

Summary
So far, opposing outcomes have been reported following neonatal apex resection in mice, questioning the validity of this injury model to investigate regenerative mechanisms. We performed a systematic evaluation, up to 180 days after surgery, of the pathophysiological events activated upon apex resection. In response to cardiac injury, we observed increased cardiomyocyte proliferation in remote and apex regions, neovascularization, and local fibrosis. In adulthood, resected hearts remain consistently shorter and display permanent fibrotic tissue deposition in the center of the resection plane, indicating limited apex regrowth. However, thickening of the left ventricle wall, explained by an upsurge in cardiomyocyte proliferation during the initial response to injury, compensated cardiomyocyte loss and supported normal systolic function. Thus, apex resection triggers both regenerative and reparative mechanisms, endorsing this injury model for studies aimed at promoting cardiomyocyte proliferation and/or downplaying fibrosis.
Keywords: neonatal apex resection, cardiac regeneration, cardiac injury response, cardiomyocyte proliferation, fibrosis, cardiac fibroblasts, extracellular matrix, neovascularization, stereology
Graphical Abstract
Highlights
-
•
Apex resection triggers fibrosis, neovascularization, and cardiomyocyte proliferation
-
•
Permanent fibrotic deposition is confined to the apex
-
•
Injured hearts display morphometric alterations but regain functional competence
-
•
Cardiomyocyte proliferation is sufficient to compensate tissue loss by resection
In this article, Nascimento and colleagues demonstrate that neonatal apex resection stimulates cardiomyocyte proliferation and permanent scarring in the apex. Newly formed cardiomyocytes compensate muscle loss by resection, and resected hearts recover functional competence in adulthood. These findings endorse this model for studies aiming to block cardiac fibrosis and/or favoring CM proliferation.
Introduction
Cardiovascular diseases are the leading cause of death worldwide (Mozaffarian et al., 2016), which is largely attributed to the limited regenerative capacity of the mammalian heart. However, this paradigm has been challenged by studies describing a developmental window for heart regeneration in mammals. Although the first reference to mammalian heart regeneration was provided by Mario Robledo in 1956 (Robledo, 1956), this concept only became robustly supported in 2011 after Sadek and colleagues demonstrated that murine hearts, when subjected to apex resection during the first day of life, were able to reestablish the myocardial tissue through activation of cardiomyocyte (CM) proliferation (Porrello et al., 2011). This report proposed that, like lower vertebrates (Flink, 2002, Poss et al., 2002), the mammalian heart holds an intrinsic ability to regenerate from injury, which could be exploited for therapeutic purposes. This work also set the stage for several studies identifying regulators of CM proliferation (Aurora et al., 2014, Bassat et al., 2017, D'Uva et al., 2015, Heallen et al., 2013, Mahmoud et al., 2013) or describing distinct injury models (Darehzereshki et al., 2015, Haubner et al., 2012, Jesty et al., 2012, Lavine et al., 2014, Porrello et al., 2013, Uygur and Lee, 2016).
In 2014, controversy arose when another laboratory, using the same injury model, reported substantial scarring in the resected apex region, which was accompanied by impaired neovascularization and CM proliferation (Andersen et al., 2014a). Further work by this group showed that fibrotic tissue deposition in the resected region was definitive, and that cardiac function was permanently reduced as animals developed dilated cardiomyopathy, with left ventricle (LV) chamber dilation and wall thinning (Andersen et al., 2016).
The reason for these contrasting findings is still unclear (Andersen et al., 2014b, Kotlikoff et al., 2014, Sadek et al., 2014), although technical variations such as injury severity were shown to impact the formation of scar tissue upon apex resection (Bryant et al., 2015) and cryoinjury (Darehzereshki et al., 2015). Thus, the ability of neonatal hearts to regenerate after apex resection seems to be dependent on small technical variations which at present hampers progress in the field.
Additional difficulties are related to the evaluation of the fibrotic scar (Andersen et al., 2014a, Bryant et al., 2015) and the fact that the assessment of CM proliferation, the most important hallmark for cardiac regeneration, is technically challenging. In particular, CM proliferation in mammals can be easily mistaken for CM binucleation that initiates around birth (reviewed in Zebrowski et al., 2016), an event characterized by karyokinesis without cytokinesis, leading to CM terminal differentiation (Soonpaa et al., 1996). Of note, studies addressing the response to apex resection in neonatal mice have either focused on functional assessment or histopathological alterations (Andersen et al., 2014a), but longitudinal studies examining cardiac remodeling, function, and the precise tissue response to injury have not yet been performed.
Since there are antagonizing reports and because tissue regeneration and repair often activate overlapping mechanisms (Aurora et al., 2014, Tidball, 2011), we evaluated both tissue responses following neonatal apex resection. Hence, a systematic assessment of the pathophysiological events occurring over 180 days after surgery was performed. We found that apex resection triggers both regenerative (e.g. neovascularization and CM proliferation) and reparative (e.g. cardiac fibrosis) mechanisms which result in an adult heart with more CMs, benign adaptive cardiac remodeling and restored systolic function.
Results
Establishment of Neonatal Apex Resection Injury Model
One-day-old (P1) C57BL/6 mice were subjected to apical resection by thoracotomy using the exposure of the LV lumen as a standardized method for resection size (Mahmoud et al., 2014, Porrello et al., 2011). Sham controls underwent the same surgical procedure without resection of the apex (no cardiac manipulation of any kind).
Immediately after the surgical procedure, the survival rate of apex-resected animals was equivalent to sham controls (∼87%). However, maternal cannibalization reduced survival at 24 hr post-surgery to 61% and 82% for injury and sham groups, respectively.
Hearts Are Not Fully Restored at the Histological Level
The ventricular surface area and heart length/body weight ratio were significantly reduced upon apical resection by ∼11% and ∼14%, respectively (Figures 1A–1C), similar to other reports (Andersen et al., 2014a, Porrello et al., 2011). Representative sampling of the heart (Figure S1A) and histological characterization by Masson's trichrome (MT) staining (Figures 1A and S1B) revealed that apex resection induced noticeable cardiac remodeling, with impact on heart morphology and local deposition of fibrotic tissue (Figures 1B–1D). From 7 to 21 days after surgery, apex regrowth was evident as myocardial tissue was observed below the resection site. Alongside, the myocardial tissue in the innermost part of the apex where the lumen was exposed, was surrounded by a subepicardial scar (Figures 1A and S1B). Higher magnification revealed that the scar undergoes a maturation process, which starts with a scattered deposition of collagenous material and matures to a fibrillar appearance, occupying most of the central apex wall from 60 days after resection onward (Figures 1A1–1A4, S1B1, S1B2, and S1C). In contrast, areas surrounding the central apex region displayed myocardial integrity.
