

Abstract
Lipoic acid is an essential cofactor for mitochondrial metabolism and is synthesized de novo using intermediates from mitochondrial fatty-acid synthesis type II, S-adenosylmethionine and iron–sulfur clusters. This cofactor is required for catalysis by multiple mitochondrial 2-ketoacid dehydrogenase complexes, including pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and branched-chain ketoacid dehydrogenase. Lipoic acid also plays a critical role in stabilizing and regulating these multienzyme complexes. Many of these dehydrogenases are regulated by reactive oxygen species, mediated through the disulfide bond of the prosthetic lipoyl moiety. Collectively, its functions explain why lipoic acid is required for cell growth, mitochondrial activity, and coordination of fuel metabolism.
Keywords: lipoic acid, cell metabolism, tricarboxylic acid cycle (TCA cycle) (Krebs cycle), redox regulation, mitochondria, oxidation-reduction (redox)
Introduction
Lipoic acid (6,8-dithiooctanoic acid) was first identified by Reed and co-workers (1, 3) and Jukes and co-workers (2) in the 1950s, and over the last several decades much has been learned about the chemical properties and biological functions of this unique cofactor. The disulfide bond within the molecule provides a source of reductive potential that is required for catalysis by mitochondrial 2-ketoacid dehydrogenases and participates in stabilization and redox-dependent regulation of these multienzyme complexes. These functions make lipoic acid essential for cell growth, oxidation of carbohydrates, amino acids, and other fuels, and regulation of mitochondrial redox balance. Herein, we discuss the intricacies of lipoic acid synthesis in various organisms, the role lipoylation plays in 2-ketoacid dehydrogenase activities and regulation, and how these enzymes are influenced by reactive oxygen species through this covalently bound lipoic acid cofactor.
Lipoic acid synthesis
Lipoic acid (LA)2 metabolism has been intensely investigated by Reed (1), Cronan (4, 5), and others through genetic and biochemical studies using prokaryotes and Saccharomyces cerevisiae (6). Homologous enzymes have been identified in plants (7, 8) and mammalian systems (9). That being said, distinct differences in LA metabolism among various organisms are relevant to the regulation of 2-ketoacid dehydrogenases. LA metabolism in Escherichia coli consists of a de novo biosynthetic pathway and a salvage pathway capable of using exogenous LA as a substrate. Both pathways lead to the covalent modification of the ϵ-amino group on conserved lysine residues within the lipoyl domain of the E2 subunits of 2-ketoacid dehydrogenase complexes (Fig. 1A) (4).
Figure 1.

Structures, enzymes, and reaction mechanisms of lipoic acid metabolism. A, de novo lipoic acid synthesis and salvage pathways in E. coli. B, lipoic acid metabolic pathway in S. cerevisiae and Homo sapiens. C, orthologous enzymes associated with lipoic acid metabolism in each organism.
The biosynthetic pathway in E. coli is initiated by an octanoyltransferase enzyme (octanoyl-(ACP:protein)-protein N-octanoyltransferase), LipB, that transfers octanoic acid derived from mitochondrial fatty-acid synthesis type II (FASII) from the acyl carrier protein (ACP) to the lipoyl domain of a target enzyme. In the first step of LA biosynthesis, LipB transfers octanoate directly onto the lipoyl domain on the E2 subunit of 2-ketoacid dehydrogenases (Fig. 1A, reaction B1) (10–14). E. coli tolerates loss of this enzyme when exogenous lipoate is present through activity of the salvage pathway (discussed below), but in the absence of exogenous lipoate, growth is suppressed, and no lipoylation of pyruvate dehydrogenase (PDH-E2) or α-ketoglutarate dehydrogenase (OGDH-E2) is observed (15).
In the second step of LA biosynthesis, two sulfur atoms are inserted at the C6 and C8 positions by an iron–sulfur lipoic acid synthase enzyme, LipA (Fig. 1A, reaction B2) (15–17). This step is similar to biotin synthesis in that the reaction mechanism requires sulfur donation from a [4Fe-4S] cluster within the enzyme and reductive cleavage of S-adenosylmethionine (SAM) to generate 5′-deoxyadenosyl 5′-radicals that remove a hydrogen atom from C6 and C8 and facilitate insertion of the sulfur atoms (5, 18, 19). The requirements for this enzyme are similar to that of LipB in that E. coli can utilize the LA salvage pathway to compensate for loss of LipA in lipoate-containing media (15). Importantly, donation of sulfur from the [4Fe-4S] cluster within the enzyme originally suggested that LipA may be a self-sacrificing protein rather than performing true enzyme catalysis (5). This had been supported by the small amount of LA generated per molecule of LipA in E. coli, which could be enhanced by co-expression of LipA with iron–sulfur cluster proteins (5, 20). However, recently, the E. coli iron–sulfur cluster proteins NfuA and IscU were shown to reinstall the [4Fe-4S] cluster of LipA to facilitate additional turns of the enzyme (21). This mechanism is consistent with deficiencies in NFU1 resulting in phenotypes associated with lipoic acid deficiency and suggests that this mechanism may be conserved in mammals (22).
The LA salvage pathway in E. coli consists of a single lipoyl-protein ligase enzyme, LplA, that first conjugates exogenous lipoic acid to an adenylate intermediate (lipoyl-AMP) followed by ligation to the lipoyl domain of E2 subunits and H-protein (Fig. 1A, reaction S1) (23–25). LplA can use both lipoic acid and octanoate to modify E2 subunits in intact cells, with the latter substrate requiring the activity of LipA to insert sulfur atoms and generate the lipoic acid moiety directly on target enzymes (23, 26). E. coli can tolerate the loss of this enzyme, but loss of LplA and LipB results in no lipoylated proteins, whereas loss of LplA and LipA results in the accumulation of octanoyl proteins (26). These data indicate that there are two distinct LA metabolism pathways in E. coli that ensure growth in both lipoate-containing and -deficient environments (11, 26).
LA metabolism in S. cerevisiae is different from E. coli primarily in that these two pathways are interdependent and cannot fully compensate for one another (27), and lipoylation of PDH-E2 (Lat1) and OGDH-E2 (Kgd2) requires initial lipoylation of the H-protein of the glycine cleavage system (Gcv3) (Fig. 1B, reaction 1) (6). This pathway is similar to Bacillus subtilis, where the octanoyltransferase in S. cerevisiae, Lip2, transfers octanoate (derived from FASII) to Gcv3, and this step is required for lipoylation of Lat1 and Kgd2 (6). Importantly, activity of the glycine cleavage system (GCS) is not required for downstream lipoylation events because loss of other GCS subunits has no impact on Lat1 or Kgd2 lipoylation (6). The lipoate synthase enzyme in S. cerevisiae is Lip5, which functions similarly to LipA in E. coli through iron–sulfur cluster-mediated insertion of the disulfide at C6 and C8 (Fig. 1B, reaction 2) (28). Yeast cannot tolerate the loss of Lip2 or Lip5 unless grown in medium containing ethanol and succinate, which bypasses Kgd2 in the TCA cycle (6).
