for 3 hours at 37C. Viral DNA was recovered by phenol:chloroform extraction and ethanol precipitation. Viral genomes were quantified by qPCR using the SYBR green PCR master mix (Life Technologies) with primers specific to the IE1 gene (Forward primer: 5′-TCGTTGCAATCCTCGGTCA-3′; Reverse primer: 5′-ACAGTCAGCTGAGTCTGGGA-3′). CT values were normalized to a standard curve generated from 1:5 dilutions of sorbitol-purified HCMV viral particles.
3.4. Selection of target proteins and peptides for parallel reaction monitoring (PRM) analysis
A list of 83 candidate peroxisome proteins with experimental validation of peroxisome localization in humans was collected from various sources, including the Uniprot database (Bateman et al., 2017), the PeroxisomeDB 2.0 database (Schlüter et al., 2010), and experimental localization to peroxisomes from previous proteomic studies (Gronemeyer et al., 2013; Jean Beltran et al., 2016). This set of proteins includes peroxisome proteins from major peroxisome functions: peroxisome biogenesis, lipid metabolism, antioxidant activity, isoprenoid metabolism, and other minor functions. A peroxisome peptide spectral library was generated using Skyline (v4.0) (MacLean et al., 2010) from data-dependent acquisition (DDA) MS analyses of whole cell lysates and organelle enriched fractions from previous DDA experiments (Jean Beltran et al., 2016) and additional DDA experiments performed in this study. For proteins with less than 5 peptides containing tandem mass spectra information, additional spectra were collected from the National Institute of Standards and Technology (NIST) HCD human peptide spectral library. Up to 10 unique tryptic peptides per proteins were selected for a first-pass PRM analysis in biological triplicate to determine retention times and validate the reliable detection of transition ions. The list of peptides was further filtered to include up to 3 per protein based on ion intensity and peak area reproducibility across replicates. Peptides with residues prone to modifications were generally avoided. A final list of 160 peptides across 60 peroxisome proteins was used for peroxisome proteome quantification by PRM.
3.5. Protein sample preparations for mass spectrometry (MS) analysis
For whole cell analysis, cell pellets were washed twice with PBS, scraped in PBS, and pelleted by centrifugation. Cell pellets were stored at −80°C until ready for analysis. Organelle-enriched cell fractions were collected as described previously (Jean Beltran et al., 2016). Briefly, cells were incubated in hypotonic solution (0.25M sucrose, 6mM EDTA, 120mM HEPES-NaOH; pH 7.4) and mechanically lysed through 14 μM polycarbonate membrane filters (Sterlitech Corporation). Organelle enrichment by differential centrifugation was achieved by pelleting the nucleus (1400xg, 10min), collecting the supernatant, and pelleting the cytosolic organelles by a second centrifugation (20,000xg for 30 min). Peroxisomes were further enriched by density-gradient ultracentrifugation using a discontinuous Optiprep (Axis-shield) stepped gradient in 5% intervals from 0 to 30% (v/v) Optiprep. Ultracentrifugation was performed in an SW60 Beckman rotor at 129,000xg for 3hrs at 4°C.
Whole cells and organelle-enriched fractions derived from differential centrifugation or density gradient ultracentrifugation were lysed in lysis buffer (4% SDS, 0.1M NaCl, 50mM TrisHCl, 0.5mM EDTA, pH8.0) and reduced/alkylated using 25mM TCEP solution (Thermo Fisher #77720) and 50mM 2-chloroacetamide (MP Biomedicals #ICN15495580) at 70°C for 20 min. Proteins were recovered by methanol-chloroform precipitation (Wessel and Flügge, 1984), and resuspended in 25mM HEPES (pH8.2) with MS-grade Pierce trypsin (Thermo Fisher #90057) at a 1:50 trypsin:protein weight ratio for 16hrs at 37°C. Peptides were adjusted to 1% trifluoroacetic acid (TFA) and desalted using the StageTip method (Rappsilber et al., 2007) and SDBP-RPS material (3M Analytical Biotechnologies). Peptides were washed with 0.2% TFA, and eluted (5% ammonium hydroxide, 80% acetonitrile), followed by drying completely in a SpeedVac (Thermo Fisher) and resuspension in 0.1% formic acid (FA) and 2% acetonitrile (ACN).
3.6. Peptide MS analysis
Samples were analyzed by nLC-MS in a Dionex Ultimate nRSLC coupled to a Thermo Fisher Q-Exactive HF mass spectrometer using DDA or PRM modes. Peptides were separated in a 120 minutes (DDA) or 60 minutes (PRM) reverse-phase linear gradient (2 to 27% solvent B; solvent A: 0.1% FA, solvent B: 0.1% FA/ 97% ACN) in a PepMap RSLC C19 2μm particle size, 75μm × 50cm EASYSpray column (ThermoFisher ES803) set to 50°C. For DDA mode, full scan range was set to 350-1800 m/z with a 120,000 resolution. The top 20 most intense precursors with min signal of 2.5E3 were subject to fragmentation for MS2 analysis at 15,000 resolution. Isolation window set to 1.2 m/z, normalized collision energy (NCE) to 28, and target values of 3E5 and 1E5 for full-scan MS and MS2, respectively. For PRM mode, the instrument was set to 30,000 resolution, 100ms maximum injection time, 5E5 acquisition target, 0.7 m/z isolation window, NCE to 28, and 150m/z fixed first mass.
