

Abstract
Melanosomes are tissue-specific organelles within which melanin is synthesized and stored. The melanocyte-specific glycoprotein Pmel17 is enriched in the lumen of premelanosomes, where it associates with characteristic striations of unknown composition upon which melanin is deposited. However, Pmel17 is synthesized as an integral membrane protein. To clarify its physical linkage to premelanosomes, we analyzed the posttranslational processing of human Pmel17 in pigmented and transfected nonpigmented cells. We show that Pmel17 is cleaved in a post-Golgi compartment into two disulfide-linked subunits: a large lumenal subunit, Mα, and an integral membrane subunit, Mβ. The two subunits remain associated intracellularly, indicating that detectable Mα remains membrane bound. We have previously shown that Pmel17 accumulates on intralumenal membrane vesicles and striations of premelanosomes in pigmented cells. In transfected nonpigmented cells Pmel17 associates with the intralumenal membrane vesicles of multivesicular bodies; cells overexpressing Pmel17 also display structures resembling premelanosomal striations within these compartments. These results suggest that Pmel17 is sufficient to drive the formation of striations from within multivesicular bodies and is thus directly involved in the biogenesis of premelanosomes.
INTRODUCTION
he diversity of cell function in higher eukaryotes is reflected in cell ultrastructure; unique functions are often mediated by cell-type–specific organelles. Unique organelles may arise as modifications of ubiquitous compartments through the expression of cell-type–specific structural proteins and/or protein sorting pathways. The lysosome-related organelles exemplify such cell-type–specific modifications and include the lytic granules in cytotoxic T lymphocytes and natural killer cells, azurophil granules in neutrophils, dense granules in megakaryocytes and platelets, and melanosomes in melanocytes and retinal pigmented epithelial cells (Dell'Angelica et al., 2000). Although each of these organelles shares features with lysosomes, they are ultrastructurally unique in accordance with their unique functions. Specific subsets of these organelles are disrupted in disorders such as Hermansky-Pudlak syndrome (HPS) and Chediak-Higashi syndrome (Dell'Angelica et al., 2000), suggesting that they are generated through both common and divergent biogenetic pathways. How lysosome-related organelles and their unique ultrastructural components are formed is unknown.
We have chosen to focus on the biogenesis of the melanosome as a model for a unique lysosome-related organelle. Melanosomes specialize in the biosynthesis and storage of melanins (King et al., 1995). Morphological characterization of pigmented epidermal melanocytes and melanoma cells suggests that mature, fully pigmented melanosomes (stage III and IV) develop from nonpigmented precursors (stage I and II), collectively referred to here as premelanosomes (Seiji et al., 1963). The first defining morphological characteristic of stage II premelanosomes is the appearance of intralumenal fibrillar striations. Melanin is subsequently deposited along these striations in stage III and IV melanosomes, suggesting that they serve as a matrix for sequestration of melanin (Seiji et al., 1963). Although the morphology of the striations has long been appreciated, their composition, function, and the pathways leading to their formation remain uncharacterized.
Of the integral membrane proteins shown to be specific components of melanosomal compartments, only one, Pmel17 (also referred to as gp100, ME20, and the product of the murine Silver locus), is known to be enriched in premelanosomes relative to mature melanosomes (Vennegoor et al., 1988; Kapur et al., 1992; Kikuchi et al., 1996; Lee et al., 1996; Raposo et al., 2001). Although its function is unclear, Pmel17 likely plays an important role in melanization because there is a close correlation between its expression and melanin production (Kwon et al., 1987). Pmel17 has been proposed to function in polymerization or stabilization of melanin intermediates (Chakraborty et al., 1996; Lee et al., 1996) and/or in protecting pigmented cells from toxic melanin intermediates (Kobayashi et al., 1994). Interestingly, several data indicate that Pmel17 is a component of premelanosomal striations, including its immunolocalization to the premelanosome lumen (Lee et al., 1996; Raposo et al., 2001), its identification as the target of melanosomal “matrix”-specific antibodies (Kobayashi et al., 1994), and its suggested interaction with detergent-insoluble melanin (Donatien and Orlow, 1995). Thus, by characterizing the biosynthetic and intracellular protein sorting pathways of Pmel17, it may be possible to unravel the origins, composition, and function of the premelanosomal striations. In turn, we may understand the morphogenetic processes in other lysosome-like organelles.
Human Pmel17 encodes at least two type I integral membrane proteins translated from alternatively spliced mRNAs. The proteins differ by the presence or absence of seven amino acids in the membrane-proximal region of the lumenal domain (Kwon et al., 1991; Adema et al., 1994; Kawakami et al., 1994; Maresh et al., 1994b) and are referred to here as the long and short forms of Pmel17. The short form is synthesized as an N-linked glycosylated precursor containing a 24-residue amino-terminal signal sequence (Maresh et al., 1994a) and is predicted to have a 566-residue lumenal domain, a 26-residue transmembrane domain, and a 45-residue cytoplasmic domain (Figure 1). After signal sequence removal, at least a fraction of the precursor is cleaved to produce a soluble polypeptide, originally termed ME20-S (referred to here as Mα), comprised of the amino-terminal 443 amino acids of the lumenal domain (Maresh et al., 1994a; Figure 1). Mα can be detected in supernatants from melanoma cells and melanocytes (Vennegoor et al., 1988; Vogel and Esclamado, 1988; Adema et al., 1994). However, either cleavage, Mα secretion, or both is likely inefficient, because Pmel17 accumulates at steady state in early stage melanosomes as judged by immunostaining and subcellular fractionation with the use of antibodies that recognize Mα (Vennegoor et al., 1988; Kapur et al., 1992; Kikuchi et al., 1996; Lee et al., 1996; Raposo et al., 2001). Furthermore, although Pmel17-derived products are frequently perceived to be non–membrane-associated components of the melanosomal matrix, it has not been determined whether they remain tethered to the premelanosomal membrane via protein–protein interactions. Finally, the fate of the carboxy-terminal 194–201 amino acids of Pmel17 remaining after Mα release is unknown.
Figure 1.

Schematic diagram of Pmel17 and proposed processed forms. The nomenclature for the bands P1, P2, Mα, and Mβ reflects their precursor/product relationships; P1 is the initial precursor, P2 is the processed precursor, and Mα and Mβ are the mature forms (see text). Antibody recognition sites are shown for HMB50 and αPEP13h; the precise recognition site for HMB50 is unknown. N-linked glycans (predicted from consensus sequences) are indicated as branched structures, and potential processing is indicated by increased branching. Disulfide-linkage of Mα and Mβ is indicated by a line; cysteine residues capable of disulfide bond formation are indicated by black circles. Amino acid 25 is the first residue of the proprotein after signal sequence removal. “598” and “623” denote the first and last residues of the transmembrane domain (tm). Assignment of amino acid 467 as the C terminus of Mα is tentative and based on Maresh et al. (1994b). ? indicates the exact N-terminus of Mβ is unknown. Cyto, cytoplasmic domain.
In hopes of better understanding the role of Pmel17 in melanization, we analyzed the posttranslational processing and localization of human Pmel17 in pigmented and nonpigmented cells. Our results support a role for Pmel17 and multivesicular bodies (MVBs) in creating the unique architecture of the premelanosome.