The inability of Lip2 and Lip5 mutants to grow on media supplemented with lipoic acid was the first indication that an independent LA salvage pathway did not exist in S. cerevisiae (29). However, identification of an LplA homolog in yeast, Lip3, and the observation that lipoylation of Gcv3 was maintained in Lip3 mutants indicated that Lip3 functions downstream of Lip2 (Fig. 1B, reaction 3). In vitro studies have demonstrated that Lip3 is an octanoyltransferase, using octanoyl-CoA or octanoyl-Gcv3 as a substrate, but the enzyme lacks the ability to utilize lipoate plus ATP to generate the adenylate intermediate seen with LplA (27). Expression of E. coli LplA in yeast strains lacking Lip2 or Lip5 completely rescues growth in the presence of lipoate indicating that the lack of a true LA salvage pathway in S. cerevisiae is an aspect of the differential activities of LplA and Lip3 (27). Questions as to the true substrate of Lip3 in vivo are still unanswered from these studies. Does Lip3 transfer an octanoyl moiety from Gcv3 to Lat1 and Kgd2, where Lip5 then generates the lipoyl moiety, or does Lip3 act as a lipoyltransferase, transferring a lipoyl moiety from Gcv3? The maintenance of Gcv3 lipoylation in Lip3 mutant strains indicates that octanoyl-Gcv3 is a substrate for Lip5 and that either Lip2 does not transfer an octanoyl moiety from ACP to Lat1 and Kgd2 or that Lip5 cannot act on octanoyl-Lat1/Kgd2. This is particularly interesting because expression of yeast Lip3 in E. coli ΔlipB ΔlplA allows for growth in the presence of octanoate but not lipoate. Furthermore, in octanoate-containing media, E. coli ΔlipB ΔlplA expressing Lip3 has lipoylated PDH and OGDH (27). As well, lipoylation was enhanced in the presence of octanoate to a greater degree than with lipoate and on PDH more so than OGDH (27). These data suggest that in an E. coli system, Lip3 is an octanoyltransferase and that LipA can act on octanoyl-PDH/OGDH, but this has not been demonstrated in the native cellular environment of Lip3.
Although less well-understood in mammalian systems, the LA biosynthetic pathway in mice and humans is carried out by an octanoyltransferase ortholog of LipB/Lip2 and a lipoic acid synthase ortholog of LipA/Lip5 known as LIPT2 and LIAS, respectively (Fig. 1C) (30–32). Deficiencies in either of these enzymes, as well as disruptions in mitochondrial FASII or iron–sulfur biogenesis, result in diminished lipoylation of PDH and OGDH and ultimately impaired mitochondrial function (30, 33, 34). LIPT1, the lipoyltransferase ortholog in mammals, is similar to LIP3 in S. cerevisiae in that it lacks the ability to generate an activated lipoyl-AMP intermediate and therefore is thought to be downstream of LIPT2 (9, 30, 35). There has been a report identifying a mammalian lipoic acid-activating enzyme that could activate exogenous lipoic acid (36); however, this function was ultimately attributed to the mitochondrial medium-chain acyl-CoA synthetase (ACSM1) (37, 38). This enzyme can utilize both the (R)- and (S)-enantiomers of LA and primarily uses GTP to activate the natural (R)-lipoic acid, but so far there has been no substantial evidence to support that this enzyme functions in LA metabolism in vivo (36). This is consistent with the inability for exogenous LA to rescue defects in cells derived from LIAS-deficient patients, embryonic lethality in LIAS-deficient mice, or to ameliorate symptoms in patients with this disease (22, 30, 32). Taken together, this suggests that mammalian LA metabolism is similar to S. cerevisiae where LIPT2 transfers octanoate from ACP to the H-protein of GCS, LIAS inserts sulfur atoms into the octanoyl group on H-protein, and LIPT1 transfers the lipoyl group from the H-protein to E2 subunits. It is unclear at present whether LIPT1 is an octanoyltransferase, a lipoyltransferase, or both, as its true in vivo substrate remains to be clarified.
Lipoic acid as a cofactor for mitochondrial 2-ketoacid dehydrogenases
As discussed above, the function of LA metabolism is to provide an essential, enzyme-bound cofactor for mitochondrial 2-ketoacid dehydrogenases and the GCS. The 2-ketoacid dehydrogenases include pyruvate dehydrogenase (PDH), α-ketoglutarate dehydrogenase (OGDH), branched-chain ketoacid dehydrogenase (BCKDH), and 2-oxoadipate dehydrogenase (OADH) (30, 39). These multienzyme complexes are composed of three independent subunits that undergo coupled reactions facilitated by the LA cofactor (Fig. 2) (40). The E1 subunit, which provides substrate specificity to the multienzyme complex, utilizes the cofactor thiamine pyrophosphate (TPP) to decarboxylate the substrate generating an acyl-TPP intermediate followed by reductive acylation of the lipoyl group on the E2 subunit. The E2 subunit functions as a dihydrolipoyl acyltransferase that transfers the acyl group to CoA, generating an acyl-CoA and dihydrolipoamide. The E3 subunit is a dihydrolipoamide dehydrogenase (DLD) using FAD to oxidize dihydrolipoamide, regenerating the disulfide bond for use in subsequent rounds of catalysis. The E3 subunit oxidizes FADH2 back to FAD using NAD+ producing NADH in the process (40) (Fig. 2A). This activity is similar for PDH, OGDH, and BCKDH, where the E1 and E2 subunits are specific to the decarboxylation substrate and the E3 protein subunit is shared among all three complexes (4). The activity of 2-OADH has not been investigated to the degree of these other complexes, but elevated 2-oxoadipate has been reported in patients with DLD deficiency, suggesting that this complex uses the same DLD subunit shared by other 2-ketoacid dehydrogenases (41).
Figure 2.
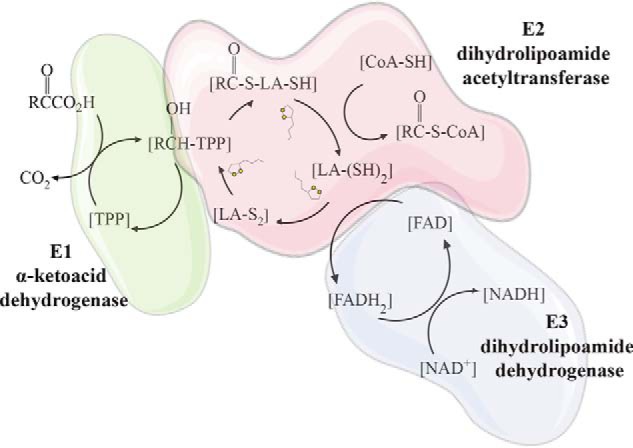
Reaction mechanisms of mitochondrial 2-ketoacid dehydrogenases and redox-dependent regulation. 2-Ketoacid dehydrogenase complexes consist of three enzyme subunits that use coupled reactions to decarboxylate a 2-ketoacid substrate and produce a CoA ester. The E1 subunit is a 2-ketoacid decarboxylase that uses a covalently bound thymine pyrophosphate (TPP) cofactor to decarboxylate the 2-ketoacid substrate followed by reductive acylation of a lipoyl moiety on the E2 subunit. The E2 subunit is a dihydrolipoamide acyltransferase that transfers the acyl intermediate from the E1 subunit to CoA generating an acyl-CoA and dihydrolipoamide. The E3 subunit is a dihydrolipoamide dehydrogenase that uses FAD to oxidize the lipoyl group on the E2 subunit for subsequent rounds of catalysis and generates NADH through coupled oxidation–reduction reactions of FADH2 and NAD+.