Figure 1.

Hearts Do Not Fully Regenerate the Resected Apex
(A) Representative MT-stained sections of apex-resected and sham controls following surgery. Sections exhibiting myocardial disruption and/or cardiac fibrosis are highlighted by a dashed line. Scale bars, 2 mm. High-magnification images of the injury site (A1, 0 day; A2, 21 days; A3, 60 days; and A4, 180 days) show collagen (blue staining) from 21 days onward. Scale bars, 250 μm.
(B and C) Ventricular surface area (from left to right, n = 8, 10, 8, 14, 3, 7, 4, 4) (B) and heart length/ body weight (BW) (from left to right, n = 11, 11, 8, 10, 4, 7, 4, 4) (C) of resected and sham-operated hearts were determined in MT-stained paraffin sections at 0, 7, 14, 21, 60, and 180 days post-surgery.
(D and E) Injury extension (from left to right, n = 8, 12, 7, 4) (D) and ventricular and scar volume (from left to right, n = 19, 14, 7, 4) (E) were calculated on MT-stained paraffin sections. All values are presented as means ± SD. See also Figure S1 for representative MT sections at 7 and 14 days after surgery, data on heart weight, heart to BW ratio, and percentage of fibrosis in the apex.
Ventricular surface area was restored at 7 days after injury (Figure 1B), and resected hearts weighed the same or were even heavier than intact hearts (Figures S1D and S1E). However, injured hearts were consistently shorter throughout the study, indicating that heart growth was mainly achieved through short-axis expansion (Figure 1C). The extent of injury, estimated by determining the percentage of sections with cardiac damage (i.e., myocardial disruption or cardiac fibrosis), decreased over time, which correlates with an actual reduction in scar volume (Figures 1D and 1E). Overall, these results indicate that resected hearts remain shorter, have permanent scarring, and do not fully regenerate.
Resected Hearts Are Functionally Competent
Echocardiograms at 21, 60, 120, and 180 days following surgery confirmed that resected ventricles underwent severe morphological alterations (i.e., long-axis shortening and short-axis widening) (Figure 2A). However, in contrast to previous reports (Andersen et al., 2016), our data showed that short-axis expansion occurred due to a thickening of the anterior and posterior wall of the LV (Figures 2B and 2C) and left ventricular chamber dilation (Figure 2D). Because LV enlargement is concurrent to a reduction in heart length (Figure 2E), the eccentricity index of injured hearts was significantly increased (Figure 2F). End-diastolic volume of injured hearts was increased at 21 days, but normalized from 60 days onward (Figure 2G). Importantly, no differences were detected in the ejection fraction (Figure 2H), stroke volume (Figure 2I) or cardiac output (Figure 2J) across groups, thus showing that the systolic function of injured hearts was not significantly impaired. Regarding diastolic function, the ratio between LV early (E) to late (A) filling velocities was marginally decreased at 180 days, suggesting that incomplete relaxation and increased filling pressures might be present in resected hearts (Figure 2K). This can be explained by increased myocardial stiffness caused by the scarring, wall thickening, and/or alterations in ventricular chamber geometry. Accordingly, myocardial performance index (also known as Tei index), which comprises both systolic and diastolic parameters, showed a tendency to be increased in resected hearts (Figure 2L) at the expense of longer relaxation times (data not shown). Electrocardiogram (ECG) tracing of injured hearts was similar to control animals (Figure 2M), which was confirmed by the absence of any significant modification in the evaluated ECG parameters (Table S1).
Figure 2.

Injured Hearts Display Morphological Alterations but Exhibit Normal Systolic Function
(A) Representative echocardiographic parasternal long-axis view 180 days post-surgery.
(B–G) End-diastolic left ventricle anterior wall thickness (LVAW), left ventricle posterior wall thickness (LVPW), left ventricle internal diameter (LVID), heart length, eccentricity index, and heart volume.
(H–J) Ejection fraction (EF), stroke volume and cardiac output determined by Simpson's method.
(K and L) Ratio between left ventricular early (E) to late (A) filling velocities and myocardial performance index (also known as Tei index) (from left to right, n = 7, 9, 22, 24, 18, 15, 18, 14).
(M) Representative ECG signal traces at 180 days post-surgery (n = 10, 6; 180 days sham, 180 days injury). See also Table S1 for detailed quantification of ECG parameters. All values are presented as means ± SD.
These findings demonstrate that, despite consistent LV chamber enlargement and wall thickening, accompanied by a reduced heart length, resected hearts are functionally competent.
Short-Term Response to Cardiac Injury Involves Extracellular Matrix Remodeling and Fibroblast Activation
Apex amputation was evident by the disruption of the expression pattern of sarcomeric-α-actinin (s-α-Actinin) and laminin (LN) (Figure S2). Forty-eight hours after resection, the apex region was marked by the deposition of fibronectin (FN) and tenascin-C (TN-C), and by an abundant CD45+ immune cell infiltrate (Figures 3A and S2, arrowheads). Myofibroblasts (α-SMA+ cells) also started to appear at the injury site at this point (Figure S2, arrows). Resolution of the inflammatory phase was evident at 7 days post-injury, whereas myofibroblasts became more frequent (Figures 3A and S2). At 21 days post-resection, α-SMA was restricted to smooth muscle cells in vessels, as observed in sham hearts. Of note, TN-C remained elevated in the border between the resection and the adjacent myocardium (Figure S2). Confocal microscopy analysis further showed that FN and TN-C were produced by CD45+ hematopoietic cells during the inflammatory phase (48 hr post-surgery) and by α-SMA+ myofibroblasts during inflammatory resolution (7 days post-surgery) (Figure 3A). Although largely overlapping, TN-C expression was restricted to the interface between the healthy and damaged myocardium, whereas FN displayed a more ubiquitous deposition, suggesting different biological roles (Figure 3A).
Figure 3.

Apex Resection Triggers Local ECM Remodeling and Fibroblast Activation
(A) Representative confocal images of CD45/TN-C/FN, α-SMA/TN-C/FN, and s-α-Actinin/TN-C/FN immunolabelling at the injury site 48 hr (n = 4) and 7 days post-surgery (n = 4). Arrowhead, CD45+ hematopoietic cells; arrows, α-SMA+ myofibroblasts; dashed segment, resection line. Scale bars, 30 μm. See also Figure S2 for detailed analysis of cellular and extracellular matrix (ECM) remodeling following injury.
