

Abstract
Mycobacterium tuberculosis (Mtb) infection is initiated in the distal airways, but the bacteria ultimately disseminate to the lung interstitium. Although various cell types, including alveolar macrophages (AM), neutrophils, and permissive monocytes, are known to be infected with Mtb, the initially infected cells as well as those that mediate dissemination from the alveoli to the lung interstitium are unknown. In this study, using a murine infection model, we reveal that early, productive Mtb infection occurs almost exclusively within airway-resident AM. Thereafter, Mtb-infected, but not uninfected, AM localize to the lung interstitium through mechanisms requiring an intact Mtb ESX-1 secretion system. Relocalization of infected AM precedes Mtb uptake by recruited monocyte-derived macrophages and neutrophils. This dissemination process is driven by non-hematopoietic host MyD88/IL-1R inflammasome signaling. Thus, IL-1-mediated crosstalk between Mtb-infected AM and non-hematopoietic cells promotes pulmonary Mtb infection by enabling infected cells to disseminate from the alveoli to the lung interstitium.
Keywords: Alveolar macrophages, IL-1, pulmonary tuberculosis, lung, innate immunity, granuloma
Graphical Abstract
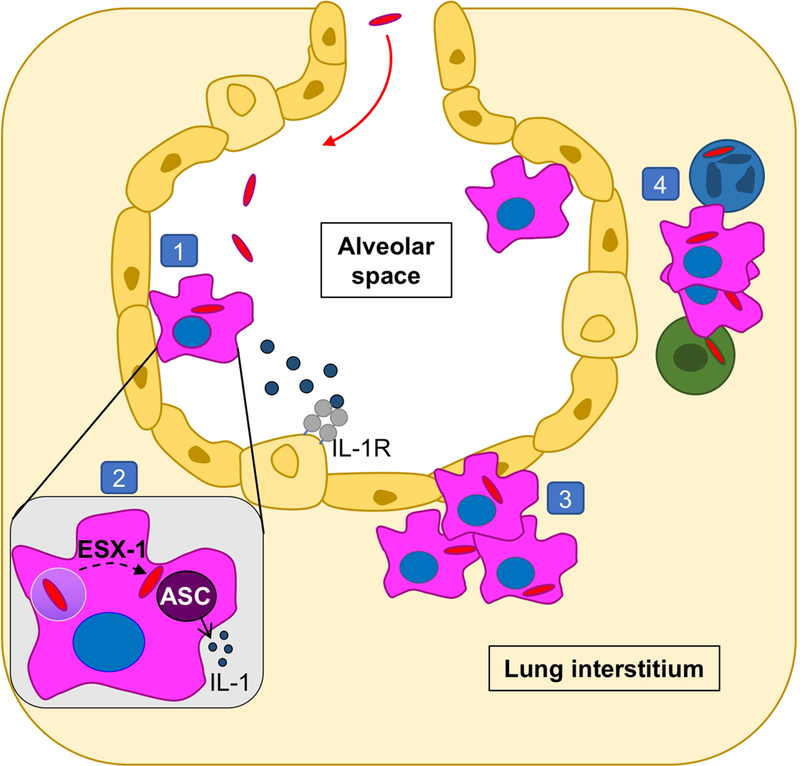
eTOC:
Using a mouse model, Cohen et al demonstrate that early Mycobacterium tuberculosis infection predominantly targets alveolar macrophages (AM). Infected AM re-localize from the alveolar space to lung interstitium, preceding bacterial dissemination into migratory myeloid populations. Re-localization requires bacterial ESX-1 and host IL-1R, suggesting dual implications for establishing infection and immunity.
Introduction
The lung is the portal of Mycobacterium tuberculosis (Mtb), as transmission occurs upon inhalation of aerosolized bacilli coughed from the lungs of infected individuals (Pai et al., 2016). The initial interaction between bacilli and lung immune cells is crucial to determine whether infection results in bacterial eradication, containment and asymptomatic infection, or unbridled replication and active disease. However, little is known about these early host-pathogen interactions.
It has long been assumed, but not definitively shown, that alveolar macrophages (AM) are the first cells to phagocytize Mtb upon inhalation (Philips and Ernst, 2012; Srivastava et al., 2014). In the M. marinum zebrafish model, microbicidal resident macrophages in the hindbrain are the first cells to harbor mycobacteria. Subsequently, chemokines induced by mycobacterial cell wall lipids recruit circulating monocytes that are more permissive to bacterial replication, and mycobacterial transfer into these cells promotes dissemination (Cambier et al., 2017; Cambier et al., 2014). However, how these events transpire within the mammalian lung remains unclear. In the mouse TB model, the earliest timepoint at which cell types harboring Mtb has been assessed is 14 days post-aerosol infection, at which point Mtb was reported to be equally distributed between AM, neutrophils, and dendritic cells (DC) (Wolf et al., 2007). Eventually chronic infection is established in the lung interstitium, at which time Mtb has been reported to reside primarily within DC (Gonzalez-Juarrero and Orme, 2001; Wolf et al., 2007). However, how Mtb infection transitions from alveoli to the lung interstitium is currently unknown.
Here we sought to define the cellular and molecular pathways governing the initial dissemination of Mtb to the lung interstitium. Our findings reveal that AM, a cellular population typically thought to reside exclusively within alveoli, actually ferry Mtb into the interstitium, a process that depends on the ESX-1 secretion system. We show that IL-1 signaling, a pathway known to be critical for immunity against TB, plays an unexpected role in facilitating this dissemination.
Results
AM are the major target of Mtb early post-infection
To investigate the early kinetics of Mtb dissemination in the mammalian lung, we measured mCherry fluorescence of lung leukocytes in mice infected with H37Rv Mtb expressing mCherry (Mtb-mCherry). To detect infected cells immediately post-exposure, mice were given a high aerosolized dose of Mtb-mCherry (1200–2500 CFU), and lungs were collected at 48 h and 7 days post-infection (p.i.) for microscopy. At 48 h, both alveolar macrophages (AM) and epithelial cells (EC) infected with single bacilli were detected by microscopy, although infected EC were less common (Figure 1A, S1H, and S1I). However, while Mtb was still observed in AM 7 days p.i., at which point there were multiple bacilli per cell (Figure 1B), Mtb could not be detected in EC, suggesting that AM represent the major reservoir for productive infection. Infected leukocytes were next examined by flow cytometry using an adaptation of recently devised gating strategies to identify pulmonary myeloid populations (Figure S1A-G) (Guilliams et al., 2016; Plantinga et al., 2013). Consistent with our microscopy findings, both infected AM and EC were uncommon 48 h following high dose infection, but mCherry+ AM were more readily detected than mcherry+ EC over naive controls (Figure S1J-K). Furthermore, by day 8 p.i., AM comprised all of the few mCherry+ cells that could be identified with a standard low dose and comprised ~80% of the infected cells by day 11 (Figure S1L). As infection progressed, however, Mtb was also identified in cell populations recruited from the periphery (Figure 1B). By 3 weeks p.i., while total numbers of infected AM plateaued, there was a vast increase in numbers of infected neutrophils (PMN, CD11b+Ly6G+) and monocyte-derived cells (MC, CD64+CD11c+MHCII+) (Figure 1C, S1E, and S1M). We observed very little Mtb within interstitial macrophages (IM, CD64+CD11clow) or conventional DC (cDC), contrary to previous studies implicating cDC as a major reservoir of infection (Wolf et al., 2007) (Figure 1B, Figure S1F). This discrepancy can be attributed to the finding that infected CD11b+CD11c+ cells previously identified as cDC express CD64, a marker absent on cDC but expressed by macrophages and recruited MC (Figure S1N) (Guilliams et al., 2016; Langlet et al., 2012; Plantinga et al., 2013).