3.7. Informatic analysis of MS data
Tandem MS spectra collected from DDA mode were analyzed by Proteome Discoverer v2.1 (ThermoFisher). A processing workflow containing the SEQUEST algorithm was used to search tandem MS spectra against a UniProt-SwissProt human database and common contaminants, generating peptide spectrum matches. Percolator was used to calculate matches q-values based on reverse sequence database searches. The resulting MSF files were imported into Skyline with a cut-off score of 0.95 and filtered for spectra matching the list of peroxisome proteins and peptide subset. RAW files containing MS2 spectra collected in PRM mode were imported into Skyline to extract product ion chromatograms (XICs) and calculate peak areas. The top three most intense coeluting product XICs were used for peak area quantification. Peak areas were normalized to peptides used as loading controls from H2B1A (LLLPGELAK) and H2A1A (AGLQFPVGR). A third peptide from α-tubulin (ISVYYNEATGGK), a protein commonly used for loading control in HCMV time course experiments, was monitored as an extra control for loading.
Normalized peak areas were exported for data analysis in R (v3.4.3) and RStudio (v1.1.423). Normalized peptide peak areas were used to determine coefficient of variation across samples. Relative protein-level abundances were determined as follows. Normalized peak areas were divided by the value in mock, and these ratios were averaged across peptides from each single protein. Heatmaps were created using the package pheatmap (v1.0.8). Hierarchical clustering for these heatmaps was done using euclidean distances and complete linkage method. To determine clusters from temporal profiles, first protein abundances relative to mock were converted to z-scores (mean-centered with unit standard deviation). The package ppclust (v.0.1.1) was used to cluster the z-score values with the fuzzy C-means algorithm (number of clusters = 4).
3.8. Microscopy
An inverted fluorescence confocal microscope (Nikon Ti-E) equipped with a Yokogawa spinning disc (CSU-21), digital CMOS camera (Hamamatsu ORCA-Flash TuCam), and precision microscope stage (Piezo) was used for all microscopy work. An environmental control chamber was used for live-cell imaging experiments to maintain a 37° C and 5% CO2 environment. Z-stacks were acquired with 0.2μm steps throughout the cell depth using a Nikon 100X Plan Apo objective for analysis of peroxisome morphology and colocalization and a Nikon 60X Plan Apo objective for all other experiments. For long-term peroxisome imaging, Z-stacks were acquired at 50ms exposure per step to limit laser exposure to <10 seconds per cell. Image acquisition was automated for sequential imaging of 20 individual cells over an 8-hour period. Time points in which cells moved out of the field of view were discarded.
3.9. Transfection for expression of fluorescent tags
MRC5 cells were seeded in 35mm glass-bottom microscope dishes (MatTek) at 1.6×105 cells/dish and transfected 24 hours later. A 1:3 ratio of μg plasmid DNA to μl X-tremeGENE HP DNA transfection reagent (Sigma-Aldrich) were mixed in 200μl Opti-MEM (LifeTechnologies) and incubated at room temperature in for 20 minutes. Transfection mix was added dropwise to cells in 1.8mL of Opti-MEM and allowed to incubate at 37° C for 6 hours. Cells were then rinsed in PBS, placed in 2mL complete medium, and allowed to recover for 18 hours prior to imaging.
The following amounts of plasmid DNA were used: 200ng eGFP-Peroxisomes-0 (gift from Michael Davidson, AddGene #54501) and 700ng mito-BFP (gift from Gia Voeltz, AddGene #49151).
3.10. Western blotting
Cells collected for western blot analysis were lysed in lysis buffer. Protein concentration was measured by Pierce BCA assay (ThermoFisher). Ten μg were reduced with 2-mercaptoethanol and subjected to SDS-PAGE. Proteins were transferred to a PVDF membrane, and the membranes were blocked in blocking buffer (5% milk in TBS), primary antibody in blocking buffer with 0.2% Tween, and secondary antibody in blocking with 0.2% Tween and 0.01% SDS. Fluorescent signal from secondary antibodies was detected using the Odyssey CLX system (Li-cor). ImageStudio v4.0 (Li-cor) was used for band intensity quantification.
