

ABSTRACT
Human cytomegalovirus (HCMV) is a widespread human pathogen that causes asymptomatic infection in healthy individuals but poses a serious threat to immunocompromised patients. During the late phase of HCMV infection, the viral capsid is transported to the cytoplasmic viral assembly center (cVAC), where it is enclosed by the tegument protein layer and the viral envelope. The cVAC consists of circularly arranged vesicles from the trans-Golgi and endosomal networks. The HCMV gene UL35 encodes ppUL35 and its shorter form, ppUL35A. We have previously shown that the UL35 gene is involved in HCMV assembly, but it is unknown how UL35 proteins regulate viral assembly. Here we show that sorting nexin 5 (SNX5), a component of the retromer and part of the retrograde transport pathway, interacts with UL35 proteins. Expression of wild-type proteins but not mutants defective in SNX5 binding resulted in the cellular redistribution of the cation-independent mannose-6-phosphate receptor (CI-M6PR), indicating that UL35 proteins bind and negatively regulate SNX5 to modulate cellular transport pathways. Furthermore, binding of UL35 proteins to SNX5 was required for efficient viral replication and for transport of the most abundant HCMV glycoprotein B (gB; gpUL55) to the cVAC. These results indicate that ppUL35 and ppUL35A control the localization of the essential gB through the regulation of a retrograde transport pathway. Thus, this work is the first to define a molecular interaction between a tegument protein and a vesicular transport factor to regulate glycoprotein localization.
IMPORTANCE Human cytomegalovirus is ubiquitously present in the healthy population, but reactivation or reinfection can cause serious, life-threatening infections in immunocompromised patients. For completion of its lytic cycle, human cytomegalovirus induces formation of an assembly center where mature virus particles are formed from multiple viral proteins. Viral glycoproteins use separate vesicular pathways for transport to the assembly center, which are incompletely understood. Our research identified a viral structural protein which affects the localization of one of the major glycoproteins. We could link this change in glycoprotein localization to an interaction of the structural protein with a cellular protein involved in regulation of vesicle transport. This increases our understanding of how the virus intersects into cellular regulatory pathways to enhance its own replication.
KEYWORDS: UL35, cytomegalovirus, glycoprotein B, sorting nexin 5
INTRODUCTION
Human cytomegalovirus (HCMV) is a significant clinical pathogen with a prevalence of nearly 70% in the human population. Whereas infection is asymptomatic for healthy individuals, infection of immunologically naive or immunocompromised individuals may cause life-threatening disease. HCMV belongs to the Betaherpesvirinae subfamily of herpesviruses (1) and, like all herpesviruses, establishes a lifelong persistence. The virion consists of an envelope with viral glycoprotein complexes, an icosahedral capsid enclosing the linear DNA genome of about 235 kb, and the intervening tegument layer. Currently, there are approximately 38 identified HCMV tegument proteins (2). The tegument proteins fulfill multiple functions during the replication cycle, such as immune modulation, trans-activation, anti-apoptosis, or virion maturation (2, 3). The major components of the tegument are the phosphoproteins ppUL32 (pp150), ppUL82 (pp71), and ppUL83 (pp65).