Figure 1. See also Figure S1. AM are the major target of Mtb early post-infection.
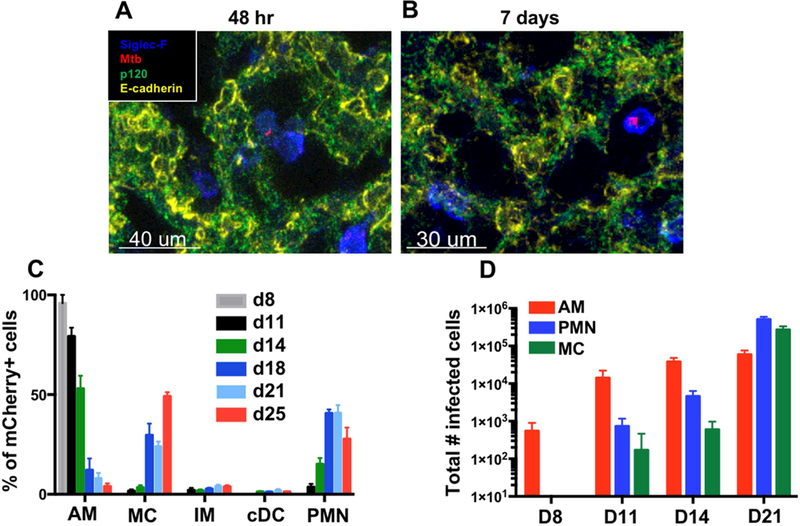
(A,B) Imaging of infected alveolar macrophages (AM, SiglecF+) from lung sections collected at (A) 48 h (7 sections, n=5 mice) or (B) D7–10 (4 sections, n=3 mice). (C) Composition of mCherry+ lung leukocytes over a timecourse of infection (3 pooled experiments, n=3–11 mice/timepoint). (D) Total numbers of infected AM, neutrophils (PMN), and monocyte-derived cells (MC) in the lung. Shown is a representative experiment (n=3–4 mice/timepoint) performed 3 times. The data are presented as means ± SEM.
Mtb-infected AM are found in the lung interstitium in the context of early cellular aggregates
To better understand how Mtb is transferred from resident AM to recruited MC, we next sought to visualize the spatial organization of Mtb-infected cells within the lung during early stages of dissemination. Interestingly, even within a single lobe, distinct foci of infection could be observed as early as 15 days p.i. Furthermore, each focus displayed distinct patterns of cellular organization that represent different stages of infection (Figure 2). For example, some foci exhibited exclusive Mtb infection of AM (panel I), others showed infected AM co-localized with uninfected and infected MC (panel II-IV), whereas still others were comprised primarily of infected MC with only a few infected AM in the periphery (panel V). This heterogeneity is consistent both with recent observations in non-human primates showing that each nidus of pulmonary Mtb infection undergoes a different pattern of progression that results in distinct outcomes (Gideon et al., 2015; Lin et al., 2014; Martin et al., 2017) as well in zebrafish showing that the fate of granulomas differs even as early as 4–11 days p.i., when only innate immunity is in play (Adams et al., 2011). By 19 days p.i., the lesions contained higher numbers of Mtb bacilli that were harbored almost exclusively within MC aggregates; Mtb-infected AM were rarely observed (panel VI).
Figure 2. See also Figure S2. Mtb-infected AM are found in the lung interstitium in the context of early cellular aggregates.
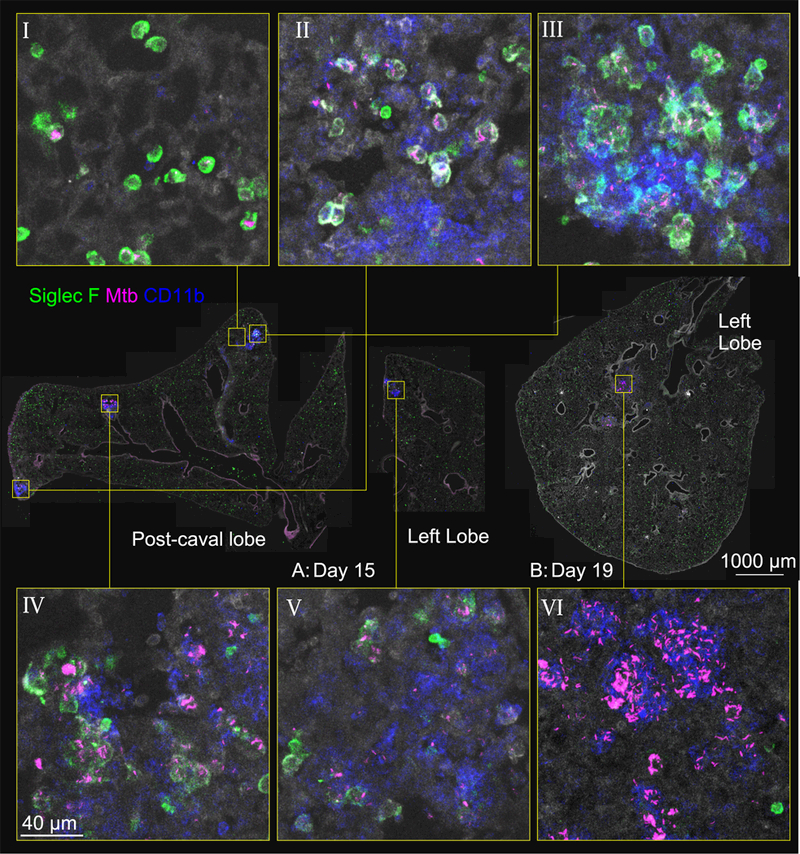
Representative immunofluorescence for Siglec F and CD11b at D15 (panels I-V) and D19 (panel VI) p.i. Zoom-in panels demonstrate infectious foci in various stages of progression: infected AM in alveolar sacs (panel I); co-localization of infected AMs with MC, with infection predominantly in AM (panel II); clustering of AM, with increased infection of MC and apparent interstitial localization of these cells (panels III, IV); progression of infection to MC, with AM pushed to the periphery (panel V). At D19, nearly all infected cells are MC (panel VI). Lung architecture is shown by autofluorescence (gray).