The following antibody dilutions were used for primary incubation: 1:200 for mouse anti-pp28 (clone 10B4 gift from Tom Shenk (Silva et al., 2003)), 1:100 for mouse anti-IE1 (clone 1B12 gift from Tom Shenk (Zhu et al., 1995)), 1:100 for mouse anti-pUL26 (clone 7H1-5 gift from Tom Shenk (Munger et al., 2006)), 1:100 for mouse anti-pp65 (clone 8F5 (Nowak et al., 1984)), 1:5000 for mouse anti-α-tubulin (ThermoFisher T6199), 1:2000 for rabbit anti-PEX14 (Abcam ab183885), 1:500 rabbit anti-GNPAT (Proteintech 14931-1-AP). The following antibodies at 1:10000 dilutions were used for secondary incubations: Alexa Fluor 680 goat anti-rabbit IgG (ThermoFisher A-21109) and IRDye 800CW goat anti-mouse (Li-cor 926-32210).
3.11. Immunofluorescence
For all immunofluorescence experiments, cells were fixed in 4% paraformaldehyde (PFA) for 15 minutes, then washed with PBS and permeabilized in cold methanol at −20° C for 20 minutes. Uninfected cells were blocked in 10% goat serum in PBS + 0.01% Triton (PBT) for 30 minutes, and infected cells were blocked in 10% goat serum with 5% human serum in PBT for 30 minutes. Cells were incubated in primary antibodies for 2 hours, washed with PBT, incubated in secondary antibodies for 45 minutes, washed with PBT, and rinsed in PBS. For peroxisome counting experiments, MRC5 cells were grown on glass coverslips in 2mL wells and mounted with ProLong Diamond antifade (ThermoFisher) on glass slides after staining. For CRISPR, morphology, and MFF/FIS1 imaging, cells were grown in glass-bottom microscope dishes (MatTek), then stored and imaged in 2mL TBS after staining. For MFF/FIS1 colocalization experiments, cells were first transfected with eGFP-Peroxisomes-0 and mito-BFP, as described above.
Antibodies were diluted in blocking buffer as follows: 1:500 for rabbit anti-Pex14 (Abcam ab183885), 1:40 for mouse anti-pp28 (clone 10B4 gift from Tom Shenk (Silva et al., 2003)), 1:40 for mouse anti-IE1 (clone 1B12 gift from Tom Shenk (Zhu et al., 1995)), 1:5000 for rabbit anti-ICP0 (Virusys Corporation H1A027-100), 1:300 for mouse anti-ICP4 (Abcam ab6514), 1:1500 for mouse anti-MFF (SantaCruz SC-398617), 1:2000 for rabbit anti-FIS1 (ThermoFisher PA5-22142), 1:1000 anti-NBR1 (Cell Signaling D2E6), 1:2000 for goat anti-rabbit IgG highly cross-adsorbed AlexaFluor 568 (ThermoFisher A-11036), 1:2000 for goat anti-mouse highly cross-adsorbed AlexaFluor 568 (ThermoFisher A-11019), and 1:2000 for goat anti-mouse IgG highly cross-adsorbed AlexaFluor 633 (ThermoFisher A-21052). Sequential staining was used for Mff co-visualization with IE1 or UL99.
3.12. Peroxisome counting, morphology analysis, and colocalization
Peroxisomes in a cell were counted with the Nikon NIS-Elements AR v5.0 imaging software. Image Z-stacks were deconvolved with the 3D Deconvolution tool, and the 3D Spot Detection tool was used to detect peroxisomes (50-300 contrast, 1:2 Z-axis elongation, 0.5 μm diameter spot, and variable size spots). For analysis of counts to be used for inference, cells were transfected with EGFP-Peroxisome-0 as described above and imaged for 8 hours at 5-minute intervals. Counts were excluded from the final dataset if cells moved out of frame.
To analyze peroxisome morphology, the ImageJ (National Institutes of Health) analysis tool 3D Objects Counter was used to threshold and measure surface area, volume, and mean length of each peroxisome, with a minimum size requirement of 10 consecutive voxels (at a resolution of 0.065 μm/pixel).
To analyze MFF and FIS1 colocalization with peroxisomes and mitochondria, maximum intensity projections were generated from z-stack images and the ImageJ analysis tool Coloc 2 (J Schindelin) was used to threshold and analyze colocalization between channels via Spearman’s correlation analysis with 100 Costes randomizations and PSF adjustment of 3.29.
To analyze the number of NBR1-positive peroxisomes, images were manually inspected and peroxisomes that were in contact with NBR1 puncta were marked as NBR1-positive.
3.13. Modeling of peroxisome biogenesis from count time series data
Our model is based on a stochastic model for yeast organelle abundance proposed by (Mukherji and O’Shea, 2014), which models the temporal evolution of the number of organelles n, or more specifically the probability that the number equals n at time t, p(n, t), in terms of the rates of four processes governing n (de novo generation kd, fission kf, fusion kfus and degradation γ). Here, in the case of human peroxisomes, fusion is neglected as this process does not occur, so that the following three processes are considered:
-
a)de novo peroxisome generation from ER vesicles, kd:
-
b)growth and fission of pre-existing peroxisomes, kf:
-
c)degradation through pexophagy, γ:
These stochastic processes are combined to generate the evolution equation for p(n, t):
| (1) |
The relative importance of the de novo/fission/degradation processes in a cell with n peroxisomes is given by kd : kf × n : γ × n. This is because the number of fission and degradation events depend on the number of peroxisomes present, as opposed to de novo events. In the following we designate the rates of the model by θ=[kd, kf, γ].