MATERIALS AND METHODS
Cells, Culture Conditions, and Transfections
The highly pigmented melanoma cell line MNT-1 (a gift of Dr. V. Hearing, National Cancer Institute [NCI], Bethesda, MD), the pigmented melanoma cell line 1011-mel (a gift of Dr. S. Topalian, NCI), and primary human foreskin melanocytes (a gift of Dr. M Herlyn, Wistar Institute, Philadelphia, PA) were cultured as described (Raposo et al., 2001). HeLa and murine NIH-3T3 cells were maintained in DME (Life Technologies Inc., Rockville, MD [LTI]) supplemented with 10% heat-inactivated FBS (Atlanta Biologics, Norcross, GA) and antibiotics. The pigmented murine cell line melan-a (a gift of Dr. V. Hearing) was maintained in RPMI 1640 (LTI), 10% heat-inactivated FBS, 0.1 mM β-mercaptoethanol, and 200 nM tetradecanoyl phorbol acetate. HeLa cells were transiently transfected with pCI-Pmel17 with the use of either calcium phosphate precipitation as described (Marks et al., 1996) or Fugene (Roche Biochemicals, Indianapolis, IN). pCI-Pmel17 encodes the long form of Pmel17 and was a gift from Dr. W. Storkus (University of Pittsburgh, Pittsburgh, PA). For pulse-chase studies, 8 μg specific plasmid DNA was used per 10-cm dish; for Western blotting, 2 μg/well of a 6-well dish; and for immunofluorescence microscopy (IFM) and immunoelectron microscopy (IEM), 80–100 ng/well of a 6-well dish, unless noted. In some cases, carrier DNA (empty vector) was added. Cells were analyzed 2 d posttransfection.
Antibodies and Electron Dense Probes
HMB50, a mouse MAb to Pmel17 (Esclamado et al., 1986), was a gift of Dr. C. Figdor (University Hospital, Nijmegen, the Netherlands). αPEP13h, similar to the previously described αPEP13 (Kobayashi et al., 1994), is a rabbit antiserum raised against a peptide (CPIGENSPLLSGQQV-CO2H) corresponding to the carboxy-terminal 15 residues of human Pmel17. The antiserum was generated by Genemed Synthesis (San Francisco, CA) and affinity purified with the use of SulfoLink beads (Pierce, Rockford, IL) coupled to the peptide. Anti-lamp-1, a rabbit antiserum, was a gift of M. Fukuda (Scripps Research Institute, San Diego, CA). Anti-CD63 (clone CLB-gran/12), a mouse MAb, was from Caltag Laboratories (Burlingame, CA). Alkaline-phosphatase–conjugated goat anti-rabbit Ig was from Amersham Pharmacia Biotech (Piscataway, NJ). LRSC- and FITC-conjugated secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). Protein A gold conjugates were purchased from Dr. J. W. Slot (Utrecht Medical School, Utrecht, The Netherlands).
Metabolic Labeling and Immunoprecipitation
Cells were harvested, starved for methionine and cysteine, pulse-labeled for 30 min (unless noted) with [35S]methionine/cysteine labeling mix, chased with excess methionine and cysteine for the indicated periods of time, and washed with PBS as described (Marks et al., 1996). In some cases, the culture media from the chase incubations were collected and clarified by low-speed centrifugation. Where indicated, 10 mM N-ethylmaleimide was added to the final PBS wash to inhibit artifactual disulfide bond formation. Brefeldin A (BFA, 1 μg/ml; Roche Biochemicals) was added during the starvation, pulse, and chase incubations, where indicated. 1% DMSO, 50 mM ammonium chloride (NH4Cl), 100 nM bafilomycin A1 (BafA1; Sigma Chemical, St. Louis, MO), 5 mM methionine methyl ester (MME; Sigma), or a cocktail of lysosomal protease inhibitors at 100 μg/ml each (leupeptin, Roche Biochemicals; E64, Sigma; and pepstatin A, Roche Biochemicals) were added to the chase medium where indicated.
Immunoprecipitations and treatments with protein N-glycanase F (endoF) and endoglycosidase H (endoH) were performed as previously described (Berson et al., 2000). Briefly, cell lysates prepared with 1% Triton X-100 or culture media brought to 1% Triton X-100 were precleared with protein A–Sepharose beads preadsorbed to isotype-matched nonspecific antibodies, immunoprecipitated, and, sometimes, treated with endoF or endoH. Where indicated, 20 mM iodoacetamide (Sigma) was added to the lysates to prevent post-lysis disulfide bond formation. Eluted proteins were fractionated by SDS-PAGE with the use of 10% acrylamide gels and detected using a Molecular Dynamics Storm 860 PhosphorImager and Imagequest software (Amersham-Pharmacia Biotech) as described.
Western Blotting
Western blotting was done as described (Berson et al., 2000). Briefly, proteins were fractionated by SDS-PAGE with the use of 10% acrylamide gels, transferred to polyvinylidene difluoride membranes, and probed with αPEP13h. Bound antibody was detected with the use of alkaline-phosphatase–conjugated goat anti-rabbit Ig and ECF (Amersham Pharmacia Biotech) and visualized as described.
Immunofluorescence Microscopy
Cells grown on coverslips were fixed with 2% formaldehyde in PBS, permeabilized with saponin, and stained with unlabeled primary antibodies and FITC- and LRSC-conjugated secondary antibodies as described (Marks et al., 1995). Cells were analyzed on a Leica DM IRBE microscope (Deerfield, IL). Digital images were collected with the use of a Hamamatsu ORCA CCD camera (Malvern, PA) and analyzed and processed with the use of Improvision OpenLab (Lexington, MA) and Adobe Photoshop software (San Jose, CA).
Immunoelectron Microscopy
MNT-1 and HeLa cells were fixed with a mixture of 2% paraformaldehyde and 0.2% glutaraldehyde in 60 mM PIPES, 25 mM HEPES, 2 mM MgCl2, 10 mM EGTA pH 6.9. Fixed cells were embedded in 10% gelatin and blocks were infused with 2.3 M sucrose as described (Raposo et al., 1997). Ultrathin cryosections were single- or double-immunogold labeled as described (Slot et al., 1991) with the use of antibodies and protein A coupled to 10 or 15 nm gold (PAG10 and PAG15) as indicated in the legends to the figures. Where indicated, ultrathin cryosections were retrieved with a mixture of methylcellulose and uranyl acetate (Liou et al., 1996) for better preservation of membranes.
RESULTS
Identification of Processed Forms of Pmel17
The highly pigmented human melanoma cell line MNT-1 was used as a model system to study the biosynthetic properties of Pmel17. The melanosomal structures within MNT-1 cells are morphologically similar to those in untransformed, eumelanin-synthesizing melanocytes (Raposo et al., 2001). Reverse transcriptase-coupled PCR analysis indicated that MNT-1 cells express both the long and short forms of Pmel17 (unpublished results); we will refer to these products collectively as Pmel17. We compared observations in MNT-1 cells with those obtained in transiently transfected, nonmelanocytic HeLa cells expressing only the long form of Pmel17. Cells were pulse labeled with [35S]methionine/cysteine for 30 min and then chased for various periods of time. To identify products of posttranslational processing derived from both the N- and C-termini of Pmel17, cell lysates and culture media were then immunoprecipitated in parallel with HMB50 (specific for the lumenal domain) and αPEP13h (specific for the cytoplasmic domain; see Figure 1). Immunoprecipitated proteins were analyzed by SDS-PAGE and phosphorimaging analysis.
Both HMB50 and αPEP13h precipitated four proteins designated P1, P2, Mα, and Mβ, from detergent lysates of MNT-1 and Pmel17-transfected HeLa cells (Figure 2, A–C). P1 migrated as a 97-kDa protein and was the most intense band after the pulse. In addition, small amounts of P2 (Mr 128,000), Mα (Mr 85,000), and Mβ (Mr 28,000) were also immunoprecipitated after the pulse from MNT-1 cell lysates. P2 and Mα were slightly smaller in HeLa cells than in MNT-1 cells, likely due to glycosylation differences. The intensity of P1 decreased after a 1-h chase, concurrent with an increase in the amount of immunoprecipitated P2, Mα, and Mβ. Although P1 ran as a discrete band, P2, Mα, and Mβ, particularly in MNT-1 cells, exhibited a more diffuse mobility characteristic of heterogeneous modifications to N- and/or O-linked glycans that occur in the Golgi complex. These observations together suggested that P1 is the precursor to P2 and perhaps to Mα and Mβ as well.