Further examination revealed that day 15-infected AM in more advanced inflammatory foci did not reside in classical alveolar air pockets, but formed clusters within the lung interstitium (Figure 2; panels II-V), consistent with observations of macrophage aggregation within days of infection in the zebrafish M. marinum model (Davis et al., 2002). These clusters were distinct from induced bronchus-associated lymphoid tissues (iBALT) in that they did not localize to bronchioles (Video S1) and were initially formed without contribution of T and B cells (data not shown). This clustering may represent local AM proliferation as Mtb infection led to increased BrdU uptake by AM as early as 13 days p.i.
(Figure S2A). Among infected AM, those in the interstitial space incorporated more BrdU than their airway-resident counterparts, suggesting that this proliferative effect may primarily occur post-translocation (Figure S2B). Thus, by 15 days post-aerosol exposure, foci of infection containing proliferating, infected AM can be found in the lung interstitium, indicating that these AM have re-located from the airspaces, generating lung aggregates that precede subsequent granuloma formation.
Intratracheal antibody administration enables tracking and quantification of Mtb-infected AM residing in the lung airways
To further study the factors that promote the interstitial localization of Mtb-infected AM, we speculated that we could distinguish cells resident in the airspace from those in the interstitium by administering an intratracheal antibody, which should only label alveolar-localized cells. To test this, anti-CD45.2 antibodies were administered intratracheally to Mtb-infected mice, and lungs were harvested for microscopy. While some peripheral regions of the lung failed to label with the antibody (data not shown), the vast majority of AM were efficiently labeled (Figure 3A). Furthermore, infected AM that seemed to be located in the interstitium were indeed spared from antibody labeling, whereas uninfected AM residing within alveolar air sacs labeled positively (Figure 3B). Upon analysis of 20-um Z-stacks, we further showed that most unlabeled, Mtb-infected AM were fully surrounded by interstitial lung tissue (Figure 3C,Video S2), whereas antibody-labeled AM, which were primarily uninfected, were in contact with air in the alveolar pocket on at least one side (Figure 3D, Video S3).
Figure 3. See also Figure S3. Intratracheal antibody administration enables tracking and quantification of airway-resident AM upon Mtb infection.
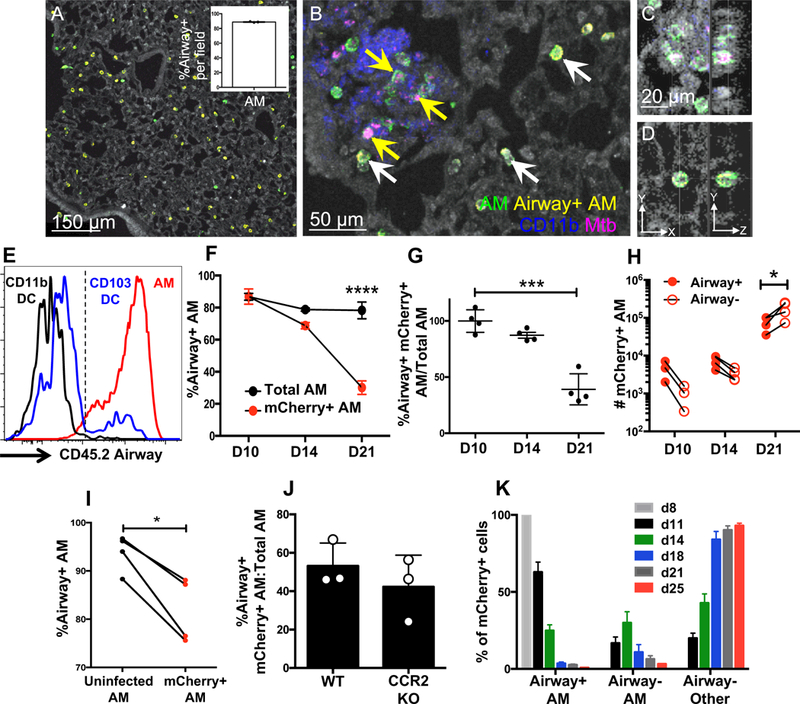
(A) Representative CD45.2 airway labeling of D15 p.i. B6 mice in an Mtb-free region with healthy lung architecture (autofluorescence). The inset shows the percentage of CD45.2+ AM over 3 independent fields. (B) A representative D15 Mtb-infected lung region, showing infected AM that are airway- and interstitial (yellow arrows) alongside airway+ uninfected AM in an alveolar pocket (white arrows). (C,D) Z-stack of D15 airway- (C) and airway+ (D) AM. (E) Airway labeling of AM, CD11b+ DC, and CD103+ DC in naive B6 mice by FACS. (F) Airway labeling of mCherry+ and total AM at indicated timepoints (n=4 mice/timepoint). (G) The data are plotted as ratios of mCherry+ over total AM to normalize for labeling efficiency. (H) Total numbers of airway+ and airway- infected AM over time (n=3 mice/timepoint). (I) Total airway+ AM were sorted from the lungs of D13 p.i. mice (n=10 mice) and adoptively transferred intratracheally into infection-matched congenic (B6.SJL) recipients (n=1 or 2 mice/group). Shown is the airway labeling of AM 3–5 days post-transfer from 3 independent experiments (each dot represents one mouse receiving AM pooled from 10 mice). (J) Airway labeling of infected AM in B6 and CCR2−/− mice D15 p.i. (n=3 mice/group). (K) Distribution of airway+ AM, airway- AM, and airway- other leukocytes among lung mCherry+ cells over time (3 pooled experiments, n=3–11 mice/timepoint). Multiple group comparisons were performed by 2-way ANOVA (F and H) and single group comparisons by unpaired t-test (G and J) or paired t-test (I). The data are presented as means ± SEM. All experiments were performed at least 2–3 times.