(B) Cardiac fibroblasts were isolated and prospectively identified by CD140a, CD90, and SCA-1 expression throughout ontogeny (see Figure S4), 7 days post-resection (n = 3) and sham surgery (n = 6). CD140a+CD90+SCA-1− (purple/SP1), CD140a+CD90−SCA-1− (blue/SP2), and CD140a+CD90+SCA-1+ (gray/SP3) fibroblasts were FACS-sorted (see Figure S5 for sorting strategy).
(C) Transcriptomic profiling of SP1 to SP3 subpopulations isolated from injured (n = 5) and sham hearts (n = 4) was performed by real-time qPCR. Expression levels were normalized against Gapdh. All values are presented as means ± SEM. See also Figures S4 and S5 for detailed phenotypic characterization of cardiac fibroblasts throughout ontogeny and sorting strategy, respectively.
We profiled hematopoietic cells (CD45+) infiltrating the apex and remote regions by flow cytometry at 2, 5, and 7 days after surgery. Single-cell suspensions were stained with antibodies to distinguish total myeloid cells (CD11b+), macrophages (CD11b+F4/80+), monocytes (CD11b+F4/80−), neutrophils (CD11b+Gr-1+), T lymphocytes (CD3+), and B lymphocytes (CD45R+) (Figure S3A). At 2 days after surgery, an influx of CD45+ cells was observed in the apex, paralleled by a reduction in the remote LV (Figures S3B and S3C). The rate of hematopoietic cells in the myocardium of apex-resected hearts was comparable with those observed in sham-operated controls at 7 days post-surgery, corroborating our immunofluorescence data. The immune infiltrate was mainly composed of myeloid cells. At the initial stages of injury-response (i.e., 2 days) neutrophils, monocytes, and macrophages infiltrated the myocardium at the apex, although only macrophages remained elevated up to 5 days post-surgery. No differences were observed regarding the percentage of T lymphocytes between groups, whereas B lymphocytes were significantly decreased 2 days post-injury (Figures S3B and S3C). Additional staining of myeloid populations allowed discrimination of CD11b+F4/80+Ly6Clo macrophages, CD11b+F4/80+Ly6Chi macrophages, CD11b+F4/80−Ly6Clo monocytes, and CD11b+F4/80−Ly6Chi inflammatory monocytes, as reported previously (Aurora et al., 2014). Of note, CD11b+F4/80+Ly6Chi macrophages and CD11b+F4/80−Ly6Chi monocytes were predominantly increased at the injury site, but not in the remote myocardium, during early inflammatory response (Figure S3D). In turn, CD11b+F4/80+Ly6Clo macrophages were increased at 5 days after injury, similar to what was reported following myocardial infarction at P1 (Aurora et al., 2014).
Cardiac fibroblasts are master mediators of myocardial remodeling following injury (reviewed in Souders et al., 2009). However, studies determining the cardiac fibroblast surface signature during the neonatal period are lacking. Considering the heterogeneity of this cell type, we discriminated fibroblast populations, from embryonic day 17 to adulthood (Figure S4), through the expression of the three well-reported markers of cardiac fibroblasts, i.e., CD90 (Thy-1), CD140a (PDGFRα), and SCA-1 (Furtado et al., 2014, Hudon-David et al., 2007, McQualter et al., 2009, Smith et al., 2011), following exclusion of non-viable cells (PI+), endothelial cells (CD31+), hematopoietic cells (CD45+), and erythrocytes (TER-119+). These results demonstrated that fibroblasts express the mesenchymal marker CD140a (≥85%) at all stages analyzed, either in the presence or absence of CD90 (Figure S4). Throughout time these fibroblasts also acquire the expression of SCA-1 and, thus, in the adult, the most abundant subsets are CD140a+CD90−SCA-1+ (Figure S4, green) and CD140a+CD90+SCA-1+ (Figure S5, gray), as reported previously (Furtado et al., 2014).
We applied the same strategy to injured and sham-operated animals 7 days post-surgery (Figure 3B). Three main populations were identified: CD140a+CD90−SCA-1− (blue; injury: 34.71% ± 2.24%; sham: 32.50% ± 2.72%), CD140a+CD90+SCA-1− (purple; injury: 36.53% ± 0.95%; sham: 40.29% ± 1.85%), and CD140a+CD90+SCA-1+ (gray; injury: 21.26% ± 1.15%; sham: 17.77% ± 1.40%). A minor population was also found expressing CD90 only (red; injury: 3.79% ± 0.94%; sham: 6.34% ± 1.17%), whereas SCA-1 was mainly confined to cells also expressing the other surface markers (Figure 3B). No statistically significant differences were detected in the relative percentages of each population between injured animals and sham-operated controls (Figure 3B). This might be explained by the use of the whole heart and/or by variability created during the isolation procedure, which may mask small differences in cell numbers in the apex. Indeed, immunofluorescence results showed a fibroblast-mediated response to injury largely confined to the lesion site (Figures 3A and S2).
The activation of most abundant populations was assessed by real-time qPCR (sorting strategy detailed in Figure S5). The CD140a+CD90+SCA-1− (purple, SP1) subpopulation, which is particularly frequent in earlier developmental periods (Figure S4), responded to apex resection by upregulating genes associated with scarring (Col1a1, Col3a1, Tgfb1, and Tgfb3), CM proliferation (Fn1, Tbx20, Igf1, Igf2, and Fstl1), and neovascularization (Vegfa) (Figure 3C). In contrast, CD90+CD140a+SCA-1+ (gray, SP3) cells, a subpopulation upsurging after birth (Figure S4), shows a repair-associated phenotype, with increased expression of Col1a1, Col3a1, Tgfb1, and Tgfb3 (Figure 3C). CD140a+CD90−SCA-1− (blue, SP2) cells, more frequent in earlier developmental periods (Figure S4), were not responsive to injury, although they exhibited high expression levels of most evaluated transcripts (Figure 3C). These findings indicate that development-associated fibroblasts support the regenerative response, whereas fibroblast subsets abundant in adulthood signal mostly toward scar formation.
CM Proliferation Is Induced upon Cardiac Injury and Leads to Newly Formed CMs
The activation of CM proliferation after apex resection has been a matter of intense debate (Zebrowski et al., 2016). At 7 days post-injury, immunostaining for histone H3 phosphorylated at serine 10 (PH3) and s-α-Actinin highlighted cycling CMs that were distributed throughout the myocardium. Quantification of PH3+ CMs in the myocardium below the papillary muscle and in isolated CMs from the same region showed that injured hearts displayed increased rates of mitotic CMs (PH3+) in comparison with sham-operated hearts (Figures 4A and 4B). Moreover, CMs undergoing mitosis show sarcomere disruption, which is essential for effective cell division (Figures 4C and S6A; Movies S1, S2, and S3). These findings were further supported by a significant increase in CMs displaying Aurora B kinase in the nucleus and in the cleavage furrow, of the border zone and remote myocardium, of resected hearts, indicating efficient cytokinesis (Figures 4D and 4E). Close to the injury, mitotic CMs were found predominantly near FN, but not TN-C. In accordance, CD29 (β1 integrin), which forms an integral part of several integrins involved in cell-matrix interactions, was found in most CMs (Ieda et al., 2009) (Figure S6).
