


Abstract
One of the most intensively studied chromatin binding factors is HP1α. HP1α is associated with silenced, heterochromatic regions of the genome and binds to H3K9me3. While H3K9me3 is necessary for HP1α recruitment to heterochromatin, it is becoming apparent that it is not sufficient suggesting that additional factors are involved. One candidate proposed as a potential regulator of HP1α recruitment is the linker histone H1.4. Changes to the underlying make-up of chromatin, such as the incorporation of the histone variant H2A.Z, has also been linked with regulating HP1 binding to chromatin. Here, we rigorously dissected the effects of H1.4, H2A.Z and H3K9me3 on the nucleosome binding activity of HP1α in vitro employing arrays, mononucleosomes and nucleosome core particles. Unexpectedly, histone H1.4 impedes the binding of HP1α but strikingly, this inhibition is partially relieved by the incorporation of both H2A.Z and H3K9me3 but only in the context of arrays or nucleosome core particles. Our data suggests that there are two modes of interaction of HP1α with nucleosomes. The first primary mode is through interactions with linker DNA. However, when linker DNA is missing or occluded by linker histones, HP1α directly interacts with the nucleosome core and this interaction is enhanced by H2A.Z with H3K9me3.
INTRODUCTION
Eukaryotic DNA is packaged into a condensed structure called chromatin. Chromatin is built from nucleosomes—approximately 147 base pairs of DNA wrapped almost twice around an octamer of histone proteins. The folding and compaction of chromatin into complex higher-order structures impacts all processes that require access to the DNA (1). As such, eukaryotic genomes are partitioned into distinct functional chromatin domains that vary in accessibility and carry distinct histone modification signatures and chromatin-associated proteins (2–6). One critical chromatin partition is constitutive heterochromatin: it is essential for chromosome segregation, silencing repetitive DNA elements and maintaining genome stability (7,8). The hallmarks of constitutive heterochromatin are trimethylated histone H3 lysine 9 (H3K9me3) and its cognate binding partner, Heterochromatin Protein 1 (HP1).
HP1 proteins are conserved in organisms ranging from fission yeast to humans. All HP1 proteins have a tripartite structure consisting of an N-terminal chromodomain, a central positively-charged hinge region, and a C-terminal chromoshadow domain. The chromodomain recognizes H3K9me3, and this interaction forms the basis of the current paradigm of how HP1 is recruited and maintained at heterochromatin (9–11). Interestingly, many eukaryotes have multiple HP1 isoforms that can each bind H3K9me3 - mammals have three, HP1α, HP1β and HP1γ (12–14). However, these isoforms often have distinct chromatin distribution patterns (15–17), indicative of different functions. Thus, factors outside the chromodomain-H3K9me3 interaction appear important for directing the chromatin-binding activity and function of different HP1 isoforms.
Both the hinge region and the chromoshadow domain of HP1 proteins have been reported to interact with a number of targets, including DNA/RNA (18,19), linker histone H1 (20,21), the globular domain of H3 (22,23), and other HP1 molecules (24–26). Furthermore, changes to the underlying make-up of chromatin, such as the incorporation of the histone variant H2A.Z, have also been linked with regulating HP1 binding to chromatin (27–29). Indeed, H2A.Z and H3K9me3 can co-exist in the same nucleosome (27) and it appears H2A.Z can substitute for H3K9me3 at heterochromatin depending on the physiological context (30,31). Consequently, there is much scope for modulating HP1-chromatin interactions.
A relatively underexplored element in HP1 binding to chromatin is that of the linker histones. Linker histones are structurally and functionally distinct from the four core histones and bind externally to the nucleosome core and flanking linker DNA (32). There are at least 11 linker histone subtypes in mammals and they are broadly distributed across the genome, including in constitutive heterochromatin (33–35). The classical view is that linker histones are simply structural elements of chromatin. However, it is increasingly clear they have specific and dynamic regulatory roles (36). A number of studies have connected HP1 proteins with linker histones, in particular mammalian HP1α and linker histone H1.4 (20,21,37). Yet, the impact of linker histones on HP1 binding to nucleosomes and the relationship with other heterochromatic elements, such as H3K9me3 and H2A.Z, is not well-understood.
Previously, we developed a robust recombinant expression system for the production of human linker histone H1.4, and we can efficiently incorporate this purified H1.4 into nucleosomes and nucleosome arrays in vitro (38). Here, we have used this system to explore the effect of H1.4 on interactions between HP1α and nucleosomes in a number of contexts in vitro. We find that H1.4 impedes the binding of HP1α to nucleosomes and nucleosome arrays and that H3K9me3 alone is unable to overcome this impediment. However, when H3K9me3 is combined with H2A.Z, a partial reversal of this H1.4-mediated binding inhibition is observed. Significantly, we also find that histone H2A.Z is able to enhance binding of HP1α to nucleosome arrays in a manner similar to H3K9me3, thus demonstrating for the first time that H2A.Z can indeed act as a functional substitute for H3K9me3 in chromatin.
MATERIALS AND METHODS
Expression and purification of recombinant histone H1.4
The construction of the human H1.4 expression plasmid and the subsequent expression and purification of the protein have been described in detail in (38).
Expression and purification of recombinant wild-type HP1α and HP1α3KA
The pET15b expression plasmids containing HP1α and HP1α3KA were transformed into Escherichia coli Rosetta2 (DE3) (Novagen) cells and spread onto LB-agar containing 50 μg/ml ampicillin (amp) and 34 μg/ml chloramphenicol (cml). After overnight incubation at 37°C, a single colony was picked, and used to inoculate 10 ml LB liquid media containing amp (50 μg/ml) and cml (34 μg/ml) and incubated at 37°C for ∼8 h. The starter culture was used to inoculate 1 l of ZYP-5052 auto-induction media (39) containing amp (50 μg/ml) and cml (34 μg/ml) in a 2 l flask. The culture was grown at 37°C overnight. The cells were collected by centrifugation at 5000 g for 12 min at 4°C, washed once with 25 ml cold PBS, and then the cell pellet was resuspended in ∼25 ml lysis buffer (20 mM Tris pH 7.5, 350 mM NaCl, 0.05% (v/v) β-mercaptoethanol, 20 mM imidazole, 0.1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, 1 μM pepstatin, 1 μM bestatin, 1 μM E-64) and either frozen and stored at −20°C or processed as follows. Cells were lysed by thawing cell suspensions then adding lysozyme (50 μg/ml) and DNaseI (10 μg/ml), followed by three cycles of freeze/thawing in liquid N2 or the addition of 1× BugBuster (10× stock; MerckMillipore) reagent. The suspension was then incubated on ice for 20 min, followed by sonication on ice for 3–5 min, cycling on for 10 s and off for 20 s, at 30% power on a Branson sonifier. The lysate was centrifuged at 35 000 g for 25 min at 4°C, and the supernatant was carefully decanted and applied to a 2.5 ml Ni-NTA resin column pre-equilibrated in lysis buffer and allowed to flow under gravity. The flow-through was reapplied to the column, followed by washing with ∼10 column volumes of lysis buffer. The protein was eluted from the column in lysis buffer containing 250 mM imidazole. The eluate was exchanged to 20 mM Tris pH 7.5, 350 mM NaCl, 0.05% (v/v) β-mercaptoethanol and the sample treated with 30 units thrombin (Sigma) overnight at room temperature to remove the N-terminal hexahistidine-tag. The cleaved protein was then purified by anion exchange chromatography using 1 ml HiTrap Q cartridges (GE Healthcare) in 20 mM Tris pH 8.5 and 1 mM DTT over a 0–1 M NaCl gradient. Fractions containing full-length HP1α proteins were collected and concentrated in 10 kDa MWCO centrifugal concentrators and the protein purified further via gel filtration on a Superdex200 10/300 column (GE Healthcare) in 20 mM Tris pH 7.5, 250 mM NaCl, 1 mM DTT. HP1α-containing fractions were pooled and concentrated in 10 kDa MWCO centrifugal concentrators to a final concentration of 500 μM. Glycerol was added to a final concentration of 10% (v/v) and the protein aliquoted, snap frozen, and stored at −80°C. Protein concentration was determined by absorbance at 280 nm and using an extinction coefficient of 23.16 mM−1 cm−1.
Production of recombinant histones and histone octamers
Histone octamers were assembled using standard protocols from purified recombinant histones (40,41), either as unlabelled proteins (for mononucleosomes) or containing AlexaFluor488-labelled H2A (for arrays). AlexaFluor488 was attached to H2A via a single cysteine residue introduced at position 120, as described previously in (42). Labelling efficiency was ∼65–70%. The labelled octamers were mixed at a 1:4 ratio with unlabelled octamers such that only ∼2 nucleosomes per array were labelled with AlexaFluor488. H3K9cme3 and H3K9cme0 were produced as described in (43). Octamers were stored at −20°C in 10 mM Tris pH 7.5, 1 mM EDTA, 1.5 M NaCl, 40% glycerol, 1 mM DTT.
In vitro nucleosome and nucleosome array assembly
Nucleosome (12N200) arrays were assembled on a 2400 bp DNA fragment consisting of twelve 200 bp tandem repeats that each contain the 147 bp 601 nucleosome positioning sequence (44) and 53 bp flanking DNA. The array fragment was propagated in a pUC18 vector, using standard methods. The array fragment was excised from the pUC18 vector backbone via digestion with EcoRV, and the backbone DNA cut into smaller fragments using DraI and HaeII enzymes. The array fragment was then separated on a 1× TAE 1.3% (w/v) agarose gel, the band excised and the DNA extracted using electroelution. The purified DNA fragment was then concentrated by ethanol precipitation and resuspended in 10 mM Tris pH 8.5 and stored at −20°C.
