

Abstract
The peptide‐substrate‐binding (PSB) domain of collagen prolyl 4‐hydroxylase (C‐P4H, an α2β2 tetramer) binds proline‐rich procollagen peptides. This helical domain (the middle domain of the α subunit) has an important role concerning the substrate binding properties of C‐P4H, although it is not known how the PSB domain influences the hydroxylation properties of the catalytic domain (the C‐terminal domain of the α subunit). The crystal structures of the PSB domain of the human C‐P4H isoform II (PSB‐II) complexed with and without various short proline‐rich peptides are described. The comparison with the previously determined PSB‐I peptide complex structures shows that the C‐P4H‐I substrate peptide (PPG)3, has at most very weak affinity for PSB‐II, although it binds with high affinity to PSB‐I. The replacement of the middle PPG triplet of (PPG)3 to the nonhydroxylatable PAG, PRG, or PEG triplet, increases greatly the affinity of PSB‐II for these peptides, leading to a deeper mode of binding, as compared to the previously determined PSB‐I peptide complexes. In these PSB‐II complexes, the two peptidyl prolines of its central P(A/R/E)GP region bind in the Pro5 and Pro8 binding pockets of the PSB peptide‐binding groove, and direct hydrogen bonds are formed between the peptide and the side chains of the highly conserved residues Tyr158, Arg223, and Asn227, replacing water mediated interactions in the corresponding PSB‐I complex. These results suggest that PxGP (where x is not a proline) is the common motif of proline‐rich peptide sequences that bind with high affinity to PSB‐II.
Keywords: collagen, extracellular matrix protein, protein‐peptide interactions, proline‐rich peptide, prolyl 4‐hydroxylase, calorimetry, X‐ray crystallography
Abbreviations
- PRS
proline‐rich peptide segment
- PRD
PRS recognizing domain
- PPII‐helix
poly‐l‐proline type‐II helix
- SH3
Src‐homology 3
- EVH1
Ena/VASP homology 1
- Hyp
4‐hydroxyproline
- C‐P4H
collagen prolyl 4‐hydroxylase
- PDI
protein disulphide isomerase
- SAXS
small‐angle X‐ray scattering
- PSB
peptide‐substrate‐binding domain of C‐P4H
- TPR
tetratricopeptide repeat
- DD
double domain construct of C‐P4H including the N‐terminal dimerization domain and the middle PSB domain of the α subunit
- (PPG)n
peptide with n Pro‐Pro‐Gly triplet repeats
- (P)9
polyproline peptide with nine prolines
- SLS
static light scattering
- CD
circular dichroism
- ITC
isothermal titration calorimetry
- PAG
PPG‐PAG‐PPG peptide
- PRG
PPG‐PRG‐PPG peptide
- PEG
PPG‐PEG‐PPG peptide
- PDB
Protein Data Bank
- SEC
size‐exclusion chromatography
- RI
refractive index
- ESRF
European Synchrotron Radiation Facility
Introduction
Proline‐rich peptide segments (PRSs) are commonly found at the interfaces of protein–protein interaction sites.1, 2, 3 The protein domains that bind such PRSs, referred to as PRS‐recognizing domains (PRDs) are involved in a wide range of functions. Typically, PRSs adopt a poly(l‐proline) type‐II helical conformation (PPII‐helix)4 when bound to their target protein. Typically, PRSs bind with their proline pyrrolidine rings stacked to the aromatic side chains of aromatic residues. Two binding motifs have been identified, being the PxxP motif recognized for example by the Src‐homology 3 (SH3) and Ena/VASP homology 1 (EVH1) domains and the xPPx motif recognized for example by the WW and GYF domains.1
Collagens are proline‐rich proteins having PRS of repeating ‐X‐Y‐Gly‐ triplets, where X and Y are commonly proline and 4‐hydroxyproline (Hyp), respectively.5, 6 The formation of Hyp is a posttranslational process catalyzed by collagen prolyl 4‐hydroxylases (C‐P4Hs) (EC 1.14.11.2).7, 8, 9 C‐P4Hs are α2β2 tetrameric enzyme complexes of 240 kDa in size. The α subunit contains the active site (in its C‐terminal half) and the β subunit is identical to protein disulphide isomerase (PDI).10 In mammals, three α subunit isoforms exist, each of them being complexed with the same β subunit into functional α2β2 C‐P4H tetramers.7 Out of those three isoforms, C‐P4H‐I and C‐P4H‐II are well‐characterized.11 C‐P4H‐I is the most abundant isoform and it is found in all tissues studied, whereas C‐P4H‐II is enriched in chondrocytes, osteoblasts and endothelial cells.11 Small‐angle X‐ray scattering (SAXS) studies have shown that the four subunits in C‐P4H‐I are assembled into a rod‐shaped molecule in solution.12
C‐P4H enzymes have a small PRD in the middle of their α subunit (preceding the C‐terminal catalytic region). This domain, referred to as peptide‐substrate‐binding (PSB) domain, is around 100 amino acid residues long, being a helical protein composed of five helices that fold into two repeats of the tetratricopeptide repeat (TPR) motif plus a C‐terminal extra helix.13, 14, 15 The recombinant PSB domains of C‐P4H‐I (PSB‐I) and C‐P4H‐II (PSB‐II) have similar peptide‐binding properties as the corresponding full‐length enzymes.13, 14 They bind procollagen mimetics with multiple Pro‐Pro‐Gly triplets [(PPG)n], and typically the affinity of the peptide‐PSB complex increases with increasing chain length of the (PPG)n peptide.13, 14 Surface plasmon resonance experiments show that the affinity of PSB‐I/C‐P4H‐I for (PPG)10 (Kd is 30 μM/20 μM) is at least five times higher than the corresponding affinity of PSB‐II/C‐P4H‐II (Kd is 290 μM/100 μM).14 Also, PSB‐I binds tightly poly(l‐proline),13 which is the competitive inhibitor of C‐P4H‐I, whereas PSB‐II has much weaker affinity,14 agreeing with the notion that polyproline is not an effective inhibitor for C‐P4H‐II.7 Crystal structures of PSB‐I, being part of a larger construct including also the N‐terminal dimerization domain of the α(I) subunit (referred to as the double domain construct, DD), have been determined, complexed with (PPG)3 and a polyproline peptide with nine prolines [(P)9].16 These structures show that both peptides bind with the same directionality in an extended PPII‐helix conformation in the peptide‐binding groove. Like in other PRD‐PRS complexes,1 there are several tyrosines lining the peptide‐binding groove (Figs. 1 and 2). The mode of binding of (PPG)3 and (P)9 to PSB‐I differs from the interactions of other PRDs with PRSs, by (i) lacking a central tryptophan residue interacting with the peptidyl prolines and by (ii) having several water‐mediated hydrogen bonds between the peptide and the PSB domain (instead of direct hydrogen bonds). Similarly, to the other PRD‐PRS complexes, these peptides are bound in PPII‐helix conformation to PSB‐I.16 In both peptide complex structures of PSB‐I, the interactions extend over all residues of the nine‐residue long peptides. However, the two most C‐terminal triplets interact more tightly and their Y‐position prolines, referred to as Pro5 and Pro8 in the (PPG)3 complex structure (Fig. 2), anchor the peptide into its binding groove.16
Figure 1.
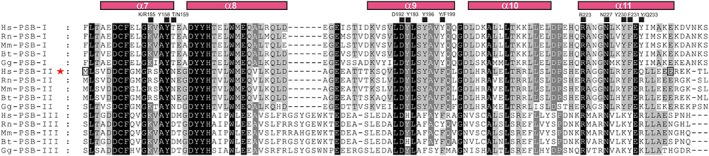
Alignment of PSB domain sequences (residues Phe144‐Ser244 of human C‐P4H‐I) from various species. The alignment includes the PSB domains from the three C‐P4H α subunit isoforms of human (Hs‐PSB‐I, Hs‐PSB‐II, Hs‐PSB‐III), rat (Rn‐PSB‐I, Rn‐PSB‐II, Rn‐PSB‐III), mouse (Mm‐PSB‐I, Mm‐PSB‐II, Mm‐PSB‐III), bovine (Bt‐PSB‐I, Bt‐PSB‐II, Bt‐PSB‐III), and chicken (Gg‐PSB‐I, Gg‐PSB‐II, Gg‐PSB‐III). The helices of the PSB domain (α7–α11) are numbered according to the full‐length C‐P4H‐α(I) subunit secondary structure16 and highlighted above the sequences (magenta). The residues in the peptide‐binding groove that interact with the bound peptide are highlighted by filled black squares and numbered according to human C‐P4H‐I α(I) subunit amino acid sequence numbering. The human PSB‐II sequence is highlighted with a red star and the first (Met142) and the last (Glu236) residues of the PSB‐II construct used in this study are highlighted by open squares.