Figure 4.

Activation of CM Proliferation and Not Binucleation Generates a Heart with Increased CM Number upon Apex Resection
(A and B) Quantification of PH3 expression by CMs in the area between papillary muscles and the apex (A) (n = 10, 10; 7 days sham, 7 days injury) and of isolated CMs from the same region (B) (n = 12, 11; 7 days sham, 7 days injury).
(C) Representative confocal image and respective orthogonal views of a CM expressing PH3 and clearly displaying sarcomere disassembly. Scale bar, 20 μm. See also Movies S1, S2, and S3.
(D) Quantification of Aurora B expression in CMs in regions close to the apex and remote myocardium (n = 3, 3; 7 days sham, 7 days injury).
(E) Representative image of Aurora B expression in proliferating CMs at 7 days post-surgery (n = 3). Scale bar, 20 μm.
(F) Assessment of CM nucleation in the apex region at 7 days post-surgery and representative images (n = 5, 4; 7 days sham, 7 days injury).
(G) Ratio between round-shaped (immature) and rod-shaped (mature) CMs in mononucleated and multinucleated subpopulations of CMs isolated from the apex (n = 6, 5; 7 days sham, 7 days injury).
(H) Three EdU pulses (days 1, 6, and 8) were given to trace proliferating CMs. Hearts were harvested 14 days post-surgery.
(I) Representative confocal images of PCM-1+EdU+ CM 14 days post-surgery. Scale bars, 40 μm.
(J) Quantification of EdU incorporation in PCM-1+ CMs in the apical and remote myocardium (n = 4, 7; 14 days sham, 14 days injury).
(K) CM nucleus density at 14 days post-surgery (n = 4, 7; 14 days sham, 14 days injury).
(L) Representative confocal image of COLL IV expression in the myocardium, which allowed the delineation of CM cell boundaries and nucleation evaluation (n = 4, 7; 14 days sham, 14 days injury). Scale bar, 30 μm.
(M) Stereological estimate of the total number of CMs in the myocardium (n = 4, 7; 14 days sham, 14 days injury). All values are presented as means ± SD.
The loss of CM proliferative potential is commonly attributed to the progression to a terminally differentiated phenotype characterized by the presence of a mature sarcomeric apparatus and binucleation (Collesi et al., 2008). To assess whether increased rates of mitosis correlated with higher rates of CM proliferation rather than multinucleation, we assessed the number of nuclei and morphometric parameters of CMs isolated from the apex by imaging flow cytometry. Injured hearts showed less multinucleation and a significant increase of mononucleated and round-shaped CMs, attesting successful cell-cycle progression and cell division (Figures 4F and 4G).
Next, we applied stereology to establish whether the proliferative upsurge observed upon apex resection impacts on total CM numbers. The mice were treated with the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU), and the frequency of EdU incorporation in thick tissue sections was determined by co-labeling with PCM-1, a marker of CM nuclei (Figure 4H). The number of EdU+ CMs was greater in resected hearts relative to sham-operated controls (Figures 4H–4J), further supporting an increment in CM proliferation after injury. Although CM proliferation was observed throughout the LV, the frequency of EdU+ CMs was more pronounced in regions proximal to the apex, compared with remote regions (Figure 4J). In accordance, the density of myocyte nuclei was found to be significantly increased in the apex regions (Figure 4K). Combination of collagen type IV (COLL IV), to delineate cell boundaries, with PCM-1 immunolabelling, showed that CM binucleation in injured (95.7% ± 0.93%) and sham-operated hearts (95.98% ± 0.86%) was similar at 14 days after surgery (Figure 4L).
Taking multinucleation into account, we determined the density of CMs in the tissue. The volume of the LV was calculated based on morphometric analysis of longitudinal tissue sections (Figure S1A) and corrected for tissue shrinkage during fixation and processing for paraffin embedding. The total number of LV CMs in resected hearts, calculated based on CM density in the remote region, was significantly higher relative to sham-operated controls (Figure 4M). These findings show that the injury-activated CM proliferation burst, although more abundant in the apex, is also extended to remote regions, overcompensating the amount of CM loss after resection.
Resected Hearts Display Increased Capillary Density and No Signs of CM Hypertrophy or Edema
To evaluate the status of the capillary network, endothelial cells (CD31+) were quantified 60 days after surgery. The number of CD31+ endothelial cells was significantly increased in injured hearts in comparison with sham-operated animals both in the apex and in the remote myocardium (Figures 5A and 5B).
Figure 5.

Resected Hearts Display Increased Coronary Vasculature but Not Hypertrophy or Edema
(A and B) Capillary density at 60 days post-surgery (i.e., number of CD31+ endothelial cells per unit area) (n = 6, 7; 60 days sham, 60 days injury). Scale bars, 10 μm.
(C and D) CM cross-sectional area at 60 and 180 days post-surgery (n = 6, 6, 4, 4; 60 days sham, 60 days injury, 180 days sham, 180 days injury). Scale bars, 25 μm.
(E and F) Area of isolated CMs at 60 and 180 days post-surgery (n = 3, 5, 4, 4; 60 days sham, 60 days injury, 180 days sham, 180 days injury). Scale bar, 100 μm.
(G) Volume of isolated CMs at 60 days post-surgery (n = 4, 6 sham, injury).
(H) Representative images of 3D segmentation of CMs. Scale bars, 50 μm.
(I) Myocardial edema was assessed by myocardial water content quantification (i.e., percentage of myocardial weight that is lost after desiccation) 180 days post-surgery (n = 10, 6; 180 days sham, 180 days injury). All values are presented as means ± SD.
To further establish whether hypertrophy was also contributing for LV wall expansion, CM size was assessed in tissue sections and isolated CMs. No significant alterations were detected in the cross-sectional area of CMs over time and in different ventricular regions, apart from the vicinity of fibrotic tissue, where CMs had a larger cross-sectional area at 180 days post-surgery (Figures 5C and 5D). Corroborating these findings, no significant differences were found in the area and volume of isolated CMs, suggesting that this hypertrophic behavior is residual and mainly localized in the apex (Figures 5E–5H). Furthermore, as edema could also explain the increase in myocardial volume, through the accumulation of fluid in the extracellular interstitial space, we examined the myocardial water content at 180 days post-surgery, but no differences were detected between the two groups (Figure 5I). Overall, these results further support that the LV wall thickening in resected hearts results from an increase in the number of CMs and is thus a consequence of injury-activated proliferation.