Mononucleosomes were assembled on AlexaFluor488-labelled DNA fragments that corresponded to the 147 bp 601 nucleosome positioning sequence (44) alone (N147) or the 601 sequence flanked by 28 bp of additional DNA on either side (N203). The fragments were produced by performing 96 parallel PCR reactions in 96-well plates using one AlexaFluor488-labelled and one unlabelled primer and MyTaq DNA polymerase (Bioline; as per the manufacturer's instructions). The PCR products were purified on native PAGE gels; 0.5× Tris–borate–EDTA (TBE; 44.5 mM Tris-Borate, 1 mM EDTA) 5% (w/v) acrylamide. The desired product bands were excised and the DNA extracted via electroelution in 0.5× TBE at room temperature for 30–60 min at 70–100 V. The DNA was concentrated by ethanol precipitation, resuspended in 10 mM Tris pH 8.5 and stored at −20°C.
Assembly of the 12N200, N203 and N147 DNA fragments into the various nucleosome arrays/mononucleosomes (±H1.4) was performed by salt-gradient dialysis, as we have described in (38). Mononucleosome assembly reactions were analysed on 0.2 × TBE 5% (w/v) native PAGE gels run at 150 V at 4°C for 45–60 min. Nucleosome array assemblies were analysed on 0.2× TBE 0.8% (w/v) agarose gels run at 110 V for 90 min at 4°C. The gels were visualized by scanning for fluorescence AlexaFluor488 on a Typhoon™ FLA9000 laser scanner.
Electrophoretic mobility shift assays (EMSAs)
Dilutions of HP1α (0–20 μM) and HP1α3KA (0–20 μM for array binding and 0–30 μM for mononucleosome binding) were incubated with AlexaFluor488-labelled 12N200 (±H1.4) arrays (4.7 nM; effective nucleosome concentration is 56 nM) or mononucleosomes (50 nM; N203, N203+H1.4, N147) in gel shift buffer (10 mM Tris pH 7.5, 25 mM NaCl, 25 mM KCl, 0.2 mM DTT, 4% (w/v) Ficoll 400) on ice for 20–30 min. Mononucleosome binding reactions were loaded onto 5% (w/v) acrylamide:bisacrylamide (49:1) gels and electrophoresed in 0.2× TBE at 150 V for 90 min at 4°C. Nucleosome array binding reactions were loaded onto 0.8% (w/v) agarose gels and electrophoresed in 0.2× TBE at 70 V for 180 min at 4°C. All gel images were acquired on an FLA9000 laser scanner (GE Healthcare) scanning for AlexaFluor488 signal.
Quantitation of gels was performed by measuring the amount of unbound (mononucleosome or nucleosome array) signal in each gel lane using ImageJ (45). Normalization across replicate experiments was performed as follows: the signal in the mononucleosome/nucleosome array-only lane (i.e. the 0 μM HP1α binding reaction) was set as the 100% unbound signal in each individual experiment. The fraction unbound for each binding condition in an experiment was then normalized relative to this signal. These values were then converted to fraction bound by subtracting them from one. Normalized data sets from replicate experiments were then fit simultaneously using the concatenated fit option in MicroCal OriginPro 2017 with the in-built Hill equation, which is of the form:
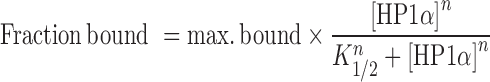 |
where ‘max. bound’ is the maximum level of binding, and represents the upper asymptote of the binding isotherm (upper limit of one); K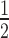 is the concentration of HP1α at half-maximal binding and is indicative of the relative affinity constant; n is the Hill coefficient. Supplementary Table S1 provides an overview of the results for each experiment.
is the concentration of HP1α at half-maximal binding and is indicative of the relative affinity constant; n is the Hill coefficient. Supplementary Table S1 provides an overview of the results for each experiment.
RESULTS
Linker histone H1.4 interferes with binding of HP1α to mononucleosomes
Linker histones have been reported to interact with both the chromodomain and the hinge region of HP1α (20,21,37). However, these studies did not examine these interactions in the context of nucleosomes. Thus, we wanted to investigate how linker histone H1.4 affects the binding of HP1α to nucleosomes.
First, we assembled mononucleosomes, either in the absence or presence of a stoichiometric amount of linker histone H1.4 (Figure 1A). These nucleosomes contained a core 147 bp 601 positioning sequence (44) flanked by 28 bp of linker DNA on either side, yielding a 203 bp symmetric nucleosome (N203); these nucleosomes were labelled with AlexaFluor488 on one end. We chose 28 bp of flanking DNA because shorter linker DNA lengths have been demonstrated to decrease the affinity of linker histones for nucleosomes (46). Figure 1A shows that H1.4-bound nucleosomes have an increased electrophoretic mobility (Figure 1A, lane 2) compared to the linker histone-free species (Figure 1A, lane 1). This is consistent with the linker histone interacting with and constraining the linker DNA, resulting in a more compact particle.
Figure 1.

Linker histone H1.4 decreases affinity but not cooperativity of HP1α binding to mononucleosomes. (A) Native PAGE analysis of symmetric 203 bp mononucleosomes (N203) assembled in the absence (lane 1) or presence (lane 2) of a stoichiometric amount of linker histone H1.4. (B) EMSA analysis of HP1α (0–20 μM; 0.65× dilutions series) binding to N203 mononucleosomes. (C) EMSA analysis of HP1α binding to 147 bp nucleosome core particles (N147). HP1α concentrations are the same as those used in (B). (D) Quantitative analysis of EMSA experiments as shown in (B and C). Data (symbols) are the average of 3–4 replicates and error bars represent the S.E. Lines represent fits to the data using the Hill equation as described in the ‘materials and methods’.
Next, we performed electrophoretic mobility shift assays (EMSAs) using these two different nucleosome species (N203 and N203+H1.4), incubating them with increasing concentrations of HP1α (noting that HP1α exists as a dimer, Figure 1B). Given the reports of a positive and direct interaction between HP1α and linker histones (20,21,37), we anticipated the presence of H1.4 would enhance the interaction between HP1α and nucleosomes. Surprisingly, this was not the case. HP1α shifts N203 nucleosomes at much lower concentrations when H1.4 is absent (indicating a higher relative affinity); compare lanes 2–7 in the upper and lower panel of Figure 1B. Despite this apparent decrease in binding affinity in the presence of H1.4, HP1α forms a more discrete complex with the H1.4-associated nucleosomes; suggesting that either H1.4 alters the interaction to yield a more stable HP1α–nucleosome complex, or that the number of available binding sites for HP1α decreased in the presence of H1.4.
It is well-documented that HP1α is able to interact with DNA (18,19,26). One possible explanation for the reduced affinity of HP1α for H1.4-associated nucleosomes is that the linker histone inhibits HP1α from accessing the linker DNA arms of the nucleosome. To examine this, we assembled 147 bp nucleosomes that lack linker DNA (N147), and assessed binding of HP1α by EMSA (Figure 1C). The binding of HP1α to N147 compared to both N203 and N203+H1.4 nucleosomes is visibly different, and displays a lower apparent affinity (Figure 1C).
To gain a more detailed picture of these differences, we generated binding curves from replicate EMSA experiments and fitted these with the Hill equation (Figure 1D) to determine the relative affinity (K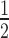 ) and cooperativity (Hill coefficient) of the HP1α interactions. These data are presented in Supplementary Table S1 (which summarizes data from the ∼140 individual EMSA experiments conducted in this study). This analysis reveals that HP1α binding to N147 (K
) and cooperativity (Hill coefficient) of the HP1α interactions. These data are presented in Supplementary Table S1 (which summarizes data from the ∼140 individual EMSA experiments conducted in this study). This analysis reveals that HP1α binding to N147 (K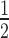 = 2.29 ± 0.36 μM) is almost 4-fold weaker than to N203 (K
= 2.29 ± 0.36 μM) is almost 4-fold weaker than to N203 (K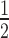 = 0.58 ± 0.02 μM) and 2-fold weaker than to N203+H1.4 (K
= 0.58 ± 0.02 μM) and 2-fold weaker than to N203+H1.4 (K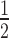 = 1.26 ± 0.04 μM). Interestingly, HP1α exhibits cooperative binding to N203 in both the absence and presence of H1.4 (Hill coefficients = 1.90 ± 0.10 and 1.88 ± 0.11, respectively), but not to N147 (Hill coefficients = 1.21 ± 0.16). These data indicate that the interactions between HP1α and the linker DNA are important for nucleosome binding. However, the effect of H1.4 on HP1α nucleosome binding is not equivalent to the complete loss of linker DNA, suggesting that H1.4 only partially occludes linker DNA accessibility in a mononucleosome.
= 1.26 ± 0.04 μM). Interestingly, HP1α exhibits cooperative binding to N203 in both the absence and presence of H1.4 (Hill coefficients = 1.90 ± 0.10 and 1.88 ± 0.11, respectively), but not to N147 (Hill coefficients = 1.21 ± 0.16). These data indicate that the interactions between HP1α and the linker DNA are important for nucleosome binding. However, the effect of H1.4 on HP1α nucleosome binding is not equivalent to the complete loss of linker DNA, suggesting that H1.4 only partially occludes linker DNA accessibility in a mononucleosome.