In this work, we considered temporal count data acquired for K cell replicates via fluorescent microscopy. We denote the measured peroxisome count in cell replicate 1 ≤ k ≤ K at time by and all the measured data (i.e. all time points and replicates) by . We assumed a Normal error model with a replicate specific standard deviation so that the measured count is obtained from the true (hidden) count, , following , i.e.
| (2) |
We modelled the cell to cell variations of the rates by assuming that the rates each follow a Gamma distribution between cells. Each rate has its own rate parameter and shape parameter: kd ~ Gamma (αkd, βkd), kf ~ Gamma(αkf, βkf) and γ ~ Gamma(αγ, βγ). This ensures that the rates are always positive and can provide a good approximation of normally distributed rates.
For each rate, we can equivalently reformulate the parameters of the gamma distribution in terms of the rates mean and standard deviations, e.g. in the case of which are easier to more useful and easier to interpret.
3.14. Bayesian inference of peroxisome biogenesis rates
We described the method for inference of rates that regulate peroxisome number in (Galitzine et al., 2018), to which the reader is referred for more details. We want to obtain the distribution of the rate means and standard deviations, e.g. μkd, σkd, as well as the error standard deviation deviations. We denote all the parameters to be inferred by α = [μkd, σkd, μkf, σkf, μγ, σγ, σ1, σ2, …, σK]. A Bayesian formulation of the inference problem is thus:
| (3) |
Since no prior information about the rates is known besides their positivity, we use a flat uninformative prior p(α) = 1 for all parameters.
We sample α from p(α|D) in Eq. (3) with a Metropolis Hasting method (Wilkinson, 2011). The distribution functions of the rate means and standard deviations are then directly obtained from the samples.
Sampling Eq. (3) necessitates the calculation of the likelihood of the data p(D|α). The overall likelihood of the data is calculated by assuming that both the replicates and the consecutive increments of each replicate are statistically independent:
| (4) |
where is obtained by solving Eq. (1), with the error model of Eq. (2) and p(θ|α) is the product of the three rate gamma distributions.
We obtain the likelihood of each individual replicate in Eq. (4) with a particle filter method (Doucet and Johansen, 2009). In such a method, computational particles are propagated following Eq. (1) along each replicate time trace. At each measurement time, they are resampled according to the normal error model of Eq. (2). Finally, the likelihood of a replicate is obtained by taking the product of particle weight sums at each measurement time.
3.15. CRISPR-mediated knockouts
CRISPR guide RNA sequences designed for knockout of human PEX3, PEX19, PEX11B, PEX7, or non-targeting negative control (from Origene pCas-Scramble) were cloned into and delivered by the LentiCRISPR v2 vector (Addgene plasmid 52961) (Table S4) from Feng Zhang (Sanjana et al., 2014). A total of 1 × 105 MRC5 cells were transduced with lentivirus (2 days) and selected with puromycin (1 μg/ml) for 4 days. Cells were cultured for an additional week to allow depletion of protein targets before performing experiments.
3.16. siRNA-mediated knockdowns
siRNA duplexes sequences were purchased from Sigma-Aldrich for GNPAT (#1, 5′-CAAGGUACCUCUCAAUGUU[dT][dT]-3′; and #2, 5′-GGCUUAUGCUCCAGCACAU[dT][dT]-3′) or negative control (MISSION siRNA Universal Negative Control #1; Sigma-Aldrich #SIC001). Transfections were performed using Lipofectamine RNAiMAX Transfection Reagent (ThermoFisher Scientific) as per manufacturer protocol. A total of 0.5 × 105 MRC5 cells were transfected with 10pg siRNA duplex and 1.5μl transfection reagent in Opti-MEM. Cells were recovered after 16 hours using full medium and either immediately infected or processed for WB analysis at 48 hours post transfection.
3.17. Plasmalogen quantification by MS
Phospholipids were isolated for MS analysis as previously described(Ivanova et al., 2007). A total of 1×106 cells were incubated in serum-free DMEM for 24 hours prior to processing. Cells were recovered by scrapping in 0.8ml of ice-cold 1:1 0.1N HCl:MetOH (v/v) and transferred to a microcentrifuge tube. A total of 200ng odd-carbon chain internal standard PE 17:0/17:0 (Avanti Polar Lipids, Inc.; No. 830756) and 0.4ml ice-cold chloroform were added. The sample was vortexed and centrifuged (5 min, 18,000xg). The organic phase was collected, solvent evaporated (Speedvac SC110A, ThermoSavant), and dissolved in 50μl methanol prior to injection of 20μl for LC-MS analysis.