Discussion
Sadek and colleagues reported that apex-resected murine hearts at P1 displayed regenerative capacity (Porrello et al., 2011). Others have thereafter described a lack of histological restoration, with formation of a fibrotic scar, ultimately leading to LV dilation and reduced systolic function (Andersen et al., 2014a, Andersen et al., 2016). We performed a detailed evaluation of cellular and histo-functional alterations up to 180 days after resection and showed a well-orchestrated response in which a combined action of regenerative and reparative mechanisms is triggered, culminating in myocardial growth alongside fibrosis deposition in the innermost part of the apex. Therefore, our results are neither compatible with full regeneration (Porrello et al., 2011) nor exclusively with repair (Andersen et al., 2014a, Andersen et al., 2016), rather showing an intermediate response which likely explains the opposing reports in the field.
As previously debated, the methodology employed during surgery and in the assessment of the extent of regeneration is determinant for obtaining reliable results and outcomes (Andersen et al., 2014b, Sadek et al., 2014). Similar to other authors (Mahmoud et al., 2014, Porrello et al., 2011), we used LV cavity exposure as a standardized method for the resection. The resultant injury, assessed by measuring the ventricular surface area, as in Porrello et al., was ∼11% of the LV and therefore less severe than the 15% reported in their study.
By quantifying (1) the incidence of sarcomere-disassembly associated with PH3, (2) EdU+ CMs in a pulse-chase experiment, and (3) Aurora B kinase in the nuclei and cleavage furrow of CMs, we showed that CMs of resected hearts exhibit increased mitotic rates, accompanied by cytokinesis. During the first week of post-natal life in mice the majority of CMs binucleate, exit from the cell cycle, and continue to grow in size (Soonpaa et al., 1996). Several lines of evidence support that proliferating CMs are mononucleated and that binucleation marks an end to cell-cycle activity during development (Bersell et al., 2009, Paradis et al., 2014). By analyzing isolated CMs by imaging flow cytometry we found that, 7 days following apex resection, CM binucleation is reduced and accompanied by an increase in the pool of mononucleated round-shaped CMs, further supporting that, in response to injury, CMs undergo karyokinesis and cytokinesis. At 14 days post-resection the levels of binucleation were already comparable with sham-operated animals, meaning that newly formed CMs follow a normal maturation progression. These results contrast with a decrease in CM mitotic activity after injury observed by Andersen et al., in which a single EdU injection was administered at the onset of surgery, most likely missing the CM proliferative burst. In a recent publication, apex resection was shown to promote a transient acceleration in binucleation, an event no longer noticeable at P8 (Zebrowski et al., 2017). According to this study, a lower number of CMs would be expected in hearts subjected to apex amputation, which was not addressed by the authors. Herein, total CM numbers, as estimated by stereology, were increased in resected hearts compared with sham controls, indicating that the majority of injury-induced CM cell-cycle activity results in proliferation, not binucleation. In the work by Zebrowski and colleagues, isolated CMs were identified by sarcomeres in phase contrast, an approach that may be adequate to identify mature (rod-shaped) CMs, but is inefficient for the identification of immature (round-shaped) CMs with less-developed sarcomeres. By bypassing the use of a CM-specific marker, the authors most likely overestimated binucleation, particularly in resected hearts, which display, according to our results, higher rates of round-shaped mononucleated CMs.
Cardiac fibroblasts are typically activated following injury, and their role in directing pathological disease remodeling is well recognized. Nonetheless, while adult fibroblasts induce hypertrophy, fetal fibroblasts promote CM proliferation and thus contribute to ventricular chamber enlargement in late development (Ieda et al., 2009). By combining the expression of three cell surface markers, we discriminated the main fibroblast subsets at different developmental stages and after cardiac injury. In response to injury, the fibroblast population, abundant at younger ages (CD140a+CD90+SCA-1−), upregulated genes associated with the production of extracellular matrix, neovascularization, and secreted factors known to activate CM proliferation. In contrast, the fibroblast subset that is more frequent during adulthood (CD140a+CD90+SCA-1+) mainly upregulated transcripts associated with fibrotic mechanisms. Altogether, these findings suggest that neonatal fibroblasts may display an intermediate phenotype between fetal and adult fibroblasts, potentially able to support both fibrogenesis and regeneration.
Earlier studies on neonatal apex resection lacked functional longitudinal studies in which the same heart is analyzed over time. Herein we paralleled the histological analysis with detailed functional evaluation by echocardiography. In contrast to Porrello et al. (2011), we observed the formation of fibrotic tissue in all analyzed hearts, which, in most cases, matured to a full-thickness scar as reported by others (Andersen et al., 2014a, Bryant et al., 2015). These contrasting observations are most likely a result of incomplete histological sampling in the former study, as shown previously (Andersen et al., 2014b). In our work, we increased sampling robustness by analyzing serial sections of the whole heart. Interestingly, while the heart weight and volume increased over time, as during normal heart growth, the scar volume diminished, indicating that the affected region was at least partially replaced by new myocardium. However, injured hearts remained shorter throughout the study, and thus the removed apex was not fully restored.
Andersen et al. (2016) performed a long-term analysis of ventricular function, using F-18-fluorodeoxyglucose positron emission tomography (FDG-PET). In this study, abnormal systolic function, short-axis enlargement and local wall thinning were demonstrated in resected hearts 180 days post-surgery, leading to the conclusion that animals developed dilated cardiomyopathy. Herein, although a tendency for reduced systolic function was evident 21 days post-surgery, no statistical differences were observed between groups over 180 days, indicating that injured hearts became functionally competent at the systolic level. Widening of resected hearts occurred through chamber expansion and LV wall thickening, which is not compatible with dilated cardiomyopathy, rather resembling an eccentric hypertrophy phenotype. The distinct results obtained from our work and Andersen et al. (2016) may reflect differences in the methodology used. While FDG-PET is particularly valuable for assessing metabolic activity, the spatial resolution of the equipment (∼1 mm) falls short to precisely quantify LV wall thickness compared with the high-resolution echocardiography (∼50 μm) (Gargiulo et al., 2012) used in our study. Moreover, a reduction in wall thickness was only significantly different in 1 of the 20 analyzed ventricular segments, which hardly explains a dilated cardiomyopathy phenotype. The authors also missed to demonstrate progressive loss of function, typically found in small animal models of this disease (Camacho et al., 2016).