The hinge domain of HP1α drives association with nucleosomes
Next, we wanted to confirm that the interactions between HP1α and the linker DNA is central for nucleosome binding. The hinge region (Figure 2A) that links the N-terminal chromodomain and C-terminal chromoshadow domain of HP1α is known to bind DNA and is important for interactions with nucleosomes (19,47). It contains a series of conserved basic residues (Figure 2A) and mutation of these basic residues has previously been shown to affect HP1α−DNA/chromatin interactions (19,26,47). We expressed and purified a triple lysine to alanine hinge-region mutant of HP1α (HP1α3KA; Figure 2A) and confirmed this mutation affects the DNA-binding activity of HP1α (Supplementary Figure S1B).
Figure 2.

The basic-rich hinge region of HP1α is critical for nucleosome binding. (A) Schematic of HP1α domain organization; the N-terminal chromo-domain is shown in light blue, the C-terminal chromoshadow domain in dark blue, and the hinge domain as a grey rippled line. Below, a multiple sequence alignment of the hinge region of HP1α proteins from distinct animal species. UniProt/UniRef accession numbers and organisms are shown on the left. The alignment is coloured according to the ClustalX colour scheme utilized in Jalview (68). The three black dots above the sequence indicate the sites of mutation in the triple lysine-to-alanine mutant HP1α3KA. (B) Binding isotherms (symbols) derived from quantitative EMSAs and corresponding fits (solid lines) for HP1α3KA binding to different mononucleosome species (black = N203; red = N203 + H1.4; purple = N147). Data are the average of a minimum of three replicate experiments and error bars represent the S.E. (C) Bar graph comparing relative affinity (K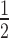 ) values derived from the fits for HP1α3KA (grey bars) in (B) and wild-type HP1α (WT; blue bars) in Figure 1D. The corresponding mononucleosome species are shown along the bottom and are the same as those defined in Figure 1.
) values derived from the fits for HP1α3KA (grey bars) in (B) and wild-type HP1α (WT; blue bars) in Figure 1D. The corresponding mononucleosome species are shown along the bottom and are the same as those defined in Figure 1.
We performed quantitative EMSA experiments (Figure 2B) with HP1α3KA and the same three mononucleosome species (N203, N203+H1.4, and N147) used for the experiments in Figure 1. Consistent with previous reports (19,47), HP1α3KA has a significantly diminished capacity for binding nucleosomes (Figure 2C). Further, the relative affinity of HP1α3KA for N203, N203+H1.4 and N147 is very similar (K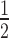 = 9.83 ± 0.42; 12.10 ± 0.83; 10.62 ± 1.75 μM, respectively). This differs from wild-type HP1α, which has strong preference for binding N203 mononucleosomes (Figure 1D). This means the HP1α3KA mutation disproportionately affects binding to nucleosomes with more available linker DNA; the affinity for N203 nucleosomes is reduced more than for N203+H1.4, which in turn is affected more than N147 nucleosomes (17-, 10- and 5-fold decrease relative to wild-type HP1α, respectively; Figure 2C). However, the interaction of HP1α3KA with N147 is still substantially weakened, suggesting there is a component to the hinge-mediated interaction that involves the nucleosome core. Clearly, the hinge region plays a critical role in HP1α binding to nucleosomes and this is mainly driven through interactions with the linker DNA.
= 9.83 ± 0.42; 12.10 ± 0.83; 10.62 ± 1.75 μM, respectively). This differs from wild-type HP1α, which has strong preference for binding N203 mononucleosomes (Figure 1D). This means the HP1α3KA mutation disproportionately affects binding to nucleosomes with more available linker DNA; the affinity for N203 nucleosomes is reduced more than for N203+H1.4, which in turn is affected more than N147 nucleosomes (17-, 10- and 5-fold decrease relative to wild-type HP1α, respectively; Figure 2C). However, the interaction of HP1α3KA with N147 is still substantially weakened, suggesting there is a component to the hinge-mediated interaction that involves the nucleosome core. Clearly, the hinge region plays a critical role in HP1α binding to nucleosomes and this is mainly driven through interactions with the linker DNA.
Linker histones significantly inhibit binding of HP1α to nucleosome arrays
The action of linker histones extends beyond the single nucleosome, with a primary function to modulate higher-order conformations across multiple nucleosomes. In addition, HP1 proteins have been demonstrated to have enhanced interactions with multi-nucleosome substrates compared to mononucleosomes (25,27,47). Thus, we next examined the effect of linker histones on the binding of HP1α to nucleosome arrays. For this, we used arrays consisting of twelve 200 bp repeats, each containing a core 601 nucleosome positioning sequence (12N200). To simplify in-gel visualization and quantitation in our EMSA experiments, we produced arrays containing H2A site-specifically labelled with AlexaFluor488, as we have described previously (38). To minimize any possible adverse effects due to the AlexaFluor488 moiety, we diluted our AlexaFluor488-H2A histone octamer preparations with unlabelled histone octamers thus limiting the number of labelled nucleosomes per 12N200 array to approximately two.
Comparable to the mononucleosome experiments (Figure 1), we observed that the binding of HP1α to 12N200 arrays is significantly diminished when the arrays are assembled with a stoichiometric amount of H1.4 (Figure 3A). Quantitation of this effect (Figure 3B; Supplementary Table S1) reveals that the affinity of HP1α for the 12N200 arrays (K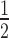 = 0.71 ± 0.02 μM) is reduced 3-fold when H1.4 is present (12N200+H14; K
= 0.71 ± 0.02 μM) is reduced 3-fold when H1.4 is present (12N200+H14; K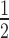 = 2.15 ± 0.23 μM). Yet, more striking is the fact that binding of HP1α to the H1.4-saturated arrays is non-cooperative (Hill coeff. = 1.32 ± 0.13), whereas strong positive cooperativity is exhibited on the linker histone-free arrays (Hill coeff. = 3.52 ± 0.29). This differs from the behaviour on N203 mononucleosomes where some cooperativity is observed irrespective of whether H1.4 is present or not (Figure 1B; Supplementary Table S1). On the other hand, binding of HP1α to N147 mononucleosomes is non-cooperative (Figure 1C, D). This suggests that H1.4 limits linker DNA accessibility in the 12N200 arrays more so than in N203 mononucleosomes and that the mode of HP1α binding to 12N200+H1.4 arrays is most similar to the mode of binding to N147 mononucleosomes, namely core particles that lack linker DNA.
= 2.15 ± 0.23 μM). Yet, more striking is the fact that binding of HP1α to the H1.4-saturated arrays is non-cooperative (Hill coeff. = 1.32 ± 0.13), whereas strong positive cooperativity is exhibited on the linker histone-free arrays (Hill coeff. = 3.52 ± 0.29). This differs from the behaviour on N203 mononucleosomes where some cooperativity is observed irrespective of whether H1.4 is present or not (Figure 1B; Supplementary Table S1). On the other hand, binding of HP1α to N147 mononucleosomes is non-cooperative (Figure 1C, D). This suggests that H1.4 limits linker DNA accessibility in the 12N200 arrays more so than in N203 mononucleosomes and that the mode of HP1α binding to 12N200+H1.4 arrays is most similar to the mode of binding to N147 mononucleosomes, namely core particles that lack linker DNA.
Figure 3.

Linker histones inhibit binding of HP1α to nucleosome arrays. (A) EMSA analysis of HP1α (0–20 μM; 0.75 × dilution series) binding to 12 × 200 × 601 (12N200) nucleosome arrays (4.7 nM; effective nucleosome concentration is ∼ 56 nM) assembled in the absence (upper) or presence (lower) of recombinant linker histone H1.4. (B) Quantitative analysis of EMSA experiments as shown in (A). Data are the average of three replicates and error bars represent the standard error (s.e.). Lines represent fits to the data using the Hill equation as described in the ‘materials and methods’. (C) EMSA-derived binding isotherms for HP1α3KA binding to 12N200 arrays in the absence (hollow circles) or presence of H1.4 (black diamonds). Lines represent fits to the data as per (B). (D) EMSA analysis of HP1α (0–20 μM; 0.75× dilution series) binding to 12N200 nucleosome arrays assembled in the presence of recombinant linker histone H1.0 (12N200 + H1.0).
We then assessed the binding of HP13KA to the 12N200 arrays (± H1.4). HP13KA binds 12N200 arrays more weakly than wild-type HP1α (Figure 3C; Supplementary Table S1), approximately 8-fold less for 12N200 (K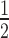 = 5.62 ± 0.26) and 7-fold less for 12N200+H1.4 (K
= 5.62 ± 0.26) and 7-fold less for 12N200+H1.4 (K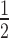 = 14.29 ± 5.58). These data also show that HP1α3KA has a definite preference for the linker histone-free arrays, binding 12N200 ∼2.6-fold more strongly than 12N200+H1.4. This is different to the behaviour of HP1α3KA on mononucleosomes, where it binds both the N203 and N203+H1.4 mononucleosomes with similar affinity (Figure 2B, C). This suggests that there is an additional component to the HP1α-array interaction that does not participate when interacting with mononucleosomes. This interaction(s) most likely involves connections that span multiple nucleosomes, and it is not directly dependent on the hinge region. This hinge independent interaction is disrupted by the presence of H1.4, since both wild-type HP1α and HP1α3KA display a 2.5–3-fold reduction in binding affinity for 12N200+H1.4 arrays compared to 12N200 arrays (Supplementary Table S1).