Figure 2.
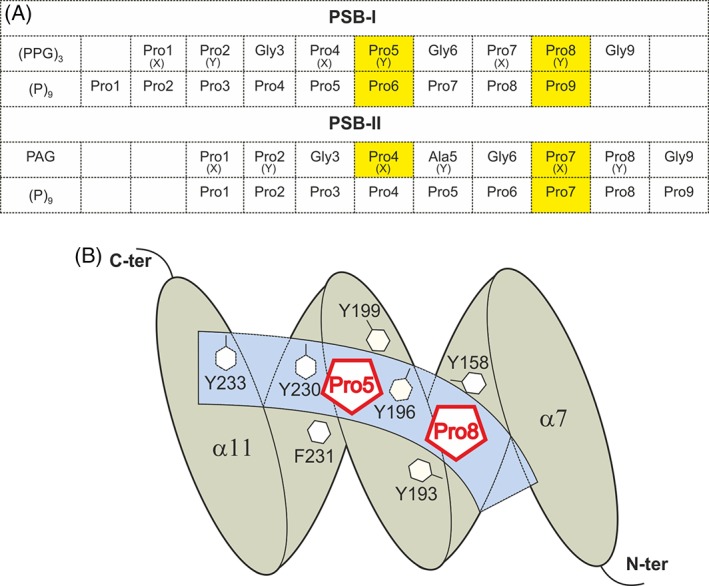
The mode of binding of proline‐rich peptides to PSB‐I and PSB‐II. (A) The residue‐numbering scheme of the bound peptides in the different peptide complex structures of PSB‐I16 and PSB‐II (this study). The (PPG)3 mode of binding to PSB‐I has identified two proline binding pockets, being the Pro5 and Pro8 binding pocket. For each peptide, the proline residues that are bound in the Pro5 and Pro8 pockets are highlighted in yellow. For each peptide, all nine residues have been built in the electron density map and the residues of each peptide are numbered from 1 to 9. (X) and (Y) identify the X‐ and Y‐positions of the corresponding ‐X‐Y‐G‐triplet. (B) Schematic drawing showing the peptide‐binding groove (light blue) with respect to the five helices of the TPR fold of PSB‐I (gray), as known from binding studies with (PPG)3. 16 The Pro5 and Pro8 binding pockets are shown. The side chains of the six aromatic residues lining the Pro5 and Pro8 pockets, as well as Tyr233 at the C‐terminal end of the PSB peptide‐binding groove, are highlighted. The first and the last α helix (α7, α11), and the N‐ and C‐termini of the PSB fold are labeled.
The exact role of the PSB domain in the C‐P4H reaction mechanism is currently unknown.9, 12 Its sequence characteristics are correlated with the different substrate binding properties of C‐P4H‐I and C‐P4H‐II.13, 14, 15 The affinity and the mode of peptide‐binding of human PSB‐II have been studied here, using biophysical characterization methods and X‐ray crystallography, and are compared with the corresponding PSB‐I properties. The results reveal a new mode of binding of PRSs to the PSB domain, in which the PRS does not have the PPII‐helix conformation and in which direct protein‐peptide hydrogen bonds with the conserved Tyr158, Arg223, and Asn227 side chains are critically important. The anchoring interactions of the prolines of the second and third triplet of the bound peptide with these side chains are described.
Results
Recombinant PSB‐II is an α helical and monomeric protein
PSB‐II was expressed and purified to homogeneity using a construct of residues Met142‐Glu236 of human C‐P4H‐II (corresponding to PSB‐I amino acids Met144‐Glu238, (Fig. 1). A His‐tag precedes the PSB‐II sequence such that the N‐terminal sequence of the construct is Met‐His6‐Met‐Leu‐Ser‐ (Fig. 1). According to static light scattering (SLS) measurements, PSB‐II is a monodisperse and monomeric protein with an absolute molecular weight of 12 kDa (Supporting Information, Fig. S1). Circular dichroism (CD) spectroscopy studies indicate that this construct is well‐folded with mainly (~75%) α‐helical structure [Supporting Information, Fig. S2(A,B)]. The melting temperature (Tm) is 60.3°C based on its CD melting curve [Supporting Information, Fig. S2(C)].
Calorimetric binding studies with proline‐rich peptides
The binding affinities of PSB‐II for several nine‐residue proline‐rich peptides were studied with isothermal titration calorimetry (ITC), including (PPG)3 and (P)9. Of the latter peptides, the structures of the corresponding PSB‐I complexes (PSB‐I being part of DD‐I) are also known.16 In contrast to PSB‐I, having high affinity for (P)9 (Kd = 9.8 μM) and (PPG)3 (Kd = 143.5 μM),16 the affinity of (P)9 and (PPG)3 for PSB‐II is low and nondetectable, respectively, as measured in these ITC experiments (Table 1, Supporting Information, Fig. S3). In addition, the affinities of the PPG‐PAG‐PPG (PAG), PPG‐PRG‐PPG (PRG), and PPG‐PEG‐PPG (PEG) peptides were determined. The affinities of these three peptides were studied because in the procollagen sequence, after Hyp (34%), the most common residues in the Y‐position are alanine (18%) and arginine (13%).6 Glutamate is rarely found in the Y‐position (2%), but it is a common residue (13%) in the X‐position.6 Interestingly, the ITC experiments showed that the PAG, PRG, and PEG peptides bind with significant affinity to PSB‐II with Kd values of 19.7 μM, 6.8 μM, and 29.2 μM, respectively (Table 1, Supporting Information, Fig. S3). The binding of these peptides to PSB‐II is enthalpy driven (Fig. 3). The much more favorable binding enthalpy term for the PRG and PEG peptides, as compared to the PAG peptide, is to a great extent compensated by a more unfavorable binding entropy term.
Table 1.
Calorimetric Binding Studies of PSB‐II with the Proline‐Rich Peptides
| Peptide | Kd (μM) | ΔH (kcal/mol) | −TΔS (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|---|---|
| (PPG)3 | NDa | |||
| (P)9 | >1500b | |||
| PPG‐PAG‐PPG | 19.7 ± 0.7 | −6.6 ± 0.1 | 0.10 ± 0.03 | −6.45 ± 0.02 |
| PPG‐PRG‐PPG | 6.8 ± 1.0 | −8.7 ± 0.8 | 1.64 ± 0.07 | −7.05 ± 0.09 |
| PPG‐PEG‐PPG | 29.2 ± 0.7 | −9.3 ± 0.5 | 3.08 ± 0.28 | −6.19 ± 0.03 |
Binding could not be detected.
Kd could not be determined accurately due to weak affinity.
Figure 3.
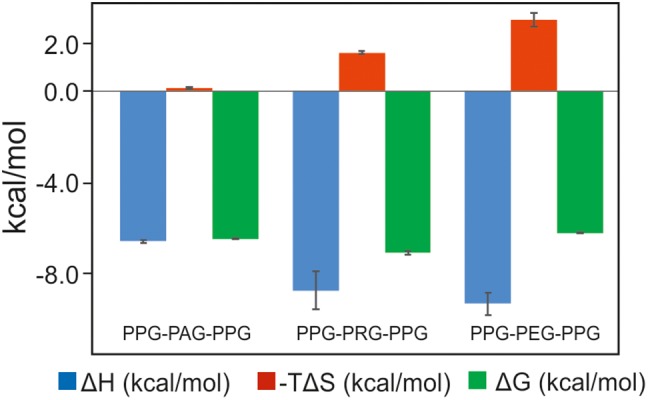
The thermodynamic fingerprint (ΔH, −TΔS, ΔG) of the binding to PSB‐II of the PAG, PRG, and PEG peptides. The error bars are also shown.
