

SUMMARY
BK polyomavirus (BKV) frequently causes nephropathy (BKVN) in kidney transplant recipients (KTRs). BKV has also been implicated in the etiology of bladder and kidney cancers. We characterized BKV variants from two KTRs who developed BKVN followed by renal carcinoma. Both patients showed a swarm of BKV sequence variants encoding non-silent mutations in surface loops of the viral major capsid protein. The temporal appearance and disappearance of these mutations highlights the intra-patient evolution of BKV. Some of the observed mutations conferred resistance to antibody-mediated neutralization. The mutations also modified the spectrum of receptor glycans engaged by BKV during host cell entry. Intriguingly, all observed mutations were consistent with DNA damage caused by antiviral APOBEC3 cytosine deaminases. Moreover, APOBEC3 expression was evident upon immunohistochemical analysis of renal biopsies from KTRs. These results provide a snapshot of in-host BKV evolution and suggest that APOBEC3 may drive BKV mutagenesis in vivo.
Keywords: Human polyomavirus, carcinoma, BKPyV, JCV, JCPyV, APOBEC, APOBEC3, urothelial, bladder
eTOC blurb
BK polyomavirus causes nephropathy in kidney transplant patients. Peretti et al. show that, during the development of nephropathy, BKV acquires mutations that confer a selective advantage to the virus. The sequence changes are suggestive of a role for cellular APOBEC3 DNA cytosine deaminases in driving BKV mutagenesis in vivo.
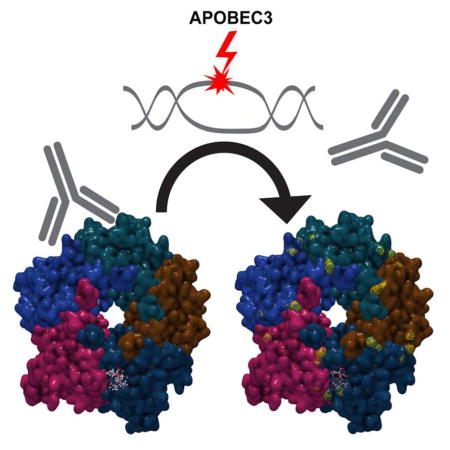
INTRODUCTION
BK polyomavirus (BKV) infections are typically acquired early in life, likely through an oral or respiratory route. Although the virus establishes chronic asymptomatic infection in the epithelial cells of the urinary tract, in immunosuppressed individuals it can replicate to pathogenic levels. BKV causes post-transplant nephropathy (BKVN) in up to 10% of kidney transplant recipients (KTRs)(Bennett et al., 2012; Rinaldo et al., 2013).
Recent findings suggest that BKV genotypes Ib2 and IV are commonly associated with the development of BKVN (Krautkramer et al., 2009; Schmitt et al., 2014; Schwarz et al., 2016). It appears that virus in the donor kidney can “hitchhike” and replicate to high levels in recipients who initially lack neutralizing antibody responses to BKV genotypes present in the graft (Pastrana et al., 2012; Scadden et al., 2017; Schmitt et al., 2014; Schwarz et al., 2016; Solis et al., 2018).
Mounting evidence suggests that BKV can play a causal role in cancers of the urinary tract, particularly in the KTR population. Epidemiological data show an increased risk of kidney and bladder cancer in KTRs (Frascà et al., 2015; Tillou and Doerfler, 2014) and large T antigen (LT)-positive high-grade urinary tract tumors have been reported in these patients (Kenan et al., 2015; Kenan et al., 2017; Papadimitriou et al., 2016; Yan et al., 2016). In recent studies, KTRs with a clinical history of BKV replication after transplantation have a significantly increased risk of developing bladder carcinomas relative to matched BKV-negative KTRs (Gupta et al., 2018; Liu et al., 2017; Rogers et al., 2017).
Polyomaviruses require sialylated glycan receptors for infectious entry into cells (O’Hara et al., 2014), and the BC loop on the apical surface of the VP1 major capsid protein appears to be a critical determinant of glycan receptor specificity (DeCaprio and Garcea, 2013; O’Hara et al., 2014). In addition to dictating the cell-surface glycans used for infectious entry, mutations in VP1 apical surface loops can also allow polyomaviruses to escape from antibody-mediated neutralization (Pastrana et al., 2013; Ray et al., 2015).
APOBEC3 (A3) proteins are a family of ssDNA cytosine deaminases that provide innate antiviral defenses by catastrophically mutating the genomes of DNA-based viruses, including retroviruses, parvoviruses and herpesviruses (Harris and Dudley, 2015; Salter et al., 2016; Siriwardena et al., 2016; Stavrou and Ross, 2015). Human APOBEC3B (A3B) preferentially deaminates cytosines in a TCA trinucleotide context, with less efficient deamination of other contexts (broadly represented as YCD in IUPAC code) (Burns et al., 2013). Evidence from in vitro models (Hoopes et al., 2017) and tumor sequencing data (Burns et al., 2013; Roberts et al., 2013) suggests that nuclear DNA cytosine deamination and erroneous repair can result in substitution of the original cytosine with any base.
A recent report has shown that BKV LT expression up-regulates A3B in cultured cells (Verhalen et al., 2016). The study also showed that BKV genome sequences are relatively deficient in preferred A3B target sites. These findings are reminiscent of previous reports pointing towards a complex reciprocal interplay between A3B and cancer-associated human papillomavirus (HPV) types (Vieira et al., 2014; Warren et al., 2015a; Warren et al., 2015b).
In this study, we examine the biology of BKV variants found in two case studies of KTRs who developed BKVN followed by clear cell renal cell carcinoma (ccRCC). Our results suggest a model in which BKV uses host A3B to acquire beneficial mutations that allow the virus to escape neutralizing antibodies and shift the spectrum of cell-surface glycans the virus engages for infectious entry.
RESULTS
BKV sequence variants from two KTRs who developed nephropathy and renal carcinoma
Previously described sequence-unbiased polyomavirus detection methods (Peretti et al., 2015) were applied to serum and urine specimens from two KTRs (described in detail in STAR Methods). The samples were collected at roughly the time the patients were diagnosed with BKVN. Deep sequencing of rolling circle amplification (RCA) products showed that both subjects had BKV genotype IVc2 in their serum and urine. The dominant sequence identified in Patient 1 (Pt1) was identical to isolate FIN-2 (GenBank: AB260033) except for two point differences (G2677A and T3823C), while isolate LAB-33 (GenBank: AB301097) was dominant in Patient 2 (Pt2). The analysis of next-generation sequencing (NGS) data did not reveal reads with similarity to other BKV-like sequences or to JC polyomavirus (JCV, a close relative of BKV).
In addition to the inferred wild-type (wt) VP1 protein sequence, which was identical in the two patients, we detected a total of 12 non-silent mutations in the VP1 coding region at varying abundances (as reflected in relative read counts). The mutations were mostly localized to the BC loop (VP1 amino acids 57–89)(Dugan et al., 2007). Many of these mutations have previously been observed in a BKV genotype I background and sporadically reported in BKV-IV isolates from the urine of KTRs (Boldorini et al., 2009; Krautkramer et al., 2009; Tremolada et al., 2010). VP1 mutations were evident in both serum and urine from Pt1, but only in urine from Pt2. Table 1 summarizes the mutations with their estimated relative abundance based on read counts. Nearly all reads covering the BC loop from Pt1 carried at least one non-silent mutation, suggesting that fully wt VP1 sequences were very rare or completely absent around the time of BKVN diagnosis. Intriguingly, all of the 12 observed VP1 mutations are consistent with the A3B signature, with deamination affecting the antisense DNA strand. The TCW-to-TKW mutational signature is strikingly reminiscent of the dominant pattern of mutations in bladder cancers (Burns et al., 2013).
Table 1. VP1 mutations revealed by unbiased viromics deep sequencing of samples collected near the time of BKV nephropathy diagnosis.