= 14.29 ± 5.58). These data also show that HP1α3KA has a definite preference for the linker histone-free arrays, binding 12N200 ∼2.6-fold more strongly than 12N200+H1.4. This is different to the behaviour of HP1α3KA on mononucleosomes, where it binds both the N203 and N203+H1.4 mononucleosomes with similar affinity (Figure 2B, C). This suggests that there is an additional component to the HP1α-array interaction that does not participate when interacting with mononucleosomes. This interaction(s) most likely involves connections that span multiple nucleosomes, and it is not directly dependent on the hinge region. This hinge independent interaction is disrupted by the presence of H1.4, since both wild-type HP1α and HP1α3KA display a 2.5–3-fold reduction in binding affinity for 12N200+H1.4 arrays compared to 12N200 arrays (Supplementary Table S1).
Next, we investigated whether these effects were specific to H1.4 or were a more general property of linker histones. To this end, we assembled our 12N200 arrays in the presence of an alternative linker histone, H1.0. We chose H1.0 as it is significantly sequence-divergent from H1.4 (∼37% sequence identity), as well as being biologically distinct; unlike H1.4, H1.0 is expressed independently of replication and is predominantly found in terminally differentiated cells (36). EMSAs (Figure 3D) using the 12N200+H1.0 arrays showed that HP1α displays the same interaction pattern as with 12N200+H1.4 arrays (Figure 3A; lower), indicating the effects on HP1α binding are not linker histone subtype-specific.
Linker histone stoichiometry modulates HP1α binding to nucleosome arrays
The experiments conducted thus far have all been in the context of a stoichiometric amount of linker histone, that is, each nucleosome is associated with one linker histone molecule. However, in cells this may not always be the case. Numerous studies have measured total linker histone stoichiometry in different cellular systems, with estimates ranging from ∼0.5–1.3 linker histone molecules per nucleosome (reviewed in (48)). We therefore explored whether varying linker histone stoichiometry can affect HP1α binding to nucleosome arrays.
We assembled a series of 12N200 arrays with differing H1.4-to-nucleosome ratios (1.26, 1.02, 0.78, 0.54, 0.27 and 0; Figure 4A) and then performed EMSAs with HP1α (Figure 4B). It should be noted that the H1.4-to-nucleosome ratio of 1.26 represents the fully saturated nucleosome array, as we have described previously (38), whereas the 1.02, 0.78, 0.54, and 0.27 ratios are approximately equivalent to 80, 60, 40 and 20% of the saturating amount of H1.4, respectively.
Figure 4.

Linker histone stoichiometry modulates HP1α binding to nucleosome arrays. (A) Representative native agarose gel of 12N200 arrays assembled with increasing amounts of linker histone H1.4. The apparent number of H1.4 molecules per nucleosome is displayed along the top of the gel. Sizes of select DNA ladder bands in lane 1 are indicated on the left. The band in lane 2 corresponds to the naked 12N200 DNA. (B) Representative EMSAs of HP1α (0–20 μM; 0.56x dilution series) binding to nucleosome arrays with varying levels of H1.4 as shown in (A). (C) Quantitative analysis of EMSA experiments as shown in (B). Data (symbols) are the average of four replicates and error bars represent the s.e. Lines represent fits to the data using the same binding model as in Figures 1 and 3. (D) Relative affinity (K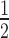 ; left axis, solid diamonds, black line) and cooperativity (Hill coefficient; right axis, hollow circles, dashed line) values extracted from each of the fits shown in (C).
; left axis, solid diamonds, black line) and cooperativity (Hill coefficient; right axis, hollow circles, dashed line) values extracted from each of the fits shown in (C).
These experiments show that there is an apparent gradual weakening of HP1α binding to the nucleosome arrays as the H1.4-level increases. Quantitative analysis of these EMSAs (Figure 4C, D) reveals that both the affinity of the interaction and binding cooperativity undergo non-linear transitions as H1.4 is varied. At low levels of H1.4 saturation (less than ∼50%) only modest weakening of HP1α binding (relative to the affinity for the H1.4-free array) is observed; ∼1.6-fold increase in K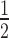 at 0.54 H1.4 per nucleosome (Figure 4D). However, at the very next increment (0.78 H1.4 per nucleosome), a much greater increase in K
at 0.54 H1.4 per nucleosome (Figure 4D). However, at the very next increment (0.78 H1.4 per nucleosome), a much greater increase in K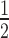 (∼2.8-fold) occurs, with further increases as the array becomes saturated with H1.4 (Figure 4D). Similarly, the positive cooperativity of binding is unaffected at the lowest ratio of H1.4 (0.27 H1.4 per nucleosome), but then a transition to a non-cooperative interaction occurs as the level of H1.4 is increased across the range of 0.54–1.02 H1.4 per nucleosome (Figure 4D). These data show that variations in linker histone stoichiometry are able to modulate the binding of HP1α to nucleosome arrays.
(∼2.8-fold) occurs, with further increases as the array becomes saturated with H1.4 (Figure 4D). Similarly, the positive cooperativity of binding is unaffected at the lowest ratio of H1.4 (0.27 H1.4 per nucleosome), but then a transition to a non-cooperative interaction occurs as the level of H1.4 is increased across the range of 0.54–1.02 H1.4 per nucleosome (Figure 4D). These data show that variations in linker histone stoichiometry are able to modulate the binding of HP1α to nucleosome arrays.
H3K9me3 and the histone variant H2A.Z both enhance binding of HP1α to nucleosome arrays
Given the established association between HP1α and the constitutive heterochromatin-associated histone modification H3K9me3, we next investigated whether this modification has any effect on the linker histone-mediated modulation of HP1α binding. We employed site-specific cysteine alkylation to install a trimethylated lysine analogue at the H3K9 position (H3K9cme3; Supplementary Figure S2A and B) (43,49), as has been used previously to study Swi6- and HP1β-chromatin interactions (12,25). We also produced the non-methylated equivalent, H3K9cme0, and confirmed that the methylation procedure had no non-specific effect on HP1α array binding (Supplementary Figure S2C and D). We then assembled 12N200 arrays containing H3K9cme3 (± H1.4) (Supplementary Figure S3A and B) and performed EMSA analysis with HP1α (Figure 5A).
Figure 5.

Methylation of H3K9 and H2A.Z modulate the binding of HP1α to nucleosomes. (A) EMSA analysis of HP1α (0–20 μM; 0.75× dilution series) binding to H3K9cme3 12N200 nucleosome arrays (4.7 nM; effective nucleosome concentration is ∼56 nM) assembled in the absence (upper) or presence (lower) of recombinant linker histone H1.4. (B) Quantitative analysis of EMSA experiments as shown in (A). Data are the average of three replicates and error bars represent the s.e. Lines represent fits to the data using the same binding model used in previous figures. For comparison, the positions of the K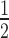 values for HP1α binding to unmodified 12N200 arrays in the absence (0.71 μM) or presence (2.15 μm) of H1.4 are indicated by the dashed grey lines (as determined in the fits in Figure 3B; flanking area shaded in light grey indicate the s.e.). (C) EMSA analysis of HP1α (0–20 μM; 0.75× dilution series) binding to 12N200 nucleosome arrays containing histone variant H2A.Z, conditions are identical to those in (A). (D) Quantitative analysis of EMSA experiments as shown in (C). Analysis is identical to that described in (B). (E) Bar graph comparing relative affinity (K
values for HP1α binding to unmodified 12N200 arrays in the absence (0.71 μM) or presence (2.15 μm) of H1.4 are indicated by the dashed grey lines (as determined in the fits in Figure 3B; flanking area shaded in light grey indicate the s.e.). (C) EMSA analysis of HP1α (0–20 μM; 0.75× dilution series) binding to 12N200 nucleosome arrays containing histone variant H2A.Z, conditions are identical to those in (A). (D) Quantitative analysis of EMSA experiments as shown in (C). Analysis is identical to that described in (B). (E) Bar graph comparing relative affinity (K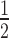 ) values derived from quantitative of EMSAs of HP1α binding to 12N200 ± H1.4 that contain either unmodified histones (blue), H3K9cme3 (grey), H2A.Z (red), or both H2A.Z and H3K9cme3 (purple). Data are derived from a minimum of three replicates for each binding experiment. Errors bars represent the S.E. (F) Bar graph comparing relative affinity (K
) values derived from quantitative of EMSAs of HP1α binding to 12N200 ± H1.4 that contain either unmodified histones (blue), H3K9cme3 (grey), H2A.Z (red), or both H2A.Z and H3K9cme3 (purple). Data are derived from a minimum of three replicates for each binding experiment. Errors bars represent the S.E. (F) Bar graph comparing relative affinity (K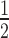 ) values derived from quantitative of EMSAs of HP1α binding to different mononucleosome substrates (N203, N203 + H1.4, and N147) that contain either unmodified histones (blue), H3K9cme3 (grey), H2A.Z (red), or both H2A.Z and H3K9cme3 (purple). Data are derived from a minimum of three replicates for each binding experiment. Errors bars represent the S.E.
) values derived from quantitative of EMSAs of HP1α binding to different mononucleosome substrates (N203, N203 + H1.4, and N147) that contain either unmodified histones (blue), H3K9cme3 (grey), H2A.Z (red), or both H2A.Z and H3K9cme3 (purple). Data are derived from a minimum of three replicates for each binding experiment. Errors bars represent the S.E.