Importantly, lipoylated 2-ketoacid dehydrogenases play a key role in carbon entry into the TCA cycle. Therefore, dysfunction in these enzymes may produce aberrant mitochondrial metabolism that can be deleterious (Fig. 3). Inborn errors have been reported in each of these enzyme complexes with mutations having been reported in all of the subunits.
Figure 3.

Mitochondrial 2-ketoacid dehydrogenases and the TCA cycle. Mitochondrial lipoylated enzymes individually contribute to pathways that generate products that can participate in the TCA cycle. Inborn errors in these dehydrogenases can be deleterious, with clinical symptoms including developmental delay (PDH and BCKDH), encephalopathy (OGDH), and microcephaly (2-OADH). Deficiencies in these enzymes can accumulate metabolites, including pyruvate and lactate (PDH), α-ketoglutarate and 2-hydroxyglutarate, branched-chain amino acids and their corresponding 2-ketoacids, and 2-oxoadipic acid. Deficiencies in lipoic acid metabolism can phenocopy multiple simultaneous 2-ketoacid dehydrogenase deficiencies and can limit the incorporation of carbon into the TCA cycle from various sources.
Pyruvate dehydrogenase deficiency is rare, but hundreds of cases have been reported, most involving mutations in the X-linked gene encoding the E1-α1 subunit (PDHA1). Severe mutations are devastating in boys but cause a more variable spectrum of severity in females due to random X-inactivation. Broadly, symptoms of PDH deficiency include lactic acidosis, hypotonia, seizures, ataxia, and developmental delay. Treatment for PDH deficiency is limited; the use of a ketogenic diet and in some cases treatment with dichloroacetate or thiamine can provide improvement of clinical symptoms (42).
α-Ketoglutarate dehydrogenase deficiency has been reported less frequently than PDH deficiency, but the primary clinical manifestations are similar, including developmental delay, ataxia, hypotonia, and in some cases encephalopathy (42). The elevation of α-ketoglutarate levels may also be associated with an elevation in 2-hydroxyglutarate, a potential epigenetic regulator. It is currently unknown whether 2-hydroxyglutarate levels contribute to pathology in α-ketoglutarate dehydrogenase deficiency (43, 44).
Deficiency in the E1 or E2 subunit of branched-chain ketoacid dehydrogenase is also known as maple syrup urine disease. This disorder includes a classic presentation of a sweet, maple-syrup like odor and an elevation of plasma branched-chain amino acids and 2-ketoacids in urine. In the severe neonatal-onset form, patients display metabolic decompensation and neurological distress, which may be severe and result in neonatal coma. There are also acute and intermittent forms that may be late-onset and consist of recurrent metabolic decompensation episodes; as well, the chronic and progressive form presents with hypotonia and developmental delay. Treatment of maple syrup urine disease involves titrating dietary branched-chain amino acid content to avoid excessive exposure while maintaining sufficient levels to support normal development (42).
2-Oxoadipic aciduria has only been reported in about 20 individuals with half of them being asymptomatic. Those individuals that do show symptoms may display psychomotor retardation and hypotonia. Mutations in dehydrogenase E1 and transketolase domain containing 1 (DHTKD1) are thought to be causative in these cases (42).
Mutations have also been reported in DLD, LIAS, LIPT2, and LIPT1. These disorders have overlapping phenotypes, including lactic acidosis, developmental delay, and seizures. Collectively, dysfunction in these enzymes falls into a larger class of mitochondrial disease known as Leigh syndrome. LIAS patients may be distinguished from LIPT1 patients through presentation of nonketotic hyperglycinemia associated with dysfunction in the GCS. An excellent review of the clinical manifestations of these disorders was recently described by Mayr et al. (30). Critical to developing treatments for these disorders is intimate knowledge of the regulation of 2-ketoacid dehydrogenase complexes and how their activities may differ in various nutritional states and in individual tissues.
Lipoic acid and ROS generation from mitochondrial 2-ketoacid dehydrogenases
Dihydrolipoamide dehydrogenase
The ability of the shared E3 subunit to regenerate oxidized lipoic acid for further catalysis in 2-ketoacid dehydrogenase complexes is controlled by the NAD+/NADH ratio within the mitochondria (45). When the availability of NAD+ is diminished, FADH2 within the E3 subunit can readily be oxidized by O2 generating a semiquinone (FADH•) and superoxide (O2˙̄) (46, 47). The semiquinone may then equilibrate with the reduced lipoyl residue generating a thioyl radical within the core complex. This can lead to inactivation of the complex due to reactions between a thioyl radical on the E2 subunit and the catalytic region of the E1 subunit (48). This can been seen as a regulatory inactivation to inhibit the catabolism of specific substrates, like pyruvate or α-ketoglutarate when NAD+ is depleted or when ROS production alters the redox balance of the mitochondria (49). It is unclear the degree to which this occurs on each 2-ketoacid complex individually. There is evidence that the E3 subunit may be involved in redox regulation independent of its activities within the 2-ketoacid dehydrogenase complexes (45). Specifically, the flavin can participate in regenerating thioredoxin 2 (Trx2) (50, 51) and has a structure similar to glutathione reductase (52), but it is unclear whether these functions require the entire 2-ketoacid dehydrogenase complex or whether the E3 subunit dissociates from the complex for this function.
α-Ketoglutarate dehydrogenase
The OGDH complex facilitates the decarboxylation of α-ketoglutarate (2-oxoglutarate) to form succinyl-CoA and NADH. This complex in mammalian systems is distinct in that the E2 core subunit (dihydrolipoyl succinyltransferase) lacks the E1- and E3-binding domains in the prokaryotic protein, suggesting that the overall structure of this complex is organized differently in mammals (53). Multiple studies have demonstrated that the OGDH complex can produce superoxide and H2O2, and many of these studies focus on the activity of the E3 subunit of the complex (46, 48, 54). Experiments in skeletal muscle mitochondria show that OGDH is responsible for a significant portion of mitochondrial superoxide production that is related to the concentrations of α-ketoglutarate and free CoA (55). Additionally, the lipoyl moiety has been shown to be glutathionylated in vivo, which is thought to be a mechanism to reversibly inactivate the complex, preventing ROS-dependent inactivation of the E1 subunit (56, 57). It is not known whether this glutathionylation is enzyme-mediated or occurs spontaneously, but the glutathione modification can be removed through the actions of glutathione reductase 2 (Grx2) (58). Although glutathionylation of the lipoyl moiety prevents oxidative damage to the E1 component, it also maintains a reduced flavin on the E3 subunit, and this may contribute to oxidative damage by E3-mediated ROS production (58). Thioredoxin 2 has been shown to protect OGDH from oxidative damage (49–51), and it has been suggested that glutathionylation of the lipoyl moiety provides the opportunity for the E3 subunit to participate in redox regulation through ROS scavenging or regeneration of thioredoxin (51). Finally, OGDH has been shown to interact with complex I of the electron transport chain, which allows for direct supply of NADH to the NADH-oxidation site of complex I (Fig. 4A) (59–61). Because glutathionylation of complex I is also regulated by Grx2 (62, 63), it has been suggested that glutathionylation may dissociate the interaction between OGDH and complex I to limit further electron entry into the electron transport chain until redox homeostasis is reached (Fig. 4B) (64). This is particularly important because OGDH activity regulates the rate of NADH production of downstream enzymes within the TCA cycle. Collectively, these findings indicate that OGDH is a key source of ROS in the mitochondria, but it may also play a key role in redox balance and regulation through multiple mechanisms.