To assess the mechanisms underlying this altered AM localization in a quantitative manner, we used flow cytometry to distinguish cells in the air space from those in the lung interstitium. Consistent with the imaging, flow cytometric analysis of uninfected mice also showed that AM effectively labeled with intratracheally administered anti-CD45.2 antibody. The specificity of the airway labeling was supported by the finding that a subset of CD103+ DC, which are known to extend dendrites into alveoli to sample luminal antigens (Daniels et al., 2016; Leepiyasakulchai et al., 2013), also labeled positively for CD45.2, while CD11b+ DC, which are distributed diffusely throughout the lung interstitium (Daniels et al., 2016; Sung et al., 2006), were CD45.2-negative (Figure 3E). We controlled for background autofluorescence and non-specific staining by showing that AM and CD103+ DC did not label with antibodies against CD45.1, an alloantigen that is not expressed by C57BL/6 mice (Figure S3A,B). Indeed, by this method, we found that only AM and CD103+ DC showed measurable airway localization (Figure S3C,D). Overall, these data support the use of flow cytometry-based airway labeling to assess the localization of myeloid cells in lung airspaces.
Localization of Mtb-infected AM to the lung interstitium precedes dissemination to other myeloid cells
Using this airway labeling, we sought to quantitate the frequencies of Mtb-infected AM in the airspace vs. the interstitium at various times after aerosol infection with Mtb-mCherry. At day 10 p.i., both the mCherry+ and total AM populations predominantly labeled with the airway-administered antibody, resulting in an airway labeling value of ~100% when presented as a ratio of infected AM to total AM, suggesting global AM localization to the alveolar airspaces early p.i. (Figure 3F,G). Consistent with the interstitial localization of infected AM observed by microscopy (Figure 2B,C), by day 14 p.i., mCherry+ AM became resistant to the airway label compared to uninfected AM; by day 21 p.i., the majority of infected AM failed to label, resulting in a labeling ratio of ~40%, and interstitial infected AM outnumbered alveolar infected AM. In contrast, uninfected AM continued to bind the airway antibody at a constant rate of ~80% over the timecourse (Figure 3F-H), supporting the imaging observations that only Mtb-infected AM localize to the interstitium. Importantly, this was not due to downregulation of CD45 in infected AM (Figure S3E) or selective cell death among airway+ AM (Figure S3F). Because these experiments evaluated single timepoints rather than following the fate of individual cells over time, we considered the possibility that infected AM were simply passing bacteria to another interstitial cell type that had upregulated AM surface markers. To investigate this possibility, total airway-positive AM (mCherry+ and mCherry-) were sorted from the lungs of infected mice 15 days p.i. and adoptively transferred intratracheally into infection-matched recipients. Five days post-transfer, recipient mice were administered an airway antibody intratracheally, and the labeling pattern of transferred mCherry- and mCherry+ AM was evaluated. Consistent with the timecourse experiments, infected donor AM were more resistant to the airway antibody than their uninfected donor counterparts (Figure 3I). Although AM are rarely replenished by circulating monocytes at steady-state (Guilliams et al., 2013; Hashimoto et al., 2013), they can contribute to the AM pool following insults, such as irradiation or macrophage ablation (Landsman and Jung, 2007). To investigate whether AM infected with Mtb derive from circulating monocytes that subsequently enter the alveolar space, we examined the localization of Mtb-infected AM in CCR2−/− mice in which monocytes fail to egress from the bone marrow and are therefore substantially reduced in the lung (Figure S3G,H). At D15 p.i., the interstitial localization of Mtb-infected AM was unaffected by CCR2 deficiency, arguing against the notion that the infected AM derived from circulating monocytes (Figure 3J). Thus, Mtb-infected AM that originate in the airway can translocate to the interstitium and do so with a greater propensity than their uninfected counterparts.
Next, we assessed how the interstitial relocalization of Mtb-infected AM correlates with bacterial dissemination to other cell types. While all of the infected cells were airway-positive AM at day 8 p.i., by day 14, the infected AM were almost evenly split between airway-positive and airway-negative, and this timepoint corresponded with the earliest detection of bacteria in other myeloid compartments in the interstitium (Figure 3K). By day 18 p.i. and later, there were more airway-negative AM than airway-positive AM amongst Mtb-infected cells, but the clear majority of infected cells were other airway-negative myeloid cells, including MC and neutrophils. These data are consistent with the observation of large infected MC infiltrates by day 19 p.i. (Figure 2, panel VI) and suggest that the localization of infected AM to the lung interstitium precedes bacterial dissemination to other cell types, representing the first step in establishing foci of infection at this site.
Lung interstitial localization of Mtb-infected AM requires inflammasome signaling on non-hematopoietic cells
We next utilized the flow cytometry assay to address the mechanisms of altered AM localization. MyD88 is required for the innate immune response to Mtb infection, and upstream IL-1R signaling has been implicated as a major component of this dependence (Fremond et al., 2007; Fremond et al., 2004; Mayer-Barber et al., 2010). Consistent with this, Mtb-infected AM significantly upregulated pro-IL-1β levels compared to uninfected AM after 15 days of infection (Figure S4A,B). To ask whether these pathways are involved in the altered localization of Mtb-infected AM to the lung interstitium, airway labeling of infected AM in MyD88−/− and IL-1R−/− mice was evaluated 19 days p.i.
Interestingly, mCherry+ AM from both MyD88−/− and IL-1R−/− mice showed significantly enhanced levels of airway labeling compared to WT mice (Figure 4A), supporting a role for MyD88/IL-1R signaling in the lung interstitial localization of Mtb-infected AM. Inflammasome activation is a well described mechanism of IL-1 production involving the recruitment of caspases to cleave IL-1 from its pro form into its active form (Latz et al., 2013). Mtb is a potent activator of the inflammasome, which contributes, at least partially, to IL-1 production during TB (Koo et al., 2008; Mayer-Barber et al., 2010; Mishra et al., 2010). Consistent with the role of IL-1R signaling in mediating interstitial AM localization, Mtb-infected AM from mice deficient for ASC(Subramanian et al., 2013), the inflammasome adaptor protein that links pattern recognition and caspase recruitment and that is required for inflammasome-dependent IL-1 production (Martinon et al., 2002), localized more to the airways (~60% airway-positive) than WT AM (~40% airway-positive) (Figure 4B).
Figure 4. See also Figure S4. Lung interstitial localization of Mtb-infected AM requires inflammasome signaling on non-hematopoietic cells.