When H3K9cme3 is incorporated into the 12N200 arrays HP1α binding is enhanced relative to the unmodified counterpart (compare Figure 5A with Figure 3A). This is both in terms of the HP1α concentration at which ‘shifting’ of the 12N200 band occurs (compare lanes 2–4 in the upper panels of Figures 3A and 5A) and also in the ‘sharpness’ of the band of the shifted complex—the final H3K9cme3 12N200-HP1α complex appears as a much sharper homogeneous band in the gel (compare lanes 15–17 in the upper panels of Figures 3A and 5A). This indicates that H3K9cme3 may also ‘lock-in’ HP1α at its preferred binding location.
Quantitative analysis (Figure 5B) shows H3K9cme3 enhances HP1α binding to the 12N200 array ∼2.5-fold relative to the unmodified array (H3K9cme3 K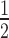 = 0.31 ± 0.02 versus unmodified K
= 0.31 ± 0.02 versus unmodified K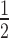 = 0.71 ± 0.02; Supplementary Table S1). However, this enhancement only occurs in the absence of H1.4. When H1.4 is incorporated into the H3K9cme3 12N200 arrays (Figure 5A, lower panel), HP1α binding is inhibited with the relative affinity of HP1α for the H3K9cme3 H1.4-saturated arrays (K
= 0.71 ± 0.02; Supplementary Table S1). However, this enhancement only occurs in the absence of H1.4. When H1.4 is incorporated into the H3K9cme3 12N200 arrays (Figure 5A, lower panel), HP1α binding is inhibited with the relative affinity of HP1α for the H3K9cme3 H1.4-saturated arrays (K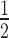 = 2.30 ± 0.29) being comparable to the unmodified counterpart (K
= 2.30 ± 0.29) being comparable to the unmodified counterpart (K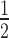 = 2.15 ± 0.23; Figure 5B). Therefore, H3K9cme3 cannot overcome the H1.4-mediated inhibition of HP1α binding to 12N200 arrays.
= 2.15 ± 0.23; Figure 5B). Therefore, H3K9cme3 cannot overcome the H1.4-mediated inhibition of HP1α binding to 12N200 arrays.
We have previously reported that the histone variant H2A.Z, like HP1α, is an important component of pericentric heterochromatin (28) and can enhance the binding of HP1α to chromatin in vitro (27). Thus, we investigated whether the incorporation of H2A.Z into our nucleosome arrays could influence HP1α binding when H1.4 is present (Supplementary Figure S3A and B). In these experiments, H2A.Z only accounts for 80% of the H2A in the arrays. This is because the fluorescent label (AlexaFlour488) is incorporated into our arrays via maleimide linkage to a site-specific cysteine mutant of H2A (H2AT120C), which we then mix one-to-four with unlabelled H2A, as described above. In this instance, we replaced the unlabelled H2A with unlabelled H2A.Z. However, this scenario is perhaps a better reflection of the in vivo situation where interspersion of core histones and histone variants occurs (50,51).
Incorporation of H2A.Z into the 12N200 arrays results in a significant enhancement of HP1α binding compared to the unmodified arrays (compare Figures 5C and 3A). Results also show that band shifting occurs at lower concentrations and a sharper homogeneous final complex band is observed (compare Figures 3A and 5C, lanes 15–17, upper panels). Interestingly, H2A.Z increases the relative affinity of HP1α for the linker histone-free 12N200 arrays ∼2.5-fold (K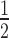 = 0.28 ± 0.01; Figure 5D, Supplementary Table S1), which is the same as H3K9cme3. However, when H1.4 is present H2A.Z has little effect, and HP1α binding to the array is still significantly inhibited (Figure 5C and D). Again, this is similar to the observations made on H3K9cme3 12N200 arrays. These data indicate that H2A.Z is able to functionally mimic the effect of H3K9me3 on the binding of HP1α to nucleosome arrays.
= 0.28 ± 0.01; Figure 5D, Supplementary Table S1), which is the same as H3K9cme3. However, when H1.4 is present H2A.Z has little effect, and HP1α binding to the array is still significantly inhibited (Figure 5C and D). Again, this is similar to the observations made on H3K9cme3 12N200 arrays. These data indicate that H2A.Z is able to functionally mimic the effect of H3K9me3 on the binding of HP1α to nucleosome arrays.
Notably, the HP1α3KA hinge mutant is still unable to bind to H3K9cme3 or H2A.Z 12N200 arrays. The relative affinity of HP1α3KA for the H3K9cme3 (K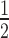 = 7.56 ± 0.29) or H2A.Z (K
= 7.56 ± 0.29) or H2A.Z (K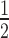 = 5.73 ± 0.0.64) 12N200 arrays is comparable to the unmodified arrays (K
= 5.73 ± 0.0.64) 12N200 arrays is comparable to the unmodified arrays (K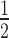 = 5.62 ± 0.26; Supplementary Table S1). The same trend is also observed when H1.4 is present in the arrays. Thus, the hinge region of HP1α is required for the proper binding of HP1α to H3K9cme3 and H2A.Z nucleosome arrays.
= 5.62 ± 0.26; Supplementary Table S1). The same trend is also observed when H1.4 is present in the arrays. Thus, the hinge region of HP1α is required for the proper binding of HP1α to H3K9cme3 and H2A.Z nucleosome arrays.
Concerning the affinity of histone H1.4 to H2A.Z-containing arrays, two studies have produced conflicting observations. In one study, H2A.Z inhibited the binding of histone H1 (52) whereas in the second study, H2A.Z did not (53). Our results are consistent with the latter study where histone H1 binds equally well to canonical arrays, H2A.Z-containing arrays and arrays modified with H3K9me3 (Figure 4, Supplementary Figure S3A and B).
Combination of H3K9me3 and H2A.Z partially reverses H1.4-mediated inhibition of HP1α binding to nucleosome arrays
H2A.Z and H3K9me3 are both components of constitutive heterochromatin and can co-exist in the same nucleosomes (27,28,54). Therefore, we investigated whether H2A.Z and H3K9me3 could work cooperatively to enhance HP1α binding. We produced 12N200 arrays containing both H2A.Z and H3K9cme3 (± H1.4) and performed quantitative EMSAs with HP1α. The relative affinity of HP1α for H2A.Z/H3K9cme3 12N200 arrays (K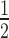 = 0.23 ± 0.01 μM) was not significantly different to arrays containing H2A.Z or H3K9cme3 alone (Figure 5E; Supplementary Figure S4 and Supplementary Table S1). However, when the H2A.Z/H3K9cme3 arrays are saturated with H1.4, there is a marked increase (approximately two-fold) in HP1α binding affinity over the other H1.4-saturated arrays (K
= 0.23 ± 0.01 μM) was not significantly different to arrays containing H2A.Z or H3K9cme3 alone (Figure 5E; Supplementary Figure S4 and Supplementary Table S1). However, when the H2A.Z/H3K9cme3 arrays are saturated with H1.4, there is a marked increase (approximately two-fold) in HP1α binding affinity over the other H1.4-saturated arrays (K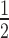 = 1.15 ± 0.09 μM; Figure 5E; Supplementary Figure S3 and Supplementary Table S1). Furthermore, a similar trend is observed with the affinities of the HP1α3KA mutant: enhanced binding only occurs on the H1.4-saturated H2A.Z/H3K9cme3 12N200 arrays, although, the overall binding affinity of HP1α3KA (K
= 1.15 ± 0.09 μM; Figure 5E; Supplementary Figure S3 and Supplementary Table S1). Furthermore, a similar trend is observed with the affinities of the HP1α3KA mutant: enhanced binding only occurs on the H1.4-saturated H2A.Z/H3K9cme3 12N200 arrays, although, the overall binding affinity of HP1α3KA (K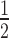 = 6.04 ± 0.70 μM) is still much weaker than wild-type HP1α (K
= 6.04 ± 0.70 μM) is still much weaker than wild-type HP1α (K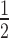 = 1.15 ± 0.09 μM; Supplementary Table S1). Thus, the combination of H2A.Z/H3K9cme3 enhances the interaction of HP1α with nucleosome arrays only when saturated with H1.4, and this enhancement appears independent of the hinge region of HP1α.
= 1.15 ± 0.09 μM; Supplementary Table S1). Thus, the combination of H2A.Z/H3K9cme3 enhances the interaction of HP1α with nucleosome arrays only when saturated with H1.4, and this enhancement appears independent of the hinge region of HP1α.
Interestingly, while the H2A.Z/H3K9cme3 combination partially restores HP1α binding affinity to the H1.4-saturated arrays, there is no re-establishment of the positive cooperative binding of HP1α (Supplementary Table S1). This suggests the combination of H2A.Z/H3K9cme3 enhances binding affinity to individual nucleosomes in the array, but does not affect HP1α interactions that bridge multiple nucleosomes.
To investigate this further, we returned to examining HP1α binding to mononucleosomes. We assembled N203, N203+H1.4 and N147 mononucleosomes with either H3K9cme3, H2A.Z, or the H2A.Z/H3K9cme3 combination and performed quantitative EMSAs with HP1α (Supplementary Figure S5A–C, Figure 5F, Supplementary Table S1). Unlike 12N200 arrays, the incorporation of H3K9cme3, H2A.Z, or the H2A.Z/H3K9cme3 combination in N203 mononucleosomes has no effect on the binding of HP1α either in the presence or absence of H1.4—the relative affinities are all approximately the same as the unmodified counterpart nucleosomes (Figure 5F). Further, the HP1α3KA mutant binds each of the H2A.Z, H3K9cme3 or H2A.Z/H3K9cme3 N203 mononucleosome species poorly, and displays no clear preferences (Supplementary Table S1). This is consistent with HP1α-linker DNA interactions being the dominant mechanism upon which HP1α binds to these mononucleosomes (see Figures 1 and 2). Therefore, the positive effect of H3K9cme3 and H2A.Z on HP1α binding to 12N200 arrays is not preserved in mononucleosomes with linker DNA.