Figure 4.
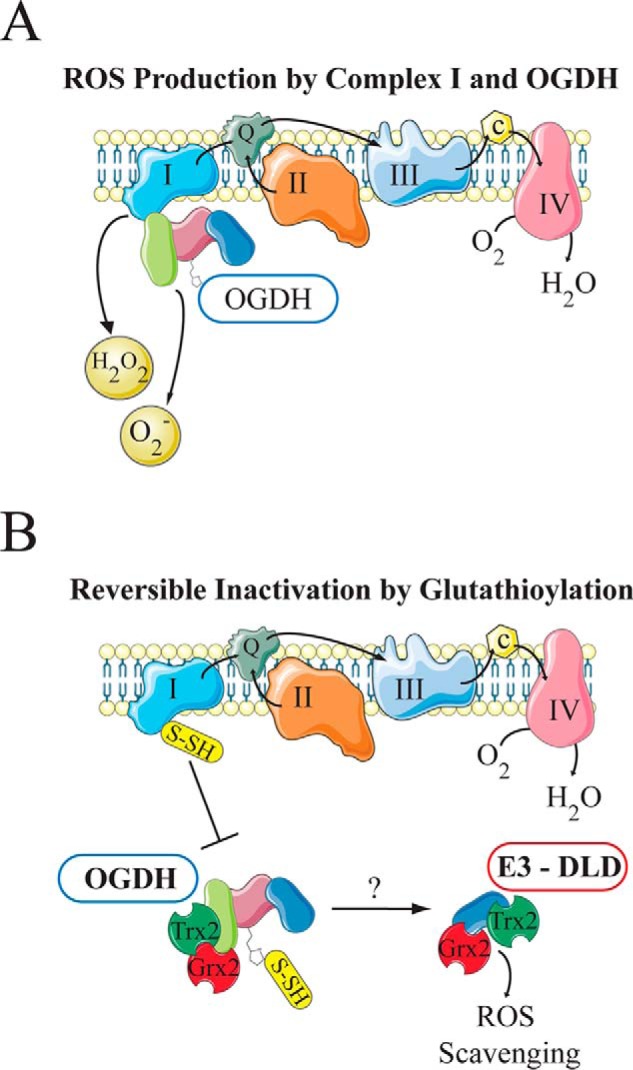
Regulation of OGDH by reversible glutathionylation. A, 2-oxoglutarate dehydrogenase (OGDH) interacts with complex I of the mitochondrial electron transport chain, and both complexes can generate ROS. B, when ROS levels increase, both complexes are glutathionylated, which is thought to dissociate the interaction between the two complexes and reversibly inactivate OGDH. The OGDH complex is protected from oxidative damage by thioredoxin (Trx2), and the glutathionylation is regulated by glutathione reductase (Grx2). Glutathionylation of the lipoyl moiety on the E2 subunit of OGDH may allow for ROS scavenging by the E3 subunit through interactions with Grx2 and Trx2.
Pyruvate dehydrogenase complex
The pyruvate dehydrogenase complex has been extensively studied as a primary regulatory node for nutrient oxidation in various metabolic states. This enzyme complex irreversibly decarboxylates pyruvate to generate acetyl-CoA and CO2 and is coordinated by three phosphorylation sites on the E1 subunit regulated by a set of kinases (PDKs) and phosphatases (PDP), which interact with lipoyl domains on the E2 subunit and E3-binding protein (65). The PDH complex structure is somewhat different from OGDH in that it includes an E3-binding protein that also contains a lipoyl domain. As well, the PDH complex has stable interactions with PDK and PDP (66, 67). The E3-binding protein lipoyl domain does undergo lipoylation; however, this lipoylation is not thought to be involved in catalysis but undergoes reductive acetylation as a regulatory mechanism for PDK and PDP (66, 68, 69). As with OGDH, PDH produces superoxide and H2O2, although there is contention around the relative contributions of the E3 and E1 subunits in vivo (55, 70). PDH has also been shown to be glutathionylated on the E2 subunit, similar to OGDH S-glutathionylation (70). This glutathionylation can increase ROS generation from the E3 subunit because FADH2 cannot be oxidized by the lipoic acid moiety and reduces O2 by one electron to produce superoxide. Importantly, glutathionylation decreases ROS production when pyruvate is being oxidized, and depletion of glutathione leads to increased ROS production from PDH (71). PDH has also been shown to interact with Grx2 suggesting that the reversible glutathionylation regulates PDH through similar mechanisms to OGDH (70). This common mechanism appears to play a critical regulatory role for mitochondrial metabolism under different nutritional states and likely in various tissues. Finally, recent studies have suggested that Sirtuin 4 (SIRT4) can regulate PDH activity through lipoamidase activity that cleaves the lipoyl moiety from the E2 component (72). This study did not investigate SIRT4 regulation of the other mitochondrial 2-ketoacid dehydrogenases, but these complexes did associate with SIRT4 in their immunoaffinity purification experiment. Subsequent studies demonstrated that this mechanism was conserved in prokaryotic systems and that OGDH and the glycine cleavage system were also targets of the SIRT4 ortholog, CobB (73). Another study also implicated SIRT4 in regulation of leucine catabolism indicating that BCKDH is likely an additional target of this regulation (74).
Branched-chain ketoacid dehydrogenase
The branched-chain ketoacid dehydrogenase complex is downstream of the branched-chain aminotransferase enzyme, which catalyzes the breakdown of valine, leucine, and isoleucine to the ketoacids, 3-methyl-2-oxobytanoate (KIV), 4-methyl-2-oxopentanoate (KIC), and (S)-3-methyl-2-oxopentanoate (KMV), respectively (Fig. 2B) (75). BCKDH then decarboxylates these ketoacids releasing CO2 and generating an acyl-CoA. This complex has a similar structure and regulation to PDH with stably associated kinase and phosphatases (76, 77). Structural analysis of BCKDH demonstrated that the phosphorylation loop of the E1 subunit undergoes a disordered-to-ordered conformational change that regulates binding of the lipoyl group on the E2 subunit near the catalytic site of the E1 subunit. In the dephosphorylated (active) state, the lipoyl group is positioned near the active site of the E1 subunit to facilitate reductive acylation immediately after the decarboxylation reaction. During the phosphorylated (inactive) state, the ordered conformation of the phosphorylation loop prevents lipoyl domain positioning near the E1 active site (78). Subsequent studies demonstrated a similar role for the lipoyl domain in the E2 subunit that involves a substrate gating mechanism. The E2 subunit undergoes a conformational change upon CoA binding, which allows for the lipoyl-acyl intermediate to enter the acyltransferase catalytic site (79). This mechanism prevents the reduced lipoamide from entering the E2 catalytic site when free CoA levels are reduced. BCKDH has been shown to produce superoxide and H2O2 to a lesser degree than OGDH and PDH (55). The overall details of redox regulation of BCKDH are less clear compared with the other 2-ketoacid dehydrogenases with most of the literature focused on the E3 subunit that is shared by the other complexes.