Trinucleotide contexts of the mutations are reported according to the VP1 antisense DNA strand. For each mutation, the relative abundance of reads (read count for the variant over the total number of reads spanning the relevant nucleotide position) is indicated as a percentage.
| Patient 1 | Patient 2 | ||||
|---|---|---|---|---|---|
| mutation (aminoacid) | mutation (trinucleotide) | abundance (serum) | abundance (urine) | abundance (serum) | abundance (urine) |
| E55Q | TCT→TGT | 9.0% | - | - | - |
| D60Ha | TCT→TGT | 7.6% | 4.0% | - | 5.7% |
| D62Na | TCA→TTA | 30.2% | 42.0% | - | 11.3% |
| D62Ya | TCA→TAA | - | - | - | 3.3% |
| R69Ka | TCT→TTT | - | - | - | 2.4% |
| E73Ka | TCA→TTA | 24.7% | 21.4% | - | - |
| E73Qa | TCA→TGA | - | - | - | 3.1% |
| D77Ha | TCA→TGA | 5.5% | 5.1% | - | - |
| D77Na | TCA→TTA | - | - | - | 25.4% |
| D336H | TCA→TGA | - | - | - | 3.5% |
| D336Y | TCA→TAA | - | - | - | 2.8% |
| G337C | CCA→CAA | - | - | - | 9.6% |
aminoacid substitutions mapping to the BC loop of VP1
To gain insight into VP1 sequences throughout the development and resolution of BKVN, we performed Sanger sequencing on VP1-specific PCR products obtained from other serum/urine samples collected at different time points. Figure 1 summarizes how VP1 mutations appear to have evolved over time. Variants were detected in the form of “split peaks” in sequencing chromatograms. Discernible split peaks confirmed on both DNA strands were only observed for the major mutations within the BC loop (D62N and E73K for Pt1, D62N and D77N for Pt2). When present, mutations were found together with the wt counterpart, which always seemed to predominate (based on the relative heights of sequencing peaks). In both subjects, only wt VP1 was observed at early time points, with mutations appearing around the time of BKVN diagnosis. Mutations were not detectable in later serum samples from Pt1, suggesting that the mutants were cleared and replaced with wt strains after the resolution of BKVN. The final serum samples of both individuals did not yield any PCR products, as expected in BKVN resolution, where a dramatic reduction in urine and plasma BKV DNA load typically occurs (Costa and Cavallo, 2012). Sanger sequencing revealed only wt VP1 sequences in serum from Pt2, consistent with NGS results. Samples collected prior to transplantation were also available from both subjects, but all PCR amplification attempts failed, as expected for non-immunosuppressed individuals.
Figure 1. Evolution of major VP1 BC loop mutations over time.

Filled arrowheads refer to time points analyzed by PCR amplification and subsequent Sanger sequencing. Empty triangles represent single time points investigated by unbiased viromics deep sequencing (reported in more detail in Table 1). Green bars indicate the time of renal cancer diagnosis.
Beyond VP1 mutations, the deep sequencing revealed other variations across the genome of the patients’ BKV isolates at the time of BKVN (Table S1). A majority of the additional point mutations were consistent with the A3B signature.
BKV neutralization serology
We next set out to use neutralization serology to address two hypotheses. First, we hypothesized that Pt1 and Pt2 acquired their nephropathic BKV-IV infections from their respective kidney donors. Second, we hypothesized that some of the observed non-silent mutations in VP1 might impart greater fitness to the virus by allowing escape from the recipients’ neutralizing antibody responses. This scenario, in which individual point mutations allow the virus to evade antibody-mediated neutralization, would be reminiscent of recent studies of JCV (Geoghegan et al., 2017; Jelcic et al., 2015; Ray et al., 2015).
Initially, we performed neutralization assays using wt BKV genotype I (subtype Ia) and BKV genotype IV (subtype IVc2) reporter pseudoviruses, as previously described (Pastrana et al., 2012; Pastrana et al., 2013). As shown in Figure 2A, donor sera efficiently neutralized both BKV-I and BKV-IV. This suggests that, in both cases, kidney donors were co-infected with BKV-I and BKV-IV. In contrast, both recipients were seronegative for BKV-IV neutralization and only modestly seropositive for BKV-I neutralization prior to transplantation. After transplantation, both patients seroconverted for BKV-IV. These findings suggest that both recipients acquired donor-derived de novo BKV-IV infections that presumably “hitchhiked” in the engrafted kidneys.
Figure 2. BKV neutralization serology.

(A) BKV-I and BKIV-IV neutralization serology in patients and matched donors. Patient sera collected at different time points before and after transplant (x-axis) and donor sera were serially diluted and tested using BKV reporter pseudovirus-based neutralization assays. Neutralization assays were performed on 293TT cells using pseudoviruses representative of wild type BKV-Ia and BKV-IVc2 isolates.
(B) Neutralization patterns of patient-cognate VP1 BC loop variants. Patient sera harvested at different time points after transplant (x-axis) and donor sera were serially diluted and tested using neutralization assays specific to the indicated patient-cognate variants. The results shown were obtained from experiments performed on ART cells. Neutralizing titers (EC50 values) for each serum are shown (y-axis). Horizontal dashed lines mark an arbitrary cutoff (1:100) for detectable neutralizing activity.
Reporter pseudoviruses representing the inferred wt or patient-specific mutant VP1 (BC loop) sequences observed in NGS were tested on three different cell lines known to support infectious entry by BKV and JCV pseudoviruses (293TT, ART and SFT) (Pastrana et al., 2013; Ray et al., 2015) and on primary renal proximal tubular epithelial (RPTE) cells (Low et al., 2004; Moriyama and Sorokin, 2009; Verhalen et al., 2016). The mutant pseudoviruses were somewhat less infectious than the wt pseudovirus on all cell lines, with the exception of RPTE cells (Figure S1, Tables S2 and S3).
Figure 2B shows neutralization profiles for each patient at different time points after transplantation. Interestingly, Pt1-cognate mutants exhibited relative resistance to neutralization when tested on ART cells. In particular, E73K and D77H pseudoviruses were neutralized, respectively, ~90-fold and ~70-fold less efficiently than the wt pseudovirus. A similar pattern was also observed on the other cell lines, with some variability in relative neutralizing titers against wt and mutant pseudoviruses (Figure S2). The results confirm the hypothesis that, as we have previously reported for JCV (Ray et al., 2015), individual point mutations in VP1 surface loops can confer relative resistance to serum antibody-mediated neutralization.
Differences in neutralization of wt versus mutant pseudoviruses were not as pronounced in Pt2, with D77N being the most resistant (neutralized ~8-fold less efficiently than the wt counterpart). For this subject, a very late serum sample (2.4 years post-transplant) was available. The later sample showed a greater neutralizing potency than the earlier time points, indicating that the patient eventually mounted a robust broadly cross-neutralizing humoral response after the resolution of BKVN.
VP1 mutations alter cell-surface receptor engagement
The fact that BKV-IV VP1 mutations mapping to the receptor-binding BC loop were associated with poorer infectivity compared to the wt strain on some cell lines led us to wonder whether the tested mutants might engage different infectious entry pathways. To test the dependence of patient-specific mutant pseudoviruses on sialylated glycan receptors, we used 3Fax-PeracetylNeu5Ac (3Fax), a previously described inhibitor of sialyltransferases that blocks synthesis of sialylated glycans (Geoghegan et al., 2017; Rillahan et al., 2012). 3Fax-treated SFT cells showed dramatic resistance to all tested BKV variants (Figure S3A). The results indicate that the patients’ BC loop mutants, like wt BKV-IV, remain dependent on sialylated glycans for infectious entry.
To explore the possibility that mutant BKV-IV strains selectively associate with a different range of sialylated glycans than the wt strain, we performed hemagglutination assays using red blood cells (RBCs) from a variety of animal species. The experiment rests on the assumption that different animals display different spectra of sialylated glycans on the surface of their RBCs. We have previously found that different BKV genotypes show differential ability to agglutinate RBCs from different animals (Pastrana et al., 2013). While wt BKV pseudovirions agglutinated goose RBCs, they did not agglutinate sheep RBCs. In contrast, each of the tested mutant pseudovirions agglutinated both goose and sheep RBCs. This indicates that the BC loop mutants are able to engage a different spectrum of cell surface glycans than the wt pseudovirus (Figure S3B).
BKV sequences in additional KTRs with ongoing BKV replication
To obtain a more general picture of BKV genotypes and VP1 mutations in additional patients, we performed a PCR/NGS analysis using multiple pairs of BKV/JCV consensus primers on DNA extracted from blood or urine collected from a cohort of 24 KTRs with ongoing BKV replication. All of the patients were treated with a reduced immunosuppression regimen as soon as they were diagnosed with BKV viremia and none went on to develop frank BKVN, with the exception of one subject who suffered from biopsy-proven nephropathy.
Overall, PCR amplification was successful in 16 of 24 plasma/serum samples and 12 of 23 urine samples. BKV genotypes and VP1 mutations observed in NGS analysis of PCR products, along with their estimated relative abundance, are listed in Table S4. The deep sequencing revealed a high prevalence of genotype Ib2 (14 plasma/serum samples and 9 urine samples) and genotype IV (8 plasma samples and 4 urine samples). Interestingly, the only observed non-silent surface (BC) loop mutation (E73Q, genotype Ib2) with an A3B signature was detected in the single patient with full-blown BKVN. In summary, non-silent surface loop mutations consistent with A3B damage were found in a total of 3/3 BKVN patients but not in any of 23 patients diagnosed only with BKV viremia.