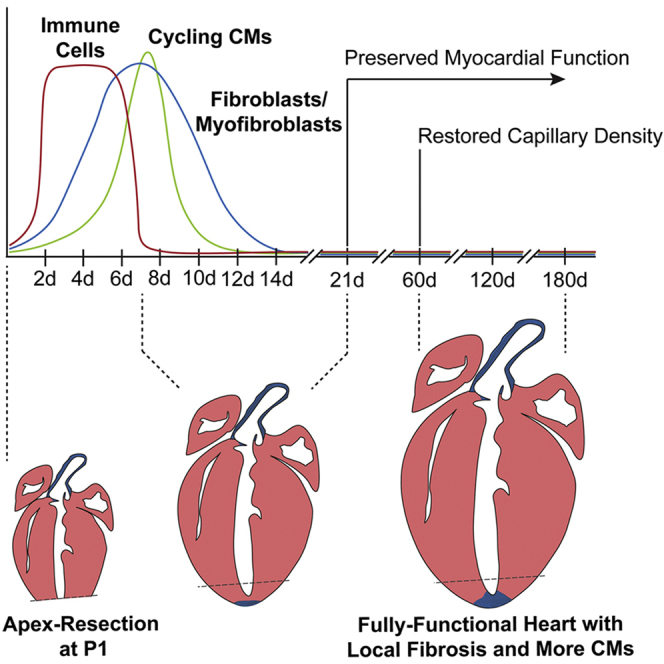
Pertinent questions have been recently raised regarding the value of neonatal apex resection to identify factors important for adult heart regeneration/repair. Although we cannot put forward conclusions on the response elicited by other injury models, our results indicate that P1 hearts subjected to mechanical trauma are capable of undergoing neomyogenesis and neovascularization, despite local fibrosis (Figure 6). In our view, these findings endorse this injury model as particularly valuable for studies aiming to block fibrosis, in favor of conditions stimulating CM proliferation and functional cardiac regeneration.
Figure 6.

Proposed Model of the Biological Response Elicited by Neonatal Apex Resection
Apex resection promotes local infiltration of inflammatory cells in the first 48 hr, which leads to the deposition of a transient FN and TN-C-rich ECM. At 7 days post-injury, rates of CM proliferation are increased throughout the left ventricular myocardium and cardiac fibroblasts are activated at the injury site. These cellular dynamics result in a thickening of left ventricle walls, de novo vessel formation and deposition of a permanent fibrotic scar at the midpoint of the injured area. Long-term evaluation showed preserved systolic function, shortened long-axis and thicker left ventricle, without hypertrophy and edema.
Experimental Procedures
Supplemental Experimental Procedures are available with a detailed description of our experimental procedures.
Animals and Apex Resection Injury Model
All animal work was approved by the IBMC-INEB (Instituto de Biologia Molecular e Celular–Instituto de Engenharia Biomédica) Animal Ethics Committee, and by the Direcção Geral de Veterinária (permit 022793), and is in conformity with Directive 2010/63/EU. Humane endpoints were followed in accordance with the OECD Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation.
P1 C57BL/6 mice were anesthetized by hypothermia for ∼3 min. Animals were laid in lateral decubitus position, the skin cut, the thorax was opened in the fourth intercostal space and the ventricular apex was resected (removing a minimal amount of tissue but ensuring lumen exposure). Subsequently, the ribs and skin were sutured and animals were exposed to heat produced by an infrared lamp to revert anesthesia and returned the progenitor. Sham-operated animals underwent the same procedure with the exception of apex resection.
Histological Characterization
At 0, 7, 14, 21, 60, 120, and 180 days after surgery, apex-resected and sham-operated hearts were harvested and processed for paraffin-embedding (see Supplemental Experimental Procedures for detailed information). Hearts were sectioned longitudinally in 3 μm sections and sampled according to Figure S1A. Subsequent histological MT staining was used to assess apex re-growth, fibrosis deposition, and injury extension.
Immunofluorescence
Immunofluorescence was performed in cryosections (0 day, 48 hr, 5 days, 7 days, 14 days, and 21 days after surgery), in histological sections (60 days post-surgery), and in cytospins of isolated CMs; as detailed in the Supplemental Experimental Procedures. Assembly, merging, and contrast/brightness adjustments were performed in Fiji v2.0.0-rc-54/.51h. software.
Flow Cytometry and Fluorescence-Activated Cell Sorting
Single-cell suspensions were prepared following digestion of cardiac tissue with crude collagenase (C2139, Sigma-Aldrich). Cells were incubated with fluorescence-conjugated antibodies or with the respective isotype controls for 30 min on ice and protected from light. Nonviable cells were excluded by adding 0.5% propidium iodide (P4170, Sigma-Aldrich) to cell suspension prior to analysis in a FACSCanto II cytometer (BD Biosciences). Data analysis was performed in FlowJo VX software. The three most frequent fibroblast subsets were isolated by FACSAria (BD Biosciences), as described in Supplemental Experimental Procedures and in Figure S5. See also Figure S3 for detailed characterization of the inflammatory infiltrate.
Real-Time qPCR
Total RNA was isolated from FACS-sorted cells using the RNeasy Mini Kit (QIAGEN). cDNA was obtained resorting to PrimeScript RT reagent Kit (Takara) and pre-amplified using SsoAdvanced PreAmp Supermix (Bio-Rad). Real-time qPCR were performed by using iQ SYBR Green Supermix (Bio-Rad) and according to the iQ5 Real-Time PCR Detection System (Bio-Rad). Primer sequences and temperature cycles are described in Supplemental Experimental Procedures. mRNA expression was defined as primer efficiency to the power of the difference in threshold cycle values between the reference (Gapdh) and the genes of interest.
EdU Pulse-Chase Assay
Neonates were subcutaneously injected with 25 μg/g of animal of EdU (C10640, Sigma-Aldrich) immediately after surgery and on days 5 and 7 after surgery. Hearts were harvested at 14 days post-surgery and LV subdivided in apex and remote myocardium, each sampled in 2 mm fragments before cryosectioning (40 μm). EdU incorporation was detected using the Click-iT reaction cocktail in accordance with manufacturer instructions (C10640, Sigma-Aldrich) followed by PCM-1 immunolabelling and DAPI counterstain. Images were acquired in a Leica TCS SP5 laser scanning confocal microscope. Tissue fragments (8–10) from both regions were quantified per animal with assistance of FIJI v2.0.0-rc-54/.51h and IMARIS 8.4.1. The total number of CMs was estimated by adapting the two-step NV × VREF method (Alkass et al., 2015), where NV is an estimate of PCM-1 density and VREF is the volume of the myocardium at 14 days post-surgery. Myocardial volume was determined by multiplying the area of the myocardium in each histological longitudinal section by the distance separating adjacent sections in 14 days post-surgery hearts. This value was corrected for tissue shrinkage during fixation and processing for paraffin embedding.