The behaviour of HP1α changes on N147 mononucleosomes. The incorporation of H2A.Z into N147 nucleosomes results in a 1.5-fold increase in the relative binding affinity of HP1α over the unmodified N147 nucleosomes, whereas H3K9cme3 elicits no change (Figure 5F; Supplementary Table S1). Most significantly, when H3K9cme3 is combined with H2A.Z, a ∼2.5-fold increase in HP1α binding affinity is observed (relative to unmodified or H3K9cme3 N147 nucleosomes). This result parallels that of HP1α binding to the H1.4-saturated 12N200 H2A.Z/H3K9cme3 arrays (Figure 5E) and is consistent with the H2A,Z/H3K9cme3 combination enhancing binding to individual nucleosome core particles. These results also indicate that 12N200+H1.4 arrays essentially behave like linker DNA-free N147 mononucleosomes.
Taken together, these data suggest that there are two alternative modes of interaction of HP1α with nucleosomes. The first mode is through interactions with linker DNA. The second mode involves HP1α binding directly to the nucleosome core region when linker DNA is not available (either missing or occluded by linker histones). This second interaction mode can be promoted by H2A.Z and further enhanced by H3K9me3.
DISCUSSION
A paradigm for the histone code hypothesis is the recruitment of HP1 by H3K9me3 via an increased affinity between the chromodomain and the H3K9me3 mark. In this study we used a defined in vitro system to thoroughly dissect the effects of H3K9me3 and H2A.Z, as well as the linker histone H1.4 on the nucleosome binding activity of HP1α in arrays, mononucleosomes and nucleosome core particles. To our knowledge, this has not been done before. The major findings are that: (i) HP1α does not display a higher affinity for H3K9me3-containing mononucleosomes or core particles because our data indicates that the primary binding mode of HP1 with chromatin is through its interaction with linker DNA, (ii) HP1α does display a higher affinity for H3K9me3-containing nucleosomes in the context of arrays. Based on the data presented here, we suggest that the function of the H3K9me3 mark is to appropriately orientate or position HP1α on the nucleosome to facilitate HP1α- HP1α cooperative interactions within the array, (iii) H2A.Z can functionally substitute for H3K9me3 in the recruitment of HP1α, (iv) H1.4 has a negative effect on nucleosome and multi-nucleosome array binding by HP1α. However, this effect can be modulated by varying linker histone stoichiometry or by combining H3K9me3 with H2A.Z and (v) the ability of H3K9me3 with H2A.Z to enhance the binding of HP1α to arrays and nucleosome core particles suggests that this mode of binding is independent of linker DNA but is via a direct interaction with the nucleosome core. Taken togther, these data provide new insights into mechanisms that may regulate the recruitment and maintenance of HP1α at constitutive heterochromatin and clearly argues that the current paradigm of how HP1 is recruited to chromatin needs to be revisited.
The reports of positive interactions between linker histones and HP1α (20,21,37) prompted us to investigate the interaction of these two factors on nucleosomes. Surprisingly, we found that H1.4 has a negative effect on HP1α binding to mononucleosomes (Figure 1) and this effect is amplified on nucleosome arrays (Figure 3). One possibility to explain this is that the linker histone blocks interactions of HP1α with the linker DNA of the nucleosome. Current structural data indicates that linker histones occupy a significant proportion of at least one of the linker DNA arms of the nucleosome (32).Our data, along with that of others (18,19,47), clearly shows that HP1α-DNA interactions are critical for HP1α binding to nucleosomes, and the hinge region of HP1α is a key element in these interactions (Figure 2). Our experiments on mononucleosomes (Figure 1) reveal a trend where HP1α-nucleosome interactions are strongest on nucleosomes with more accessible linker DNA: N203 is the most accessible state, N203+H1.4 an intermediate state, and N147 represents an inaccessible state. This suggests that HP1α acts as a ‘promiscuous’ or ‘opportunistic’ DNA binder, and there is simply increased opportunity for binding nucleosomes with more accessible linker DNA. Subsequent to this opportunistic DNA binding, additional interactions between HP1α and the nucleosome core may contribute to stable complex formation. This is consistent with recent single-molecule data that points towards a similar mechanism (55).
Interestingly, our data also shows that the inhibitory effect of H1.4 on HP1α binding is greater in the context of nucleosome arrays (Figure 3). This is despite there being a similar relative amount of linker DNA per nucleosome in the N203 mononucleosomes and the 12N200 arrays; there are two 28 bp linker arms in the mononucleosomes, whereas each nucleosome in the array has the equivalent of two 26.5 bp linker arms. This suggests that H1.4 is acting via additional mechanisms at the level of nucleosome arrays. One simple explanation is that linker DNA in the array is generally less accessible owing to the steric and conformational restraints that come with being connected to other nucleosomes (56), and that linker histones induce compaction. Therefore, even though H1.4 may occupy a similar relative amount of linker DNA in both arrays and mononucleosomes, the remaining available DNA in an array is likely more difficult for HP1α to access. In addition to H1.4 reducing the affinity of HP1α for arrays, we also observe a negation of the positive cooperativity of binding (Figure 3). This effect is not observed in N203 mononucleosomes (Figure 1), where a similar level of cooperative binding occurs on both N203 and N203+H1.4. We conclude that H1.4 inhibits HP1α binding to arrays by both limiting access to linker DNA and preventing cooperative HP1α- HP1α interactions within the array (depicted in Figure 6).
Figure 6.

A model for the interaction of HP1α with chromatin and the role of H2A.Z, H3K9me3 and histone H1. (A) HP1α (green boomerangs) binds chromatin promiscuously to a nucleosomal array because of its strong affinity for DNA. On the other hand, the incorporation of H3K9me3 facilitates the proper orientation of HP1α on the nucleosome (via the H3K9me3-chromodomain interaction) to promote cooperative HP1α-HP1α interactions along the array. (B) H2A.Z mimics H3K9me3 in promoting cooperative HP1α-HP1α interactions via HP1α–acidic patch interactions (red circle on the face of the nucleosome). Note that the incorporation of H2A.Z promotes array compaction (represented by the zigzag conformation. (C) Histone H1 (which also promotes array compaction) inhibits both the affinity of binding of HP1α to nucleosomes and cooperative HP1α-HP1α interactions. H2A.Z and H3K9me3 can partially overcome the reduced HP1α binding affinity but cannot restore cooperative HP1α-HP1α interactions.
The non-linear relationship between the level of H1.4 saturation and changes in HP1α binding (Figure 4) supports an argument against a simple model of one-to-one binding-site competition where a pre-bound linker histone occupies or blocks access to an HP1α binding site. Instead, it points towards a model in which an H1.4-induced conformation in the nucleosome array is incompatible with productive cooperative binding of HP1α. Cooperative binding to arrays likely requires multivalent interactions that bridge nucleosomes, as has been reported for the fission yeast HP1 homologue Swi6 (25), and multivalent interactions have also been implicated in mammalian HP1α binding (57). Consistent with this, we show that HP1α interactions involving more than one nucleosome are important for H3K9cme3 or H2A.Z to enhance HP1α binding, as they each elicit a maximum effect in the context of 12N200 arrays (Figure 5). This effect is then abrogated by the introduction of H1.4 into the 12N200 array, further supporting the notion that the linker histone is likely disrupting trans-nucleosomal cooperative interactions of HP1α.
Genome-wide data that map the genomic locations of linker histones and HP1 in humans and flies show that while both linker histones and HP1 are clearly present in heterochromatin, there is not a strong enrichment of linker histones in HP1 heterochromatic domains (4,33,34). These data, along with our observations (Figure 4), suggest linker histone stoichiometry may act to regulate the distribution and/or dynamics of HP1α on chromatin: where low to intermediate levels of linker histone would be permissive to HP1α binding and the spreading of heterochromatin, while high levels of linker histone would destabilize HP1α-chromatin interactions and possibly even create a barrier to heterochromatin spreading. More complex layers of regulation may also be involved through the modification of specific linker histones. For example, trimethylation of lysine 26 in H1.4 (H1.4K26me3) has been shown to interact with HP1α (20) and may act like an H3K9me3 mimic in the absence of H3K9me3 (58). However, the functional significance of this modification in the context of HP1α binding in vivo is still unclear.
Perhaps one of the most significant findings of this study is that H2A.Z can mimic the effects of H3K9me3 in enhancing HP1α binding to nucleosome arrays (Figure 5). There has been a long-standing connection made between HP1 proteins and H2A.Z across a number of organisms, suggesting a conserved functional relationship between these two proteins (27,29,30). Our data provides further strength to this connection and suggests that H2A.Z could indeed act as a functional replacement for H3K9me3. Previously we showed that compared to H2A, the increased affinity of HP1α for a H2A.Z-containing nucleosome was due to its extended acidic patch (27).