APOBEC3 expression patterns in renal tissues
The fact that BKV infection specifically up-regulates A3B in primary cultured cells (Verhalen et al., 2016) suggests A3B-mediated editing as the most parsimonious explanation for the observed VP1 mutations. Nevertheless, related enzymes APOBEC3A/C/H exhibit similar preference for TC deamination targets and cell-wide (including nuclear) expression patterns (Salter et al., 2016). Induction of several A3 family members (including A3B) has been observed in various types of hematopoietic cells (HIV infection), hepatocytes (hepatitis virus infection) and cervical keratinocytes (HPV infection) in response to inflammatory cytokines that are up-regulated during viral infections (Siriwardena et al., 2016; Stavrou and Ross, 2015). Since A3-type mutations were only observed in the setting of full-blown BKVN but not in BKV viremic subjects, we wondered whether A3 family members might likewise be induced in the context of inflammation associated with the development of BKVN (Bennett et al., 2012; Costa and Cavallo, 2012; Rinaldo et al., 2013). To begin to address this question, we performed immunohistochemical staining on formalin-fixed paraffin-embedded (FFPE) renal biopsies from additional KTR cases using an anti-A3B monoclonal antibody that also cross-reacts with APOBEC3A (A3A) and APOBEC3G (A3G) (Leonard et al., 2015), as well as a monoclonal antibody recognizing BKV LT (Sunden et al., 2006). A3 nuclear expression was observed, to varying degrees, in biopsies that showed signs of inflammatory cell infiltration or acute drug toxicity (Figures 3B–D), but not in one biopsy that appeared to be devoid of significant pathological findings (Figure 3E). Although widespread LT staining was not observed in these samples, individual cells positive for both LT and A3 were observed in serial sections of one sample (Figures 3A and 3B). While the A3 antibody did not allow us to distinguish between A3B, A3A and A3G, the fact that the observed staining pattern was exclusively nuclear strongly implicates A3B (rather than A3A or A3G) as the most likely enzyme expressed in these renal tissues. Altogether, these results indicate that multiple pathways, including LT expression, can contribute to the up-regulation of A3B in the kidney.
Figure 3. APOBEC3 immunohistochemical analysis of renal biopsies from kidney transplant recipients.

(A and B) Serial sections from a transplanted kidney biopsy previously found to show signs of severe acute drug toxicity and occasional large T antigen positivity were stained using antibodies to large T antigen (A) and APOBEC3 (B). Selected areas are displayed at a higher magnification. Black arrows in magnified insets indicate nuclei that stained positive for both large T antigen and APOBEC3.
(C and D) Tissue sections from transplanted kidney biopsies previously found to show micro-foci of inflammatory cell infiltration with modest signs of tubulitis (C) or modest signs of acute drug toxicity (D) were stained using an antibody to APOBEC3. Representative areas of positivity are highlighted.
(E) A tissue section from a transplanted kidney biopsy previously found devoid of relevant pathological signs was stained using an antibody to APOBEC3.
Scale bars represent 100 μm in main images, 50 μm in insets.
Exome sequencing of kidney tumors
We hypothesized that BKV-induced A3 damage to the cellular genome might serve as a durable record of the past presence of the virus in a tumor. To examine this hypothesis, we performed exome sequencing on the ccRCC tumors from Pt1 and Pt2. The sequences revealed only a low level of A3-type mutations in the tumor exome, with frequencies similar to prior analyses of kidney cancer specimens from the general population (Figure S4).
DISCUSSION
Recent studies support the view that BKVN in KTRs is typically associated with donor-derived de novo infections with BKV-Ib2 or BKV-IV. Our results further reinforce this view and show that nephropathic BKV infections evolve in a way that can render the virus at least partially resistant to the patient’s nascent humoral response. The observed neutralization-escape mutations in the major capsid protein VP1 also alter the spectrum of sialylated glycans the virus engages during the infectious entry process, potentially triggering altered tissue tropism and pathogenicity. Dominant VP1 mutations detected in BKVN patients are consistent with A3 damage, most likely A3B, which the virus is known to up-regulate. Together, these findings suggest a model in which BKV has evolved to hijack A3B (thought to normally provide innate antiviral defense) to attract site-specific mutations that confer a selective advantage to the virus.
A3 proteins are only enzymatically active against ssDNA and thus have a greater tendency to damage the lagging strand during DNA replication and the sense strand (i.e., non-transcribed strand) during gene expression (Haradhvala et al., 2016; Seplyarskiy et al., 2016). Although BKV genomes are relatively depleted of A3B target motifs, there is a peak occurrence of TC target sites in the antisense (lagging) DNA strand underlying the BC loop of VP1 (Verhalen et al., 2016). Given the complex bi-directional nature of polyomavirus transcription (Carmichael, 2016), it is possible to imagine transcription-coupled damage occurring when early transcripts transit the late region. Intriguingly, many of the observed mutations create a new A3 target site on the opposing DNA strand. This raises the possibility that the virus could acquire, for example, the common E73Q mutation through A3 damage to the VP1 antisense/lagging DNA strand then later acquire a Q73E reversion mutation through A3 damage to the hypothetically more accessible VP1 sense/lagging-template DNA strand. Reversible toggling would allow for reconciliation of the observed intra-patient evolution with the extraordinarily slow rate at which polyomaviruses appear to stably accumulate genetic change over millennial time scales (Buck et al., 2016).
Our findings are strikingly reminiscent of serological studies of mutants observed in individuals suffering from a JCV-induced brain disease, progressive multifocal leukoencephalopathy (PML). PML patients appear to show neutralization “blind spots” for specific VP1 sequences found in their cerebrospinal fluid (Jelcic et al., 2015; Ray et al., 2015). Similar to the BKV VP1 mutations investigated in the current study, the most common PML-associated JCV VP1 mutations are known to alter the spectrum of glycans the virus can use for infectious entry (Geoghegan et al., 2017; Gorelik et al., 2011) and also fit the typical A3B signature. In contrast to BKV, the dominant PML-associated mutations in JCV affect the VP1 sense (lagging-template) DNA strand. In a recently reported chimeric humanized mouse model of JCV/PML, various VP1 point mutations observed at an 8-week infection time point were consistent with the A3B signature affecting both the sense and antisense DNA strands underlying VP1 surface loops (Kondo et al., 2014). Since the mice used in the study lacked adaptive immunity, the observation implies that A3-type mutations can evolve in the absence of selective pressure from neutralizing antibodies.
Under conditions of impaired T-cell function due to post-transplant immunosuppressive therapy, antibody-mediated neutralization may serve as a last line of defense against many types of viral infections. In this setting, mutations allowing evasion from neutralizing antibodies would be especially likely to confer selective advantage. The BC loop mutants detected in Pt1 exhibited relative resistance to neutralization during BKVN development. Although Pt2 showed less evidence of effective escape from antibody-mediated neutralization, the overall neutralizing titers observed for Pt2 at BKVN diagnosis were much lower than those observed for Pt1. This raises the possibility that BKV-neutralizing antibody concentrations may need to be above a certain threshold to exert significant selective pressure on the virus in the setting of BKVN development.
STAR METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chris Buck (buckc@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients and samples
Two unrelated KTRs who were diagnosed with BKVN and subsequent ccRCC were treated at the University Hospital of Novara (Italy). Patient 1 (Pt1; male) received kidney transplant at the age of 54 years, and was diagnosed with BKVN 5.2 months post-transplant and with ccRCC (native kidney; grade FG2, stage T1a) 5.6 months post-transplant. Patient 2 (Pt2; female) received kidney transplant at the age of 37 years, and was diagnosed with BKVN 9.2 months post-transplant and with ccRCC (allograft; grade FG4, stage T3b N2) 60.2 months post-transplant. In addition to ccRCC, Pt2 was diagnosed with cervical intraepithelial neoplasia (CIN) twice after kidney transplant (CIN grade 2, 4.0 months post-transplant; CIN grade 1, 60.0 months post-transplant). Prior to ccRCC diagnosis, biopsies of the nephropathic kidneys from both individuals were positive in routine diagnostic LT immunohistochemical staining. Tissue sections from the urothelial lining of the ureter of Pt1’s native kidney (which was removed due to ccRCC three weeks after BKVN diagnosis) stained positive for LT, but the tumor cells did not show detectable LT expression. Malignant cells in the ccRCC affecting Pt2’s allograft kidney (which was removed more than four years after BKVN diagnosis) also stained negative for LT. Both patients were administered Basiliximab at the time of transplant to induce immunosuppression. Subsequent maintenance treatment consisted of Sirolimus (replaced with Tacrolimus at day 15 post-transplant) + mycophenolate mofetil + prednisone for Pt1 and Tacrolimus + mycophenolate mofetil + prednisone for Pt2. At BKVN diagnosis, Pt1 started Cidofovir treatment and was also administered a methylprednisolone dose for a concomitant acute rejection. At BKVN diagnosis, Pt2 started receiving leflunomide instead of mycophenolate mofetil (Tacrolimus + leflunomide + prednisone). After excision of the native kidney (Pt1) or allograft kidney (Pt2) for ccRCC, both patients’ immunosuppressive treatments continued with prednisone only. Pt1 died from an unknown cause 6 years after removal of the native kidney. Pt2 has remained on dialysis during the 4 years since the allograft kidney was excised.