Crystal structure of the unliganded PSB‐II
PSB‐II crystallized in the P3221 space group with one molecule in the asymmetric unit, and a complete X‐ray data set was collected at 1.87 Å resolution (Table 2). The electron density maps were clearly interpretable from Leu145 to Glu236 (to simplify comparisons the PSB‐I amino acid numbering is used from here onward, the actual numbers in the PSB‐II sequence are Leu143 and Glu234, respectively). The overall TPR fold of the PSB‐II structure is very similar to that of PSB‐I. A root mean square deviation of 1.2 Å is calculated for 92 corresponding Cα atoms when superimposing (using the SSM‐option17 of COOT18) the PSB‐II structure on the PSB‐I structure (PDB code 2V5F [Protein Data Bank, http://www.rcsb.org]). The small rearrangements of the side chains of Tyr196, Arg223 and Asp192 (Fig. 4) correlate with differences in crystal contacts, being that in this structure of the PSB‐I domain, the binding groove is complexed with the C‐terminal His‐tag of a neighboring molecule.
Table 2.
Data Collection and Refinement Statistics
| Data set | PSB‐II (unliganded) | PSB‐II_(P)9 | PSB‐II_PAG | PSB‐II_PRG | PSB‐II_PEG |
|---|---|---|---|---|---|
| Data collection | |||||
| X‐ray source | Bruker X8, Oulu | Bruker X8, Oulu | ESRF, ID30A‐3 | ESRF, ID30A‐3 | ESRF, ID30A‐3 |
| Data processing software | Proteum2 | Proteum2 | XDS/Aimless | XDS/Aimless | XDS/Aimless |
| Data collection statistics | |||||
| Unit cell parameters (Å) | a = 55.45 b = 55.45 c = 71.71 α = β =90.0 γ = 120 |
a = 56.90 b = 56.90 c = 67.87 α = β = 90.0 γ = 120.0 |
a = 54.93 b = 54.93 c = 72.94 α = β = 90.0 γ = 120.0 |
a = 55.38 b = 55.38 c = 72.99 α = β = 90.0 γ = 120.0 |
a = 55.02 b = 55.02 c = 73.27 α = β = 90.0 γ = 120.0 |
| Space group | P3221 | P3221 | P3221 | P3221 | P3221 |
| Resolution range (Å) | 48.0–1.87 (1.90–1.87) |
39.9–2.00 (2.03–2.0) |
47.6–1.48 (1.51–1.48) |
48.0–1.55 (1.58–1.55) |
40.0–1.68 (1.71–1.68) |
| Vm (Å3/D) | 2.7 | 2.7 | 2.7 | 2.7 | 2.7 |
| Molecules per asymmetric unit | 1 | 1 | 1 | 1 | 1 |
| Number of observations | 157415 (2725) | 96188 (1321) | 183665 (9180) | 162871 (8338) | 124144 (6725) |
| Redundancy | 14.3 (5.2) | 10.7 (3.4) | 8.4 (8.7) | 8.4 (8.8) | 8.2 (8.8) |
| Completeness (%) | 99.9 (99.8) | 99.4 (93.9) | 100 (100.0) | 99.9 (100.0) | 99.6 (99.9) |
| I/s(I) | 15.0 (2.0) | 25.7 (2.6) | 17.6 (2.2) | 19.0 (2.0) | 21.3 (2.1) |
| Rmerge (%) | 13.9 (62.9)a | 6.1 (35.8)a | 5.1 (91.5) | 5.4 (103.5) | 3.9 (99.4) |
| Rpim (%) | 3.8 (29.8) | 1.9 (21.0) | 1.8 (32.2) | 2.0 (36.6) | 1.4 (35.4) |
| Wilson B‐factor (Å2) | 54.4 | 54.2 | 22.0 | 21.0 | 34.4 |
| Refinement statistics | |||||
| Resolution (Å) | 48.0–1.87 (1.91–1.87) | 39.9–2.00 (2.06–2.00) | 28.9–1.48 (1.52–1.48) | 48.0–1.55 (1.59–1.55) | 40.0–1.68 (1.73–1.68) |
| Rwork (%) | 18.2 (33.1) | 17.0 (23.6) | 16.3 (25.2) | 15.8 (23.3) | 18.3 (28.2) |
| Rfree (%) | 22.4 (39.9) | 21.9 (30.2) | 17.9 (26.1) | 18.0 (24.8) | 21.5 (31.9) |
| Number of reflections | 10951 | 8927 | 21721 | 19238 | 14962 |
| Number of atomsb | 939 | 955 | 960 | 971 | 894 |
| Protein | 798 | 776 | 790 | 808 | 772 |
| Peptide | – | 64 | 53 | 59 | 56 |
| Sulfate | 5 | 5 | 10 | 10 | 5 |
| DMSO | 4 | 4 | 16 | 12 | 12 |
| Glycine | 10 | – | – | – | – |
| Waters | 122 | 106 | 91 | 82 | 49 |
| Geometry statistics | |||||
| Rmsd bond length (Å) | 0.006 | 0.006 | 0.007 | 0.008 | 0.009 |
| Rmsd bond angle (°) | 0.8 | 0.9 | 1.0 | 1.0 | 1.0 |
| Average B‐factor (Å 2 ) | |||||
| All atoms | 17.4 | 23.9 | 31.5 | 31.9 | 48.3 |
| Protein | 15.2 | 21.8 | 28.5 | 28.7 | 46.5 |
| Peptide | – | 36.1 | 51.1 | 50.7 | 60.0 |
| Sulfate | 41.9 | 23.2 | 56.0 | 67.4 | 81.2 |
| DMSO | 29.5 | 44.1 | 47.6 | 56.9 | 70.9 |
| Waters | 29.3 | 31.2 | 33.1 | 42.4 | 54.1 |
| Ramachandran plot | |||||
| Favored (%) | 97.8 | 99.0 | 100.0 | 100.0 | 99.0 |
| Additionally allowed (%) | 2.2 | 1.0 | 0.0 | 0.0 | 1.0 |
| Generously allowed (%) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| PDB code | 6EVL | 6EVM | 6EVN | 6EVO | 6EVP |
Rint value calculated by the Proteum2 package.
Non‐hydrogen atoms.
Figure 4.
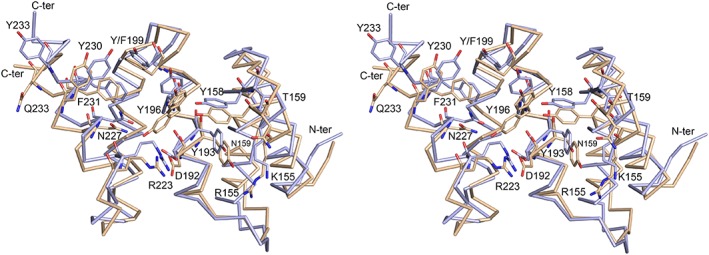
Comparison of the Cα trace of the unliganded structure of the PSB‐II domain (light brown) and the structure of the PSB‐I domain (light blue, PDB code 2V5F) (standard view, stereo). The side chains of the key residues forming the peptide‐binding groove of the PSB domain are shown. Tyr196 adopts two conformations in the unliganded structure of PSB‐II, the major conformation points to Arg223.