Figure 2.

Metabolic pulse-chase analysis of Pmel17 in melanocyte and nonmelanocyte-derived cells. (A). MNT-1 cells were metabolically pulse-labeled with [35S]methionine/cysteine for 30 min and then chased for 0, 1, or 4 h. Cell lysates (cell) or culture medium (sup) were immunoprecipitated in parallel with HMB50 (50) and αPEP13h (P), and precipitated proteins were separated by SDS-PAGE and detected by PhosphorImager analysis. Migration of MW standards (kDa) is indicated to the right, and that of relevant bands, including P1, P2, Mα, Mβ, and band X, are indicated to the left. (B) MNT-1 cells were metabolically labeled as in A except for only 10 min and then chased for 0, 30, or 60 min. Cell lysates were immunoprecipitated with HMB50 or an isotype matched negative control antibody (7G7.B6; -CTL) and immunoprecipitates were analyzed as in A. Unfilled arrowheads indicate bands that were not consistently observed in all experiments. (C) HeLa cells were transiently transfected with Pmel17. Two days posttransfection, the cells were metabolically labeled and immunoprecipitates were analyzed as in A. (D) The ratio of HMB50- or αPEP13h-immunoprecipitable Mα to Mβ at the indicated time points is shown for MNT-1 and HeLa cells. Mα/Mβ is expected to be ∼2:1 based on methionine/cysteine content. The results represent the mean and SD of three experiments. (E and F) Maturation and degradation of the intracellular forms of Pmel17 from MNT-1 (E) and HeLa (F) cells was determined by quantitating the P1, P2, Mα, and Mβ bands immunoprecipitated by HMB50 in the experiments shown in part A and C, and expressing them as a percentage of the initial total Pmel17 from the pulse sample (0 h). This is representative of at least three experiments. (G) Degradation of total Pmel17 from MNT-1 or HeLa cells was determined by summing the intensities of the P1, P2, Mα, and Mβ bands immunoprecipitated by HMB50 from either the lysates or culture medium and expressing these values as a percentage of the total cell associated Pmel17 from the pulse sample. The data are from the experiments shown in A and C, and are representative of at least three experiments.
Two experiments were done to confirm that P1 is an ER form of Pmel17 and that P2, Mα, and Mβ are generated later. First, P1 but not P2, Mα, or Mβ, was immunoprecipitated from cells pulsed for only 10 min, whereas P2 only appeared after 30 min of chase and Mα and Mβ after 1 h of chase (Figure 2B). This confirms the order of appearance predicted for a precursor/product relationship. Second, we examined the maturation state of the N-linked glycans of the immunoprecipitated proteins at various time points (Figure 3). All four proteins contained N-linked oligosaccharides, as judged by decreased Mr upon treatment with N-glycanase F (endoF), which cleaves N-linked oligosaccharides regardless of maturation state. As expected, P1 at all time points was sensitive to digestion with endoglycosidase H (endoH), which cleaves only immature, high-mannose, N-linked oligosaccharides. In contrast, P2, Mα, and Mβ were largely endoH resistant at all time points (Figure 3, although both P2 and Mα contained at least one glycan that remained endoH sensitive). This shows that P1 is limited to the ER or early Golgi, whereas P2, Mα, and Mβ arise predominantly after passage through the Golgi complex. Even after digestion with endoF, P2 migrated slower than P1, suggesting the presence of additional post-ER modifications such as complex O-linked glycosylation. Similar results were obtained in both MNT-1 (Figure 3A) and transfected HeLa cells (Figure 3C). Taken together, these results support that P1 is a precursor to P2 and that Mα and Mβ either also derive from P1 or associate with P2 after passage through the Golgi complex.
Figure 3.

Endoglycosidase sensitivity of Pmel17 in melanocyte- and nonmelanocyte-derived cells. (A) MNT-1 cells were metabolically labeled as in Figure 2A, cell lysates were immunoprecipitated with HMB50, and precipitated proteins were mock (−), endoH (H), or endoF (F) treated before loading. Asterisk denotes bands resulting from cleavage of the indicated polypeptides by endoH or endoF. Only the relevant regions of the gel are shown. (B) Metabolically pulse labeled MNT-1 cells were chased for 1, 2, 4, and 6 h, and cell supernatants from each time point were pooled and immunoprecipitated with HMB50. Precipitated proteins were mock, endoH, or endoF treated as above. The major band comigrates with Mα, as in Figure 2A. (C) HeLa cells were transiently transfected with Pmel17 and metabolically labeled as in Figure 2C, and cell lysates (cells) or supernatants (Supe) were mock, endoH, or endoF treated as above.
Mα was predominantly immunoprecipitated as a diffuse, endoH-resistant band from lysates after a 1-h chase (Figures 2, A–C, and 3). An endoH-sensitive band partially comigrating with Mα (band X) was observed in the pulse of MNT-1 cells, but not of transfected HeLa cells; band X represents the product of an alternatively spliced Pmel17 RNA produced normally by melanocytic cells, distinct from the previously described long and short forms (J. F. Berson, S. E. Nichols, D. C. Harper, and M. S. Marks, unpublished results). Band X is not the same as Mα observed at later chase times based on endoH sensitivity (Figure 3A), its failure to appear in transfected HeLa cells (Figures 2C and 3C) and its precipitability under denaturing conditions with αPEP13h. Both HMB50 and αPEP13h precipitated Mα equally well from cell lysates under native conditions (Figure 2, A and C). A small amount of an endoH-resistant band comigrating with Mα (<10% of the initially synthesized material) was also immunoprecipitated from supernatants of both MNT-1 and transfected HeLa cells after 1–4 h of chase (Figures 2, A and C, and 3, B and C). The failure to immunoprecipitate this band from supernatants with the use of αPEP13h (Figure 2, A and C) or nonspecific antibodies and the absence of coprecipitated P1, P2, and Mβ bands supports the notion that this polypeptide was secreted rather than derived from cell fragments, was directly recognized by HMB50, and lacked the cytoplasmic domain. Therefore, this band could not correspond to P1 or P2, but rather must correspond to a lumenal domain fragment of Pmel17. It is likely equivalent to the previously described secreted protein ME20-S, reported to comprise amino acids 25–467 of Pmel17 (Maresh et al., 1994a). Because this secreted polypeptide migrated identically to the cell-associated Mα in both MNT-1 and transfected HeLa cells, we conclude that they are identical and correspond to a proteolytic cleavage product of full-length Pmel17 and/or band X.
Cleavage of Pmel17 to generate Mα would be predicted to also generate a C-terminal glycopeptide consisting of the cytoplasmic and transmembrane domains and part of the lumenal domain with a predicted N-glycosylation site. This glycopeptide would have a predicted molecular weight of 24–25 kDa (excluding additional posttranslational modification), close to the Mr 27,000 Mβ observed in MNT-1 cell lysates (Figure 2, A–C). Indeed, Western blotting of cell lysates with αPEP13h identified Mβ as the C-terminal fragment of Pmel17 (Figure 4); in addition to a ∼100-kDa band that corresponds to full-length Pmel17 (P1, because it contains the αPEP13h-reactive cytoplasmic domain and is fully endoH sensitive), αPEP13h detected a Mr 27,000 band in MNT-1 cells and Pmel17-transfected HeLa cells, but not in untransfected HeLa cells. Mβ was slightly larger in transfected HeLa cells, as expected if the predominant protein made in MNT-1 cells is the short form. To verify that Pmel17 cleavage to Mα and Mβ is common among pigmented cells, we assayed for Mβ in a second human melanoma cell line, primary human melanocytes, and a mouse melanocyte cell line. In all cases αPEP13h recognized both P1 and Mβ. These data demonstrate that the generation of Mβ from Pmel17 occurs normally in pigmented cells of both human and mouse origin and is independent of cell type.
Figure 4.