Serum and urine samples (volume range 150 μl – 1 ml) harvested from the patients at different time points before and after kidney transplantation and FFPE blocks containing the respective ccRCC lesions were obtained from the pathology archive of the University Hospital of Novara. A single serum sample was also available from each of the matched kidney donors. Additionally, a series of FFPE renal tissues from different KTRs was retrieved from the same archive. The study was approved by the ethics committee at the University Hospital of Novara.
Archived plasma and urine samples from 23 BKV viremic KTRs under the care of the Saarland University Medical Center (SUMC), Homburg (Germany) were also collected. Use of the banked samples was approved by a vote from the SUMC ethics committee. In addition, a single serum sample was obtained from a KTR with biopsy-confirmed nephropathy enrolled by the Henry Ford Hospital, Detroit (USA). Written informed consent was obtained from the patient and the study was approved by the Henry Ford Hospital’s ethics committee.
Samples were anonymized prior to shipment to the National Cancer Institute, Bethesda (USA).
Cell cultures
The previously described cell lines 293TT, ART and SFT were originally generated by stable transfection of respective parental lines with linearized plasmid pTIH, carrying a SV40 large T Antigen cDNA expression cassette (Buck et al., 2004; Pastrana et al., 2013; Ray et al., 2015). 293TT cells (sex: female) were derived from the parental line 293T, which in turn was engineered from the parental human embryonic kidney line HEK-293 (Buck et al., 2004). A detailed description of 293TT and relative parental lines is available through our lab website (https://home.ccr.cancer.gov/lco/293TT.htm). 293TT cells were maintained in DMEM medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), GlutaMAX-I (Invitrogen), MEM Non-Essential Amino Acids (Invitrogen) and 250 μg/ml Hygromicin B Gold (InvivoGen). ART cells (sex: female) were derived from the parental ovarian cancer line NCI/ADR/RES (Developmental Therapeutics Program, National Cancer Institute; Bethesda, USA). SFT cells (sex: female) were derived from the parental gliosarcoma line SF-539 (Developmental Therapeutics Program, National Cancer Institute; Bethesda, USA). ART and SFT cells were maintained in RPMI 1640 medium (Invitrogen) containing 5% fetal bovine serum (Invitrogen), GlutaMAX-I (Invitrogen) and 170 μg/ml (ART) or 50 μg/ml (SFT) Hygromicin B Gold (InvivoGen).
Primary human renal proximal tubular epithelial cells (RPTE; sex of specific batch: male) (Lonza) were cultured in REGM medium with SingleQuots Bulletkit (Lonza) and passaged up to six times as previously described (Abend et al., 2007).
All cell lines were grown at 37 °C and 5% CO2 in a humidified incubator.
METHOD DETAILS
Sample processing & deep sequencing (serum/urine)
Serum and urine samples collected from Pt1 and Pt2 near the time of BKVN diagnosis were subjected to a previously reported method for enriching virions, amplifying the viral genome with bacteriophage phi29 DNA polymerase (RCA), and deep sequencing (Peretti et al., 2015). Briefly, samples were supplemented with 1 mM MgCl2, Triton X-100 to a final concentration of 1% and 5 μl of Benzonase nuclease (Sigma), and brought up to a volume of 3 ml with DPBS. Mixtures were incubated at 37°C for 40 min with occasional mixing. After incubation, samples were adjusted to pH ~7 by addition of ammonium sulfate from a 1M stock solution at pH 9. Samples were supplemented with NaCl to a final concentration of 0.85 M, rocked at room temperature for 15 min, then clarified at 1200 × g for 6 min. Clarified supernatants were loaded onto a 27-33-39% iodixanol (OptiPrep Density Gradient Medium, Sigma) step gradient and centrifuged at 50,000 rpm for 3.5 h (234,000 × g) at 16°C in a SW55Ti rotor (Beckman). The entire gradient was collected into 5–10 consecutive fractions by bottom puncture of the tubes. DNA was extracted from each fraction by incubating at 50°C for 15 min in a digest buffer consisting of 20 mM Tris pH 8, 20 mM DTT, 20 mM EDTA, 0.2% SDS, 0.2% proteinase K stock (Qiagen). Virion-derived DNA was then ethanol-precipitated and the pelleted DNA was re-dissolved in phi29 DNA polymerase Sample Buffer and subjected to RCA according to manufacturer’s instructions (Illustra TempliPhi Amplification Kit, GE Healthcare). Equal volumes of RCA products from each fraction were pooled together for each sample. Finally, each sample pool was ethanol-precipitated and re-dissolved in 55 μl of 0.2× TE buffer (Sigma).
The four final RCA samples were prepared for deep sequencing using the Nextera XT DNA Library Preparation Kit, as directed by the manufacturer (Illumina). Samples were barcoded and analyzed on the MiSeq platform using a V3 600 cycle cassette. The resulting reads for each sample were assembled into contigs using CLC Genomics Workbench v11.0 (Qiagen). Contigs were screened against GenBank using BLASTN/BLASTX algorithms for sequence assignment. For the purpose of this manuscript, only sequences with similarity to BKV or JCV were taken into account. Reads mapping to the patients’ BKV isolates as inferred from contig assembly were in the following amounts:
Pt1, serum: 24,687 of 1,133,608 (2.2%)
Pt1, urine: 56,589 of 925,524 (6.1%)
Pt2, serum: 14,186 of 1,942,608 (0.7%)
Pt2, urine: 151,391 of 1,232,590 (12.3%)
Mutations were detected analyzing re-alignments of individual reads against final contigs through the CLC Genomics Workbench Basic Variant Detection Tool. Each mutational “hotspot” was then validated and confirmed by visual examination of mapping tracks.
BKV VP1 PCR & Sanger sequencing (serum/urine)
Serum and urine samples from Pt1 and Pt2 collected at time points before or after BKVN diagnosis were processed for DNA extraction using QIAamp DNA Mini Kit (Qiagen). The extracted DNA was subjected to PCR amplification using BKV consensus primers spanning the VP1 BC loop. Visible bands were obtained with a single round of PCR amplification in the case of urine, while nested PCR amplification was necessary for serum. The following primers were used:
Pair 1 (amplicon size 534bp)
BK-VP1_For1: GCCTGTACGGGACTGTAAC
BK-VP1_Rev1: CTCCCTGCATTTCCAAG
Pair 2 (nested, amplicon size 334bp)
BK-VP1_For2: GTGCAAGTGCCAAAACTAC
BK-VP1_Rev2: GACCCTGCATGAAGGTTAAG
PCR amplification was performed using Herculase II Fusion DNA Polymerase (Agilent), and consisted of 30 cycles of 20 sec at 95°C, 20 sec at 53°C (pair 1) / 56°C (pair 2) and 30 sec at 72°C. Reactions were run on an Eppendorf Mastercycler Pro PCR thermal cycler. PCR products were gel-purified by means of QIAquick Gel Extraction Kit (Qiagen) and subjected to Sanger sequencing on an Applied Biosystems 3730xl DNA Analyzer, using both primer pairs (FW1 and BW1 for urine-derived amplicons, FW2 and BW2 for serum-derived amplicons) in separate reactions to confirm sequences on both DNA strands.