Crystallographic binding studies with proline‐rich peptides
PSB‐II cocrystallization experiments were carried out with (PPG)3 and (P)9. PSB‐II crystals were obtained (in the same condition as used for the unliganded crystals) in the presence of 5 mM (PPG)3, but the peptide was not present in the structure. However, the structure of a PSB‐II‐peptide complex, refined at 2.0 Å resolution (Table 2), and referred to as the PSB‐II_(P)9 structure, was obtained on cocrystallization with (P)9. The crystal packing of this PSB‐II_(P)9 complex crystal form is different from the unliganded PSB‐II structure although the space group is the same and the cell dimensions are similar. All nine prolines of (P)9 could be built in electron density, and the C‐terminal part of the peptide is best defined [Fig. 5, Supporting Information Fig. S4(A)]. The peptide is bound in a PPII‐helix conformation (Supporting Information, Table SI). The C‐terminal region, Pro7–Pro9, interacts tightly with the binding groove via direct hydrogen bonds and via pi‐stacking interactions. Direct hydrogen bonds are formed between the OH groups of Tyr158 and Tyr193 and the peptidyl oxygens of Pro7 and Pro8, respectively. The C‐terminal carboxylate oxygen atoms (of Pro9) are hydrogen‐bonded to the PSB‐II Arg155 and Asn159 side chains (Fig. 5) and this carboxylate group is well‐defined by its corresponding electron density [Supporting Information Fig. S4(A)], indicating that other possible modes of binding (reverse or shifted) occur at most with low occupancy. The N‐terminal prolines (Pro1–Pro3), instead, are interacting only loosely via one water‐mediated hydrogen bond to Gln233 [Fig. 5(B)]. However, this N‐terminal peptide region interacts also with symmetry‐related molecules [Fig. 5(A)]. These additional interactions, caused by the crystal packing, rationalize why it has been possible to capture this mode of binding, despite the low affinity of the peptide, as suggested by the ITC studies. There is also a water‐mediated hydrogen bond interaction between the side chain of Asn227 and the main chain oxygen of Pro5. The Pro8 pocket, occupied by the Pro7 of (P)9, is lined by PSB‐II Tyr158, Tyr193 and Tyr196, and the Arg223‐Asp192 salt bridge. Only the side chain of Tyr196 and the Arg223‐Asp192 salt bridge have different conformations when comparing the unliganded PSB‐II and PSB‐II_(P)9 structures.
Figure 5.
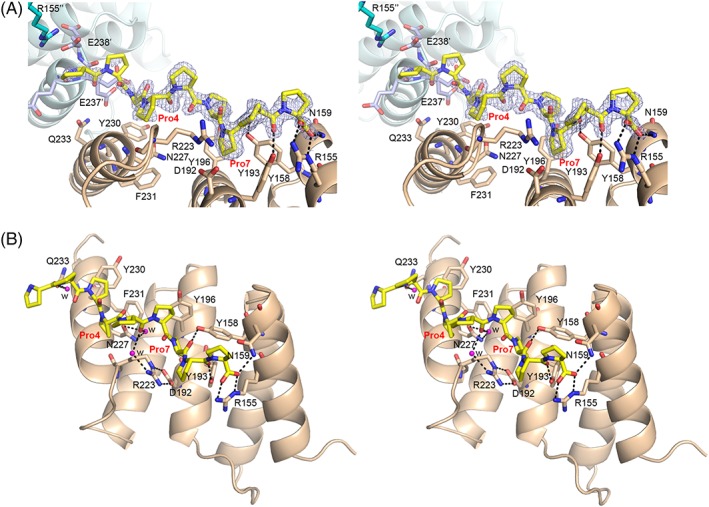
The mode of binding of (P)9 to PSB‐II. (A) Stereo view showing the (P)9 peptide (yellow sticks) bound to the PSB‐II domain (light brown ribbon, side view). The omit 2Fo−Fc electron density map, calculated after three cycles of omit refinement (leaving the peptide out of the model) and contoured at 1.0 σ, is also drawn around the (P)9 peptide. The two key prolines, Pro4 and Pro7, of the (P)9 peptide are labeled in red. The direct hydrogen bonds between the key residues of PSB‐II (shown in light brown sticks) and (P)9 are shown. Residues from two neighboring PSB‐II molecules, which also interact with the N‐terminal region of the bound (P)9 peptide, are also shown. It concerns the C‐terminal residues Glu236′‐Glu238′ (shown in light blue sticks) of the symmetry‐related molecule 1 (shown in light blue ribbon), and Arg155′′ of the symmetry‐related molecule 2. The view is rotated ~90° clockwise around the horizontal axis from the standard view. (B) Stereo view showing the PSB‐II–(P)9 interactions in detail (standard view). Three water molecules sharing hydrogen bonds with PSB‐II and with (P)9, are shown in magenta spheres. Prolines 4 and 7 of (P)9 are labeled in red.
PSB‐II was cocrystallized with PAG in very similar conditions as used for the unliganded PSB‐II and the PSB‐II_(P)9 complex. The structure of this complex, refined at 1.48 Å (Table 2), is referred to as the PSB‐II_PAG structure. The packing of the molecules is the same as the crystal packing of the unliganded PSB‐II and different from the PSB‐II_(P)9 crystal form. In this crystal form, the neighboring molecule is not interacting with the N terminus of the peptide, as seen in the PSB‐II_(P)9 crystal form. The presence and conformation of the PAG peptide are clearly defined by its electron density [Fig. 6(A) and Supporting Information, Fig. S4(B)] and all nine residues of the PAG peptide were built. The most C‐terminal PPG triplet of the PAG peptide interacts tightly with the peptide‐binding groove in such a way that the X‐position proline of the third triplet, Pro7, binds in the Pro8 pocket (Fig. 6). The carboxylate oxygens of the glycine of this triplet (Gly9, C‐terminus of the PAG peptide) are hydrogen‐bonded to the side chains of Asn159 and Arg155, suggesting, like in the PSB‐II_(P)9 complex, that other modes of binding (shifted or reversed) occur most at low occupancy. The side chains of Tyr193 and Tyr158 form direct hydrogen bonds with the PAG peptide oxygens like in the PSB‐II_(P)9 complex. However, in the PAG complex, PSB‐II Tyr193 is interacting with the C‐terminus of the peptide (Fig. 6), whereas in the PSB‐II_(P)9 complex, Tyr193 is hydrogen‐bonded to Pro8 (Fig. 5). Unlike in the PSB‐II_(P)9 complex structure, also peptide residues 3–5 interact tightly with the PSB‐II domain. The X‐position proline of the second triplet of the PAG peptide (Pro4) is bound in the Pro5 pocket lined by Tyr196, Phe/Tyr199, Tyr230 and Phe231 (Figs. 2 and 6). The conformation of the Asn227 side chain is well‐defined by the electron density map [Supporting Information Fig. S4(B)] and the mode of binding of the PAG peptide allows for the formation of direct hydrogen bonds between the OD1 and the ND2 side chain atoms of Asn227 and the peptide backbone N atom and carbonyl oxygen, respectively, of Ala5 of the middle triplet of the PAG peptide. Tyr196 is also involved in the hydrogen bond network between the PSB‐II and the PAG by forming one water and one DMSO‐mediated hydrogen bond with the PAG peptide [Fig. 6(B)]. The N‐terminal triplet of the PAG peptide is not interacting with the groove and this triplet has weak electron density in the structure [Fig. 6(A)].
Figure 6.
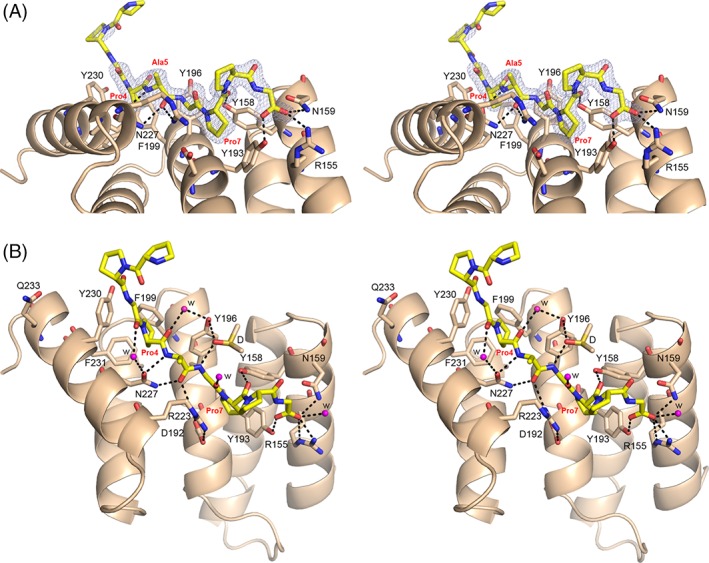
The mode of binding of PAG to PSB‐II. (A) Stereo view showing the peptide (yellow sticks) bound to the PSB‐II domain (light brown ribbon) (side view). The omit 2Fo−Fc electron density map, calculated after three cycles of omit refinement (leaving the peptide out of the model) and contoured at 1.0 σ, is also drawn around the PAG peptide. Pro4, Pro7, and Ala5 of the PAG peptide are labeled in red. The direct hydrogen bonds between the key residues of PSB‐II (shown in light brown sticks) and PAG are shown. (B) Stereo view showing the PSB‐II–PAG interactions in detail (standard view). The five waters (magenta) and DMSO (yellow) involved in the hydrogen bond network around PAG, are also shown. Pro4 and Pro7, as well as Ala5 of PAG are labeled in red.