Western blot analysis of Pmel17 processing in melanocytic and nonmelanocytic cells. Cell lysates from human melanoma lines (MNT-1 and 1011-mel), a mouse melanocyte line (melan-a), human primary foreskin melanocytes (1°FS1 and 1°FS2), and nonmelanocyte derived human (HeLa) and mouse (NIH-3T3) lines, as well as HeLa cells transiently transfected with Pmel17 (HeLa/Pmel), were fractionated by SDS-PAGE. Proteins were transferred to PVDF, probed with αPEP13h, and detected by ECF. Migration of MW standards (kDa) is indicated.
Thus, we conclude that Mα and Mβ are derived from Pmel17 by intracellular cleavage (see Figure 1). Their accumulation in cell lysates by immunoprecipitation (Figure 2) and Western blotting (Figure 4) suggests that both proteolytic products are stable intracellularly.
Intracellular Mα and Mβ Remain Associated by Disulfide Linkage
Even after a 24-h chase, the majority of Mα produced in either MNT-1 or HeLa cells was found in cell lysates; <10% of the initially synthesized material was found in cell supernatants. This suggested that Mα was actively retained within the cell, which could be readily explained by its continued association with Mβ after cleavage. Such an association is supported by the immunoprecipitation analyses shown above. In cell lysates of both MNT-1 and transfected HeLa cells, Mα and Mβ were coprecipitated at all time points in equivalent ratios by both HMB50, which recognizes secreted Mα and thus should not bind directly to Mβ, and αPEP13h, which binds to an epitope only in Mβ (Figure 2D). Furthermore, both Mα and Mβ exhibited identical rates of appearance and disappearance from cell lysates in both MNT-1 and transfected HeLa cells (Figure 2, E and F). To determine whether the Mα/Mβ association is maintained by interchain disulfide-bonds, the immunoprecipitated products of a metabolic pulse-chase of MNT-1 cells were fractionated by SDS-PAGE under reducing and nonreducing conditions (Figure 5). Cells were pretreated with alkylating agents before and during lysis to prevent artifactual disulfide bond formation. A significant fraction of all forms of Pmel17 shifted into higher molecular weight bands under nonreducing conditions, indicating that Pmel17 is largely included in disulfide-bonded oligomeric complexes. Most importantly, both Mα and Mβ completely disappeared under nonreducing conditions. In contrast, the melanosomal glycoprotein TRP1 exhibited identical migration patterns under reducing and nonreducing conditions (unpublished results), demonstrating that our conditions did not result in nonspecific disulfide bond formation. This result, in combination with the coimmunoprecipitation of Mα and Mβ, shows that Mα and Mβ remain associated through disulfide bonds.
Figure 5.
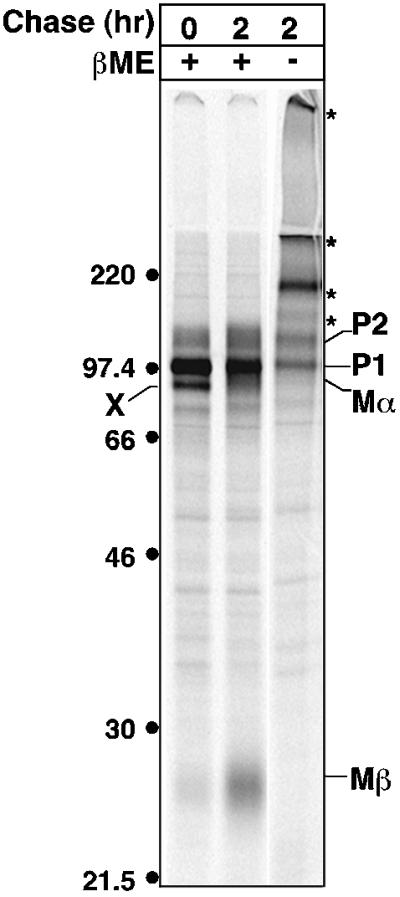
Nonreducing gel analysis of Pmel17. MNT-1 cells were metabolically labeled as in Figure 2A and chased for 0 or 2 h. N-ethylmaleimide was added to the final PBS wash of the labeling procedure. Cell lysates were prepared in the presence of iodoacetamide and immunoprecipitated with αPEP13h. Immunoprecipitated proteins were fractionated by SDS-PAGE in the presence (+) or absence (−) of β-mercaptoethanol (β-ME) and detected by PhosphorImager analysis. Asterisk indicates high MW forms of Pmel17. Migration of MW standards (kDa) is indicated to the left.
Kinetics and Localization of Pmel17 Processing and Degradation
Further analysis of the results of the pulse-chase studies and endoglycosidase treatments described above reveals that Mα and Mβ derive from proteolytic cleavage of P2 rather than P1. First, P2 was more intense than Mα and Mβ at the pulse in MNT-1 cells (Figures 2, A and B, and 3A) and preceded Mα and Mβ in appearance during the chase (Figure 2, A–C), as underscored by quantitation of band intensities (Figure 2, E and F). Second, the vast majority of Mβ and cell-associated Mα were resistant to digestion with endoH, in contrast to full-length Pmel17, which consisted of endoH-sensitive (P1) and endoH-resistant (P2) forms at all time points (Figure 3, A and C). Taken together, the data suggest that P2 represents the Golgi modified form of the P1 precursor and that cleavage of P2 generates Mα/β heterooligomers.
That N-linked oligosaccharide maturation of P1 precedes proteolytic processing suggests that Pmel17 is cleaved in a post-Golgi compartment. To verify this, we treated MNT-1 cells with BFA during a pulse-chase experiment. BFA treatment causes Golgi components to fuse with the ER, thus preventing anterograde traffic beyond the Golgi (Klausner et al., 1992). BFA treatment resulted in accumulation of a band migrating between P1 and P2 but dramatically inhibited production of Mβ, indicating that although partial maturation of the N-linked sugars proceeded, proteolytic processing was blocked (Figure 6A). A 97-kDa protein was precipitated at all time points; this band likely represents a partially Golgi-modified form of band X (Figure 2, A and B), because it was not observed in BFA-treated HeLa cells; preliminary data suggests that band X is fully capable of ER export and Golgi processing (unpublished results). These results confirm that proteolytic processing occurs from the P2 precursor in a post-Golgi compartment. To further define this compartment in MNT-1 cells, we tested the effects of treatment with NH4Cl, BafA1, or MME, reagents that neutralize the acidic environment of different intracellular compartments and thereby block cleavage by resident acid-dependent proteases. NH4Cl is expected to neutralize all acidic compartments, MME neutralizes esterase-containing compartments, such as lysosomes, and BafA1 is an inhibitor of the vacuolar H+ ATPases present in endosomal compartments. None of the treatments adversely affected maturation of P1 to P2, and comparable amounts of Mβ were generated in the control, MME-, and BafA1-treated cells at the early time points (Figure 6, B and C). However, the level of Mβ produced in NH4Cl-treated cells was greatly reduced. This indicates that Mβ generation and hence Pmel17 cleavage is inhibited by NH4Cl but not by MME. Mβ generation at the later time points may also have been blocked by BafA1, because P2 accumulated (see below). The data suggest that the protease responsible for cleaving Pmel17 is pH dependent and active in a post-Golgi, prelysosomal compartment.
Figure 6.

Effect of inhibitors on Pmel17 processing and degradation. (A) MNT-1 cells were metabolically labeled and chased as in Figure 2A, except that BFA was added to the starvation, pulse and chase media in half of the samples. Cell lysates (C) or culture medium (S) was immunoprecipitated with HMB50, and precipitated proteins were separated by SDS-PAGE and detected by PhosphorImager analysis. Shown separately are the upper and lower parts of the gel encompassing the critical regions. Migration of MW standards (kDa) is indicated to the right. (B) MNT-1 cells were metabolically labeled as in Figure 1A and chased for 0, 1, 2, 4, or 6 h in the presence of DMSO, NH4Cl, BafA1, MME, or a mix of E64, pepstatin A, and leupeptin (PIs). Cell lysates were immunoprecipitated with αPEP13h and analyzed as in A. The images on the left show the upper part of the gel including P1, P2, and Mα, and the images on the right show the lower part of the gel encompassing Mβ. The images on the right were exposed 3.5-fold longer than those on the left in order to better appreciate the differences among samples. (C) The relative intensities of Mβ in part B were quantitated and expressed in arbitrary units. A representative experiment is shown.