BKV/JCV VP1 PCR & deep sequencing (blood/urine)
Plasma and urine samples collected from a cohort of 23 KTRs with BKV viremia were subjected to DNA extraction using the NucliSENS easyMAG system (bioMérieux). DNA was also extracted from single serum and urine samples of a KTR with biopsy-proven nephropathy. Consensus primers targeting conserved segments of the polyomavirus clade encompassing BKV, JCV and SV40 were designed, with the goal of amplifying any possible sequence variants:
PBK761For: AGCACTKTTGGGGGACCTAGTTGC
PBK785For: TGTDTCTGAGGCTGCTGCTGC
PBK824For: GCTGAAATTGCTGCTGGRGAGGC
PBK740Rev: TGGCAACTAGGTCCCCCAAMAGTG
PBK4934Rev: TGGATAGATTGCTACTGCWTTGAYTGCT
BKJC-VP1A: AAGCATATGAAGATGGCCCCA
BKJC-VP1B: CTGCTCCTCAATGGATGTTGCC
BKJC-VP1C: GTACGGGACTGTAACACCTG
BKJC-VP1Drev: CTCTGGACATGGATCAAGCAC
BKJC-VP1Frev: GGAARGAAAGGCTGGATTYWG
BKJC-VP1Grev: TYAGGCCWGTWGCTGAYTTTGC
Different primer pair combinations were used, including nested and heminested PCR approaches:
PBK761For/PBK4934Rev (first round) → PBK785For/PBK4934Rev (heminested); amplicon: nearly entire genome
PBK785For/PBK740Rev (first round) → PBK824For/PBK4934Rev (nested); amplicon: nearly entire genome
BKJC-VP1B/BKJC-VP1Grev (first round) → BKJC-VP1A/BKJC-VP1Drev (nested); amplicon: entire VP1 coding region
BKJC-VP1C/BKJC-VP1Frev (first round) → BKJC-VP1A/BKJC-VP1Drev (nested); amplicon: entire VP1 coding region
For some samples, PCR products were obtained directly from the first round of PCR amplification, while other samples required a nested or heminested second round PCR reaction. All of the bands with appropriate size were subjected to gel-purification, deep sequencing and bioinformatic analysis, as described above. In cases where different primer pairs successfully generated PCR products from the same sample, all of the respective bands were analyzed and results were combined. In order to catalog VP1 variants, individual reads were mapped against single BKV reference genomes (found in healthy individuals) representative of each genotype found. Both partial variants appearing within each sample and full sequence variations compared to “normal” reference were listed as VP1 mutations.
Generation of pseudoviruses
Plasmids expressing codon-modified versions of the VP1 gene from different BKV isolates were used to produce pseudoviral particles as previously described, with minor modifications (Buck et al., 2004; Buck and Thompson, 2007; Geoghegan et al., 2017; Pastrana et al., 2012; Pastrana et al., 2013; Pastrana et al., 2009). Plasmids encoding VP1 proteins from a BKV-Ia isolate (BK-D) and a BKV-IVc2 isolate (A-66H) were already available in our lab (Pastrana et al., 2013). Plasmids encoding the different VP1 variants of the study patients’ BKV-IVc2 strains were constructed using standard overlap PCR-based mutagenesis, starting from the BKV-IVc2 (A-66H) VP1 plasmid pwB (Pastrana et al., 2013). Previously reported plasmids expressing codon-modified versions of the VP2/VP3 minor capsid protein genes (Pastrana et al., 2013) were used for all the BKV pseudoviruses produced for this study.
Briefly, 293TT cells were pre-plated 18 h in advance at 1.1 × 105 cells per cm2 of culture area in the absence of hygromicin and co-transfected 24 h later with 0.5 μg of DNA and 0.9 μl of Lipofectamine2000 Transfection Reagent (Invitrogen) per cm2 of culture area according to manufacturer’s instructions. For reporter pseudoviruses, two plasmids encoding secreted NanoLuc luciferase under the control of different promoters (phsNuc and pcsNuc) (Geoghegan et al., 2017) were also co-transfected. For transfections, a ratio of 2:1:1:1:1 for plasmids encoding VP1, VP2, VP3 and secreted NanoLuc luciferase was used, respectively. After overnight incubation, the transfection medium was replaced with fresh culture medium. Roughly 48 h after transfection, particles were harvested by trypsinizing cells, washing and resuspending pellets in 1.2 pellet volumes of DPBS supplemented with 9.5 mM MgCl2 in siliconized tubes.
Neuraminidase V (Sigma) was added to a final concentration of 2 U/ml, and cell suspensions were incubated at 37°C for 15 min. Cells were then lysed by the addition of 0.5% Triton X-100, and lysates were buffered with ammonium sulfate pH 9 to a final concentration of 25 mM. For reporter pseudoviruses, lysates were treated with 0.1% RNase Cocktail Enzyme Mix (Ambion). For pseudovirions to be used in hemagglutination assay (virus-like particles without reporter vectors), lysates were treated with 0.1% benzonase (Sigma) and 0.1% Plasmid-Safe ATP-Dependent DNase (Epicentre). After overnight capsid maturation at 37°C, mixtures were chilled on ice. For virus-like particles, NaCl at a final concentration of 0.85 M was added to the mixtures, followed by incubation on ice for 10 min. Samples were then subjected to 2–3 clarification steps, consisting of centrifugation at maximum speed for 5 min at 4°C followed by pellet resuspension in 1–2 pellet volumes of DPBS with one freeze-thaw in between (reporter pseudoviruses) or DPBS / 0.85 M NaCl (virus-like particles). The resulting pooled clarified material was purified through a 27-33-39% OptiPrep step gradient by centrifuging at 50,000 rpm for 3.5 h (234,000 × g) at 16°C in a SW55 rotor (Beckman). After ultracentrifugation, the gradient was collected into 10 consecutive fractions in siliconized tubes. For virus-like particles, gradient fractions were screened for the presence of encapsidated DNA using Quant-iT Picogreen dsDNA Reagent (Invitrogen) and for protein content using Pierce BCA Protein Assay (Invitrogen). For reporter pseudoviruses, fractions were tested for infectivity by transducing 293TT cells (pre-plated 2 h in advance in 96-well plates at 1.1 × 105 cells per cm2 of culture area) with 1 μl of each fraction and assessing the expression of EGFP (encoded by all the plasmids used in transfections) and NanoLuc reporter signal (as detailed below for neutralization assays) after 72 h. In all cases, positive fractions were pooled into siliconized tubes, and the VP1 content of each stock was determined by comparison to a bovine serum albumin standard (Bio-Rad) in Coomassie-stained SDS-PAGE gels (NuPAGE 4–12% Bis-Tris Protein Gels; Invitrogen).
Merkel cell polyomavirus (MCV) and HPV16 pseudoviruses were produced according to similar protocols as previously described (Buck and Thompson, 2007; Pastrana et al., 2009), with secreted NanoLuc luciferase reporter plasmids (phsNuc and pcsNuc) used in place of Gaussia luciferase reporter plasmids.
All plasmid maps and detailed production methods are posted in our lab website http://home.ccr.cancer.gov/Lco/.
Neutralization assays
Neutralization assays were performed in a 96-well plate format as described previously (Pastrana et al., 2012; Pastrana et al., 2013; Pastrana et al., 2009), with slight modifications.
Briefly, cells were pre-plated at a density of 3 × 104 (293TT), 7 × 103 (ART and SFT) or 1 × 104 (RPTE) cells per well in a volume of 100 μl in the absence of hygromicin and allowed to attach for 5–7 h. Pseudovirus doses ranged approximately from 50 to 1600 pg of VP1 per well, depending on the infectivity of each specific pseudovirus stock. Human serum samples were heat inactivated at 56°C for 30 min. Diluted pseudovirus stocks were combined with nine 5-fold serial dilutions of serum samples (starting dilution: 1:50) and incubated on ice for 1 h, before adding the mixture to cells in a volume of 100 μl. Pseudovirus and serum dilutions were performed in appropriate culture media. Each experiment also contained six wells of cells receiving pseudovirus stock without test serum (“no serum” control) and six wells with cells that received only culture medium (“no pseudovirus” control). After 3 days (293TT and SFT cells) or 7 days (ART and RPTE cells), 25 μl of culture supernatants were harvested and the presence of secreted NanoLuc reporter protein was determined with Nano-Glo Luciferase Assay System (Promega) according to manufacturer’s instructions, with minor modifications (only 10 μl of Nano-Glo Luciferase Assay reagent were used per 25 μl of assayed sample). Relative light units (RLUs) were read on a POLARstar Optima microplate luminometer (BMG Labtech) at a gain ranging from 2200 to 3200, depending on the specific pseudovirus stocks and cell types tested. The reciprocal 50% neutralizing dilution (EC50) for each serum was calculated using GraphPad Prism v7.0 software by fitting a variable-slope sigmoidal dose-response curve to RLU values for each serum dilution series, with top and bottom values constrained to the average values of “no serum” and “no pseudovirus” control wells respectively. Based on prior observations that sera can exert non-specific neutralizing effects at dilutions of less than 1:100, an EC50 value of 100 was set as a cutoff for detectable serum antibody-mediated neutralization (Pastrana et al., 2009).
Figure S1 and Tables S2 and S3 detail the quantitative parameters and characteristics of BKV-IV pseudovirus stocks, including VP1 amounts used in the assays and typical RLU signals observed with the different cell lines.