CM Isolation
CMs were isolated at 7, 60, and 180 days post-surgery. Neonatal CMs were isolated as described previously (Mollova et al., 2013), with the following alterations: CMs were fixed in 4% paraformaldehyde at room temperature (RT) for 2 hr and digested with 3 mg/mL collagenase type II (Worthington). Adult CMs were isolated by reperfusion in a Langendorff system (∼80 mmHg and at 3 mL/min for 25–30 min) with Liberase TM (05401127001, Roche; 13.3 μg/mL) and Trypsin (27250018, Gibco; 13.8 μg/mL), and fixed in 4% paraformaldehyde in PBS.
Imaging Flow Cytometry
Cells were permeabilized with 1× BD Perm/Wash buffer during 15 min at RT. Incubations with primary and secondary antibodies lasted 2 and 0.5 hr on ice, respectively. Antibodies were diluted in 1× BD Perm/Wash. Immediately before acquisition in ImageStreamX cells were filtered and nuclei stained with 200 μM DRAQ5 (BioStatus).
Statistical Analysis
IBM SPSS Statistics 24 was used for statistical analysis. Shapiro-Wilk test was used to evaluate normal distribution of data. Outliers were excluded from the statistical analysis. Homoscedasticity of the sample was tested by Levene's test, which defined the statistical test(s) applied. Normally distributed and homocedastic data were tested with independent sample Student's t test and one-way ANOVA for two or three or more groups, respectively. Tukey's post-hoc test for correction of multiple comparisons was performed whenever significant differences were found between three or more groups.
Non-normally distributed and/or heterocedastic data were tested with Mann-Whitney U test and Kruskal-Wallis one-way ANOVA for two or three or more groups, respectively. Again, multiple comparisons were performed when three or more groups were considered statistically different. The statistical significance level chosen for all statistical tests was p < 0.05.
Author Contributions
V.S.-P. conceived, designed, and performed the experiments, analyzed and interpreted data, and wrote the manuscript. S.C.R., T.L.L., E.D.S., F.V.-N., A.C.S., R.J.C., N.P., T.P.R., and G.D. performed the experiments and analyzed and interpreted data. S.T. and A.L.-M. interpreted data and revised the manuscript. P.P.-Ó. conceived the study and interpreted data, revised the manuscript, and provided funding. D.S.N. conceived, designed, and performed the experiments, analyzed and interpreted data, revised the manuscript, and provided funding. All authors have read and approved the manuscript.
Acknowledgments
The authors acknowledge the support of i3S scientific platforms (animal facility, ALM, HEMS, BSU, b.IMAGE, CCGEN, TraCy). The authors are thankful to current and past members of Pinto-do-Ó laboratory for the critical discussion. This work was financed by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF) (NORTE-01-0145-FEDER-000012); by European Structural and Investment Funds (ESIF), under Lisbon Portugal Regional Operational Programme and National Funds through FCT (Fundação para a Ciência e Tecnologia [Foundation for Science and Technology] [POCI-01-0145-FEDER-016385]); by INFARMED – Autoridade Nacional do Medicamento e Produtos de Saúde, I.P. (FIS-FIS-2015-01_CCV_20150630-157); and by FCT/Ministério da Ciência, Tecnologia e Inovação in the framework of the project “Institute for Research and Innovation in Health Sciences” (POCI-01-0145- FEDER-007274) and individual fellowships: SFRH/BD/111799/2015 to V.S.-P., PD/BD/127997/2016 to T.L.L., SFRH/BD/88780/2012 to A.C.S., and SFRH/BPD/80588/2011 to T.P.R.
Published: March 1, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, one table, and three movies and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.01.042.
Supplemental Information
References
- Alkass K., Panula J., Westman M., Wu T.D., Guerquin-Kern J.L., Bergmann O. No evidence for cardiomyocyte number expansion in preadolescent mice. Cell. 2015;163:1026–1036. doi: 10.1016/j.cell.2015.10.035. [DOI] [PubMed] [Google Scholar]
- Andersen D.C., Ganesalingam S., Jensen C.H., Sheikh S.P. Do neonatal mouse hearts regenerate following heart apex resection? Stem Cell Reports. 2014;2:406–413. doi: 10.1016/j.stemcr.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen D.C., Jensen C.H., Sheikh S.P. Response to Sadek et al. and Kotlikoff et al. Stem Cell Reports. 2014;3:3–4. doi: 10.1016/j.stemcr.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen D.C., Jensen C.H., Baun C., Hvidsten S., Zebrowski D.C., Engel F.B., Sheikh S.P. Persistent scarring and dilated cardiomyopathy suggest incomplete regeneration of the apex resected neonatal mouse myocardium – a 180 days follow up study. J. Mol. Cell. Cardiol. 2016;90:47–52. doi: 10.1016/j.yjmcc.2015.11.031. [DOI] [PubMed] [Google Scholar]
- Aurora A.B., Porrello E.R., Tan W., Mahmoud A.I., Hill J.A., Bassel-Duby R., Sadek H.A., Olson E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Invest. 2014;124:1382–1392. doi: 10.1172/JCI72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassat E., Mutlak Y.E., Genzelinakh A., Shadrin I.Y., Baruch Umansky K., Yifa O., Kain D., Rajchman D., Leach J., Riabov Bassat D. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature. 2017;547:179–184. doi: 10.1038/nature22978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersell K., Arab S., Haring B., Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- Bryant D.M., O'Meara C.C., Ho N.N., Gannon J., Cai L., Lee R.T. A systematic analysis of neonatal mouse heart regeneration after apical resection. J. Mol. Cell. Cardiol. 2015;79:315–318. doi: 10.1016/j.yjmcc.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho P., Fan H., Liu Z., He J.Q. Small mammalian animal models of heart disease. Am. J. Cardiovasc. Dis. 2016;6:70–80. [PMC free article] [PubMed] [Google Scholar]
- Collesi C., Zentilin L., Sinagra G., Giacca M. Notch1 signaling stimulates proliferation of immature cardiomyocytes. J. Cell Biol. 2008;183:117–128. doi: 10.1083/jcb.200806091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Uva G., Aharonov A., Lauriola M., Kain D., Yahalom-Ronen Y., Carvalho S., Weisinger K., Bassat E., Rajchman D., Yifa O. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015;17:627–638. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- Darehzereshki A., Rubin N., Gamba L., Kim J., Fraser J., Huang Y., Billings J., Mohammadzadeh R., Wood J., Warburton D. Differential regenerative capacity of neonatal mouse hearts after cryoinjury. Dev. Biol. 2015;399:91–99. doi: 10.1016/j.ydbio.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flink I.L. Cell cycle reentry of ventricular and atrial cardiomyocytes and cells within the epicardium following amputation of the ventricular apex in the axolotl, Amblystoma mexicanum: confocal microscopic immunofluorescent image analysis of bromodeoxyuridine-labeled nuclei. Anat. Embryol. 2002;205:235–244. doi: 10.1007/s00429-002-0249-6. [DOI] [PubMed] [Google Scholar]