2-Oxoadipate dehydrogenase
Catabolism of tryptophan and lysine leads to the production of 2-oxoadipate (2-OA), which is converted to glutaryl-CoA by 2-oxoadipate dehydrogenase (OADH) (80). Glutaryl-CoA has recently been shown to post-translationally modify lysine residues on mitochondrial proteins. Thus, dysfunction in this enzyme could impact mitochondrial function through mechanisms independent of 2-OA per se (81). Mutations in dehydrogenase E1 and transketolase domain containing 1 (DHTKD1) lead to accumulation of 2-OA; however, reports have indicated that OGDH can also decarboxylate 2-OA, which is just slightly larger than its conventional substrate, α-ketoglutarate. It is unclear whether DHTKD1 is associated with an independent dehydrogenase complex or whether it shares activity with OGDH and which enzyme deficiency may be causative in 2-oxoadipate aciduria. Recent studies have indicated that ROS production can be attributed to the DHTKD1 enzyme (82). Paradoxically, ROS production seems to increase when the enzyme is either suppressed or overexpressed, although the mechanisms regulating this phenomenon are not understood (83). Expression data indicate that DHTKD1 is highly expressed in liver and kidney with comparable protein levels observed in human skeletal muscle (82), indicating that these tissues would be the appropriate systems to investigate these open questions.
Concluding remarks
Lipoic acid is an often overlooked, essential cofactor for mitochondrial oxidative metabolism, which participates in catalysis and regulation of multiple enzyme complexes. Biosynthesis of LA links mitochondrial fatty-acid synthesis, SAM, and iron–sulfur cluster biosynthesis with TCA cycle oxidative capacity. This information is particularly important for diagnosis of inborn errors of metabolism because defects in lipoic acid metabolism can promote a myriad of clinical symptoms associated with dysfunction in a number of enzyme complexes (22, 30, 31, 84). Additionally, because lipoic acid plays a regulatory role, symptoms of some inborn errors may only manifest, or may worsen, in nutritional states associated with high flux through enzymes requiring lipoylation. The lack of an independent salvage pathway in humans abrogates the use of LA supplementation as a therapeutic option; thus, further investigation of how LA metabolism is regulated and functions in humans will be necessary to treat inborn errors of this pathway.
This work was supported by National Institutes of Health Grant R35 CA220449 (to R. J. D.), the Howard Hughes Medical Institute (Faculty Scholars Program), and by a gift from the Once Upon a Time Foundation. This is the fifth article in the Thematic Minireview Series “Redox metabolism and signaling.” The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LA
- lipoic acid
- FASII
- fatty-acid synthesis type II
- ACP
- acyl carrier protein
- GCS
- glycine cleavage system
- PDH
- pyruvate dehydrogenase
- OGDH
- α-ketoglutarate dehydrogenase
- BCKDH
- branched-chain ketoacid dehydrogenase
- SAM
- S-adenosylmethionine
- TCA
- tricarboxylic acid
- TPP
- thiamine pyrophosphate
- DLD
- dihydrolipoamide dehydrogenase
- PDK
- pyruvate dehydrogenase kinase
- PDP
- pyruvate dehydrogenase phosphatase
- 2-OADH
- 2-oxoadipate dehydrogenase
- 2-OA
- 2-oxoadipate
- ROS
- reactive oxygen species
- LIAS
- lipoic acid synthase.
References
- 1. Reed L. J. (2006) in Advances in Enzymology and Related Areas of Molecular Biology, pp. 319–347, John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 2. Patterson E. L., Brockman J. A. Jr., Day F. P., Pierce J. V., Macchi M. E., Hoffman C. E., Fong C. T., Stokstad E. L., and Jukes T. H. (1951) Crystallization of a derivative of protogen-B. J. Am. Chem. Soc. 73, 5919–5920 10.1021/ja01156a566 [DOI] [Google Scholar]
- 3. Reed L. J., DeBusk B. G., Gunsalus I. C., and Hornberger C. S. Jr. (1951) Crystalline α-lipoic acid; a catalytic agent associated with pyruvate dehydrogenase. Science 114, 93–94 10.1126/science.114.2952.93 [DOI] [PubMed] [Google Scholar]
- 4. Cronan J. E. (2016) Assembly of lipoic acid on its cognate enzymes: an extraordinary and essential biosynthetic pathway. Microbiol. Mol. Biol. Rev. 80, 429–450 10.1128/MMBR.00073-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cronan J. E. (2014) Biotin and lipoic acid: synthesis, attachment, and regulation. EcoSal Plus. 6, 10.1128/ecosalplus.ESP-0001-2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schonauer M. S., Kastaniotis A. J., Kursu V. A., Hiltunen J. K., and Dieckmann C. L. (2009) Lipoic acid synthesis and attachment in yeast mitochondria. J. Biol. Chem. 284, 23234–23242 10.1074/jbc.M109.015594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ewald R., Hoffmann C., Florian A., Neuhaus E., Fernie A. R., and Bauwe H. (2014) Lipoate-protein ligase and octanoyltransferase are essential for protein lipoylation in mitochondria of Arabidopsis. Plant Physiol. 165, 978–990 10.1104/pp.114.238311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kang S. G., Jeong H. K., Lee E., and Natarajan S. (2007) Characterization of a lipoate-protein ligase A gene of rice (Oryza sativa L.). Gene 393, 53–61 10.1016/j.gene.2007.01.011 [DOI] [PubMed] [Google Scholar]
- 9. Fujiwara K., Okamura-Ikeda K., and Motokawa Y. (1996) Lipoylation of acyltransferase components of α-ketoacid dehydrogenase complexes. J. Biol. Chem. 271, 12932–12936 10.1074/jbc.271.22.12932 [DOI] [PubMed] [Google Scholar]
- 10. Jordan S. W., and Cronan J. E. Jr. (1997) A new metabolic link. J. Biol. Chem. 272, 17903–17906 10.1074/jbc.272.29.17903 [DOI] [PubMed] [Google Scholar]
- 11. Jordan S. W., and Cronan J. E. (2003) The Escherichia coli lipB gene encodes lipoyl (octanoyl)-acyl carrier protein:protein transferase. J. Bacteriol. 185, 1582–1589 10.1128/JB.185.5.1582-1589.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jordan S. W., and Cronan J. E. (1997) Biosynthesis of lipoic acid and posttranslational modification with lipoic acid in Escherichia coli. Methods Enzymol. 279, 176–183 10.1016/S0076-6879(97)79021-9 [DOI] [PubMed] [Google Scholar]
- 13. Parry R. J. (1977) Biosynthesis of lipoic acid. 1. Incorporation of specifically tritiated octanoic acid into lipoic acid. J. Am. Chem. Soc. 99, 6464–6466 10.1021/ja00461a061 [DOI] [Google Scholar]