In addition to revealing the kinetics of cleavage to Mα and Mβ, the pulse-chase studies also revealed a rapid loss of all Pmel17-derived products from MNT-1 cell lysates, consistent with previous reports (Kobayashi et al., 1994). The half-life of Pmel17 in MNT-1 cells was only 2.5 h (Figure 2G). This short half-life could not be accounted for by secretion of Mα, which was inefficient (see above) or by ER-associated degradation because Pmel17 was protected in BFA treated MNT-1 cells (Figure 6A). Thus, the rapid loss of intracellular Pmel17 likely reflects either degradation or masking of epitopes in a post-Golgi compartment. To determine if Pmel17 was degraded in lysosomes, metabolically pulse-labeled cells were treated during the chase with a cocktail of lysosomal protease inhibitors. This treatment completely blocked the lysosomal proteolytic processing of cathepsin D (unpublished results), but only modestly protected P2, Mα, and Mβ from the dramatic loss observed in untreated cells (Figure 6, B and C). This level of protection was equal to that seen with MME treatment (also expected to block lysosomal acid hydrolases). In contrast, BafA1 and NH4Cl treatments potently inhibited Pmel17 loss, resulting in the accumulation of P2, and Mα and Mβ in the case of BafA1 (Figure 6, B and C). Although it is possible that the relevant proteases were not blocked by the inhibitors used, the data support the notion that the majority of intracellular Pmel17 loss is due to a pH-dependent mechanism in the TGN, early endosomal compartments, or melanosomes and not to lysosomal degradation.
It has been suggested that Pmel17 directly interacts with melanin and that this interaction obscures epitopes present on Pmel17 (Donatien and Orlow, 1995). If the loss of Pmel17 seen here occurs within melanosomes, Pmel17 would be expected to be more stable in HeLa cells, which lack melanin synthesis. This is indeed the case. The half-life of Pmel17 in HeLa cells was 4 h, compared with 2.5 h in MNT-1 cells (Figure 2G). Furthermore, although levels of intracellular Mα and Mβ were decreasing by 4 h in MNT-1 cells (Figure 2E), they were still accumulating in HeLa cells (Figure 2F). The data indicate that rapid loss of mature forms of Pmel17 is a phenomenon restricted to highly pigmented cells, consistent with Pmel17 becoming “buried” by melanin. We could not detect either full-length Pmel17 or Mβ in Triton X-100 insoluble fractions of MNT-1 cells, although this may be due to a limitation in our reagents.
Pmel17 Localizes to the Internal Membranes of MVBs
Pmel17 localizes to the lumen of premelanosomes (Lee et al., 1996; Raposo et al., 2001), but the data thus far suggest that intracellular Mα remains tethered to the integral membrane subunit, Mβ, and is thus likely membrane associated. How can one reconcile these conclusions? One possibility is that Pmel17 accesses the lumen of premelanosomes as an integral membrane protein of intralumenal vesicles (ILVs) that pinch off the limiting membrane. If this were true, then Pmel17 might be predicted to accumulate in MVBs in nonpigmented cells. To test this prediction, Pmel17 was immunolocalized in transfected HeLa cells. We first used IFM to compare the gross distribution of Pmel17 with that of lamp-1 and CD63, glycoproteins enriched in multivesicular late endosomes. Pmel17 localized to vesicular structures, nearly all of which were positive for lamp-1 (Figure 7) and CD63. However, not all lamp-1–positive structures were costained for Pmel17 in transfected cells. This indicates that Pmel17 accumulates in a subset of late endosomes and/or lysosomes and thus could potentially be localized to multivesicular late endosomes. To identify the subset, ultrathin cryosections of transfected HeLa cells were analyzed by IEM with the use of immunogold labeling. As predicted, Pmel17 localized predominantly to ILVs within MVBs (Figure 8A). Some HMB50 labeling was observed at the cell surface and in the Golgi, but the bulk of the labeling was observed over numerous ILVs of MVBs. The same structures were also labeled with antibodies to CD63, indicating that they corresponded to the Pmel17-positive structures seen by IFM (see Figure 9C). These results confirm that Pmel17 accumulates on the ILVs of MVBs.
Figure 7.

Immunofluorescence localization of Pmel17 in HeLa cells. HeLa cells transiently transfected with Pmel17 were fixed, permeabilized, and stained with HMB50 (left) and anti-lamp-1 (middle), followed by LRSC- and FITC-conjugated secondary antibodies, and cells were analyzed by IFM. (right) A merged image.
Figure 8.

Localization of Pmel17 to intralumenal vesicles of MVBs. (A) Ultrathin cryosections of HeLa cells transiently transfected with Pmel17 were immunogold labeled with HMB50 (PAG10). The cytoplasm is filled with numerous MVBs. Labeling for Pmel17 is mainly restricted to the ILVs. (B) Ultrathin cryosections of MNT-1 cells retrieved with a mixture of methylcellulose (MC) and uranyl acetate (UA). Note the presence of ILVs beneath polymerized melanin in late stage melanosomes. (C) Ultrathin cryosections of MNT-1 cells were immunogold labeled with HMB50 (PAG10). Early premelanosomal structures (stage I, stars) with no visible striations and displaying ILVs (arrows) are shown. Bars, 200 nm.
Figure 9.

Immunogold localization of Pmel17 to striations. (A) Ultrathin cryosections of MNT-1 cells were immunogold labeled with HMB50 (PAG10). Stage II premelanosomes with visible striations are shown. (B–D) Ultrathin cryosections of HeLa cells transiently overexpressing Pmel17 (cells were transfected with 2 μg DNA/6-well) were immunogold labeled with HMB50 (PAG10) (B and C) or HMB50 (PAG10) and anti-CD63 (PAG15) (D). (B) Note the presence of striated-like structures highly labeled for Pmel17 in the lumen of the MVBs (arrows). (C) Some of the striated-like structures are very well developed. (D) The ILVs and associated tubular structures contain both Pmel17 and CD63, whereas the fibrillar structures (arrows) are labeled only for Pmel17. Bars, 200 nm.
Melanosomes are morphologically and functionally distinct from multivesicular late endosomes (Raposo et al., 2001). Nevertheless, ILVs can be detected close to or underneath the striations and melanin in stage II-IV melanosomes (Figure 8B), as has been observed in other pigmented cells (Turner et al., 1975; Jimbow et al., 1979). ILVs are also detected, and perhaps formed within, electron-lucent coated endosomes that correspond to stage I premelanosomes and that serve as precursors to the striated stage II structures (Raposo et al., 2001 and Figure 8C). Importantly, although Pmel17 is enriched in stage II premelanosomes with well-formed striations (Raposo et al., 2001; see Figure 9A), Pmel17 is also present to a lesser extent within coated endosomes (Raposo et al., 2001) in which it is largely associated with ILVs (Figure 8C). A small amount of Pmel17 can also be found on small vesicles within stage II and III melanosomes. These results suggest that Pmel17 associates with ILVs found within the lumen of premelanosomes and melanosomes and particularly in compartments that serve as precursors to the striated stage II premelanosomes.