The cocrystallization experiments with the PRG and PEG peptides produced the same crystal form as with the PAG peptide and as with the unliganded PSB‐II crystal form. The structures of these complexes, refined at 1.55 Å and 1.68 Å (Table 2) and referred to as the PSB‐II_PRG and PSB‐II_PEG structures, respectively, are very similar to the PSB‐II_PAG structure and the mode of peptide binding is identical with the PAG complex, such that the C‐terminus of the peptide has well‐defined electron density whereas the N terminus is weakly bound [Supporting Information, Fig. S5(A‐D)]. In this mode of binding, the side chain of the Y‐position residue of the second triplet (being either Ala, Arg, or Glu in the PAG, PRG, and PEG structures, respectively) is pointing to the bulk solvent. In the PRG and PEG structures, the side chains of Arg5 and Glu5, respectively, are disordered and have only weak density in the refined electron density maps [Supporting Information, Fig. S5(B,D)].
Discussion
C‐P4H is a processive enzyme19 and it has been proposed that the PSB domain anchors the unhydroxylated procollagen in such a way that the catalytic domain of the α subunit can do several successive hydroxylations without being released from its substrate.12, 13 Therefore, the PSB domain is expected to have an important role in the substrate specificity of C‐P4Hs.13, 14 Indeed, the characterization of variants of mutated C‐P4H‐I tetramer and the corresponding mutated PSB domain, show that the Km values for (PPG)10 of the C‐P4H‐I tetramer variants correlate well with the affinity of this model substrate to the PSB‐I domain variants.15 The peptide‐binding studies of PSB‐II reported here highlight that PSB‐II has much weaker affinity to (P)9 and (PPG)3 as compared to PSB‐I.16 Previous studies have shown that (PPG)5 and (PPG)10 do bind to PSB‐II, although with six and nine times weaker affinity, respectively as compared to PSB‐I.14
The affinity of the homologues of (PPG)3 in which the Y‐position proline of the middle PPG triplet is changed to a non‐proline residue, like an alanine, arginine, or glutamate is dramatically increased (Table 1), being around 10 times higher than the previously reported affinity of (PPG)10 to PSB‐II.14 The structures of these complexes show that the overall, extended, mode of binding of these peptides is the same as in PSB‐I. In both structures, the peptide‐binding groove is lined by several tyrosines and by the Arg223‐Asp192 salt bridge. Importantly, also the directionality of peptide binding is the same (Fig. 7), whereas typically, PRDs can bind PRSs in both ways, from N to C or from C to N.1 The precise mode of binding of the peptides in PSB‐II differs from the PSB‐I mode of binding. The shape of the binding pockets is also different (Fig. 7), in particular at both flanking ends, such as the presence of Gln233, which is a tyrosine in PSB‐I (Fig. 1). This tyrosine interacts with the prolines of the N‐terminal triplet in the PSB‐I complexes. The presence of Gln233 at the N‐terminal end of the peptide‐binding groove in PSB‐II apparently prevents the rigid (P)9 peptide to bind deeper into the groove. The importance of this sequence difference between C‐P4H‐I and C‐P4H‐II for their substrate specificity properties have been described.13 At the C‐terminal end of the peptide, its carboxylate oxygens are hydrogen‐bonded to the side chains of Arg155 and Asn159 (Figs. 5 and 6) in the PSB‐II complexes. In PSB‐I, the corresponding residues are a lysine and a threonine, respectively, and these residues do not interact with the bound peptides in the PSB‐I complex structures.16
Figure 7.
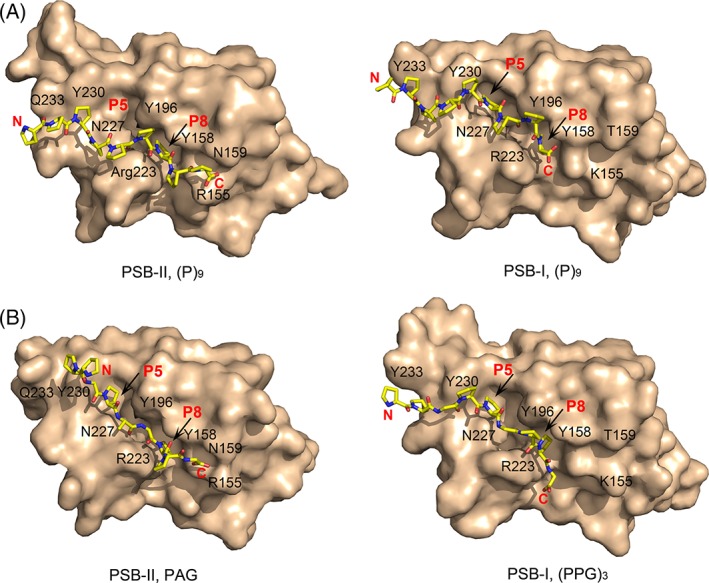
Comparison of the peptide mode of binding to PSB‐II and PSB‐I. (A) The (P)9 mode of binding to PSB‐II (left) and PSB‐I (right, PDB code 4BT9). (B) The PAG mode of binding to PSB‐II (left) and the (PPG)3 mode of binding to PSB‐I (right, PDB code 4BT9). The PSB domains are shown using the surface representation (standard view), and the bound peptides are shown with yellow stick representation. Some key residues shaping the peptide‐binding groove are labeled. Also the Pro5 and Pro8 binding pockets are indicated with red P5 and P8 labels, respectively. In each of these complexes, the full‐length nine‐residue peptide has been built.
These peptides interact with PSB‐II most strongly via their second and third triplet in such a way that the prolines in the X‐positions of the second (PAG/ PRG/ PEG) and third (PPG) triplets bind in the Pro5 and Pro8 binding pockets, respectively. This mode of binding is different from the mode of binding of (PPG)3 to PSB‐I, where the prolines in the Y‐position of the second and third triplet of (PPG)3 bind to these two pockets (Fig. 2 and Supporting Information, Fig. S6).16 Importantly, the middle triplets of the PAG, PRG, and PEG peptides bind more deeply into the groove, facilitating the formation of direct hydrogen bonds between the peptide and the side chains of Tyr158, Arg223, Asn227 (Fig. 8). The latter residues are well‐conserved in the multiple sequence alignment (Fig. 1). The direct hydrogen bond between ND2(Asn227) and the peptide oxygen of Ala5 (or Arg5 or Glu5) corresponds to the direct hydrogen bond in the SH3 and EVH1 PxxP peptide binding, which is one of the fully conserved interactions (together with the two buried prolines of the PxxP motif) that characterize the mode of binding of these peptides.1 In the PSB‐I_(PPG)3 complex structure, Asn227 participates in peptide binding only via water‐mediated hydrogen bonds (Fig. 8). The replacement of a proline in the Y‐position of the second triplet of (PPG)3 allows Ala5 (or Arg5 or Glu5) to adopt a phi/psi main chain conformation of −135°/130° (Supporting Information, Table SI), whereas the phi value of a proline is restricted to approximately −60°.2 Gly6, adopts phi/psi values of +90/−177, restricting this position to only a glycine residue, thereby suggesting that the PSB‐II binding motif is PxGP, of which the two proline residues correspond to the X‐position prolines in the ‐X‐Y‐Gly‐ collagenous repeats. This PxGP‐motif (where x is not a proline) occurs 31 times in the triple helical region (~1000 residues) of the human procollagen α1(I) chain.
Figure 8.