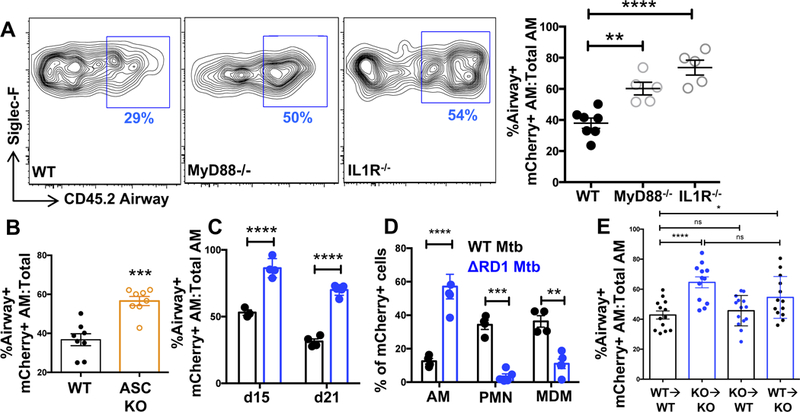
(A) Representative FACS plots and cumulative data of airway+ mCherry+ AM in B6, MyD88−/−, and IL1R−/− mice D19 p.i. (n=5 mice/group). (B) Cumulative data of airway labeling of B6 and ASC−/− mCherry+ AM D19 p.i. (n=8 mice/group from 2 pooled experiments). (C) Cumulative airway labeling of AM infected with either WT (H37Rv) or RD1-deficient Mtb (ΔRD1) at D15 and D21 p.i. (D) D21 composition of lung mCherry+ cells following H37Rv or ΔRD1 infection (n=4–5 mice/group). (E) Cumulative airway labeling data of infected AM in criss-cross IL1R−/− chimeras d19 p.i. (3 pooled experiments with 13–14 mice/group). Multiple-group comparisons were performed by 2-way ANOVA (D and E) and single group comparisons by t-test (A, B, and E). Experiments were performed at least twice and are shown either as representative data (A, C, and D) or pooled data from multiple experiments (B and E). The data are presented as means ± SEM.
IL-1 production is reliant upon the Mtb ESX-1 secretion system, suggesting that ESX-1-mediated bacterial escape from the phagosome is required for inflammasome activation (Koo et al., 2008; Mishra et al., 2010). We therefore assessed AM localization in mice infected with the H37Rv:ΔRD1 (ΔRD1) strain of Mtb that lacks a functional ESX-1 (Lewis et al., 2003) but that still retains expression of the phthiocerol dimycocerosate (PDIM) lipid that is important for Mtb virulence (data not shown). Given the importance of IL-1R in the localization of infected AM to the interstitium, we hypothesized that AM localization would differ in mice infected with H37Rv and ΔRD1 Mtb. Indeed, ΔRD1-infected AM remained predominantly airway-positive throughout infection, while, as expected, approximately half of the H37Rv-infected AM became interstitial by day 21 p.i. (Figure 4C). The failure of ΔRD1-infected AM to localize to the interstitium corresponded with an increased proportion of AM comprising the infected pool; minimal infection of MC or neutrophils was observed, supporting the hypothesis that translocation of AM to the interstitium precedes bacterial dissemination into other myeloid cells (Figure 4D).
We next generated WT:MyD88−/− mixed bone marrow chimeras (MBMC) to determine whether the relocalization of AM to the interstitium required intrinsic MyD88 expression. MBMC were infected with Mtb-mCherry, and airway localization was measured 14 and 19 days p.i. Although AM from global MyD88−/− mice were impaired in translocation to the interstitium (Figure 4A), MyD88−/− AM from MBMC exhibited equivalent airway localization as WT AM at both timepoints (Figure S4C), suggesting an AM-extrinsic role for this pathway. We further generated WT:ASC−/− MBMC to address the intrinsic role of ASC in mediating altered AM localization during Mtb infection. ASC-deficient AM in MBMC also displayed equivalent airway localization as their WT counterparts (Figure S4D), suggesting that inflammasome signaling is also extrinsic to the translocated AM. Because IL-1 is known to recruit neutrophils (Jones et al., 2005; Schmitz et al., 2005), we considered the possibility that IL-1 could be recruiting neutrophils into the airways as a mechanism of Mtb dissemination out of the AM compartment. However, although neutrophils were observed in the airspaces at d15 p.i. (Figure S4E), neutrophil depletion did not promote the translocation of infected AM to the interstitium (Figure S4F,G), arguing against this hypothesis. As shown in Fig. 3J, we likewise excluded a role for monocyte recruitment as a prerequisite of AM localization to the interstitium as there was no difference in AM translocation between WT and CCR2−/− mice. To determine if neutrophils played a compensatory role in the absence of monocytes, we further depleted neutrophils in CCR2 KO mice. Again, no difference was observed between WT, CCR2 KO, or CCR2 KO mice deficient in neutrophils (Figure S4H).
We next hypothesized that IL-1R signaling could be acting on non-hematopoietic cells, as this pathway operates through epithelial cells in other models of airway inflammation (Gasse et al., 2007). To address this, IL-1R−/− chimeras were generated in which either the hematopoietic or non-hematopoietic compartments were entirely derived from IL-1R−/− bone marrow. The expected impairment in infected AM interstitial localization was observed in the KO→KO controls compared to WT→WT mice (Figure 4E), similar to the global IL-1R−/− mice (Figure 4A). Consistent with our findings in the WT:MyD88−/− MBMC, infected AM in the KO→WT group, in which intact IL-1R is expressed only on non-hematopoietic cells, phenocopied the WT→WT controls, with significantly less airway labeling than the KO→KO group. However, chimeras with intact IL-1R expression solely in the hematopoietic compartment (WT→KO) exhibited significantly greater airway labeling than the WT→WT group and were not statistically different from the KO→KO group, indicating a role for non-hematopoietic IL-1R signaling in orchestrating the translocation of infected AM to the lung interstitium (Figure 4E).
Discussion
The lung interstitium provides a niche for bacterial persistence during chronic Mtb infection, and localization to this site from the airways is required for transmission, which occurs when bacilli from eroded granulomas are aerosolized via coughing. Although the mechanisms used by other bacterial pathogens to traverse epithelial barriers at mucosal surfaces are well-described (Ribet and Cossart, 2015), the means by which Mtb gains access to the lung interstitium has remained elusive. Our data support a model in which Mtb infection drives the selective relocalization of Mtb-infected AM across the airway epithelium into the lung interstitium via RD1-dependent inflammasome signaling.