Pmel17 Expression Drives Formation of Premelanosomal-like Striations
Immunogold-labeled cryosections of MNT-1 cells clearly show that the majority of labeling for Pmel17 within stage II premelanosomes aligns closely with the characteristic striations (Raposo et al., 2001 and Figure 9A). This suggests that after entering the lumen of premelanosomes on ILVs, Pmel17 is recruited to newly formed striations. Given the association of Pmel17 with the striations and the lack of other known markers of these structures, we hypothesized that expression of Pmel17 alone might be sufficient to induce striation formation in nonpigmented cell types. In ultrathin cryosections of transfected HeLa cells expressing low or modest levels of Pmel17, Pmel17 labeling was restricted primarily to ILVs (see Figure 8A). However, in a significant fraction of cells expressing high levels of the transgene, labeling for Pmel17 was additionally found in organized fibrillar structures adjacent to the ILVs (Figure 9, B–D). Pmel17-bound gold particles on these structures appeared to line up like beads on a string, similar to the labeling on premelanosomal striations. Although the ILVs contained both CD63 and Pmel17, the striation-like structures labeled only for Pmel17, even when both vesicles and striations were present within the same membrane-delimited compartment (Figure 9D). Thus, expression of Pmel17 alone appears to be sufficient to drive formation of striation-like structures from within MVBs, even in cells of nonmelanosomal origin. Furthermore, these structures appear to largely exclude at least some other residents of the ILVs.
DISCUSSION
The unique morphological characteristics of melanosomes, like those of other lysosome-like organelles, likely facilitate the specialized function of these organelles. The condensation of melanin within melanosomes is expected to both prevent melanin toxicity within the endosomal pathway and facilitate melanin transfer to keratinocytes. The fibrous lumenal striations that are the morphological hallmark of premelanosomes serve as the site of melanin deposition and are thus undoubtedly essential for melanin condensation. Nevertheless, very little is known about their composition or biogenesis. We show here that Pmel17, a pigment cell-specific integral membrane protein that localizes to the striations (Zhou et al., 1994; Raposo et al., 2001), is a critical biogenetic component of premelanosome striations. Despite posttranslational cleavage, the large N-terminal lumenal fragment of Pmel17 remains tethered to an integral membrane fragment in detergent extracts, implying that it maintains its association with membranes within the cell. These membranes likely correspond to ILVs of premelanosomes, on which Pmel17 accumulates before its association with striations. Finally, overexpression of Pmel17 alone was sufficient to initiate striation formation within MVBs of nonmelanocytic cells. Taken together, the data suggest that the ILVs of premelanosomes provide a scaffolding from which Pmel17 initiates striation formation. Similar mechanisms are likely to underlie the biogenetic processes by which other lysosome-like organelles are formed.
Biosynthesis and Processing of Pmel17
Previous models of Pmel17 association with the premelanosomal matrix assumed that the Pmel17 accumulating within melanosomes was not membrane associated (Zhou et al., 1994), a hypothesis that was supported by the identification of a secreted lumenal domain fragment in melanoma cell lines, melanocytes, and transfected nonmelanocytic cells (Vogel and Esclamado, 1988; Adema et al., 1994; Maresh et al., 1994a). Our data confirm that Pmel17 is posttranslationally cleaved within the lumenal domain, but show that cleavage does not result in immediate release of the lumenal fragment from membranes.
Pmel17 is synthesized as a precursor protein, designated P1 (Mr 97,000), which is posttranslationally modified to yield P2 (Mr 128,000); endoglycosidase digestions show that these modifications include both N-linked oligosaccharide maturation and additional changes, including perhaps O-linked oligosaccharide addition (Figure 3). Bands similar in size to P2 have been previously observed, if not specifically noted (Vennegoor et al., 1988; Vogel and Esclamado, 1988; Adema et al., 1994). Once formed, P2 is proteolytically processed into two subunits, the large lumenal fragment Mα (Mr 85,000) and the transmembrane domain-containing Mβ (Mr 28,000). That Mα and Mβ derive from P2 is supported by their delayed appearance relative to P2 in pulse-chase assays, their resistance to endoH, and the generation of P2 but not Mα and Mβ in BFA- or NH4Cl-treated cells (Figures 2, 3, and 6). Mα is most likely equivalent to ME20-S, a secreted form of Pmel17 previously shown to comprise amino acids 25–467 of the lumenal domain (Maresh et al., 1994a). Mβ likely corresponds to the remaining fragment, based on its apparent size, its sensitivity to endoF (indicating that it is glycosylated and therefore contains lumenal domain residues; Figure 3), and its reactivity with αPEP13h (indicating that it contains the C terminus; Figure 4). Reexamination of published data shows that bands similar in Mr to Mα and Mβ have been immunoprecipitated from cell lysates before (Vennegoor et al., 1988; Vogel and Esclamado, 1988; Adema et al., 1994), supporting our results.
Importantly, despite cleavage, only a small fraction of Mα is secreted, whereas most Mα and Mβ remain associated with each other intracellularly. This is supported by the complete coimmunoprecipitation of both Mα and Mβ by both αPEP13h and HMB50 from cell lysates (Figure 2D), by the quantitative shift of Mα and Mβ into high molecular weight complexes in nonreducing gels (Figure 5), and by their cosedimentation and coprecipitation by sucrose density gradient fractionation (unpublished data). The interpretation most consistent with these data is that Mα and Mβ form disulfide-linked heterooligomers. Therefore, cleaved or uncleaved, Mα is predominantly retained intracellularly in association with a transmembrane domain, and hence in association with membranes. Our data cannot exclude the possibility that Mα is subsequently released from Mβ into detergent-insoluble, membrane-free complexes that might underlie the striations.
Do other proteins associate with Mα and Mβ within the heterooligomers? The only additional [35S]methionine-labeled band that coprecipitated with Pmel17 was a protein from MNT-1 pulse-labeled lysates that migrated closely with mature Mα (band X). This band, found in all melanocytic cells analyzed, represents the protein product of a novel alternatively spliced RNA from the Pmel17 gene distinct from the described “short form” (unpublished results). It is not typically generated in HeLa cells transfected with Pmel17 cDNA expression vectors, explaining the absence of band X in these cells. Furthermore, unlike Mα, band X can be reimmunoprecipitated from denatured and reduced samples with antibodies increased against both the N- and C-termini of Pmel17 (unpublished data). The presence of the Pmel17 C-terminus within this band distinguishes it from the closely comigrating Mα band, which lacks this determinant. At this time, it is uncertain whether the diffuse Mα band in melanocytic cells also contains processed forms of band X. No additional labeled bands are consistently coprecipitated with Pmel17 at later time points.
The conversion of P1 to P2 appears to be rate limiting in maturation of Pmel17, because P2 fails to accumulate and endoH-sensitive full-length protein is present in MNT-1 cells even after a 4-h chase. This is in agreement with Maresh et al. (1994a), who showed that the full-length Pmel17 accumulating at steady state contains only high-mannose N-linked carbohydrates. In contrast to P2, Mβ accumulated at steady state in both MNT-1 and transfected HeLa cells. These data support the conclusion that P2 is a transient intermediate in the maturation of Pmel17 and that the Mα/Mβ complex constitutes the major species of mature Pmel17 present intracellularly at steady state. The persistence of P2 at late chase times in pulse-chase assays may therefore reflect its continued generation from P1 rather than its failure to be converted to Mα/Mβ.
Intracellular Sites of Proteolytic Processing and Degradation
We show here that Pmel17 is cleaved in a pH-dependent manner in a post-Golgi, prelysosomal compartment. This is supported by the inhibition of processing by BFA and NH4Cl and the failure to inhibit processing with the use of reagents that exclusively affect lysosomal enzyme function (MME and proteinase inhibitors). BafA1 treatment, which inhibits the vacuolar ATPase in endosomes and lysosomes, did not initially block production of Mβ, but resulted in the accumulation of P2. It is likely that BafA1 inhibits acidification required for the time-dependent loss of Pmel17 from cell lysates (see below), resulting in apparent stabilization of all Pmel17 forms; the accumulation of P2 may reflect a gradual decrease in cleavage efficiency within endosomes because of BafA1-mediated inhibition of endosomal recycling of a required protease, such as a furin-like proprotein convertase. Current efforts are under way to determine the precise site of cleavage and the protease responsible.