HP1 proteins are evicted from heterochromatin during mitosis via Aurora B-dependent phosphorylation of H3S10, which antagonises HP1 binding to H3K9me3. However, a proportion of HP1α remains stably bound to mitotic pericentric heterochromatin (59) despite H3S10 phsophorylation. Our data is consistent with a speculative model where H2A.Z is responsible for maintaining this stably bound population of HP1α, given H2A.Z is a consistent feature of pericentric heterochromatin (27,31,60,61). Furthermore, H2A.Z is actively deposited at constitutive heterochromatin during early G1 phase (31), which coincides with when the bulk of HP1 proteins are beginning to be re-established at heterochromatin after mitotic eviction (62,63), suggesting H2A.Z may also have a role to play in this re-establishment. Our data also demonstrates that H2A.Z can substitute for H3K9me3 in recruiting HP1 thus supporting the notion that this replacement process can act as a compensatory mechanism under different physiological conditions where the level of H3K9me3 is reduced in constitutive heterochromatin (30,31). Further, it has been reported that HP1 is recruited to active promoters in Drosophila melanogaster independent of H3K9me3 (64). Given that H2A.Z is located at active promoters, it is attractive to speculate that H2A.Z might be involved in this recruitment.
In addition to acting as a possible substitute for H3K9me3, we also observe that H2A.Z and H3K9me3 can cooperate to enhance HP1α binding in specific circumstances, namely in the absence of linker DNA in mononucleosomes and the presence of linker histone H1.4 in nucleosome arrays (Figure 5). We have shown previously that both H2A.Z and H3K9me3 co-exist in nucleosomes in vivo and these can be pulled down by HP1α (27). The data presented in this study suggests that the combination of these two chromatin ‘marks’ may be important to facilitate binding of HP1α to chromatin when linker histones are present. Further, these results argue that HP1α has a second mode of binding to chromatin, when linker DNA is inaccessible, it can interact with the nucleosome core, which is facilitated by a combination of H2A.Z and H3K9me3.
Recently, the structure of a HP1-H3K9me3 dinucleosome complex was solved by cryo-EM and unexpectedly, no interaction between HP1 and linker DNA was observed (65). This contradicts the data present here, and by others, where the DNA binding ability of the hinge region is critical for chromatin binding (19,26,47). The reason for this discrepancy is not known but could be due to a number of non-mutually exclusive factors. First, the strongest interaction between HP1 and a nucleosomal template occurs in arrays and moreover, HP1 induces array conformational changes which could facilitate HP1-linker DNA interactions (27). Such HP1 induced conformational changes would not occur with a dinucleosome substrate. Further, it appears that the tetranucleosome is a fundamental structural unit of chromatin and therefore important inter-nucleosomal contacts are missing in a dinucleosome. Indeed, a recent study assembled chromatin using 12 copies of the ‘601’ nucleosome positioning sequence and using smFRET, showed that HP1α stabilized nucleosome stacking within the tetranucleosome reducing chromatin accessibility (66). These observations are consistent with our conclusions. Second, the linker-DNA exposure observed in the above study used a dinucleosome substrate with a short non-physiological linker length of 15bp. A combination of such a short linker length with the fact that different linker lengths affects linker DNA trajectory and thus the relative orientation of two adjacent nucleosomes (67) might affect the ability of HP1 to interact with linker DNA. In our study, we employed a longer physiological linker length (53 bp), which gives nucleosomes the added flexibility to adjust their orientation. Third, HP1 does display rapid exchange dynamics (66) and therefore differences in sample preparation might affect whether HP1-linker DNA interactions are captured. Interestingly though, the DNA binding mutant HP1α3KA binds with a slightly higher affinity to arrays compared to mononucleosomes suggesting that HP1 does participate in cooperative interactions in the absence of linker DNA binding in an array.
Based on the findings of this study, we propose a model for the binding of HP1α to different chromatin compositions depicted in Figure 6. Being a promiscuous DNA binding protein, there are numerous DNA binding sites available to HP1α in a nucleosomal array to yield a disorganized chromatin structure. However, either in the presence of H3K9me3 (Figure 6A) or H2A.Z (Figure 6B), HP1α is ‘locked in’ to its preferred or optimal binding location, which in turn enhances cooperative HP1α-HP1α interactions. On the other hand, histone H1 inhibits both the binding of HP1α to arrays and cooperative HP1α-HP1α interactions (Figure 6C). However, a combination of both H3K9me3 and H2A.Z increases the affinity of HP1α to individual nucleosomes in linker-containing arrays but are unable to overcome linker histone mediated inhibition of cooperative HP1α-HP1α interactions (Figure 6C). In conclusion, our data builds a more complex picture of the HP1α regulatory chromatin network, which may help explain better its function and its dynamic behaviour in chromatin.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Tremethick lab for helpful discussions and Emmalene Bartlett for providing feedback on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Australian Research Council [DP1401011268]. Funding for open access charge: School Budget.
Conflict of interest statement. None declared.
REFERENCES
- 1. Luger K., Dechassa M.L., Tremethick D.J.. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair. Nat. Rev. Mol. Cell Biol. 2012; 13:436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roadmap Epigenomics C., Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Filion G.J., van Bemmel J.G., Braunschweig U., Talhout W., Kind J., Ward L.D., Brugman W., de Castro I.J., Kerkhoven R.M., Bussemaker H.J. et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010; 143:212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G.. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012; 148:458–472. [DOI] [PubMed] [Google Scholar]
- 6. The, E.P.C. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang J., Jia S.T., Jia S.. New insights into the regulation of heterochromatin. Trends Genet. 2016; 32:284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saksouk N., Simboeck E., Dejardin J.. Constitutive heterochromatin formation and transcription in mammals. Epigenet. Chromatin. 2015; 8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lachner M., O’Carroll D., Rea S., Mechtler K., Jenuwein T.. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001; 410:116–120. [DOI] [PubMed] [Google Scholar]
- 10. Bannister A.J., Zegerman P., Partridge J.F., Miska E.A., Thomas J.O., Allshire R.C., Kouzarides T.. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001; 410:120. [DOI] [PubMed] [Google Scholar]
- 11. Jacobs S.A., Khorasanizadeh S.. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002; 295:2080–2083. [DOI] [PubMed] [Google Scholar]
- 12. Hiragami-Hamada K., Soeroes S., Nikolov M., Wilkins B., Kreuz S., Chen C., De La Rosa-Velázquez I.A., Zenn H.M., Kost N., Pohl W. et al. Dynamic and flexible H3K9me3 bridging via HP1β dimerization establishes a plastic state of condensed chromatin. Nat. Commun. 2016; 7:11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mishima Y., Jayasinghe C.D., Lu K., Otani J., Shirakawa M., Kawakami T., Kimura H., Hojo H., Carlton P., Tajima S. et al. Nucleosome compaction facilitates HP1gamma binding to methylated H3K9. Nucleic Acids Res. 2015; 43:10200–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Azzaz A.M., Vitalini M.W., Thomas A.S., Price J.P., Blacketer M.J., Cryderman D.E., Zirbel L.N., Woodcock C.L., Elcock A.H., Wallrath L.L. et al. Human heterochromatin protein 1alpha promotes nucleosome associations that drive chromatin condensation. J. Biol. Chem. 2014; 289:6850–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dialynas G.K., Terjung S., Brown J.P., Aucott R.L., Baron-Luhr B., Singh P.B., Georgatos S.D.. Plasticity of HP1 proteins in mammalian cells. J. Cell Sci. 2007; 120:3415–3424. [DOI] [PubMed] [Google Scholar]
- 16. Kwon S.H., Workman J.L.. The changing faces of HP1: From heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription. Bioessays. 2011; 33:280–289. [DOI] [PubMed] [Google Scholar]
- 17. Greil F., van der Kraan I., Delrow J., Smothers J.F., de Wit E., Bussemaker H.J., van Driel R., Henikoff S., van Steensel B.. Distinct HP1 and Su(var)3-9 complexes bind to sets of developmentally coexpressed genes depending on chromosomal location. Genes Dev. 2003; 17:2825–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao T., Heyduk T., Allis C.D., Eissenberg J.C.. Heterochromatin protein 1 binds to nucleosomes and DNA in vitro. J. Biol. Chem. 2000; 275:28332–28338. [DOI] [PubMed] [Google Scholar]
- 19. Meehan R.R., Kao C.F., Pennings S.. HP1 binding to native chromatin in vitro is determined by the hinge region and not by the chromodomain. EMBO J. 2003; 22:3164–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daujat S., Zeissler U., Waldmann T., Happel N., Schneider R.. HP1 binds specifically to Lys26-methylated histone H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1 binding. J. Biol. Chem. 2005; 280:38090–38095. [DOI] [PubMed] [Google Scholar]
- 21. Hale T.K., Contreras A., Morrison A.J., Herrera R.E.. Phosphorylation of the linker histone H1 by CDK regulates its binding to HP1alpha. Mol. Cell. 2006; 22:693–699. [DOI] [PubMed] [Google Scholar]
- 22. Lavigne M., Eskeland R., Azebi S., Saint-Andre V., Jang S.M., Batsche E., Fan H.Y., Kingston R.E., Imhof A., Muchardt C.. Interaction of HP1 and Brg1/Brm with the globular domain of histone H3 is required for HP1-mediated repression. PLos Genet. 2009; 5:e1000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smothers J.F., Henikoff S.. The HP1 chromo shadow domain binds a consensus peptide pentamer. Curr. Biol. 2000; 10:27–30. [DOI] [PubMed] [Google Scholar]