A Shimadzu UFLC system coupled to a Thermo LTQ XL mass spectrometer was used for LC-MS analysis in positive-ion mode. Phospholipids were separated in a 60-minute stepped linear reverse-phase gradient (solvent B set to 0% 5min, 30% 15 min, 50% 60min) using an ACE C18 colum(μm, 75mm × 1mm) at 45°C. Solvent composition for buffer A (2:2:1 acetonitrile:methanol:water (v/v/v) supplemented with 0.1% formic acid and 0.028% ammonia) and buffer B (isopropanol supplemented with 0.1% formic acid and 0.028% ammonia). Scan range was set to 680-820 m/z with a 60,000 resolution for MS1 in the FT and 200-800 m/z for MS2 in the IT. Instrument was set to perform a full MS scan followed by isolation and collision induced dissociation (CID) for MS2 acquisition from an inclusion list containing m/z for phosphatidylethanolamine (PE) plasmalogen precursor [M+H]+ (Table S3) and for PE 17:0/17:0 internal standard (m/z = 720.5). Isolation window set to 1.9 m/z, normalized collision energy (NCE) to 32, acquisition Q of 0.250, and activation time of 30ms. Raw data files were imported into skyline for analysis. Two characteristic fragment ions were used to confirm the plasmalogen species (Table S3), as described in (Nakanishi et al., 2009; Zemski Berry and Murphy, 2004). If an isomeric plasmalogen species was observed to co-elute, the major species is reported, and minor species are mentioned in Table S3. The 579.4 m/z fragment ion was used to identify PE 17:0/17:0 internal standard. Precursor XIC areas under the curve were used for quantification and were normalized to 17:0 PE.
4. Quantification and statistical analysis
Statistical analysis was performed using the software Prism v5.04. Bar graphs and horizontal lines in scatter plots indicate means. Error bars indicate standard error of the mean, unless noted in the figure legend. The number and type of replicates and the type of statistical tests can be found in each figure legend. Statistical significance is indicated as asterisks in figures as (*) for P<0.05, (**) for P<0.01, and (***) for P<0.001.
5. Data and software availability
The MS proteomic RAW files have been deposited to the ProteomeXchange Consortium via the PRIDE (Deutsch et al., 2017) partner repository with the dataset identifier PXD009321. The MS RAW files for lipid analysis have been deposited to the EMBL-EBI MetaboLights database with the identifier MTBLS652, and can be accessed with the URL (http://www.ebi.ac.uk/metabolights/MTBLS652). Skyline files have been deposited in Panorama Public (Sharma et al., 2014) for the peroxisome proteome quantification (https://panoramaweb.org/peroxisomehcmv.url) and for the plasmalogen quantification (https://panoramaweb.org/plasmalogenhcmv.url).
Supplementary Material
Accurate quantification of peroxisomes in 3D reconstructions. Related to Figure 2. Cell was stained using anti-PEX14 (grey) to label peroxisomes, and an image z-stack was used to render a 3D reconstruction. Colored spheres indicate automatically detected peroxisomes.
Peroxisomes are spherical and uniform in uninfected human fibroblasts. Related to Figure 5. Video displaying a 3D rendered uninfected cell. Peroxisomes are labelled with PEX14 antibody, z-stack steps were 0.2μm, and images were acquired at 100X.
Peroxisomes exhibit irregular, flattened morphology late in HCMV infection. Related to Figure 5. Video displaying a 3D rendered cell late in HCMV infection (120 hpi). Peroxisomes are labelled with PEX14 antibody, z-stack steps were 0.2μm, and images were acquired at 100X.
Quantification of peroxisome proteins throughout the time course of HCMV infection. Related to Figure 1. (A) Protein quantitative values from three averaged biological replicates normalized to the uninfected (mock) sample. Temporal cluster classification using fuzzy c-means is shown, as in Fig S2. (B) Peroxisome protein quantification during PFA treatment or during infection with UV-irradiated virus.
Inference results for peroxisome biogenesis rates in uninfected HFs. Related to Figure 4. Mean and standard deviation inferred rates kd, kf*N, and γ*N in three biological replicate experiments. N is the average peroxisome number per cell in each dataset.
Quantification of plasmalogen species in HCMV infected samples. Related to Figure 7. (A) Species quantified and the MS2 fragments monitored for identification. Other minor isomeric species detected are shown. (B) Quantification of plasmalogens between infected (120 hpi) and infected samples. Mean and standard error of the mean (SEM) of 3 biological replicates normalized to uninfected (mock). (C) Quantification of plasmalogen species during infection of PEX3 KO, PEX11B KO, or CRISPR control cell lines. Mean and SEM of 3 biological replicates normalized to uninfected (mock) CRISPR control.