Infectivity assay with 3Fax-treatment
Treatment of SFT cells with silayltransferase inhibitor 3Fax-PeracetylNeu5Ac (3Fax; EMD Millipore) was conducted with 200 μM 3Fax or the equivalent volume of DMSO (mock treatment) in standard SFT culture medium as previously described (Geoghegan et al., 2017). After 72 h, treated cells were trypsinized and re-seeded in 96-well plates at a density of 5 × 103 cells per well in medium supplemented with 200 μM 3Fax or DMSO. After several hours, pseudoviruses were applied to the cells in approximate dose ranges of 100–1600 pg of VP1 per well, according to the infectivity of each specific stock. After 72 h, 25 μl of each culture supernatant were harvested and the presence of secreted NanoLuc reporter protein was determined as described above. RLU values were used to calculate the percent inhibition of transduction of 3Fax-treated cells relative to mock-treated cells. Two independent quintuplicate experiments were performed.
Hemagglutination assay
Sodium citrate-preserved animal blood was purchased from Lampire Biological Laboratories. Red blood cells (RBCs) were rinsed three times in chilled PBS by centrifugation at 2,000 × g for 5 min at 4°C. RBCs were then pelleted and resuspended at 5% (v/v) in chilled PBS. Two-fold serial dilutions of each patient-specific BKV-IVc2 pseudovirion stock spanning a range of 20,000 ng/ml to 10 pg/ml prepared in a volume of 40 μl were added to 10 μl of the diluted 5% RBC suspension in a round-bottom 96-well plate and mixed. Mixtures were allowed to settle at 4°C for 2 h. Four independent experiments were performed.
Immunohistochemical analysis of renal tissues
Consecutive 4-μm thick sections obtained from FFPE renal tissues were processed for immunohistochemical detection of polyomavirus LT and A3B. FFPE samples showing miscellaneous histological characteristics (variable degrees of inflammation, variable signs of acute drug toxicity, variable LT positivity) were obtained from different KTRs. Sections were deparaffinized and endogenous peroxidase activity was blocked with 0.3% H2O2 in 1× TBS. Antigen unmasking was performed using a 30 minute steam incubation in citrate buffer pH 6. Slides were then incubated in Background Sniper blocking solution (Biocare Medical) for 15 minutes at room temperature. For LT staining, slides were incubated overnight at 4°C with mouse monoclonal antibody PAb2003, which was generated against JCV LT and cross-reacts with BKV LT (Sunden et al., 2006), at a dilution of 1:8000 in 10% blocking solution-1× TBS. For A3B staining, slides were incubated overnight at 4°C with an in-house-developed rabbit monoclonal antibody (10-87-13), generated against A3B and cross-reacting with A3A and A3G (Leonard et al., 2015), at a dilution of 1:1000 in 10% blocking solution-1× TBS. After one wash in 1× TBS, slides were incubated in Polymer Kit (Post Primary Block + Polymer) Novocastra Novolink (Leica Biosystems) per manufacturer’s protocols. Immunostaining was performed by incubation of the slides in diaminobenzidine (DAB Substrate Buffer with Stabilizer; Covance), monitoring the enzymatic reaction under a conventional microscope. Finally, slides were counterstained with hematoxylin for 1 minute, dehydrated and mounted with Fisher Chemical Permount Mounting Medium (Fisher Scientific). Images were acquired using digital slide scanner TissueScope LE (Huron Digital Pathology).
Exome sequencing of kidney tumor specimens
FFPE blocks containing the ccRCC lesions of Pt1 and Pt2 were sectioned and subjected to routine hematoxilyn-eosin staining to guide the collection of normal and tumor tissue cores. One tumor core area from Pt1 and four tumor core areas with different histology from Pt2 were processed. DNA was extracted from each tissue core using DNAeasy Blood & Tissue Kit (Qiagen) and subjected to OncoVar exome sequencing, as previously described (Killian et al., 2014; Wang et al., 2014). Somatic point mutations were analyzed using muTect v2.0 software (Cibulskis et al., 2013). Mutational signature analyses were performed as previously described using the R package SomaticSignatures (Gehring et al., 2015). Significant enrichment of A3-signature mutations was calculated using methods detailed by Chan and colleagues (Chan et al., 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Results relative to infectivity assay with 3Fax treatment and hemagglutination shown in Figure S3 are expressed as the mean calculated from two (3Fax experiment) or four (hemagglutination) independent experiments, with error bars signifying SEM. Typical luminometric signals for the BKV reporter pseudovirus stocks reported in Table S3 represent the mean calculated from ten replicate values ± SEM. EC50 values in neutralization assays were calculated using GraphPad Prism v7.0 software as detailed above. All sequence analysis was conducted using CLC Genomics Workbench v11.0 software (Qiagen). Kidney tumor exome sequencing analysis was performed using the software and methods described above.
DATA AND SOFTWARE AVAILABILITY
The raw deep sequencing data for Pt1 and Pt2 have been deposited in the Sequence Read Archive (SRA) under the accession number SRP139959.
ADDITIONAL RESOURCES
Description: https://home.ccr.cancer.gov/Lco/protocols.asp
Supplementary Material
i) Regulatory region rearrangements. In BKV, as well as in other polyomaviruses, the regulatory region directs bi-directional gene transcription and genome replication, and is known to undergo complex rearrangements that convert it from “archetype” (basal transmissible form of the virus associated with subclinical infection) to stereotypical “rearranged” variants associated with viremia and disease progression (Emami et al., 2015; Olsen et al., 2009). A great majority of regulatory region sequences in both individuals’ serum/urine at the time of BKVN appeared to represent the archetype arrangement. Examples of lower-frequency regulatory region rearrangements are shown according to the arbitrary convention of dividing the regulatory region sequence into 5 functional blocks (O143, P68, Q39, R63, S63) (Markowitz and Dynan, 1988; Moens et al., 1995). Non-tandem repeats of identical sequence blocks are color-coded. Rearrangements were detected using the InDels and Structural Variants tool of the CLC Genomics Workbench software (Qiagen).
ii) Point mutations and small insertions/deletions. In addition to VP1 mutations (listed in main text, Table 1), NGS analysis revealed point mutations and small insertions/deletions affecting different genomic regions and often introducing aminoacid changes in viral proteins of both subjects’ isolates. A majority of the point mutations fit the same TCW-to-TKW pattern observed in VP1. Mutations were detected using the Basic Variant Detection tool of the CLC Genomics Workbench software (Qiagen). Numbering for nucleotide positions is set with the start base of large T antigen open reading frame (lying on the negative DNA strand) as wrap point. For each mutation, the relative abundance of reads (read count for the variant over the total number of reads spanning the relevant nucleotide position) is indicated as a percentage.
iii) Genomic rearrangements. NGS analysis also revealed examples of strains carrying extensive rearrangements (e.g. large insertions, deletions, inversions etc.) often involving a large segment of the viral genome, that presumably represent defective variants resulting from high multiplicity viral replication (Kovar, 1991). Rearrangements were detected using the InDels and Structural Variants tool of the CLC Genomics Workbench software (Qiagen). Numbering for nucleotide positions is set with the start base of large T antigen open reading frame (lying on the negative DNA strand) as wrap point.
Highlights.