AM are not typically thought to leave their alveolar niche, but here we report that Mtb-infected AM can traverse the airway epithelium and gain access to the interstitium. Consistent with previous observations of decreased bacterial loads upon AM depletion (Huang et al., 2018; Leemans et al., 2001), these findings indicate that AM play a crucial role in the establishment of disease by serving as a vehicle to transport Mtb into the interstitium, a site where transfer to Mtb-replication permissive monocytes may occur (Cambier et al., 2017; Cambier et al., 2014). It has long been assumed, but never definitively shown, that AM are the first cells to be infected by Mtb. Here we find that both AM and EC are infected within 48 h of Mtb infection, consistent with previous data that Mtb is capable of infecting EC in vitro, albeit at a very low frequency (Bermudez et al., 2002; Scordo et al., 2016). It is possible that our flow cytometry assay underestimated the true infection level of EC, as few EC were recovered from the lungs. However, using image analysis of tissue sections, we observed Mtb replication only in AM by 7 days p.i. Thus, while both AM and EC can be infected, productive infection appears to only occur in AM, and EC may play a limited role in propagating Mtb infection, which is surprising considering EC vastly outnumber AM in the lung. A role for epithelial IL-1R signaling in early granuloma generation is supported by the observation that MyD88/IL-1R−/− mice have impaired granuloma formation two weeks p.i. (Fremond et al., 2007; Juffermans et al., 2000). Conversely, intratracheally-administered IL-1-coated beads can directly induce granuloma formation in mouse lungs (Kasahara et al., 1988). Inflammasome activation and IL-1 signaling on lung epithelial cells have been shown to increase alveolar permeability, resulting in the influx of immune cells, such as neutrophils, into the airways (Barry et al., 2013; Ding et al., 2017; LeibundGut-Landmann et al., 2011), and our results suggest that this increased permeability may be bi-directional, enabling infected AM to translocate across the epithelium into the interstitium. While it remains unclear if this process is specific to Mtb infection, it is tempting to speculate that access to the interstitium may be a more general function of AM, potentially as a surveillance mechanism to ensure that luminal antigens are sampled and delivered to immune effector cells in the lung interstitium. Future studies are needed to explore whether AM relocalize to the lung interstitium in other inflammatory settings.
The role of MyD88 in macrophage mobilization has been previously examined in the context of mycobacterial infection. Previous studies in zebrafish found that macrophage recruitment is accelerated by phenolic glycolipid (PGL) expression on the bacterial cell surface, which does not depend on ESX-1 or MyD88 (Cambier et al., 2017; Cambier et al., 2014). In contrast, we show here that both MyD88/IL1R and ESX-1 are required for infected AM to egress the alveolar space, underscoring the idea that AM localization to the lung interstitium is a distinct process from macrophage recruitment. This conclusion is further supported by our finding that AM re-localization occurs using H37Rv, a PGL-negative Mtb strain, and in CCR2−/− mice.
Combined, our results suggest that Mtb infection of the lung interstitium is initiated when a subpopulation of Mtb-infected AM undergoes ESX-1-mediated inflammasome activation and IL-1 release. The finding that ASC signaling is extrinsic to the infected AM indicates that Mtb-infected AM that reach the interstitium are not the cells that undergo inflammasome activation, but rather may translocate in response to IL-1 produced by other airway-resident AM. Thus, our findings suggest a model that granuloma formation is initiated when surviving Mtb-infected AM relocalize to the lung interstitium and proliferate to form aggregates, setting the stage for bacterial dissemination to recruited MC and neutrophils. Because many current vaccine efforts aim to curb Mtb infection prior to the establishment of granulomas, understanding how pulmonary infection is initiated may inform new avenues to achieve this goal.
Materials and Methods
Contact for reagent and resource sharing.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kevin Urdahl (kevin.urdahl@cidresearch.org).
Experimental model and research details.
Mice.
C57BL/6, CCR2−/−, and IL1R−/− mice were purchased from the Jackson Laboratories (Bar Harbor, ME). ASC−/− mice were a generous gift from Dr. Naeha Subramanian (Institute for Systems Biology, Seattle, WA), and MyD88−/− mice were generously provided by Dr. Alan Aderem (CIDR, Seattle, WA). All mice were housed and maintained in specific pathogen-free conditions at the Center for Infectious Disease Research (CIDR), and all experiments were performed in compliance with the CIDR Animal Care and Use Committee. Male and female mice between the ages of 8–12 weeks of age were used for all experiments.
Method details.
Aerosol infections.
Most aerosol infections were performed with a stock of wildtype H37Rv Mtb transformed with an mCherry reporter plasmid driven by the pMSP12 promoter (Cosma et al., 2004). Some infections used an Mtb strain with a deletion of the virulence determinant RD1 region (ΔRD1), provided by Dr.David Sherman (CIDR, Seattle, WA), transformed with the same pMSP12 mCherry expression plasmid. To perform standard dose aerosol infections, mice were enclosed in a Glas-Col aerosol infection chamber, and ~50–100 CFU were deposited directly into their lungs. For high-dose infections, 1200–2500 CFU were deposited. In order to confirm the infectious dose, two mice in each infection were immediately sacrificed and their lung homogenates plated onto 7H10 plates for CFU enumeration.
Intratracheal airway and intravenous labeling.
To label airway-resident cells, mice were deeply anesthetized with 25% isoflurane diluted in propylene glycol (Fisher Scientific). Once anesthetized, mice were suspended vertically, their tongues were gently pulled aside with forceps to expose the trachea, and 0.25 μg of antibody diluted in 50μl of PBS was pipetted directly into the airway. The tongues were continued to be held with the forceps as the mice inhaled the antibody mixture and for several deep breaths following inhalation to ensure the antibody entered the airway. Mice were returned to their cages for 30 min of in vivo antibody incubation prior to euthanasia by cervical dislocation. Antibodies used for airway labeling include CD45.2 PE-Cy7, CD45.1 PE-Cy7, CD11c BV650, CD11c APC, or SiglecF PE. In some experiments, an intravenous CD45.2 antibody (0.2 μg, FITC) was infused 5 min prior to sacrifice to exclude intravascular monocytes.
Lung single cell suspensions.
At the indicated times post-infection, mouse lungs were excised and lightly homogenized in HEPES buffer containing Liberase Blendzyme 3 (70 μg/ml; Roche) and DNaseI (30 μg/ml; Sigma-Aldrich) using a gentleMacs dissociator (Miltenyi Biotec). The lungs were then incubated for 30 min at 37°C and then further homogenized a second time with the gentleMacs. The homogenates were filtered through a 70 μm cell strainer, pelleted for RBC lysis with RBC lysing buffer (Thermo), and resuspended in FACS buffer (PBS containing 2.5% FBS and 0.1% NaN3).
Antibody staining.
Single cell suspensions were first washed in PBS and then incubated with 50 μl Zombie Aqua viability dye (BioLegend) for 10 min at room temperature in the dark. Viability dye was immediately quenched by the addition of 100 μl of a surface antibody cocktail diluted in 50% FACS buffer/50% 24G2 Fc block buffer using saturating levels of antibodies. Surface staining was performed for 20 min at 4°C. Then, the cells were washed once with FACS buffer and fixed with 2% paraformaldehyde for 1 h prior to analyzing on an LSRII flow cytometer (BD Biosciences). Cell death was evaluated by Sytox Green staining following the manufacturer’s recommendations (Thermo).
Imaging.