- 14. White R. H. (1980) Stable isotope studies on the biosynthesis of lipoic acid in Escherichia coli. Biochemistry 19, 15–19 [DOI] [PubMed] [Google Scholar]
- 15. Reed K. E., and Cronan J. E. (1993) Lipoic acid metabolism in Escherichia coli: sequencing and functional characterization of the lipA and lipB genes. J. Bacteriol. 175, 1325–1336 10.1128/jb.175.5.1325-1336.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parry R. J., and Trainor D. A. (1978) Biosynthesis of lipoic acid. 2. Stereochemistry of sulfur introduction at C-6 of octanoic acid. J. Am. Chem. Soc. 100, 5243–5244 10.1021/ja00484a073 [DOI] [Google Scholar]
- 17. Miller J. R., Busby R. W., Jordan S. W., Cheek J., Henshaw T. F., Ashley G. W., Broderick J. B., Cronan J. E. Jr., and Marletta M. A. (2000) Escherichia coli LipA is a lipoyl synthase: in vitro biosynthesis of lipoylated pyruvate dehydrogenase complex from octanoyl-acyl carrier protein. Biochemistry 39, 15166–15178 10.1021/bi002060n [DOI] [PubMed] [Google Scholar]
- 18. Wang S. C., and Frey P. A. (2007) S-Adenosylmethionine as an oxidant: the radical SAM superfamily. Trends Biochem. Sci. 32, 101–110 10.1016/j.tibs.2007.01.002 [DOI] [PubMed] [Google Scholar]
- 19. Vanden Boom T. J., Reed K. E., and Cronan J. E. (1991) Lipoic acid metabolism in Escherichia coli: isolation of null mutants defective in lipoic acid biosynthesis, molecular cloning and characterization of the E. coli lip locus, and identification of the lipoylated protein of the glycine cleavage system. J. Bacteriol. 173, 6411–6420 10.1128/jb.173.20.6411-6420.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kriek M., Peters L., Takahashi Y., and Roach P. L. (2003) Effect of iron–sulfur cluster assembly proteins on the expression of Escherichia coli lipoic acid synthase. Protein Expr. Purif. 28, 241–245 10.1016/S1046-5928(02)00680-0 [DOI] [PubMed] [Google Scholar]
- 21. McCarthy E. L., and Booker S. J. (2017) Destruction and reformation of an iron–sulfur cluster during catalysis by lipoyl synthase. Science 358, 373–377 10.1126/science.aan4574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baker P. R. 2nd., Friederich M. W., Swanson M. A., Shaikh T., Bhattacharya K., Scharer G. H., Aicher J., Creadon-Swindell G., Geiger E., MacLean K. N., Lee W.-T., Deshpande C., Freckmann M.-L., Shih L.-Y., Wasserstein M., et al. (2014) Variant nonketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 137, 366–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morris T. W., Reed K. E., and Cronan J. E. (1994) Identification of the gene encoding lipoate-protein ligase A of Escherichia coli. Molecular cloning and characterization of the lplA gene and gene product. J. Biol. Chem. 269, 16091–16100 [PubMed] [Google Scholar]
- 24. Green D. E., Morris T. W., Green J., Cronan J. E. Jr., and Guest J. R. (1995) Purification and properties of the lipoate protein ligase of Escherichia coli. Biochem. J. 309, 853–862 10.1042/bj3090853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reed L. J., Leach F. R., and Koike M. (1958) Studies on a lipoic acid-activating system. J. Biol. Chem. 232, 123–142 [PubMed] [Google Scholar]
- 26. Morris T. W., Reed K. E., and Cronan J. E. (1995) Lipoic acid metabolism in Escherichia coli: the lplA and lipB genes define redundant pathways for ligation of lipoyl groups to apoprotein. J. Bacteriol. 177, 1–10 10.1128/jb.177.1.1-10.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hermes F. A., and Cronan J. E. (2013) The role of the Saccharomyces cerevisiae lipoate protein ligase homologue, Lip3, in lipoic acid synthesis. Yeast 30, 415–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sulo P., and Martin N. C. (1993) Isolation and characterization of LIP5. A lipoate biosynthetic locus of Saccharomyces cerevisiae. J. Biol. Chem. 268, 17634–17639 [PubMed] [Google Scholar]
- 29. Marvin M. E., Williams P. H., and Cashmore A. M. (2001) The isolation and characterisation of a Saccharomyces cerevisiae gene (LIP2) involved in the attachment of lipoic acid groups to mitochondrial enzymes. FEMS Microbiol. Lett. 199, 131–136 10.1111/j.1574-6968.2001.tb10663.x [DOI] [PubMed] [Google Scholar]
- 30. Mayr J. A., Feichtinger R. G., Tort F., Ribes A., and Sperl W. (2014) Lipoic acid biosynthesis defects. J. Inherit. Metab. Dis. 37, 553–563 10.1007/s10545-014-9705-8 [DOI] [PubMed] [Google Scholar]
- 31. Habarou F., Hamel Y., Haack T. B., Feichtinger R. G., Lebigot E., Marquardt I., Busiah K., Laroche C., Madrange M., Grisel C., Pontoizeau C., Eisermann M., Boutron A., Chrétien D., Chadefaux-Vekemans B., et al. (2017) Biallelic mutations in LIPT2 cause a mitochondrial lipoylation defect associated with severe neonatal encephalopathy. Am. J. Hum. Genet. 101, 283–290 10.1016/j.ajhg.2017.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yi X., and Maeda N. (2005) Endogenous production of lipoic acid is essential for mouse development. Mol. Cell. Biol. 25, 8387–8392 10.1128/MCB.25.18.8387-8392.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith S., Witkowski A., Moghul A., Yoshinaga Y., Nefedov M., de Jong P., Feng D., Fong L., Tu Y., Hu Y., Young S. G., Pham T., Cheung C., Katzman S. M., Brand M. D., et al. (2012) Compromised mitochondrial fatty-acid synthesis in transgenic mice results in defective protein lipoylation and energy disequilibrium. PLoS ONE 7, e47196–15 10.1371/journal.pone.0047196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heimer G., Kerätär J. M., Riley L. G., Balasubramaniam S., Eyal E., Pietikäinen L. P., Hiltunen J. K., Marek-Yagel D., Hamada J., Gregory A., Rogers C., Hogarth P., Nance M. A., Shalva N., Veber A., et al. (2016) MECR mutations cause childhood-onset dystonia and optic atrophy, a mitochondrial fatty-acid synthesis disorder. Am. J. Hum. Genet. 99, 1229–1244 10.1016/j.ajhg.2016.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fujiwara K., Okamura-Ikeda K., and Motokawa Y. (1994) Purification and characterization of lipoyl-AMP:N ϵ-lysine lipoyltransferase from bovine liver mitochondria. J. Biol. Chem. 269, 16605–16609 [PubMed] [Google Scholar]
- 36. Fujiwara K., Takeuchi S., Okamura-Ikeda K., and Motokawa Y. (2001) Purification, characterization, and cDNA cloning of lipoate-activating enzyme from bovine liver. J. Biol. Chem. 276, 28819–28823 10.1074/jbc.M101748200 [DOI] [PubMed] [Google Scholar]