Oligonucleotides used for cloning of sgRNA sequence into lentiCRISPRv2. Related to STAR Methods.
Highlights.
HCMV infection temporally upregulates peroxisome biogenesis and lipid metabolism proteins
Peroxisome numbers increase late in infection and are required for HCMV/ HSV-1 replication
HCMV infection alters peroxisome morphology to increase the membrane-to-lumen ratio
Increased peroxisome biogenesis enhances plasmalogen synthesis to help virus replication
Acknowledgments
We thank the Princeton University Microscopy and Mass Spectrometry Facilities for support. We are grateful for funding from the NIH (GM114141) and a Mallinckrodt Scholar Award to IMC, a Harold W Dodds Fellowship to PMJB, and the NINS Strategic International Research Exchange Promotion Program to YH.
Footnotes
Declaration of Interest
The authors declare that there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahlemeyer B, Gottwald M, and Baumgart-Vogt E (2012). Deletion of a single allele of the Pex11 gene is sufficient to cause oxidative stress, delayed differentiation and neuronal death in mouse brain. Dis. Model. Mech 5, 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Martin MJ, O’Donovan C, Magrane M, Alpi E, Antunes R, Bely B, Bingley M, Bonilla C, Britto R, et al. (2017). UniProt: The universal protein knowledgebase. Nucleic Acids Res 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman NE, and Moser AB (2012). Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta - Mol. Basis Dis 1822, 1442–1452. [DOI] [PubMed] [Google Scholar]
- Di Cara F, Sheshachalam A, Braverman NE, Rachubinski RA, and Simmonds AJ (2017). Peroxisome-Mediated Metabolism Is Required for Immune Response to Microbial Infection. Immunity 93–106. [DOI] [PubMed] [Google Scholar]
- Chahal JS, and Flint SJ (2012). Timely synthesis of the adenovirus type 5 E1B 55-kilodalton protein is required for efficient genome replication in normal human cells. J. Virol 86, 3064–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CS, and Gould SJ (1999). Identification of a common PEX1 mutation in Zellweger syndrome. Hum. Mutat 14, 45–53. [DOI] [PubMed] [Google Scholar]
- Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, et al. (2017). The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 45, D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit E, Boulant S, Zhang Y, Lee ASY, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, et al. (2010). Peroxisomes Are Signaling Platforms for Antiviral Innate Immunity. Cell 141, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet A, and Johansen AM (2009). A tutorial on particle filtering and smoothing: fifteen years later. Handb. Nonlinear Filter 12, 656–704. [Google Scholar]
- Galitzine C, Jean Beltran PM, Cristea IM, and Vitek O (2018). Statistical inference of peroxisome dynamics In Research in Computational Molecular Biology, (Paris, France: ), pp. 54–74. [Google Scholar]
- Gandre-Babbe S, and Bliek AM van der (2008). The Novel Tail-anchored Membrane Protein Mff Controls Mitochondrial and Peroxisomal Fission in Mammalian Cells. Mol. Biol. Cell 19, 2402–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronemeyer T, Wiese S, Ofman R, Bunse C, Pawlas M, Hayen H, Eisenacher M, Stephan C, Meyer HE, Waterham HR, et al. (2013). The Proteome of Human Liver Peroxisomes: Identification of Five New Peroxisomal Constituents by a Label-Free Quantitative Proteomics Survey. PLoS One 8, e57395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua R, andKim PK (2016). Multiple paths to peroxisomes: Mechanism of peroxisome maintenance in mammals. Biochim. Biophys. Acta - Mol. Cell Res 1863, 881–891. [DOI] [PubMed] [Google Scholar]
- Itzkovitz B, Jiralerspong S, Nimmo G, Loscalzo M, Horovitz DDG, Snowden A, Moser A, Steinberg S, andBraverman N (2012). Functional characterization of novel mutations in GNPAT and AGPS, causing rhizomelic chondrodysplasia punctata (RCDP) types 2 and 3. Hum. Mutat 33, 189–197. [DOI] [PubMed] [Google Scholar]
- Ivanova PT, Milne SB, Byrne MO, Xiang Y, andBrown HA (2007). Glycerophospholipid Identification and Quantitation by Electrospray Ionization Mass Spectrometry. Methods Enzymol 432, 21–57. [DOI] [PubMed] [Google Scholar]
- Jean Beltran PM, Mathias RA, andCristea IM (2016). A Portrait of the Human Organelle Proteome In Space and Time during Cytomegalovirus Infection. Cell Syst 3, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp S, Wei H-M, Lu J-F, Braiterman LT, McGuinness MC, Moser AB, Watkins PA, andSmith KD (1998). Gene redundancy and pharmacological gene therapy: Implications for X-linked adrenoleukodystrophy. Nat. Med 4, 1261–1268. [DOI] [PubMed] [Google Scholar]