Mice were infected with H37Rv Mtb-mCherry by aerosol and intratracheally administered 0.25 μg CD45.2 Alexa-700 antibody 30 minutes prior to euthanasia at 48 hours, 15 days or 19 days post-infection. The trachea was then exposed, the lungs were inflated with 1 ml Cytofix fixative solution (BD Biosciences) via a 25-gauge needle, and the trachea was then tied off with suture thread to prevent leakage of fixative. The lungs were carefully excised from the chest cavity and submerged in 20 ml Cytofix fixative solution for 22 hours at 4Ό. Lungs were then washed twice in PBS a nd dehydrated in 30% sucrose for at least 24 hours prior to embedding in OCT and rapid freezing in a methylbutane-dry ice slurry. Sections (20 μm) were stained with SiglecF BV421 and CD11b APC-F750 or SiglecF BV421, E-cadherin Alexa647, and p120 Alexa488 overnight at room temperature and coverslipped with Fluoromount G mounting media (Southern Biotec). To image thick tissue sections, we cut 300 μm sections on a cryostat, then performed tissue-clearing as described previously (Li et al., 2017). Briefly, we rehydrated sections in TRIS buffer, then blocked for 24 hours. Staining with antibodies was performed for 48 hours at 37°C. Tissues were prepared for clearing in 0.2% Triton X-100 and 0.5% 1-thioglycerol. Finally, the refractive index of the tissue was equalized by incubation with 86% Histodenz, 22% n-methylacetamide, and 0.2% Triton X-100 for 24 hours at room temperature. Sections were then cover-slipped. Images were acquired on a Leica SP8X confocal microscope, compensated for fluorophore spillover using LAS X (Leica), and analyzed with Imaris (Bitplane).
Bone marrow chimeras.
For WT:MyD88−/− and WT:ASC−/− mixed bone marrow chimeras, WT CD45.1/2 F1 mice were lethally irradiated with 1000 rads and reconstituted with a 1:1 mixture of either CD3-depleted (Miltenyi Biotec) B6.SJL:MyD88−/− or B6.SJL:ASC−/− bone marrow; B6.SJL mice are congenically marked with CD45.1, and both knockout mouse strains express CD45.2. For criss-cross chimeras, WT B6.SJL (CD45.1) and IL1R−/− mice were lethally irradiated with 1000 rads, and the hematopoietic compartment was reconstituted either with the same bone marrow, for WT into WT and KO into KO controls, or with the opposite bone marrow, for WT into KO and KO into WT experimental groups.
Alveolar macrophage adoptive transfers.
C57BL/6 (n=10) and B6.SJL (n=1 or 2) mice were infected with H37Rv Mtb-mCherry by aerosol. On day 15 post-infection, C57BL/6 mice were intratracheally administered 0.25 μg SiglecF PE antibody 30 min prior to euthanasia. Single-cell lung suspensions were prepared, and CD19+ cells were depleted with Magnisort negative selection beads (Thermo). Airway+ alveolar macrophages were sorted based on SiglecF PE expression and autofluorescence on a FACSAria sorter (BD) in a BSL-3 facility. Sorted cells were then adoptively transferred intratracheally into infection-matched or naive B6.SJL mice. Due to cell yield limitations, alveolar macrophages from ten donor mice were transferred into only one or two recipients. Five days post-transfer, recipient B6.SJL mice were intratracheally administered 0.25 μg CD11c BV650 antibody, and lungs were harvested 30 min later. Transferred alveolar macrophages were enriched by CD45.2 positive selection pull-downs using anti-APC magnetic beads and LS columns (Miltenyi Biotec) followed by flow cytometric analysis of airway labeling.
Neutrophil depletion.
C57BL/6 mice were infected with H37Rv Mtb-mCherry by aerosol. On days 5, 7, 9, 11, and 13 post-infection, mice were intraperitoneally injected with 200 μg anti-1A8 antibody diluted in PBS (BioXcell). Mice were euthanized and lungs harvested 15 days post-infection. To ensure depletion, lungs were stained for 1A8, CD11b, and Gr-1 to identify neutrophils. Depletion efficiency was ~90–95%.
BrdU incorporation.
BrdU (2 mg) was injected introperitoneally into D13 or 15 Mtb-infected C57BL/6 mice 24 h prior to harvesting lungs for flow cytometric analysis using the BD BrdU Flow Kit (BD Biosciences).
Quantification and statistical analysis.
Flow data were analyzed using FlowJo V9 software (Tree Star) and presented as means ± SEM. Statistical analysis and graphical representation of data were done using GraphPad Prism v6.0 software. At least 3–5 mice were used per group in each experiment, as indicated in the respective figure legends, and all experiments were performed at least 2–3 times. Investigators were not blinded to the analysis, and randomization was not applicable to these studies. Due to technical constraints with the intratracheal antibody administration technique, mice occasionally did not receive a sufficient label for analysis. In such cases, mice were excluded if the overall labeling efficiency of alveolar macrophages was less than 50%. Statistical significance was determined using a t-test or ANOVA, as indicated in the figure legends. For p-values, * p≤0.05, ** p≤0.01, *** p≤0.001, **** p≤0.0001.
Supplementary Material
Related to Figure 2. Clusters of infected AM form independently of bronchioles.
Related to Figure 3. Infected AM reside in the lung interstitium.
Related to Figure 4. Uninfected AM reside in the alveolar lumen.
Highlights:
Early M. tuberculosis infection primarily targets lung alveolar macrophages (AM)
Mtb-infected AM selectively re-localize from the airways to the lung interstitium
AM interstitial localization precedes Mtb dissemination to other immune cell types
Re-localization of infected AM is dependent on Mtb ESX-1 and host IL-1R signaling
Acknowledgments
We thank A. Pagan and L. Ramakrishnan for comments on the manuscript. This study was supported by the National Institute of Health grants 1R01AI134246 (KU), 1R01AI076327 (KU), U19AI13597 (KU), 1K22AI108628–01A1 (MG), 5F32AI126703 (SC), and T32AI007509–16 (SC).