BKV variants sequenced from kidney transplant patients during development of nephropathy
BKV major capsid protein acquires surface loop mutations over time
Mutations confer resistance to neutralizing antibodies and modify glycan receptor usage
Mutational signatures match APOBEC3 activity and renal biopsies are positive for APOBEC3
Acknowledgments
This work was funded by the NIH Intramural Research Program and supported by grants from San Paolo Company (grant CSP 2014 to C.B.) and Italian Association for Cancer Research (AIRC; grant IG 2016 to M.G.). Salary support for A.P. was provided by the Italian Foundation for Cancer Research (FIRC; Fellowship for Abroad 2013). We gratefully acknowledge Antonio Amoroso (University of Turin, Italy) for providing pre-transplantation and donor sera for Pt1 and Pt2. Salary support for G.J.S. is provided by a NFS Graduate Research Fellowship (00039202). R.S.H. is an investigator of the Howard Hughes Medical Institute (Minneapolis, USA).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, A.P. and C.B.B.; Methodology, D.V.P. and C.B.B.; Investigation, A.P., E.M.G., D.V.P., G.J.S., E.K.L., V.B., J.K.K., J.Z., M.P., and P.S.M.; Formal Analysis, A.P., E.S.L., D.W., P.C.F., G.J.S., R.S.H., J.K.K., J.Z., M.P., and P.S.M.; Resources, S.S., P.F., M.S., S.L., M.R., C.B., M.G., R.B., and C.B.B.; Writing – Original Draft, A.P. and C.B.B.; Writing – Review & Editing, all authors; Supervision, C.B.B.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bennett SM, Broekema NM, Imperiale MJ. BK polyomavirus: emerging pathogen. Microbes Infect. 2012;14:672–683. doi: 10.1016/j.micinf.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldorini R, Allegrini S, Miglio U, Paganotti A, Veggiani C, Mischitelli M, Monga G, Pietropaolo V. Genomic mutations of viral protein 1 and BK virus nephropathy in kidney transplant recipients. J Med Virol. 2009;81:1385–1393. doi: 10.1002/jmv.21520. [DOI] [PubMed] [Google Scholar]
- Buck CB, Pastrana DV, Lowy DR, Schiller JT. Efficient intracellular assembly of papillomaviral vectors. J Virol. 2004;78:751–757. doi: 10.1128/JVI.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck CB, Thompson CD. Production of papillomavirus-based gene transfer vectors. Curr Protoc Cell Biol. 2007;Chapter 26(Unit 26) doi: 10.1002/0471143030.cb2601s37. [DOI] [PubMed] [Google Scholar]
- Buck CB, Van Doorslaer K, Peretti A, Geoghegan EM, Tisza MJ, An P, Katz JP, Pipas JM, McBride AA, Camus AC, et al. The Ancient Evolutionary History of Polyomaviruses. PLoS Pathog. 2016;12:e1005574. doi: 10.1371/journal.ppat.1005574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael GG. Gene Regulation and Quality Control in Murine Polyomavirus Infection. Viruses. 2016;8 doi: 10.3390/v8100284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, Kim J, Kwiatkowski DJ, Fargo DC, Mieczkowski PA, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47:1067–1072. doi: 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C, Cavallo R. Polyomavirus-associated nephropathy. World J Transplant. 2012;2:84–94. doi: 10.5500/wjt.v2.i6.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio JA, Garcea RL. A cornucopia of human polyomaviruses. Nat Rev Microbiol. 2013;11:264–276. doi: 10.1038/nrmicro2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan AS, Gasparovic ML, Tsomaia N, Mierke DF, O’Hara BA, Manley K, Atwood WJ. Identification of amino acid residues in BK virus VP1 that are critical for viability and growth. J Virol. 2007;81:11798–11808. doi: 10.1128/JVI.01316-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami A, Yaghobi R, Moattari A, Baseri Salehi M, Roozbeh J. Noncoding Control Region Pattern of BK Polyomavirus in Kidney Transplant Patients With Nephropathy. Exp Clin Transplant. 2015;15:150–156. doi: 10.6002/ect.2014.0230. [DOI] [PubMed] [Google Scholar]
- Frascà GM, Sandrini S, Cosmai L, Porta C, Asch W, Santoni M, Salviani C, D’Errico A, Malvi D, Balestra E, et al. Renal cancer in kidney transplanted patients. J Nephrol. 2015;28:659–668. doi: 10.1007/s40620-015-0219-8. [DOI] [PubMed] [Google Scholar]
- Gehring JS, Fischer B, Lawrence M, Huber W. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics. 2015;31:3673–3675. doi: 10.1093/bioinformatics/btv408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan EM, Pastrana DV, Schowalter RM, Ray U, Gao W, Ho M, Pauly GT, Sigano DM, Kaynor C, Cahir-McFarland E, et al. Infectious entry and neutralization of pathogenic JC polyomaviruses. Cell Rep. 2017;21:1169–1179. doi: 10.1016/j.celrep.2017.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, Reid C, Testa M, Brickelmaier M, Bossolasco S, Pazzi A, Bestetti A, Carmillo P, Wilson E, McAuliffe M, et al. Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change its receptor specificity. J Infect Dis. 2011;204:103–114. doi: 10.1093/infdis/jir198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G, Kuppachi S, Kalil RS, Buck CB, Lynch CF, Engels EA. Treatment for Presumed BK Polyomavirus Nephropathy and Risk of Urinary Tract Cancers among Kidney Transplant Recipients in the United States. Am J Transplant. 2018;18:245–252. doi: 10.1111/ajt.14530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, Rheinbay E, Kim J, Maruvka YE, Braunstein LZ, et al. Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair. Cell. 2016;164:538–549. doi: 10.1016/j.cell.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Dudley JP. APOBECs and virus restriction. Virology. 2015;479–480:131–145. doi: 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes JI, Hughes AL, Hobson LA, Cortez LM, Brown AJ, Roberts SA. Avoidance of APOBEC3B-induced mutation by error-free lesion bypass. Nucleic Acids Res. 2017;45:5243–5254. doi: 10.1093/nar/gkx169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelcic I, Combaluzier B, Jelcic I, Faigle W, Senn L, Reinhart BJ, Stroh L, Nitsch RM, Stehle T, Sospedra M, et al. Broadly neutralizing human monoclonal JC polyomavirus VP1-specific antibodies as candidate therapeutics for progressive multifocal leukoencephalopathy. Sci Transl Med. 2015;7:306ra150. doi: 10.1126/scitranslmed.aac8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenan DJ, Mieczkowski PA, Burger-Calderon R, Singh HK, Nickeleit V. The oncogenic potential of BK-polyomavirus is linked to viral integration into the human genome. J Pathol. 2015;237:379–389. doi: 10.1002/path.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenan DJ, Mieczkowski PA, Latulippe E, Cote I, Singh HK, Nickeleit V. BK Polyomavirus Genomic Integration and Large T Antigen Expression: Evolving Paradigms in Human Oncogenesis. Am J Transplant. 2017;17:1674–1680. doi: 10.1111/ajt.14191. [DOI] [PubMed] [Google Scholar]
- Killian JK, Miettinen M, Walker RL, Wang Y, Zhu YJ, Waterfall JJ, Noyes N, Retnakumar P, Yang Z, Smith WI, Jr, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014;6:268ra177. doi: 10.1126/scitranslmed.3009961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Windrem MS, Zou L, Chandler-Militello D, Schanz SJ, Auvergne RM, Betstadt SJ, Harrington AR, Johnson M, Kazarov A, et al. Human glial chimeric mice reveal astrocytic dependence of JC virus infection. J Clin Invest. 2014;124:5323–5336. doi: 10.1172/JCI76629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovar H. A detailed analysis of duplications appearing during early, high multiplicity infections with polyoma virus. J Gen Virol. 1991;72:1943–1952. doi: 10.1099/0022-1317-72-8-1943. [DOI] [PubMed] [Google Scholar]
- Krautkramer E, Klein TM, Sommerer C, Schnitzler P, Zeier M. Mutations in the BC-loop of the BKV VP1 region do not influence viral load in renal transplant patients. J Med Virol. 2009;81:75–81. doi: 10.1002/jmv.21359. [DOI] [PubMed] [Google Scholar]
- Leonard B, McCann JL, Starrett GJ, Kosyakovsky L, Luengas EM, Molan AM, Burns MB, McDougle RM, Parker PJ, Brown WL, et al. The PKC/NF-kappaB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 2015;75:4538–4547. doi: 10.1158/0008-5472.CAN-15-2171-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chaudhry MR, Berrebi AA, Papadimitriou JC, Drachenberg CB, Haririan A, Alexiev BA. Polyomavirus Replication and Smoking Are Independent Risk Factors for Bladder Cancer After Renal Transplantation. Transplantation. 2017;101:1488–1494. doi: 10.1097/TP.0000000000001260. [DOI] [PubMed] [Google Scholar]
- Low J, Humes HD, Szczypka M, Imperiale M. BKV and SV40 infection of human kidney tubular epithelial cells in vitro. Virology. 2004;323:182–188. doi: 10.1016/j.virol.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Markowitz RB, Dynan WS. Binding of cellular proteins to the regulatory region of BK virus DNA. J Virol. 1988;62:3388–3398. doi: 10.1128/jvi.62.9.3388-3398.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens U, Johansen T, Johnsen JI, Seternes OM, Traavik T. Noncoding control region of naturally occurring BK virus variants: sequence comparison and functional analysis. Virus Genes. 1995;10:261–275. doi: 10.1007/BF01701816. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Sorokin A. BK virus (BKV): infection, propagation, quantitation, purification, labeling, and analysis of cell entry. Curr Protoc Cell Biol. 2009;Chapter 26(Unit 26) doi: 10.1002/0471143030.cb2602s42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara SD, Stehle T, Garcea R. Glycan receptors of the Polyomaviridae: structure, function, and pathogenesis. Curr Opin Virol. 2014;7:73–78. doi: 10.1016/j.coviro.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Olsen GH, Hirsch HH, Rinaldo CH. Functional analysis of polyomavirus BK non-coding control region quasispecies from kidney transplant recipients. J Med Virol. 2009;81:1959–1967. doi: 10.1002/jmv.21605. [DOI] [PubMed] [Google Scholar]