- Koyuncu E, Purdy JG, Rabinowitz JD, andShenk T (2013). Saturated Very Long Chain Fatty Acids Are Required for the Production of Infectious Human Cytomegalovirus Progeny. PLoS Pathog 9, e1003333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Sumpter R, Zou Z, Sirasanagandla S, Wei Y, Mishra P, Rosewich H, Crane DI, andLevine B (2017). Peroxisomal protein PEX13 functions in selective autophagy. EMBO Rep 18, 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Baumgart E, Morrell JC, Jimenez-Sanchez G, Valle D, andGould SJ (2002). PEX11β deficiency is lethal and impairs neuronal migration but does not abrogate peroxisome function. Mol. Cell. Biol 22, 4358–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu STH, Sharon-Friling R, Ivanova P, Milne SB, Myers DS, Rabinowitz JD, Brown HA, andShenk T (2011). Synaptic vesicle-like lipidome of human cytomegalovirus virions reveals a role for SNARE machinery in virion egress. Proc. Natl. Acad. Sci 108, 12869–12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodhi IJ, andSemenkovich CF (2014). Peroxisomes: A nexus for lipid metabolism and cellular signaling. Cell Metab 19, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohner K, Balgavy P, Hermetter A, Paltauf F, andLaggner P (1991). Stabilization of non-bilayer structures by the etherlipid ethanolamine plasmalogen. BBA - Biomembr 1061, 132–140. [DOI] [PubMed] [Google Scholar]
- Lorizate M, Sachsenheimer T, Glass B, Habermann A, Gerl MJ, Kräusslich HG, andBrügger B (2013). Comparative lipidomics analysis of HIV-1 particles and their producer cell membrane in different cell lines. Cell. Microbiol 15, 292–304. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, andMacCoss MJ (2010). Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhães AC, Ferreira AR, Gomes S, Vieira M, Gouveia A, Valença I, Islinger M, Nascimento R, Schrader M, Kagan JC, et al. (2016). Peroxisomes are platforms for cytomegalovirus’ evasion from the cellular immune response. Sci. Rep 6, 26028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manicklal S, Emery VC, Lazzarotto T, Boppana SB, andGupta RK (2013). The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev 26, 86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Acebes MA, Merino-Ramos T, Blazquez A-B, Casas J, Escribano-Romero E, Sobrino F, andSaiz J-C (2014). The Composition of West Nile Virus Lipid Envelope Unveils a Role of Sphingolipid Metabolism in Flavivirus Biogenesis. J. Virol 88, 12041–12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Tamura S, Furuki S, Miyata N, Moser A, Shimozawa N, Moser HW, Suzuki Y, Kondo N, andFujiki Y (2003). Mutations in novel peroxin gene PEX26 that cause peroxisome-biogenesis disorders of complementation group 8 provide a genotype-phenotype correlation. Am. J. Hum. Genet 73, 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S, andO’Shea EK (2014). Mechanisms of organelle biogenesis govern stochastic fluctuations in organelle abundance. Elife 3, e02678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger J, Yu D, andShenk T (2006). UL26-deficient human cytomegalovirus produces virions with hypophosphorylated pp28 tegument protein that is unstable within newly infected cells. J. Virol 80, 3541–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntau AC, Mayerhofer PU, Paton BC, Kammerer S, andRoscher AA (2000). Defective Peroxisome Membrane Synthesis Due To Mutations in Human PEX3 Causes Zellweger Syndrome, Complementation Group G. Am. J. Hum. Genet 67, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, Ogiso H, andTaguchi R (2009). Qualitative and Quantitative Analyses of Phospholipids by LC-MS for Lipidomics In Lipidomics, (Humana Press; ), pp. 287–313. [DOI] [PubMed] [Google Scholar]
- Nowak B, Sullivan C, Sarnow P, Thomas R, Bricout F, Nicolas JC, Fleckenstein B, andLevine AJ (1984). Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 132, 325–338. [DOI] [PubMed] [Google Scholar]
- Phuyal S, Skotland T, Hessvik NP, Simolin H, Øverbye A, Brech A, Parton RG, Ekroos K, Sandvig K, andLlorente A (2015). The ether lipid precursor hexadecylglycerol stimulates the release and changes the composition of exosomes derived from PC-3 cells. J. Biol. Chem 290, 4225–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian G, Fan W, Ahlemeyer B, Karnati S, andBaumgart-Vogt E (2015). Peroxisomes in different skeletal cell types during intramembranous and endochondral ossification and their regulation during osteoblast differentiation by distinct peroxisome proliferator-activated receptors. PLoS One 10, e0143439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, andIshihama Y (2007). Protocol for micro-purification, enrichment, prefractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, andZhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter A, Real-Chicharro A, Gabaldón T, Sánchez-Jiménez F, andPujol A (2010). PeroxisomeDB 2.0: An integrative view of the global peroxisomal metabolome. Nucleic Acids Res 38, D800–D805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader M, andFahimi HD (2006). Peroxisomes and oxidative stress. Biochim. Biophys. Acta - Mol. Cell Res 1763, 1755–1766. [DOI] [PubMed] [Google Scholar]