Footnotes
Declaration of interests. The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, and Ramakrishnan L (2011). Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145, 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry KC, Fontana MF, Portman JL, Dugan AS, and Vance RE (2013). IL-1 alpha signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J Immunol 190, 6329–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez LE, Sangari FJ, Kolonoski P, Petrofsky M, and Goodman J (2002). The efficiency of the translocation of Mycobacterium tuberculosis across a bilayer of epithelial and endothelial cells as a model of the alveolar wall is a consequence of transport within mononuclear phagocytes and invasion of alveolar epithelial cells. Infect Immun 70, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier CJ, O’Leary SM, O’Sullivan MP, Keane J, and Ramakrishnan L (2017). Phenolic Glycolipid Facilitates Mycobacterial Escape from Microbicidal Tissue-Resident Macrophages. Immunity 47, 552–565 e554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, Cosma CL, and Ramakrishnan L (2014). Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 505, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma CL, Humbert O, and Ramakrishnan L (2004). Superinfecting mycobacteria home to established tuberculous granulomas. Nat Immunol 5, 828–835. [DOI] [PubMed] [Google Scholar]
- Daniels NJ, Hyde E, Ghosh S, Seo K, Price KM, Hoshino K, Kaisho T, Okada T, and Ronchese F (2016). Antigen-specific cytotoxic T lymphocytes target airway CD103+ and CD11b+ dendritic cells to suppress allergic inflammation. Mucosal Immunol 9, 229–239. [DOI] [PubMed] [Google Scholar]
- Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, and Ramakrishnan L (2002). Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity 17, 693–702. [DOI] [PubMed] [Google Scholar]
- Ding X, Jin S, Tong Y, Jiang X, Chen Z, Mei S, Zhang L, Billiar TR, and Li Q (2017). TLR4 signaling induces TLR3 up-regulation in alveolar macrophages during acute lung injury. Sci Rep 7, 34278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, Jacobs M, Ryffel B, and Quesniaux VF (2007). IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol 179, 1178–1189. [DOI] [PubMed] [Google Scholar]
- Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, and Ryffel B (2004). Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest 114, 1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, et al. (2007). IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest 117, 3786–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, Maiello P, Rutledge T, Marino S, Fortune SM, et al. (2015). Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS Pathog 11, e1004603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Juarrero M, and Orme IM (2001). Characterization of murine lung dendritic cells infected with Mycobacterium tuberculosis. Infect Immun 69, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, and Lambrecht BN (2013). Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med 210, 1977–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, Van Gassen S, Chen J, Poidinger M, De Prijck S, et al. (2016). Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 45, 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al. (2013). Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Nazarova EV, Tan S, Liu Y, and Russell DG (2018). Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med 215, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Simms BT, Lupa MM, Kogan MS, and Mizgerd JP (2005). Lung NF-kappaB activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J Immunol 175, 7530–7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juffermans NP, Florquin S, Camoglio L, Verbon A, Kolk AH, Speelman P, van Deventer SJ, and van Der Poll T (2000). Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J Infect Dis 182, 902–908. [DOI] [PubMed] [Google Scholar]
- Kasahara K, Kobayashi K, Shikama Y, Yoneya I, Soezima K, Ide H, and Takahashi T (1988). Direct evidence for granuloma-inducing activity of interleukin-1. Induction of experimental pulmonary granuloma formation in mice by interleukin-1-coupled beads. Am J Pathol 130, 629–638. [PMC free article] [PubMed] [Google Scholar]
- Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, and Brown EJ (2008). ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol 10, 1866–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsman L, and Jung S (2007). Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J Immunol 179, 3488–3494. [DOI] [PubMed] [Google Scholar]
- Langlet C, Tamoutounour S, Henri S, Luche H, Ardouin L, Gregoire C, Malissen B, and Guilliams M (2012). CD64 expression distinguishes monocyte-derived and conventional dendritic cells and reveals their distinct role during intramuscular immunization. J Immunol 188, 1751–1760. [DOI] [PubMed] [Google Scholar]
- Latz E, Xiao TS, and Stutz A (2013). Activation and regulation of the inflammasomes. Nat Rev Immunol 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemans JC, Juffermans NP, Florquin S, van Rooijen N, Vervoordeldonk MJ, Verbon A, van Deventer SJ, and van der Poll T (2001). Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J Immunol 166, 4604–4611. [DOI] [PubMed] [Google Scholar]
- Leepiyasakulchai C, Taher C, Chuquimia OD, Mazurek J, Soderberg-Naucler C, Fernandez C, and Skold M (2013). Infection rate and tissue localization of murine IL-12p40-producing monocyte-derived CD103(+) lung dendritic cells during pulmonary tuberculosis. PLoS One 8, e69287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeibundGut-Landmann S, Weidner K, Hilbi H, and Oxenius A (2011). Nonhematopoietic cells are key players in innate control of bacterial airway infection. J Immunol 186, 3130–3137. [DOI] [PubMed] [Google Scholar]
- Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, and Sherman DR (2003). Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J Infect Dis 187, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Germain RN, and Gerner MY (2017). Multiplex, quantitative cellular analysis in large tissue volumes with clearing-enhanced 3D microscopy (Ce3D). Proc Natl Acad Sci U S A 114, E7321–E7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, Sacchettini J, Fortune SM, and Flynn JL (2014). Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med 20, 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CJ, Cadena AM, Leung VW, Lin PL, Maiello P, Hicks N, Chase MR, Flynn JL, and Fortune SM (2017). Digitally Barcoding Mycobacterium tuberculosis Reveals In Vivo Infection Dynamics in the Macaque Model of Tuberculosis. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, et al. (2010). Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 184, 3326–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, and Anes E (2010). Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol 12, 1046–1063. [DOI] [PubMed] [Google Scholar]
- Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, Ginsberg A, Swaminathan S, Spigelman M, Getahun H, et al. (2016). Tuberculosis. Nat Rev Dis Primers 2, 16076. [DOI] [PubMed] [Google Scholar]
- Philips JA, and Ernst JD (2012). Tuberculosis pathogenesis and immunity. Annu Rev Pathol 7, 353–384. [DOI] [PubMed] [Google Scholar]
- Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, Vanhoutte L, Neyt K, Killeen N, Malissen B, et al. (2013). Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38, 322–335. [DOI] [PubMed] [Google Scholar]
- Ribet D, and Cossart P (2015). How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect 17, 173–183. [DOI] [PubMed] [Google Scholar]
- Schmitz N, Kurrer M, Bachmann MF, and Kopf M (2005). Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol 79, 6441–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scordo JM, Knoell DL, and Torrelles JB (2016). Alveolar Epithelial Cells in Mycobacterium tuberculosis Infection: Active Players or Innocent Bystanders? J Innate Immun 8, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Ernst JD, and Desvignes L (2014). Beyond macrophages: the diversity of mononuclear cells in tuberculosis. Immunol Rev 262, 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian N, Natarajan K, Clatworthy MR, Wang Z, and Germain RN (2013). The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung SS, Fu SM, Rose CE Jr., Gaskin F, Ju ST, and Beaty SR (2006). A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol 176, 2161–2172. [DOI] [PubMed] [Google Scholar]
- Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, and Ernst JD (2007). Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol 179, 2509–2519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figure 2. Clusters of infected AM form independently of bronchioles.
Related to Figure 3. Infected AM reside in the lung interstitium.
Related to Figure 4. Uninfected AM reside in the alveolar lumen.