- Papadimitriou JC, Randhawa P, Rinaldo CH, Drachenberg CB, Alexiev B, Hirsch HH. BK Polyomavirus Infection and Renourinary Tumorigenesis. Am J Transplant. 2016;16:398–406. doi: 10.1111/ajt.13550. [DOI] [PubMed] [Google Scholar]
- Pastrana DV, Brennan DC, Cuburu N, Storch GA, Viscidi RP, Randhawa PS, Buck CB. Neutralization serotyping of BK polyomavirus infection in kidney transplant recipients. PLoS Pathog. 2012;8:e1002650. doi: 10.1371/journal.ppat.1002650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana DV, Ray U, Magaldi TG, Schowalter RM, Cuburu N, Buck CB. BK polyomavirus genotypes represent distinct serotypes with distinct entry tropism. J Virol. 2013;87:10105–10113. doi: 10.1128/JVI.01189-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana DV, Tolstov YL, Becker JC, Moore PS, Chang Y, Buck CB. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009;5:e1000578. doi: 10.1371/journal.ppat.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretti A, FitzGerald PC, Bliskovsky V, Buck CB, Pastrana DV. Hamburger polyomaviruses. J Gen Virol. 2015;96:833–839. doi: 10.1099/vir.0.000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray U, Cinque P, Gerevini S, Longo V, Lazzarin A, Schippling S, Martin R, Buck CB, Pastrana DV. JC polyomavirus mutants escape antibody-mediated neutralization. Sci Transl Med. 2015;7:306ra151. doi: 10.1126/scitranslmed.aab1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, Dell A, Haslam SM, Paulson JC. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat Chem Biol. 2012;8:661–668. doi: 10.1038/nchembio.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldo CH, Tylden GD, Sharma BN. The human polyomavirus BK (BKPyV): virological background and clinical implications. APMIS. 2013;121:728–745. doi: 10.1111/apm.12134. [DOI] [PubMed] [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers R, Gohh R, Noska A. Urothelial cell carcinoma after BK polyomavirus infection in kidney transplant recipients: A cohort study of veterans. Transpl Infect Dis. 2017;19:e12752. doi: 10.1111/tid.12752. [DOI] [PubMed] [Google Scholar]
- Salter JD, Bennett RP, Smith HC. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem Sci. 2016;41:578–594. doi: 10.1016/j.tibs.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden JR, Sharif A, Skordilis K, Borrows R. Polyoma virus nephropathy in kidney transplantation. World J Transplant. 2017;7:329–338. doi: 10.5500/wjt.v7.i6.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt C, Raggub L, Linnenweber-Held S, Adams O, Schwarz A, Heim A. Donor origin of BKV replication after kidney transplantation. J Clin Virol. 2014;59:120–125. doi: 10.1016/j.jcv.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Schowalter RM, Pastrana DV, Buck CB. Glycosaminoglycans and sialylated glycans sequentially facilitate merkel cell polyomavirus infectious entry. PLoS Pathog. 2011;7:e1002161. doi: 10.1371/journal.ppat.1002161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz A, Linnenweber-Held S, Heim A, Framke T, Haller H, Schmitt C. Viral Origin, Clinical Course, and Renal Outcomes in Patients With BK Virus Infection After Living-Donor Renal Transplantation. Transplantation. 2016;100:844–853. doi: 10.1097/TP.0000000000001066. [DOI] [PubMed] [Google Scholar]
- Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016;26:174–182. doi: 10.1101/gr.197046.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siriwardena SU, Chen K, Bhagwat AS. Functions and Malfunctions of Mammalian DNA-Cytosine Deaminases. Chem Rev. 2016;116:12688–12710. doi: 10.1021/acs.chemrev.6b00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis M, Velay A, Porcher R, Domingo-Calap P, Soulier E, Joly M, Meddeb M, Kack-Kack W, Moulin B, Bahram S, et al. Neutralizing Antibody-Mediated Response and Risk of BK Virus-Associated Nephropathy. J Am Soc Nephrol. 2018;29:326–334. doi: 10.1681/ASN.2017050532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrou S, Ross SR. APOBEC3 Proteins in Viral Immunity. J Immunol. 2015;195:4565–4570. doi: 10.4049/jimmunol.1501504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunden Y, Suzuki T, Orba Y, Umemura T, Asamoto M, Nagashima K, Tanaka S, Sawa H. Characterization and application of polyclonal antibodies that specifically recognize JC virus large T antigen. Acta Neuropathol. 2006;111:379–387. doi: 10.1007/s00401-005-0025-9. [DOI] [PubMed] [Google Scholar]
- Tillou X, Doerfler A. Urological tumors in renal transplantation. Minerva Urol Nefrol. 2014;66:57–67. [PubMed] [Google Scholar]
- Tremolada S, Delbue S, Castagnoli L, Allegrini S, Miglio U, Boldorini R, Elia F, Gordon J, Ferrante P. Mutations in the external loops of BK virus VP1 and urine viral load in renal transplant recipients. J Cell Physiol. 2010;222:195–199. doi: 10.1002/jcp.21937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhalen B, Starrett GJ, Harris RS, Jiang M. Functional Upregulation of the DNA Cytosine Deaminase APOBEC3B by Polyomaviruses. J Virol. 2016;90:6379–6386. doi: 10.1128/JVI.00771-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira VC, Leonard B, White EA, Starrett GJ, Temiz NA, Lorenz LD, Lee D, Soares MA, Lambert PF, Howley PM, et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio. 2014;5:e02234–02214. doi: 10.1128/mBio.02234-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Thomas A, Lau C, Rajan A, Zhu Y, Killian JK, Petrini I, Pham T, Morrow B, Zhong X, et al. Mutations of epigenetic regulatory genes are common in thymic carcinomas. Sci Rep. 2014;4:7336. doi: 10.1038/srep07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CJ, Van Doorslaer K, Pandey A, Espinosa JM, Pyeon D. Role of the host restriction factor APOBEC3 on papillomavirus evolution. Virus Evol. 2015a;1 doi: 10.1093/ve/vev015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CJ, Xu T, Guo K, Griffin LM, Westrich JA, Lee D, Lambert PF, Santiago ML, Pyeon D. APOBEC3A functions as a restriction factor of human papillomavirus. J Virol. 2015b;89:688–702. doi: 10.1128/JVI.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Salama ME, Lanciault C, Matsumura L, Troxell ML. Polyomavirus large T antigen is prevalent in urothelial carcinoma post-kidney transplant. Hum Pathol. 2016;48:122–131. doi: 10.1016/j.humpath.2015.09.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
i) Regulatory region rearrangements. In BKV, as well as in other polyomaviruses, the regulatory region directs bi-directional gene transcription and genome replication, and is known to undergo complex rearrangements that convert it from “archetype” (basal transmissible form of the virus associated with subclinical infection) to stereotypical “rearranged” variants associated with viremia and disease progression (Emami et al., 2015; Olsen et al., 2009). A great majority of regulatory region sequences in both individuals’ serum/urine at the time of BKVN appeared to represent the archetype arrangement. Examples of lower-frequency regulatory region rearrangements are shown according to the arbitrary convention of dividing the regulatory region sequence into 5 functional blocks (O143, P68, Q39, R63, S63) (Markowitz and Dynan, 1988; Moens et al., 1995). Non-tandem repeats of identical sequence blocks are color-coded. Rearrangements were detected using the InDels and Structural Variants tool of the CLC Genomics Workbench software (Qiagen).
ii) Point mutations and small insertions/deletions. In addition to VP1 mutations (listed in main text, Table 1), NGS analysis revealed point mutations and small insertions/deletions affecting different genomic regions and often introducing aminoacid changes in viral proteins of both subjects’ isolates. A majority of the point mutations fit the same TCW-to-TKW pattern observed in VP1. Mutations were detected using the Basic Variant Detection tool of the CLC Genomics Workbench software (Qiagen). Numbering for nucleotide positions is set with the start base of large T antigen open reading frame (lying on the negative DNA strand) as wrap point. For each mutation, the relative abundance of reads (read count for the variant over the total number of reads spanning the relevant nucleotide position) is indicated as a percentage.
iii) Genomic rearrangements. NGS analysis also revealed examples of strains carrying extensive rearrangements (e.g. large insertions, deletions, inversions etc.) often involving a large segment of the viral genome, that presumably represent defective variants resulting from high multiplicity viral replication (Kovar, 1991). Rearrangements were detected using the InDels and Structural Variants tool of the CLC Genomics Workbench software (Qiagen). Numbering for nucleotide positions is set with the start base of large T antigen open reading frame (lying on the negative DNA strand) as wrap point.