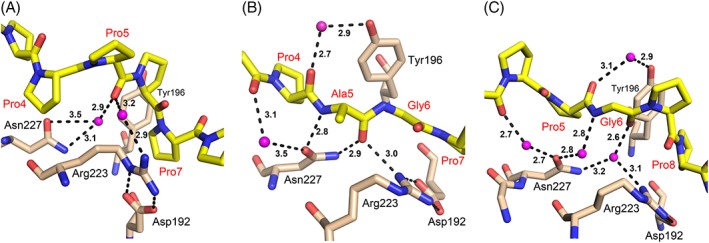
The interactions of the conserved Asn227 and Arg223‐Asp192 salt bridge in the three different modes of binding of a proline‐rich peptide to the PSB domain. (A) The loose mode of binding of (P)9 to PSB‐II, (B) The deep mode of binding of PAG to PSB‐II. (C) The water‐mediated mode of binding of (PPG)3 to PSB‐I (PDB code 4BT9). The dotted lines identify hydrogen bond interactions; the labels specify the corresponding distances (in Å).
The structures of the PSB‐II complexes with the three PxGP peptides show that the peptide‐binding groove in PSB‐II is shorter than in PSB‐I, extending only over two triplets, whereas in the PSB‐I complexes it interacts with all three triplets. The structural data also show that these relatively short, tight binding, peptides, when complexed to PSB‐II, do not adopt the PPII‐helix conformation. Further structural enzymological studies with PSB‐II, and also with PSB‐I, have been initiated to probe the importance of the PxGP motif, when embedded in longer peptides, for tight binding to the PSB domains.
Materials and Methods
Cloning, expression and purification of human PSB‐II
The PSB‐II construct was generated using standard molecular biology protocols using a codon optimized gene for the full‐length CP4H α(II) subunit (GenScript). The construct Met142‐Glu236 [corresponding to Phe144‐Glu238 of human PSB‐I (Fig. 1) was amplified from this template using a forward primer containing the NdeI site (underlined) (5′‐GAAGGAGATATACATATGCACCACCACCACCACCACATGCTGAGCGTG‐3′) and a reverse primer containing the BamHI site (underlined) followed by a STOP codon (TAA) (5′‐GAGCTCGAATTCGGATCCTTATTCTTCTTCCAGCAGCTGTTCAAAATAGCG‐3′). The amplified product was digested using NdeI and BamHI. This was then ligated into a linear (double digested) pET22b(+) plasmid (Novagen) and its sequence was verified by DNA sequencing. This N terminally His‐tagged PSB domain was expressed using Rosetta™(DE3)pLysS cells in LB medium containing 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. The cells were grown at 37°C until the OD600 reached 0.6–0.8 after which they were induced for protein expression using 0.5 mM isopropyl β‐D‐1‐thiogalactopyranoside and kept at 20°C overnight. The cells were harvested and resuspended in lysis buffer composed of 50 mM tris, 100 mM NaCl, and 100 mM glycine at pH 8.0. After sonication, the supernatant was loaded on a Talon® metal affinity resin column (Clontech Laboratories Inc.), washed with lysis buffer and eluted in the same buffer containing 250 mM imidazole. The fractions were pooled, concentrated, and loaded on a Superdex75 16/600 preparative grade size‐exclusion chromatography (SEC) column (GE Healthcare), preequilibrated with SEC buffer composed of 20 mM tris, 50 mM NaCl, and 50 mM glycine at pH 8.0. The final protein solution was concentrated to about 6 mg/mL (concentration measured with a NanoDrop 1000, Thermo Scientific), and stored in SEC buffer in 100 μL aliquot volumes at −70°C.
Static light scattering
SLS analysis of PSB‐II was carried out using the miniDAWN™ TREOS multiangle SLS detector (Wyatt Technologies) in on‐line mode. For these experiments, 100 μL of the purified PSB‐II sample was filtered (with 0.1 μm pore size) and loaded onto a Superdex200 10/300GL column (GE Healthcare) preequilibrated with SEC buffer. Before entering the SLS detector, the samples flow through a RI‐101 refractive index (RI) detector (Shodex). The RI‐signal is used to measure the concentration of the protein sample. Molecular mass, polydispersity, and other analyses were carried out using the ASTRA software (Wyatt Technologies).
Circular dichroism
The CD spectra of purified PSB‐II were obtained using a Chirascan™ CD spectrophotometer (Applied Photophysics Ltd.) in a quartz cuvette with 1 mm path length. PSB‐II was diluted to 0.1 mg/mL using filtered milli‐Q water, the final buffer conditions being 2 mM tris, pH 7.8, 5 mM glycine, and 5 mM NaCl. A wavelength scan from 180 nm–260 nm was used to analyze the secondary structure of the protein. Thermal denaturation was recorded by measuring the CD spectrum between 190 nm and 260 nm using a QUANTUM temperature controller with a 2°C step size at 1°C/min ramp rate with ±0.2°C tolerance. Data analysis was carried out using Pro‐Data Viewer (Applied Photophysics Ltd.), CDNN (http://bioinformatik.biochemtech.uni-halle.de/cdnn), and Global3 (Applied Photophysics Ltd.).
Isothermal titration calorimetry
The ITC experiments were performed using a MicroCal iTC‐200 microcalorimeter (Malvern). All the used peptides were ordered from TAG Copenhagen A/S (Denmark). In the ITC experiments, 280 μL of PSB‐II protein (25 μM) was loaded into the sample cell and 40 μL of each peptide (1 mM) was loaded into the injection syringe. PSB‐II and the peptides were dissolved in the SEC buffer. The experiments were performed according to the instructions provided by the manufacturer. Titration measurements of peptides into the buffer (without protein) were used for the baseline correction calculations. The titrations were carried out at 25°C with a stirring rate of 750 rpm. Titrations for binding were initiated by one injection of 0.4 μL followed by 3.6 μL injections every 150 s. In total 11 injections were monitored. ITC data analyses were done using the ORIGIN software (OriginLab). The first injection was excluded from integration according to the manufacturer's instructions. In particular, the affinities of the PAG and PEG peptides are relatively weak, therefore the integrated data were analyzed using the one set of sites fitting model with a fixed stoichiometry (n = 1),20 ensuring that the calculated affinities of the PAG, PRG and PEG peptides can be compared. All measurements were repeated three times and the standard deviations are shown in Table 1 and Fig. 3.
Crystallization, data collection, and structure refinement
Crystallization trials were carried out using in‐house screens21 and the experiments were done in IQ 96‐well sitting drop plates (TTP Labtech) using the mosquito LCP nanodispenser (TTP Labtech). Initial hits were obtained only at 4°C with PSB‐II in the absence of peptides using well solutions containing 100 mM MOPS, pH 7.5, 2.45 M ammonium sulfate and 10% DMSO. The protein was then subjected to an additive screen (Hampton Research) crystallization experiment using the above solution as the reference condition. Several additives improved the crystal shape, and the unliganded PSB‐II crystal (~200 μm in size) obtained with 4% (v/v) pentaerythritol ethoxylate (Hampton Research) was used for further crystallographic studies.
Cocrystallization experiments were performed with (PPG)3, (P)9, PPG‐PAG‐PPG, PPG‐PRG‐PPG, and PPG‐PEG‐PPG and at 4°C. The peptides were added to the protein solution at a final concentration of 5 mM [except (P)9, where 2.0 mM final concentration was used] and incubated for 45 min–60 min on ice before setting up the crystallization experiments, always in the cold room at 4°C. PSB‐II crystallizes in the presence of (PPG)3, (P)9, PAG, PRG, and PEG peptides in the same conditions as used for the experiments with the unliganded PSB‐II. These crystals were grown in the presence of the following additives: 10 mM copper(II)chloride dihydrate, 4% hexanediol, 3% d‐galactose, 5% PEG 400, and 10 mM EDTA respectively, of the crystallization additive screen (Hampton Research).