As in other melanocytic cells (Kobayashi et al., 1994; Donatien and Orlow, 1995), Pmel17 has a short half-life in MNT-1 cells. Although typical of rapid lysosomal degradation, the loss of Pmel17 was insensitive to inhibitors of lysosomal degradation but sensitive to agents that affect the acidity of prelysosomal compartments (Figure 6). Although we cannot rule out that our inhibitors were insufficient to specifically protect Pmel17, the longer half-life of Pmel17 in HeLa cells hints at a melanocyte-specific mechanism of loss. We thus favor the interpretation proposed by Orlow and colleagues that Pmel17 becomes trapped in detergent insoluble melanin aggregates (Donatien and Orlow, 1995). The rapid loss of Mα/Mβ from MNT-1 cell lysates may thus reflect entrapment in melanin shortly after it is generated, consistent with a cleavage event in the TGN, endosomes, or premelanosomes.
Implications of Pmel17 Membrane Association on Melanosome Biogenesis
The biochemical characterization of detergent soluble Pmel17 suggests that Mα may not dissociate from membranes, despite intralumenal cleavage. How, then, can we explain the localization of Pmel17 to the lumen (Lee et al., 1996; Raposo et al., 2001) or “matrix” (Zhou et al., 1994) of premelanosomes? IEM of MNT-1 and transfected HeLa cells revealed HMB50 labeling on ILVs found within premelanosomes and late endosomal MVBs, respectively, indicating that Mα/Mβ or P2 resides on these vesicles. Such ILVs have been previously described in melanosomes and premelanosomes (Turner et al., 1975; Jimbow et al., 1979) and were easily observed in MNT-1 cells—particularly in the coated endosomal precursor of stage II premelanosomes (Figure 8 and Raposo et al., 2001). We interpret these results to indicate that Pmel17 accesses the lumen of the newly forming premelanosome by associating with invaginating membranes that form ILVs.
Although present in ILVs in earlier compartments, Pmel17 becomes most enriched in stage II premelanosomes in close alignment with striations (Raposo et al., 2001). These striations are considered to be proteinaceous by virtue of the failure to detect membrane-like structures by EM (Maul, 1969). How, then, are the striations formed, and if they derive from MVBs, why is the underlying membrane not apparent? At least two models can be envisioned. In one, the ILVs are required as a substrate to initiate striation formation but are subsequently released, perhaps through dissociation of Mα from Mβ. Our inability to detect free Mα may reflect detergent insolubility, which may be due in part to striation formation as well as subsequent melanin deposition. A second model would suggest that Pmel17, and perhaps other proteins, coat the membrane such that it is no longer exposed for detection with the use of classical techniques. In this model, biochemical characterization of the striations would be expected to reveal the presence of lipids enriched in the ILVs. The derivation of specialized structures from MVBs is not an entirely new idea; similar biogenetic pathways have been suggested for platelet-dense granules (Youssefian and Cramer, 2000), major histocompatibility complex class II compartments (Peters et al., 1995; Raposo et al., 1996), Weibel-Palade bodies (Kobayashi et al., 2000), and cytotoxic granules (Peters et al., 1991). Our data therefore add premelanosomes to this list of MVB-derived organelles.
Surprisingly, overexpression of Pmel17 alone in nonmelanocytic HeLa cells resulted in deposition of electron-dense material in characteristic linear arrays over multivesicular compartments; the linear arrays were similar in morphology to the premelanosomal striations, albeit less well developed, and were immunolabeled for Pmel17 (Figure 9). These results suggest that expression of Pmel17 alone is sufficient to initiate striation formation. Because these results were in HeLa cells, which lack other melanocyte-specific components, we suspect that Pmel17 functions as both a structural component and a catalyst for the formation of striations; other components and perhaps melanocyte-specific sorting pathways (Raposo et al., 2001) are likely to contribute to the efficiency and morphological integrity of the striations. Thus, the results implicate Pmel17 as a major determinant in the biogenesis of the characteristic morphology of premelanosomes. These data are analogous to the induction of Birbeck granules in Langerhans cells by ectopic expression of Langerin (Valladeau et al., 2000).
Relationship of Melanosomes to Other Lysosome-like Organelles
The notion that premelanosomes originate from MVBs has important implications for the mechanisms underlying the biogenesis of unique lysosome-like organelles and for the etiology of genetic diseases that affect these organelles. As discussed above, platelet-dense granules also derive from MVBs (Youssefian and Cramer, 2000), and classical late endosomes, the precursors of lysosomes, are also multivesicular in morphology (Trowbridge et al., 1993). The localization of specific cargo proteins, such as Pmel17, to the ILVs of these structures may therefore provide a common mechanism to create novel structures within lysosome-related organelles. Interestingly, the organelles most affected by HPS are melanosomes and platelet dense granules (Swank et al., 1998; Dell'Angelica et al., 2000). Given the model for premelanosome formation presented here, it is possible that some forms of HPS are due to defects in MVB formation. Further analysis of the pathways by which the MVBs are formed and the ILVs are transformed into striations may therefore provide insight into the etiology of a class of genetic diseases.
ACKNOWLEDGMENTS
The authors thank Drs. V. Hearing, S. Topalian, M. Herlyn, W. Storkus, and C. Figdor for providing reagents used in these studies and Drs. E. Dell'Angelica, J. Bonifacino, and C. Burd for critically reading this manuscript. G.R. is indebted to D. Louvard for continuous support. This work was supported by National Institutes of Health (NIH) grant R01 EY 12207 from the National Eye Institute and American Cancer Society grant RPG-97-003-01-BE. J.F.B. was supported by NIH National Cancer Institute Training Grant T32 CA 09140 and American Cancer Society Fellowship PF-99-336-01-CIM.