- 24. Cowieson N.P., Partridge J.F., Allshire R.C., McLaughlin P.J.. Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Curr. Biol. 2000; 10:517–525. [DOI] [PubMed] [Google Scholar]
- 25. Canzio D., Chang E.Y., Shankar S., Kuchenbecker K.M., Simon M.D., Madhani H.D., Narlikar G.J., Al-Sady B.. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell. 2011; 41:67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Larson A.G., Elnatan D., Keenen M.M., Trnka M.J., Johnston J.B., Burlingame A.L., Agard D.A., Redding S., Narlikar G.J.. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature. 2017; 547:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fan J.Y., Rangasamy D., Luger K., Tremethick D.J.. H2A.Z alters the nucleosome surface to promote HP1alpha-mediated chromatin fiber folding. Mol. Cell. 2004; 16:655–661. [DOI] [PubMed] [Google Scholar]
- 28. Rangasamy D., Berven L., Ridgway P., Tremethick D.J.. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J. 2003; 22:1599–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Verni F., Cenci G.. The Drosophila histone variant H2A.V works in concert with HP1 to promote kinetochore-driven microtubule formation. Cell Cycle. 2015; 14:577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bulynko Y.A., Hsing L.C., Mason R.W., Tremethick D.J., Grigoryev S.A.. Cathepsin L stabilizes the histone modification landscape on the Y chromosome and pericentromeric heterochromatin. Mol. Cell. Biol. 2006; 26:4172–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boyarchuk E., Filipescu D., Vassias I., Cantaloube S., Almouzni G.. The histone variant composition of centromeres is controlled by the pericentric heterochromatin state during the cell cycle. J. Cell Sci. 2014; 127:3347–3359. [DOI] [PubMed] [Google Scholar]
- 32. Bednar J., Garcia-Saez I., Boopathi R., Cutter A.R., Papai G., Reymer A., Syed S.H., Lone I.N., Tonchev O., Crucifix C. et al. Structure and dynamics of a 197 bp nucleosome in complex with linker histone H1. Mol. Cell. 2017; 66:384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Braunschweig U., Hogan G.J., Pagie L., van Steensel B.. Histone H1 binding is inhibited by histone variant H3.3. EMBO J. 2009; 28:3635–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Izzo A., Kamieniarz-Gdula K., Ramirez F., Noureen N., Kind J., Manke T., van Steensel B., Schneider R.. The genomic landscape of the somatic linker histone subtypes H1.1 to H1.5 in human cells. Cell Rep. 2013; 3:2142–2154. [DOI] [PubMed] [Google Scholar]
- 35. Millan-Arino L., Islam A.B., Izquierdo-Bouldstridge A., Mayor R., Terme J.M., Luque N., Sancho M., Lopez-Bigas N., Jordan A.. Mapping of six somatic linker histone H1 variants in human breast cancer cells uncovers specific features of H1.2. Nucleic Acids Res. 2014; 42:4474–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hergeth S.P., Schneider R.. The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015; 16:1439–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weiss T., Hergeth S., Zeissler U., Izzo A., Tropberger P., Zee B.M., Dundr M., Garcia B.A., Daujat S., Schneider R.. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenet. Chromatin. 2010; 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ryan D.P., Tremethick D.J.. A dual affinity-tag strategy for the expression and purification of human linker histone H1.4 in Escherichia coli. Protein Expr. Purif. 2016; 120:160–168. [DOI] [PubMed] [Google Scholar]
- 39. Studier F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005; 41:207–234. [DOI] [PubMed] [Google Scholar]
- 40. Luger K., Rechsteiner T.J., Richmond T.J.. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 1999; 119:1–16. [DOI] [PubMed] [Google Scholar]
- 41. Fan J.Y., Gordon F., Luger K., Hansen J.C., Tremethick D.J.. The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states. Nat. Struct. Biol. 2002; 9:172–176. [DOI] [PubMed] [Google Scholar]
- 42. Silva A.P., Ryan D.P., Galanty Y., Low J.K., Vandevenne M., Jackson S.P., Mackay J.P.. The N-terminal region of chromodomain helicase DNA-binding protein 4 (CHD4) is essential for activity and contains a high mobility group (HMG) Box-like-domain that can bind Poly(ADP-ribose). J. Biol. Chem. 2016; 291:924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simon M.D., Shokat K.M.. Wu C, Allis CD. Methods in Enzymology. 2012; 512:Academic Press; 57–69. [DOI] [PubMed] [Google Scholar]
- 44. Lowary P.T., Widom J.. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998; 276:19–42. [DOI] [PubMed] [Google Scholar]
- 45. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Caterino T.L., Fang H., Hayes J.J.. Nucleosome linker DNA contacts and induces specific folding of the intrinsically disordered H1 carboxyl-terminal domain. Mol. Cell. Biol. 2011; 31:2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mishima Y., Watanabe M., Kawakami T., Jayasinghe C.D., Otani J., Kikugawa Y., Shirakawa M., Kimura H., Nishimura O., Aimoto S. et al. Hinge and chromoshadow of HP1alpha participate in recognition of K9 methylated histone H3 in nucleosomes. J. Mol. Biol. 2013; 425:54–70. [DOI] [PubMed] [Google Scholar]
- 48. Woodcock C.L., Skoultchi A.I., Fan Y.. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 2006; 14:17–25. [DOI] [PubMed] [Google Scholar]
- 49. Simon M.D., Chu F., Racki L.R., de la Cruz C.C., Burlingame A.L., Panning B., Narlikar G.J., Shokat K.M.. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007; 128:1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nekrasov M., Soboleva T.A., Jack C., Tremethick D.J.. Histone variant selectivity at the transcription start site: H2A.Z or H2A.Lap1. Nucleus. 2013; 4:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Soboleva T.A., Nekrasov M., Ryan D.P., Tremethick D.J.. Histone variants at the transcription start-site. Trends Genet. 2014; 30:199–209. [DOI] [PubMed] [Google Scholar]
- 52. Thakar A., Gupta P., Ishibashi T., Finn R., Silva-Moreno B., Uchiyama S., Fukui K., Tomschik M., Ausio J., Zlatanova J.. H2A.Z and H3.3 histone variants affect nucleosome structure: biochemical and biophysical studies. Biochemistry. 2009; 48:10852–10857. [DOI] [PubMed] [Google Scholar]
- 53. White A.E., Hieb A.R., Luger K.. A quantitative investigation of linker histone interactions with nucleosomes and chromatin. Sci. Rep. 2016; 6:19122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Greaves I.K., Rangasamy D., Devoy M., Marshall Graves J.A., Tremethick D.J.. The X and Y chromosomes assemble into H2A.Z-containing [corrected] facultative heterochromatin [corrected] following meiosis. Mol. Cell. Biol. 2006; 26:5394–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bryan L.C., Weilandt D.R., Bachmann A.L., Kilic S., Lechner C.C., Odermatt P.D., Fantner G.E., Georgeon S., Hantschel O., Hatzimanikatis V. et al. Single-molecule kinetic analysis of HP1-chromatin binding reveals a dynamic network of histone modification and DNA interactions. Nucleic Acids Res. 2017; 45:10504–10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song F., Chen P., Sun D., Wang M., Dong L., Liang D., Xu R.-M., Zhu P., Li G.. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science. 2014; 344:376–380. [DOI] [PubMed] [Google Scholar]
- 57. Kilic S., Bachmann A.L., Bryan L.C., Fierz B.. Multivalency governs HP1α association dynamics with the silent chromatin state. Nat. Commun. 2015; 6:7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ruan J., Ouyang H., Amaya M.F., Ravichandran M., Loppnau P., Min J., Zang J.. Structural basis of the chromodomain of Cbx3 bound to methylated peptides from histone h1 and G9a. PLoS One. 2012; 7:e35376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schmiedeberg L., Weisshart K., Diekmann S., Meyer Zu Hoerste G., Hemmerich P.. High- and low-mobility populations of HP1 in heterochromatin of mammalian cells. Mol. Biol. Cell. 2004; 15:2819–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Greaves I.K., Rangasamy D., Ridgway P., Tremethick D.J.. H2A.Z contributes to the unique 3D structure of the centromere. Proc Natl Acad Sci U S A. 2007; 104:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nekrasov M., Amrichova J., Parker B.J., Soboleva T.A., Jack C., Williams R., Huttley G.A., Tremethick D.J.. Histone H2A.Z inheritance during the cell cycle and its impact on promoter organization and dynamics. Nat. Struct. Mol. Biol. 2012; 19:1076–1083. [DOI] [PubMed] [Google Scholar]
- 62. Hirota T., Lipp J.J., Toh B.-H., Peters J.-M.. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005; 438:1176. [DOI] [PubMed] [Google Scholar]
- 63. Fischle W., Tseng B.S., Dormann H.L., Ueberheide B.M., Garcia B.A., Shabanowitz J., Hunt D.F., Funabiki H., Allis C.D.. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005; 438:1116–1122. [DOI] [PubMed] [Google Scholar]
- 64. Figueiredo M.L., Philip P., Stenberg P., Larsson J.. HP1a recruitment to promoters is independent of H3K9 methylation in Drosophila melanogaster. PLos Genet. 2012; 8:e1003061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Machida S., Takizawa Y., Ishimaru M., Sugita Y., Sekine S., Nakayama J.I., Wolf M., Kurumizaka H.. Structural basis of heterochromatin formation by human HP1. Mol. Cell. 2018; 69:385–397. [DOI] [PubMed] [Google Scholar]
- 66. Kilic S., Felekyan S., Doroshenko O., Boichenko I., Dimura M., Vardanyan H., Bryan L.C., Arya G., Seidel C.A.M., Fierz B.. Single-molecule FRET reveals multiscale chromatin dynamics modulated by HP1alpha. Nat. Commun. 2018; 9:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nikitina T., Norouzi D., Grigoryev S.A., Zhurkin V.B.. DNA topology in chromatin is defined by nucleosome spacing. Sci. Adv. 2017; 3:e1700957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J.. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009; 25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.