For the X‐ray experiments, crystals were frozen in the cold room by transfer into liquid nitrogen. No cryoprotectant was used for the freezing. From all PSB‐II crystals, data sets with a resolution of approximately 1.9 Å–2.1 Å were obtained, using the in‐house Microstar X8 Proteum Cu‐rotating anode X‐ray generator (Bruker) of the Biocenter Oulu protein X‐ray crystallography core facility. A synchrotron source was also used for data collection and for the PSB‐II_PAG, PSB‐II_PRG, and PSB‐II_PEG crystals higher resolution data sets were obtained at the European radiation synchrotron facility, Grenoble (ESRF), France (Table 2). The home source data were integrated and processed using the Proteum2 software package (Bruker), whereas the synchrotron data were integrated with XDS22 by using the autoPROC data processing pipeline23 at the ESRF, and then scaled with AIMLESS.24 Structure determination, structure refinement, and model validation were done using the Phenix package25 and the model building was done with COOT.18 The PSB‐I structure (PDB code 2V5F) was used as an initial search model in the molecular replacement calculations of unliganded PSB‐II by PHASER.26 The bound peptides were built in their corresponding density once the structure of the protein part had been well‐refined. A summary of the refinement and validation results is presented in Table 2. The coordinates and structure factors of the refined structures have been deposited in the Protein Data Bank with accession codes 6EVL (unliganded PSB‐II), 6EVM [PSB‐II_(P)9], 6EVN (PSB‐II_PAG), 6EVO (PSB‐II_PRG), and 6EVP (PSB‐II_PEG).
Conflict of Interest Statement
Johanna Myllyharju owns equity in Fibrogen Inc., which develops hypoxia‐inducible factor prolyl 4‐hydroxylase inhibitors as potential therapeutics. This company supports research in the J.M. group.
Supporting information
Table SI The detailed conformation of the PAG, (P)9 peptides bound to PSB‐II (this work), and (PPG)3,(P)9 peptides bound to PSB‐I16. The prolines that bind to the Pro5‐ and Pro8‐pockets are shown in italics. The parameters for the ideal PPII conformation are φ − 78°, ψ + 146° and ω +180°27.
Figure S1 The online SLS/SEC analysis of PSB‐II using the Äkta Purifier equipped with a Superdex200 10/300GL column (GE Healthcare) and a Wyatt miniDAWN TREOS for recording the SLS signal. The plot of molar mass (horizontal red line) versus elution time is shown. Also shown is the corresponding SLS signal (curved red profile) showing the PSB‐II peak, eluting with an elution volume of 17.1 mL. The molar mass of PSB‐II (12 kDa) was calculated from the refractive index (RI) and static light scattering (SLS) signals using the Astra software.
Figure S2 Secondary structure analysis of PSB‐II. (A) Far UV CD spectrum. (B) Thermal denaturation CD spectra. (C) Unfolded protein fraction profile with a calculated Tm of 60.3°C.
Figure S3 Calorimetric binding studies of PSB‐II with proline‐rich peptides. The titration curves with subtracted baselines (red) of (PPG)3 (A), (P)9 (B), PPG‐PAG‐PPG (C), PPG‐PRG‐PPG (D), and PPG‐PEG‐PPG (E) are shown. The bottom graphs display normalized integration data (black squares) and fitted curves (black lines).
Figure S4 Omit (2Fo‐Fc) electron density maps (after omit refinement) of the (P)9 and the PAG peptides, contoured at 1.0 σ. (A) The zoomed‐in view of the C‐terminal region of the (P)9‐peptide (after three cycles of omit refinement, leaving the peptide out of the model). (B) The zoomed‐in view of the middle region of the PAG‐peptide (after three cycles of omit refinement, leaving the peptide and the Asn227 residue out of the model).
Figure S5 The omit and final (2Fo‐Fc) electron density maps of respectively, the PPG‐PRG‐PPG (A, B) and PPG‐PEG‐PPG (C, D) peptides, complexed with PSB‐II (stereo, side view). The key residues of the PSB‐II are also shown. The direct hydrogen bonds between the peptide and the protein are shown as black dashed lines. (A, C) The omit (2Fo‐Fc) electron density maps for the peptides in the PSB‐II complex structures, calculated after three cycles of omit refinement steps (leaving out the peptide of the model) and contoured at 1.0 σ. (B, D) The final (2Fo‐Fc) electron density maps for the peptides (after refinement in the presence of the peptide) of the complex structures, contoured at 1.0 σ.
Figure S6 Comparison of the mode of binding of PPG‐PAG‐PPG to PSB‐II (light brown) and the mode of binding of (PPG)3 to PSB‐I (PDB code 4BT9, light blue), highlighting the deeper mode of binding of PAG to PSB‐II versus (PPG)3 to PSB‐I (stereo, standard view). The side chains of the PSB residues interacting with the peptides are labeled and shown in sticks. The PAG and (PPG)3 peptides are shown in yellow and light blue sticks, respectively. The N and C termini of both peptides, as well as the anchoring prolines Pro5 and Pro8 of the PSB‐I_(PPG)3 structure (corresponding to Pro4 and Pro7, respectively, of the PSB‐II_PAG structure), are labeled in red. The direct hydrogen bond interactions (mainly in the PSB‐II_PAG complex) and most of the water‐mediated hydrogen bond interactions in the PSB‐I_(PPG)3 complex (mainly around Asn227) are shown as black dashed lines. The sulfate ion (SO4) in the PSB‐II_PAG structure, which is bound close to the binding groove, is also shown in sticks. It can be noted that in the PAG peptide mode of binding (yellow) the N‐terminal (PPG) triplet points away from the PSB binding groove. The conformations of the side chains of Tyr196 and of the Arg223‐Asp192 salt bridge in both complexes are the same.
Acknowledgments
The expert support of the beamline scientists at ESRF ID30A‐3 has been very important. We also thank Ville Ratas and Eeva Lehtimäki for their technical contributions to the experimental work. The use of the instruments and expertise of the Biocenter Oulu proteomics and protein analysis, DNA sequencing and protein X‐ray crystallography (supported by Biocenter Finland and Instruct‐FI) core facilities, are gratefully acknowledged. This research was supported by the Sigrid Jusélius Foundation (M.K.K., R.K.W, J.M.), Academy of Finland through grants 296498 (J.M.) and Center of Excellence 2012–2017 grant 251314 (J.M.), and the Jane and Aatos Erkko Foundation (J.M.).
Jothi Anantharajan current address is Experimental Therapeutics Centre, 31 Biopolis Way, #03‐01 Nanos, 138669, Singapore.
Broader statement: Crystal structures of human PSB‐II (the peptide‐substrate‐binding domain of collagen prolyl 4‐hydroxylase type‐II, C‐P4H‐II) complexed with the low affinity (P)9 peptide and with three high affinity PPG‐PxG‐PPG (x is alanine, arginine or glutamate) peptides are compared with structures of the corresponding human PSB‐I complexes. The importance of this structural information for understanding the substrate specificity properties of C‐P4H‐I and C‐P4H‐II is discussed.