- 37. Fujino T., Takei Y. A., Sone H., Ioka R. X., Kamataki A., Magoori K., Takahashi S., Sakai J., and Yamamoto T. T. (2001) Molecular identification and characterization of two medium-chain acyl-CoA synthetases, MACS1 and the SaGene product. J. Biol. Chem. 276, 35961–35966 10.1074/jbc.M106651200 [DOI] [PubMed] [Google Scholar]
- 38. Vessey D. A., Lau E., Kelley M., and Warren R. S. (2003) Isolation, sequencing, and expression of a cDNA for the HXM-A form of xenobiotic/medium-chain fatty acid:CoA ligase from human liver mitochondria. J. Biochem. Mol. Toxicol. 17, 1–6 10.1002/jbt.10056 [DOI] [PubMed] [Google Scholar]
- 39. Reed L. J. (2001) A trail of research from lipoic acid to α-keto acid dehydrogenase complexes. J. Biol. Chem. 276, 38329–38336 10.1074/jbc.R100026200 [DOI] [PubMed] [Google Scholar]
- 40. Reed L. J. (1974) Multienzyme complexes. Acc. Chem. Res. 1974, 7, 40–46 10.1021/ar50074a002 [DOI] [Google Scholar]
- 41. Shaag A., Saada A., Berger I., Mandel H., Joseph A., Feigenbaum A., and Elpeleg O. N. (1999) Molecular basis of lipoamide dehydrogenase deficiency in Ashkenazi Jews. Am. J. Med. Genet. 82, 177–182 10.1002/(SICI)1096-8628(19990115)82:2%3C177::AID-AJMG15%3E3.0.CO%3B2-9 [DOI] [PubMed] [Google Scholar]
- 42. Saudubray J.-M., Baumgartner M. R., and Walter J. (eds) (2016) Inborn Metabolic Diseases, 4th Ed., Chapter 12, pp. 169–170, Springer-Verlag, Berlin, Heidelberg, Germany: 10.1007/978-3-662-49771-5_14 [DOI] [Google Scholar]
- 43. Intlekofer A. M., Dematteo R. G., Venneti S., Finley L. W., Lu C., Judkins A. R., Rustenburg A. S., Grinaway P. B., Chodera J. D., Cross J. R., and Thompson C. B. (2015) Hypoxia induces production of l-2-hydroxyglutarate. Cell Metab. 22, 304–311 10.1016/j.cmet.2015.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burr S. P., Costa A. S., Grice G. L., Timms R. T., Lobb I. T., Freisinger P., Dodd R. B., Dougan G., Lehner P. J., Frezza C., and Nathan J. A. (2016) Mitochondrial protein lipoylation and the 2-oxoglutarate dehydrogenase complex controls HIF1? Stability in aerobic conditions. Cell Metab. 24, 740–752 10.1016/j.cmet.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ambrus A., and Adam-Vizi V. (2017) Human dihydrolipoamide dehydrogenase (E3) deficiency: novel insights into the structural basis and molecular pathomechanism. Neurochem. Int. 2017, S0197-0186(17)30118-3 10.1016/j.neuint.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 46. Starkov A. A., Fiskum G., Chinopoulos C., Lorenzo B. J., Browne S. E., Patel M. S., and Beal M. F. (2004) Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 24, 7779–7788 10.1523/JNEUROSCI.1899-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bunik V. I., and Pavlova O. G. (1993) Inactivation of α-ketoglutarate dehydrogenase during oxidative decarboxylation of α-ketoadipic acid. FEBS Lett. 323, 166–170 [DOI] [PubMed] [Google Scholar]
- 48. Bunik V. I., and Sievers C. (2002) Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur. J. Biochem. 269, 5004–5015 10.1046/j.1432-1033.2002.03204.x [DOI] [PubMed] [Google Scholar]
- 49. Bunik V. I. (2003) 2-Oxo acid dehydrogenase complexes in redox regulation. Eur. J. Biochem. 270, 1036–1042 10.1046/j.1432-1033.2003.03470.x [DOI] [PubMed] [Google Scholar]
- 50. Bunik V., Follmann H., and Bisswanger H. (1997) Activation of mitochondrial 2-oxoacid dehydrogenases by thioredoxin. Biol. Chem. 378, 1125–1130 [DOI] [PubMed] [Google Scholar]
- 51. Bunik V., and Follmann H. (1993) Thioredoxin reduction dependent on α-ketoacid oxidation by α-ketoacid dehydrogenase complexes. FEBS Lett. 336, 197–200 [DOI] [PubMed] [Google Scholar]
- 52. Williams C. H. Jr., Arscott L. D., and Schulz G. E. (1982) Amino acid sequence homology between pig heart lipoamide dehydrogenase and human erythrocyte glutathione reductase. Proc. Natl. Acad. Sci. U.S.A. 79, 2199–2201 10.1073/pnas.79.7.2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakano K., Takase C., Sakamoto T., Nakagawa S., Inazawa J., Ohta S., and Matuda S. (1994) Isolation, characterization and structural organization of the gene and pseudogene for the dihydrolipoamide succinyltransferase component of the human 2-oxoglutarate dehydrogenase complex. Eur. J. Biochem. 224, 179–189 10.1111/j.1432-1033.1994.tb20010.x [DOI] [PubMed] [Google Scholar]
- 54. Ambrus A., Tretter L., and Adam-Vizi V. (2010) Inhibition of the α-ketoglutarate dehydrogenase-mediated reactive oxygen species generation by lipoic acid. Biochim. Biophys. Acta 1797, 57 10.1016/j.bbabio.2010.04.187 [DOI] [PubMed] [Google Scholar]
- 55. Quinlan C. L., Goncalves R. L., Hey-Mogensen M., Yadava N., Bunik V. I., and Brand M. D. (2014) The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 289, 8312–8325 10.1074/jbc.M113.545301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Applegate M. A., Humphries K. M., and Szweda L. I. (2008) Reversible inhibition of α-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry 47, 473–478 10.1021/bi7017464 [DOI] [PubMed] [Google Scholar]
- 57. Nulton-Persson A., Szweda L., and Humphries K. (2009) in Lipoic Acid, Oxidative Modification of Lipoic Acid (Packer L. and Patel M. S., eds) CRC Press, Inc., Boca Raton, FL: 10.1201/9781420045390.ch8 [DOI] [Google Scholar]
- 58. Mailloux R. J., Craig Ayre D., and Christian S. L. (2016) Induction of mitochondrial reactive oxygen species production by GSH mediated S-glutathionylation of 2-oxoglutarate dehydrogenase. Redox Biol. 8, 285–297 10.1016/j.redox.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fukushima T., Decker R. V., Anderson W. M., and Spivey H. O. (1989) Substrate channeling of NADH and binding of dehydrogenases to complex I. J. Biol. Chem. 264, 16483–16488 [PubMed] [Google Scholar]
- 60. Sumegi B., and Srere P. A. (1984) Complex I binds several mitochondrial NAD-coupled dehydrogenases. J. Biol. Chem. 259, 15040–15045 [PubMed] [Google Scholar]
- 61. Porpaczy Z., Sumegi B., and Alkonyi I. (1987) Interaction between NAD-dependent isocitrate dehydrogenase, α-ketoglutarate dehydrogenase complex, and NADH:ubiquinone oxidoreductase. J. Biol. Chem. 262, 9509–9514 [PubMed] [Google Scholar]