Abbreviations used:
- BafA1
bafilomycin A1
- BFA
brefeldin A
- HPS
Hermansky-Pudlak syndrome
- IEM
immunoelectron microscopy
- IFM
immunofluorescence microscopy
- ILVs
intralumenal vesicles
- MME
methionine methyl ester
- MVBs
multivesicular bodies
- NH4Cl
ammonium chloride
- PAG
protein A-gold
REFERENCES
- Adema GJ, de Boer AJ, Vogel AM, Loenen WAM, Figdor CG. Molecular characterization of the melanocyte lineage-specific antigen gp100. J Biol Chem. 1994;269:20126–20133. [PubMed] [Google Scholar]
- Berson JF, Frank DW, Calvo PA, Bieler BM, Marks MS. A common temperature-sensitive allelic form of human tyrosinase is retained in the endoplasmic reticulum at the nonpermissive temperature. J Biol Chem. 2000;275:12281–12289. doi: 10.1074/jbc.275.16.12281. [DOI] [PubMed] [Google Scholar]
- Chakraborty AK, Platt JT, Kim KK, Kwon BS, Bennett DC, Pawelek JM. Polymerization of 5,6-dihydroxyindole-2-carboxylic acid to melanin by the pmel 17/silver locus protein. Eur J Biochem. 1996;236:180–188. doi: 10.1111/j.1432-1033.1996.t01-1-00180.x. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica EC, Mullins C, Caplan S, Bonifacino JS. Lysosome-related organelles. FASEB J. 2000;14:1265–1278. doi: 10.1096/fj.14.10.1265. [DOI] [PubMed] [Google Scholar]
- Donatien PD, Orlow SJ. Interaction of melanosomal proteins with melanin. Eur J Biochem. 1995;232:159–164. doi: 10.1111/j.1432-1033.1995.tb20794.x. [DOI] [PubMed] [Google Scholar]
- Esclamado RM, Gown AM, Vogel AM. Unique proteins defined by monoclonal antibodies specific for human melanoma. Am J Surg. 1986;152:376–385. doi: 10.1016/0002-9610(86)90308-9. [DOI] [PubMed] [Google Scholar]
- Jimbow K, Oikawa O, Sugiyama S, Takeuchi T. Comparison of eumelanogenesis in retinal and follicular melanocytes; role of vesiculo-globular bodies in melanosome differentiation. J Invest Dermatol. 1979;73:278–284. doi: 10.1111/1523-1747.ep12531650. [DOI] [PubMed] [Google Scholar]
- Kapur RP, Bigler SA, Skelly M, Gown AM. Anti-melanoma monoclonal antibody HMB45 identifies an oncofetal glycoconjugate associated with immature melanosomes. J Histochem Cytochem. 1992;40:207–212. doi: 10.1177/40.2.1552165. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi A, Shimizu H, Nishikawa T. Expression and ultrastructural localization of HMB-45 antigen. Br J Dermatol. 1996;135:400–405. [PubMed] [Google Scholar]
- King RA, Hearing VJ, Creel DJ, Oetting WS. Albinism. In: Scriver C R, Beaudet A L, Sly W S, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, Inc.; 1995. pp. 4353–4392. [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Urabe K, Orlow SJ, Higashi K, Imokawa G, Kwon BS, Potterf B, Hearing VJ. The Pmel 17/silver locus protein. Characterization and investigation of its melanogenic function. J Biol Chem. 1994;269:29198–29205. [PubMed] [Google Scholar]
- Kobayashi T, Vischer UM, Rosnoblet C, Lebrand C, Lindsay M, Parton RG, Kruithof EK, Gruenberg J. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol Biol Cell. 2000;11:1829–1843. doi: 10.1091/mbc.11.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BS, Halaban R, Kim GS, Usack L, Pomerantz S, Haq AK. A melanocyte-specific complementary DNA clone whose expression is inducible by melanotropin and isobutylmethyl xanthine. Mol Biol Med. 1987;4:339–355. [PubMed] [Google Scholar]
- Kwon BS, Chintamaneni C, Kozak CA, Copeland NG, Gilbert DJ, Jenkins N, Barton D, Francke U, Kobayashi Y, Kim KK. A melanocyte-specific gene, Pmel 17, maps near the silver coat color locus on mouse chromosome 10 and is in a syntenic region on human chromosome 12. Proc Natl Acad Sci USA. 1991;88:9228–9232. doi: 10.1073/pnas.88.20.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ZH, Hou L, Moellmann G, Kuklinska E, Antol K, Fraser M, Halaban R, Kwon BS. Characterization and subcellular localization of human Pmel 17/silver, a 100-kDa (pre)melanosomal membrane protein associated with 5,6,-dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J Invest Dermatol. 1996;106:605–610. doi: 10.1111/1523-1747.ep12345163. [DOI] [PubMed] [Google Scholar]
- Liou W, Geuze HJ, Slot JW. Improving structural integrity of cryosections for immunogold labeling. Histochem Cell Biol. 1996;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- Maresh GA, Wang W-C, Beam KS, Malacko AR, Hellström I, Hellström KE, Marquardt H. Differential processing and secretion of the melanoma-associated ME20 antigen. Arch Biochem Biophys. 1994a;311:95–102. doi: 10.1006/abbi.1994.1213. [DOI] [PubMed] [Google Scholar]
- Maresh GA, Marken JS, Neubauer M, Aruffo A, Hellström I, Hellström KE, Marquardt H. Cloning and expression of the gene for the melanoma-associated ME20 antigen. DNA Cell Biol. 1994b;13:87–95. doi: 10.1089/dna.1994.13.87. [DOI] [PubMed] [Google Scholar]
- Marks MS, Woodruff L, Ohno H, Bonifacino JS. Protein targeting by tyrosine- and di-leucine-based signals: evidence for distinct saturable components. J Cell Biol. 1996;135:341–354. doi: 10.1083/jcb.135.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MS, Roche PA, van Donselaar E, Woodruff L, Peters PJ, Bonifacino JS. A lysosomal targeting signal in the cytoplasmic tail of the β chain directs HLA-DM to the MHC class II compartments. J Cell Biol. 1995;131:351–369. doi: 10.1083/jcb.131.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul GG. Golgi-melanosome relationship in human melanoma in vitro. J Ultrasctruct Res. 1969;26:163–176. doi: 10.1016/s0022-5320(69)90042-2. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Raposo G, Neefjes JJ, Oorschot V, Leijendekker RL, Geuze HJ, Ploegh HL. Major histocompatibility complex class II compartments in human B lymphoblastoid cells are distinct from early endosomes. J Exp Med. 1995;182:325–334. doi: 10.1084/jem.182.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters PJ, Borst J, Oorschot V, Fukuda M, Krähenbühl O, Tschopp J, Slot JW, Geuze HJ. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med. 1991;173:1099–1109. doi: 10.1084/jem.173.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Kleijmeer MJ, Posthuma G, Slot JW, Geuze HJ. Immunogold labeling of ultrathin cryosections: application in immunology. In: Herzenberg L A, Weir D, Herzenberg L A, Blackwell C, editors. Handbook of Experimental Immunology. Cambridge, MA: Blackwell Science, Inc.; 1997. pp. 1–11. [Google Scholar]
- Raposo G, Tenza D, Murphy DM, Berson JF, Marks MS. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J Cell Biol. 2001;152:809–823. doi: 10.1083/jcb.152.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, Geuze HJ. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiji M, Fitzpatrick TM, Simpson RT, Birbeck MSC. Chemical composition and terminology of specialized organelles (melanosomes and melanin granules) in mammalian melanocytes. Nature. 1963;197:1082–1084. doi: 10.1038/1971082a0. [DOI] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James D. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swank RT, Novak EK, McGarry MP, Rusiniak ME, Feng L. Mouse models of Hermansky Pudlak syndrome: a review. Pigment Cell Res. 1998;11:60–80. doi: 10.1111/j.1600-0749.1998.tb00713.x. [DOI] [PubMed] [Google Scholar]
- Trowbridge IS, Collawn JF, Hopkins CR. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu Rev Cell Biol. 1993;9:129–161. doi: 10.1146/annurev.cb.09.110193.001021. [DOI] [PubMed] [Google Scholar]
- Turner WAJ, Taylor JD, Tchen TT. Melanosome formation in the goldfish: the role of multivesicular bodies. J Ultrastruct Res. 1975;51:16–31. doi: 10.1016/s0022-5320(75)80004-9. [DOI] [PubMed] [Google Scholar]
- Valladeau J, Ravel O, Dezutter-Dambuyant C, Moore K, Kleijmeer M, Liu Y, Duvert-Frances V, Vincent C, Schmitt D, Davoust J, Caux C, Lebecque S, Saeland S. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 2000;12:71–81. doi: 10.1016/s1074-7613(00)80160-0. [DOI] [PubMed] [Google Scholar]
- Vennegoor C, Hageman P, van Nouhuijs H, Ruiter DJ, Calafat J, Ringens PJ, Rümke P. A monoclonal antibody specific for cells of the melanocytic lineage. Am J Pathol. 1988;130:179–192. [PMC free article] [PubMed] [Google Scholar]
- Vogel AM, Esclamado RM. Identification of a secreted Mr 95,000 glycoprotein in human melanocytes and melanomas by a melanocyte specific monoclonal antibody. Cancer Res. 1988;48:1286–1294. [PubMed] [Google Scholar]
- Youssefian T, Cramer EM. Megakaryocyte dense granule components are sorted in multivesicular bodies. Blood. 2000;95:4004–4007. [PubMed] [Google Scholar]
- Zhou BK, Kobayashi T, Donatien PD, Bennett DC, Hearing VJ, Orlow SJ. Identification of a melanosomal matrix protein encoded by the murine si (silver) locus using “organelle scanning.”. Proc Natl Acad Sci USA. 1994;91:7076–7080. doi: 10.1073/pnas.91.15.7076. [DOI] [PMC free article] [PubMed] [Google Scholar]