References
- 1. Ball LJ, Kühne R, Schneider‐Mergener J, Oschkinat H (2005) Recognition of proline‐rich motifs by protein‐protein‐interaction domains. Angew Chem Int Ed Engl 44:2852–2869. [DOI] [PubMed] [Google Scholar]
- 2. Bhattacharyya R, Chakrabarti P (2003) Stereospecific interactions of proline residues in protein structures and complexes. J Mol Biol 331:925–940. [DOI] [PubMed] [Google Scholar]
- 3. Opitz R, Müller M, Reuter C, Barone M, Soicke A, Roske Y, Piotukh K, Huy P, Beerbaum M, Wiesner B, Beyermann M, Schmieder P, Freund C, Volkmer R, Oschkinat H, Schmalz HG, Kühne R (2015) A modular toolkit to inhibit proline‐rich motif‐mediated protein‐protein interactions. Proc Natl Acad Sci USA 112:5011–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cubellis MV, Caillez F, Blundell TL, Lovel SC (2005) Properties of polyproline II, a secondary structure element implicated in protein‐protein interactions. Proteins 58:880–892. [DOI] [PubMed] [Google Scholar]
- 5. Shoulders MD, Raines RT (2009) Collagen structure and stability. Annu Rev Biochem 78:929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Piez KA. In: Ramachandran GN, Reddi AH, Eds, 1976. Biochemistry of collagens. New York, NY: Plenum Publishing Corporation; p. 1–44. [Google Scholar]
- 7. Myllyharju J (2003) Prolyl 4‐hydroxylases, the key enzymes of collagen biosynthesis. Matrix Biol 22:15–24. [DOI] [PubMed] [Google Scholar]
- 8. Myllyharju J (2008) Prolyl 4‐hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann Med 40:402–417. [DOI] [PubMed] [Google Scholar]
- 9. Gorres KL, Raines RT (2010) Prolyl 4‐hydroxylases. Crit Rev Biochem Mol Biol 45:106–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pihlajaniemi T, Helaakoski T, Tasanen K, Myllylä R, Huhtala M‐L, Koivu J, Kivirikko KI (1987) Molecular cloning of the β‐subunit of human prolyl 4‐hydroxylase. This subunit and the protein disulphide isomerase are products of the same gene. EMBO J 6:643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nissi R, Autio‐Harmainen H, Marttila P, Sormunen R, Kivirikko KI (2001) Prolyl 4‐hydroxylase isoenzyme I and II have different expression patterns in several human tissues. J Histochem Cytochem 49:1143–1153. [DOI] [PubMed] [Google Scholar]
- 12. Koski MK, Anantharajan J, Kursula P, Dhavala P, Murthy AV, Bergmann U, Myllyharju J, Wierenga R (2017) Assembly of the elongated collagen prolyl 4‐hydroxylase α2β2 heterotetramer around a central α2 dimer. Biochem J 474:751–769. [DOI] [PubMed] [Google Scholar]
- 13. Myllyharju J, Kivirikko KI (1999) Identification of a novel proline‐rich peptide‐binding domain in prolyl 4‐hydroxylase. EMBO J 18:306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hieta R, Kukkola L, Permi P, Pirilä P, Kivirikko KI, Kilpeläinen I, Myllyharju J (2003) The peptide‐substrate‐binding domain of human collagen prolyl 4‐hydroxylases. Backbone assignments, secondary structure, and binding of proline‐rich peptides. J Biol Chem 278:34966–34974. [DOI] [PubMed] [Google Scholar]
- 15. Pekkala M, Hieta R, Bergmann U, Kivirikko KI, Wierenga RK, Myllyharju J (2004) The peptide‐substrate‐binding domain of collagen prolyl 4‐hydroxylases is a tetratricopeptide repeat domain with functional aromatic residues. J Biol Chem 279:52255–52261. [DOI] [PubMed] [Google Scholar]
- 16. Anantharajan J, Koski MK, Kursula P, Hieta R, Bergmann U, Myllyharju J, Wierenga RK (2013) The structural motifs for substrate binding and dimerization of the α subunit of collagen prolyl 4‐hydroxylase. Structure 21:2107–2118. [DOI] [PubMed] [Google Scholar]
- 17. Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst D60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 18. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Jong L, van der Kraan I, de Waal A (1991) The kinetics of the hydroxylation of procollagen by prolyl 4‐hydroxylase. Proposal for a processive mechanism of binding of the dimeric hydroxylating enzyme in relation to the high k cat/K m ratio and a conformational requirement for hydroxylation of ‐X‐Pro‐Gly‐ sequences. Biochim Biophys Acta 1079:103–111. [DOI] [PubMed] [Google Scholar]
- 20. Wang X, Mart RJ, Webb SJ (2007) Vesicle aggregation by multivalent ligands: relating crosslinking ability to surface affinity. Org Biomol Chem 5:2498–2505. [DOI] [PubMed] [Google Scholar]
- 21. Zeelen JP, Pauptit RA, Wierenga RK, Kunau WH, Hiltunen JK (1992) Crystallization and preliminary X‐ray diffraction studies of mitochondrial short‐chain Δ3, Δ2‐enoyl‐CoA isomerase from rat liver. J Mol Biol 224:273–275. [DOI] [PubMed] [Google Scholar]
- 22. Kabsch W (2010) XDS. Acta Cryst D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G (2011) Data processing and analysis with the autoPROC toolbox. Acta Cryst D67:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Evans PR, Murshudov GN (2013) How good are my data and what is the resolution. Acta Cryst D69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Terwilliger TC, Grosse‐Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung L‐H, Read RJ, Adams PD (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst D64:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table SI The detailed conformation of the PAG, (P)9 peptides bound to PSB‐II (this work), and (PPG)3,(P)9 peptides bound to PSB‐I16. The prolines that bind to the Pro5‐ and Pro8‐pockets are shown in italics. The parameters for the ideal PPII conformation are φ − 78°, ψ + 146° and ω +180°27.
Figure S1 The online SLS/SEC analysis of PSB‐II using the Äkta Purifier equipped with a Superdex200 10/300GL column (GE Healthcare) and a Wyatt miniDAWN TREOS for recording the SLS signal. The plot of molar mass (horizontal red line) versus elution time is shown. Also shown is the corresponding SLS signal (curved red profile) showing the PSB‐II peak, eluting with an elution volume of 17.1 mL. The molar mass of PSB‐II (12 kDa) was calculated from the refractive index (RI) and static light scattering (SLS) signals using the Astra software.
Figure S2 Secondary structure analysis of PSB‐II. (A) Far UV CD spectrum. (B) Thermal denaturation CD spectra. (C) Unfolded protein fraction profile with a calculated Tm of 60.3°C.
Figure S3 Calorimetric binding studies of PSB‐II with proline‐rich peptides. The titration curves with subtracted baselines (red) of (PPG)3 (A), (P)9 (B), PPG‐PAG‐PPG (C), PPG‐PRG‐PPG (D), and PPG‐PEG‐PPG (E) are shown. The bottom graphs display normalized integration data (black squares) and fitted curves (black lines).
Figure S4 Omit (2Fo‐Fc) electron density maps (after omit refinement) of the (P)9 and the PAG peptides, contoured at 1.0 σ. (A) The zoomed‐in view of the C‐terminal region of the (P)9‐peptide (after three cycles of omit refinement, leaving the peptide out of the model). (B) The zoomed‐in view of the middle region of the PAG‐peptide (after three cycles of omit refinement, leaving the peptide and the Asn227 residue out of the model).
Figure S5 The omit and final (2Fo‐Fc) electron density maps of respectively, the PPG‐PRG‐PPG (A, B) and PPG‐PEG‐PPG (C, D) peptides, complexed with PSB‐II (stereo, side view). The key residues of the PSB‐II are also shown. The direct hydrogen bonds between the peptide and the protein are shown as black dashed lines. (A, C) The omit (2Fo‐Fc) electron density maps for the peptides in the PSB‐II complex structures, calculated after three cycles of omit refinement steps (leaving out the peptide of the model) and contoured at 1.0 σ. (B, D) The final (2Fo‐Fc) electron density maps for the peptides (after refinement in the presence of the peptide) of the complex structures, contoured at 1.0 σ.
Figure S6 Comparison of the mode of binding of PPG‐PAG‐PPG to PSB‐II (light brown) and the mode of binding of (PPG)3 to PSB‐I (PDB code 4BT9, light blue), highlighting the deeper mode of binding of PAG to PSB‐II versus (PPG)3 to PSB‐I (stereo, standard view). The side chains of the PSB residues interacting with the peptides are labeled and shown in sticks. The PAG and (PPG)3 peptides are shown in yellow and light blue sticks, respectively. The N and C termini of both peptides, as well as the anchoring prolines Pro5 and Pro8 of the PSB‐I_(PPG)3 structure (corresponding to Pro4 and Pro7, respectively, of the PSB‐II_PAG structure), are labeled in red. The direct hydrogen bond interactions (mainly in the PSB‐II_PAG complex) and most of the water‐mediated hydrogen bond interactions in the PSB‐I_(PPG)3 complex (mainly around Asn227) are shown as black dashed lines. The sulfate ion (SO4) in the PSB‐II_PAG structure, which is bound close to the binding groove, is also shown in sticks. It can be noted that in the PAG peptide mode of binding (yellow) the N‐terminal (PPG) triplet points away from the PSB binding groove. The conformations of the side chains of Tyr196 and of the Arg223‐Asp192 salt bridge in both complexes are the same.