- Sharma V, Eckels J, Taylor GK, Shulman NJ, Stergachis AB, Joyner SA, Yan P, Whiteaker JR, Halusa GN, Schilling B, et al. (2014). Panorama: A targeted proteomics knowledge base. J. Proteome Res 13, 4205–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Yu Q-C, Enquist L, andShenk T (2003). Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol 77, 10594–10605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, andAitchison JD (2013). Peroxisomes take shape. Nat. Rev. Mol. Cell Biol 14, 803–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, andMoser HW (2006). Peroxisome biogenesis disorders. Biochim. Biophys. Acta - Mol. Cell Res 1763, 1733–1748. [DOI] [PubMed] [Google Scholar]
- Sychev ZE, Hu A, DiMaio TA, Gitter A, Camp ND, Noble WS, Wolf-Yadlin A, andLagunoff M (2017). Integrated systems biology analysis of KSHV latent infection reveals viral induction and reliance on peroxisome mediated lipid metabolism. PLOS Pathog 13, e1006256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner LB, Chng C, Guan XL, Lei Z, Rozen SG, andWenk MR (2014). Lipidomics identifies a requirement for peroxisomal function during influenza virus replication. J. Lipid Res 55, 1357–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilton C, Clippinger AJ, Maguire T, andAlwine JC (2011). Human Cytomegalovirus Induces Multiple Means To Combat Reactive Oxygen Species. J. Virol 85, 12585–12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanders RJA, andWaterham HR (2006). Biochemistry of Mammalian Peroxisomes Revisited. Annu. Rev. Biochem 75, 295–332. [DOI] [PubMed] [Google Scholar]
- Wang D, Bresnahan W, andShenk T (2004). Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc. Natl. Acad. Sci 101, 16642–16647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel D, andFlügge UI (1984). A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem 138, 141–143. [DOI] [PubMed] [Google Scholar]
- Wilkinson DJ (2011). Stochastic Modelling for Systems Biology, Second Edition (CRC Press; ). [Google Scholar]
- You J, Hou S, Malik-Soni N, Xu Z, Kumar A, Rachubinski RA, Frappier L, andHobman TC (2015). Flavivirus Infection Impairs Peroxisome Biogenesis and Early Antiviral Signaling. J. Virol 89, 12349–12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Smith GA, Enquist LW, andShenk T (2002). Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol 76, 2316–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemski Berry KA, andMurphy RC (2004). Electrospray ionization tandem mass spectrometry of glycerophosphoethanolamine plasmalogen phospholipids. J. Am. Soc. Mass Spectrom 15, 1499–1508. [DOI] [PubMed] [Google Scholar]
- Zhu H, Shen Y, andShenk T (1995). Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol 69, 7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Accurate quantification of peroxisomes in 3D reconstructions. Related to Figure 2. Cell was stained using anti-PEX14 (grey) to label peroxisomes, and an image z-stack was used to render a 3D reconstruction. Colored spheres indicate automatically detected peroxisomes.
Peroxisomes are spherical and uniform in uninfected human fibroblasts. Related to Figure 5. Video displaying a 3D rendered uninfected cell. Peroxisomes are labelled with PEX14 antibody, z-stack steps were 0.2μm, and images were acquired at 100X.
Peroxisomes exhibit irregular, flattened morphology late in HCMV infection. Related to Figure 5. Video displaying a 3D rendered cell late in HCMV infection (120 hpi). Peroxisomes are labelled with PEX14 antibody, z-stack steps were 0.2μm, and images were acquired at 100X.
Quantification of peroxisome proteins throughout the time course of HCMV infection. Related to Figure 1. (A) Protein quantitative values from three averaged biological replicates normalized to the uninfected (mock) sample. Temporal cluster classification using fuzzy c-means is shown, as in Fig S2. (B) Peroxisome protein quantification during PFA treatment or during infection with UV-irradiated virus.
Inference results for peroxisome biogenesis rates in uninfected HFs. Related to Figure 4. Mean and standard deviation inferred rates kd, kf*N, and γ*N in three biological replicate experiments. N is the average peroxisome number per cell in each dataset.
Quantification of plasmalogen species in HCMV infected samples. Related to Figure 7. (A) Species quantified and the MS2 fragments monitored for identification. Other minor isomeric species detected are shown. (B) Quantification of plasmalogens between infected (120 hpi) and infected samples. Mean and standard error of the mean (SEM) of 3 biological replicates normalized to uninfected (mock). (C) Quantification of plasmalogen species during infection of PEX3 KO, PEX11B KO, or CRISPR control cell lines. Mean and SEM of 3 biological replicates normalized to uninfected (mock) CRISPR control.
Oligonucleotides used for cloning of sgRNA sequence into lentiCRISPRv2. Related to STAR Methods.