- Furtado M.B., Costa M.W., Pranoto E.A., Salimova E., Pinto A.R., Lam N.T., Park A., Snider P., Chandran A., Harvey R.P. Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ. Res. 2014;114:1422–1434. doi: 10.1161/CIRCRESAHA.114.302530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargiulo S., Greco A., Gramanzini M., Petretta M.P., Ferro A., Larobina M., Panico M., Brunetti A., Cuocolo A. PET/CT imaging in mouse models of myocardial ischemia. J. Biomed. Biotechnol. 2012;2012:541872. doi: 10.1155/2012/541872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubner B.J., Adamowicz-Brice M., Khadayate S., Tiefenthaler V., Metzler B., Aitman T., Penninger J.M. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging. 2012;4:966–977. doi: 10.18632/aging.100526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heallen T., Morikawa Y., Leach J., Tao G., Willerson J.T., Johnson R.L., Martin J.F. Hippo signaling impedes adult heart regeneration. Development. 2013;140:4683–4690. doi: 10.1242/dev.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudon-David F., Bouzeghrane F., Couture P., Thibault G. Thy-1 expression by cardiac fibroblasts: lack of association with myofibroblast contractile markers. J. Mol. Cell. Cardiol. 2007;42:991–1000. doi: 10.1016/j.yjmcc.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Ieda M., Tsuchihashi T., Ivey K.N., Ross R.S., Hong T.T., Shaw R.M., Srivastava D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell. 2009;16:233–244. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesty S.A., Steffey M.A., Lee F.K., Breitbach M., Hesse M., Reining S., Lee J.C., Doran R.M., Nikitin A.Y., Fleischmann B.K. c-kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc. Natl. Acad. Sci. USA. 2012;109:13380–13385. doi: 10.1073/pnas.1208114109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlikoff M.I., Hesse M., Fleischmann B.K. Comment on "Do neonatal mouse hearts regenerate following heart apex resection"? Stem Cell Reports. 2014;3:2. doi: 10.1016/j.stemcr.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine K.J., Epelman S., Uchida K., Weber K.J., Nichols C.G., Schilling J.D., Ornitz D.M., Randolph G.J., Mann D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA. 2014;111:16029–16034. doi: 10.1073/pnas.1406508111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud A.I., Kocabas F., Muralidhar S.A., Kimura W., Koura A.S., Thet S., Porrello E.R., Sadek H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–253. doi: 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud A.I., Porrello E.R., Kimura W., Olson E.N., Sadek H.A. Surgical models for cardiac regeneration in neonatal mice. Nat. Protoc. 2014;9:305–311. doi: 10.1038/nprot.2014.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQualter J.L., Brouard N., Williams B., Baird B.N., Sims-Lucas S., Yuen K., Nilsson S.K., Simmons P.J., Bertoncello I. Endogenous fibroblastic progenitor cells in the adult mouse lung are highly enriched in the sca-1 positive cell fraction. Stem Cells. 2009;27:623–633. doi: 10.1634/stemcells.2008-0866. [DOI] [PubMed] [Google Scholar]
- Mollova M., Bersell K., Walsh S., Savla J., Das L.T., Park S.Y., Silberstein L.E., Dos Remedios C.G., Graham D., Colan S. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA. 2013;110:1446–1451. doi: 10.1073/pnas.1214608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarian D., Benjamin E.J., Go A.S., Arnett D.K., Blaha M.J., Cushman M., Das S.R., de Ferranti S., Despres J.P., Fullerton H.J. Heart disease and stroke statistics-2016 update: a report from the American heart association. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- Paradis A.N., Gay M.S., Zhang L. Binucleation of cardiomyocytes: the transition from a proliferative to a terminally differentiated state. Drug Discov. Today. 2014;19:602–609. doi: 10.1016/j.drudis.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello E.R., Mahmoud A.I., Simpson E., Hill J.A., Richardson J.A., Olson E.N., Sadek H.A. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello E.R., Mahmoud A.I., Simpson E., Johnson B.A., Grinsfelder D., Canseco D., Mammen P.P., Rothermel B.A., Olson E.N., Sadek H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA. 2013;110:187–192. doi: 10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss K.D., Wilson L.G., Keating M.T. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- Robledo M. Myocardial regeneration in young rats. Am. J. Pathol. 1956;32:1215–1239. [PMC free article] [PubMed] [Google Scholar]
- Sadek H.A., Martin J.F., Takeuchi J.K., Leor J., Nie Y., Giacca M., Lee R.T. Multi-investigator letter on reproducibility of neonatal heart regeneration following apical resection. Stem Cell Reports. 2014;3:1. doi: 10.1016/j.stemcr.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.L., Baek S.T., Sung C.Y., Tallquist M.D. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 2011;108:e15–e26. doi: 10.1161/CIRCRESAHA.110.235531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soonpaa M.H., Kim K.K., Pajak L., Franklin M., Field L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 1996;271:H2183–H2189. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- Souders C.A., Bowers S.L., Baudino T.A. Cardiac fibroblast: the renaissance cell. Circ. Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball J.G. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 2011;1:2029–2062. doi: 10.1002/cphy.c100092. [DOI] [PubMed] [Google Scholar]
- Uygur A., Lee R.T. Mechanisms of cardiac regeneration. Dev. Cell. 2016;36:362–374. doi: 10.1016/j.devcel.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebrowski D.C., Becker R., Engel F.B. Towards regenerating the mammalian heart: challenges in evaluating experimentally induced adult mammalian cardiomyocyte proliferation. Am. J. Physiol. Heart Circ. Physiol. 2016;310:H1045–H1054. doi: 10.1152/ajpheart.00697.2015. [DOI] [PubMed] [Google Scholar]
- Zebrowski D.C., Jensen C.H., Becker R., Ferrazzi F., Baun C., Hvidsten S., Sheikh S.P., Polizzotti B.D., Andersen D.C., Engel F.B. Cardiac injury of the newborn mammalian heart accelerates cardiomyocyte terminal differentiation. Sci. Rep. 2017;7:8362. doi: 10.1038/s41598-017-08947-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.