- 62. Wu H., Xing K., and Lou M. F. (2010) Glutaredoxin 2 prevents H2O2-induced cell apoptosis by protecting complex I activity in the mitochondria. Biochim. Biophys. Acta 1797, 1705–1715 10.1016/j.bbabio.2010.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Beer S. M., Taylor E. R., Brown S. E., Dahm C. C., Costa N. J., Runswick M. J., and Murphy M. P. (2004) Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins. J. Biol. Chem. 279, 47939–47951 10.1074/jbc.M408011200 [DOI] [PubMed] [Google Scholar]
- 64. McLain A. L., Cormier P. J., Kinter M., and Szweda L. I. (2013) Glutathionylation of α-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic. Biol. Med. 61, 161–169 10.1016/j.freeradbiomed.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wieland O. H. (1983) The mammalian pyruvate dehydrogenase complex: structure and regulation. Rev. Physiol. Biochem. Pharmacol. 96, 123–170 10.1007/BFb0031008 [DOI] [PubMed] [Google Scholar]
- 66. Powers-Greenwood S. L., Rahmatullah M., Radke G. A., and Roche T. E. (1989) Separation of protein X from the dihydrolipoyl transacetylase component of the mammalian pyruvate dehydrogenase complex and function of protein X. J. Biol. Chem. 264, 3655–3657 [PubMed] [Google Scholar]
- 67. Gopalakrishnan S., Rahmatullah M., Radke G. A., Powers-Greenwood S., and Roche T. E. (1989) Role of protein X in the function of the mammalian pyruvate dehydrogenase complex. Biochem. Biophys. Res. Commun. 160, 715–721 10.1016/0006-291X(89)92492-3 [DOI] [PubMed] [Google Scholar]
- 68. Rahmatullah M., and Roche T. E. (1987) The catalytic requirements for reduction and acetylation of protein X and the related regulation of various forms of resolved pyruvate dehydrogenase kinase. J. Biol. Chem. 262, 10265–10271 [PubMed] [Google Scholar]
- 69. Neagle J., De Marcucci O., Dunbar B., and Lindsay J. G. (1989) Component X of mammalian pyruvate dehydrogenase complex: Structural and functional relationship to the lipoate acetyltransferase (E2) component. FEBS Lett. 253, 11–15 [DOI] [PubMed] [Google Scholar]
- 70. O'Brien M., Chalker J., Slade L., Gardiner D., and Mailloux R. J. (2017) Protein S-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radic. Biol. Med. 106, 302–314 10.1016/j.freeradbiomed.2017.02.046 [DOI] [PubMed] [Google Scholar]
- 71. Fisher-Wellman K. H., Gilliam L. A., Lin C.-T., Cathey B. L., Lark D. S., and Neufer P. D. (2013) Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload. Free Radic. Biol. Med. 65, 1201–1208 10.1016/j.freeradbiomed.2013.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mathias R. A., Greco T. M., Oberstein A., Budayeva H. G., Chakrabarti R., Rowland E. A., Kang Y., Shenk T., and Cristea I. M. (2014) Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625 10.1016/j.cell.2014.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rowland E. A., Greco T. M., Snowden C. K., McCabe A. L., Silhavy T. J., and Cristea I. M. (2017) Sirtuin lipoamidase activity is conserved in bacteria as a regulator of metabolic enzyme complexes. MBio 8, e01096–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Anderson K. A., Huynh F. K., Fisher-Wellman K., Stuart J. D., Peterson B. S., Douros J. D., Wagner G. R., Thompson J. W., Madsen A. S., Green M. F., Sivley R. M., Ilkayeva O. R., Stevens R. D., Backos D. S., Capra J. A., et al. (2017) SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 25, 838–855 10.1016/j.cmet.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Faure M., Glomot F., Bledsoe R., Hutson S., and Papet I. (1999) Purification and cloning of the mitochondrial branched-chain amino acid aminotransferase from sheep placenta. Eur. J. Biochem. 259, 104–111 10.1046/j.1432-1327.1999.00009.x [DOI] [PubMed] [Google Scholar]
- 76. Wynn R. M., Kato M., Machius M., Chuang J. L., Li J., Tomchick D. R., and Chuang D. T. (2004) Molecular mechanism for regulation of the human mitochondrial branched-chain α-ketoacid dehydrogenase complex by phosphorylation. Structure 12, 2185–2196 10.1016/j.str.2004.09.013 [DOI] [PubMed] [Google Scholar]
- 77. Chang C.-F., Chou H.-T., Chuang J. L., Chuang D. T., and Huang T.-H. (2002) Solution structure and dynamics of the lipoic acid-bearing domain of human mitochondrial branched-chain α-keto acid dehydrogenase complex. J. Biol. Chem. 277, 15865–15873 10.1074/jbc.M110952200 [DOI] [PubMed] [Google Scholar]
- 78. Machius M., Wynn R. M., Chuang J. L., Li J., Kluger R., Yu D., Tomchick D. R., Brautigam C. A., and Chuang D. T. (2006) A versatile conformational switch regulates reactivity in human branched-chain α-ketoacid dehydrogenase. Structure 14, 287–298 10.1016/j.str.2005.10.009 [DOI] [PubMed] [Google Scholar]
- 79. Kato M., Wynn R. M., Chuang J. L., Brautigam C. A., Custorio M., and Chuang D. T. (2006) A synchronized substrate-gating mechanism revealed by cubic-core structure of the bovine branched-chain α-ketoacid dehydrogenase complex. EMBO J. 25, 5983–5994 10.1038/sj.emboj.7601444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Danhauser K., Sauer S. W., Haack T. B., Wieland T., Staufner C., Graf E., Zschocke J., Strom T. M., Traub T., Okun J. G., Meitinger T., Hoffmann G. F., Prokisch H., and Kölker S. (2012) DHTKD1 mutations cause 2-aminoadipic and 2-oxoadipic aciduria. Am. J. Hum. Genet. 91, 1082–1087 10.1016/j.ajhg.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tan M., Peng C., Anderson K. A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G. R., Green M. F., Madsen A. S., Schmiesing J., et al. (2014) Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 19, 605–617 10.1016/j.cmet.2014.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Goncalves R. L., Bunik V. I., and Brand M. D. (2016) Production of superoxide/hydrogen peroxide by the mitochondrial 2-oxoadipate dehydrogenase complex. Free Radic. Biol. Med. 91, 247–255 10.1016/j.freeradbiomed.2015.12.020 [DOI] [PubMed] [Google Scholar]
- 83. Xu W., Zhu H., Gu M., Luo Q., Ding J., Yao Y., Chen F., and Wang Z. (2013) DHTKD1 is essential for mitochondrial biogenesis and function maintenance. FEBS Lett. 587, 3587–3592 10.1016/j.febslet.2013.08.047 [DOI] [PubMed] [Google Scholar]
- 84. Soreze Y., Boutron A., Habarou F., Barnerias C., Nonnenmacher L., Delpech H., Mamoune A., Chrétien D., Hubert L., Bole-Feysot C., Nitschke P., Correia I., Sardet C., Boddaert N., Hamel Y., et al. (2013) Mutations in human lipoyltransferase gene LIPT1 cause a Leigh disease with secondary deficiency for pyruvate and α-ketoglutarate dehydrogenase. Orphanet J. Rare Dis. 8, 192 10.1186/1750-1172-8-192 [DOI] [PMC free article] [PubMed] [Google Scholar]