Previously, the tegument protein ppUL35 was described as an important activator of the HCMV major immediate early enhancer/promoter (MIEP) in cooperation with ppUL82 (4, 5) and an important factor for virus maturation (6). The UL35 gene encodes two proteins, the full-length ppUL35 (72 kDa) and the shorter form, ppUL35A (25 kDa), which corresponds to the C terminus of ppUL35 and is translated from a separate mRNA (4). Both forms can interact physically with each other as well as with other viral and cellular proteins (5, 7), but only full-length ppUL35 is packed into the virus particles (4). Deletion of the UL35 gene led to a multiplicity of infection (MOI)-dependent replication defect of the mutant virus, indicating a role in immediate early regulation (6). Additionally, this virus mutant showed a much stronger defect in virus assembly, with a disturbed localization of ppUL82, ppUL83, and gB during the late phase. Overexpression of ppUL35, but not UL25A, induced the formation of nuclear bodies (7). Moreover, ppUL35 was shown to be able to form nuclear bodies independently from promyelocytic leukemia protein (PML) and recruited nuclear proteins, such as Daxx, SP100, and PML, into these nuclear bodies (7). On the other hand, ppUL35A inhibited nuclear body formation when coexpressed with ppUL35 (7). It is assumed that formation of ppUL35-nuclear bodies and recruitment of the above-mentioned host cell proteins support viral replication by regulating the function of these proteins. Recent studies identified DCAF1, a member of the Cul4 DNA damage response complex, to be a cellular interactor of ppUL35, but not ppUL35A (8). Studies in transiently transfected cells showed that DCAF1 is recruited into ppUL35-containing nuclear bodies. Cells overexpressing ppUL35 exhibit activation of the DNA damage repair response and accumulate in the G2 phase (8). These findings support the suggestion that ppUL35 promotes viral replication by manipulating the host cell DNA repair response during the immediate early and early phase of infection.
HCMV-infected cells are subjected to an extensive reorganization of the secretory and endosomal transport routes to create the cytoplasmic viral assembly center (cVAC), in which tegumentation and the final envelopment take place. The cVAC has a concentric arrangement of compartments derived from the Golgi apparatus, the trans-Golgi network (TGN), and endosomal vesicles (9, 10). In the two-step model of herpesvirus envelopment, the capsid migrates from the nucleus to the cVAC, receives the tegument layer, and is surrounded by the envelope after budding into vesicles of the TGN (11). Gene expression analysis of HCMV-infected cells (12, 13) and in vitro experiments (14–17) revealed the extensive influence on the cellular transport machinery by HCMV that contributes to virus maturation. Tegument proteins and posttranslational modulation of cellular proteins have been implicated in the formation of the cVAC (18, 19). Inhibition or depletion of cellular proteins or viral proteins involved in virus maturation and/or cVAC structure formation led to a reduction of infectious virus yield (14–18, 20, 21).
The main HCMV glycoprotein complexes, gcI (gB), gcII (gM [gpUL100] and gN [gpUL73]), and gcIII (gH [gpUL75], gL [gpUL115], and gO [gpUL74]), have important functions during attachment and entry of HCMV into the host cell, in immune evasion, and as determinants of host cell tropism (22, 23). One of the most abundant HCMV glycoproteins, gB, is posttranslationally processed in the endoplasmic reticulum and the Golgi apparatus (24, 25), transported along the secretory pathway to the plasma membrane, and internalized into the early endosome. The path of internalization is at present unclear, since inhibition of endocytosis with dominant negative dynamin I had no influence on virus production (26). Finally, gB is transported to cVAC vesicles, dependent on the cellular connector protein PACS-1 (27).
Sorting nexins (SNXs) are evolutionarily conserved from yeast to mammals and comprise a family of proteins involved in cargo recognition and sorting during retrograde transport from the early endosome to the Golgi apparatus (28). Mammalian genomes encode 33 SNXs (29). All SNXs share the phox homology domain (PX), which binds to phosphatidylinositol phosphates, which are characteristic for the early endosomal membrane network. SNXs can be further subdivided into the SNX-BAR subfamily, whose members contain the Bin-amphiphysin-Rvs (BAR) domain, which is able to sense highly curved membranes (30). The second subfamily comprises the PX-only SNXs, and the third subfamily consists of SNXs with different other additional protein domains. SNX1, SNX2, SNX5, SNX6, and SNX32 form the membrane deformation subcomplex of the retromer (31–33), while the cargo-selective subcomplex formed by VPS29, VPS26A/B, and VPS35 is responsible for cargo selection (34–36). The main function of the retromer complex is selecting cargo proteins for retrograde transport from early endosomes to the Golgi apparatus. Depletion of one member of the SNX subcomplex results in a disturbed recycling of certain cargo proteins, such as the cation-independent mannose-6-phosphate receptor (CI-M6PR), and an altered structure of the TGN (32, 33).
In this study, we show for the first time a physical interaction between the HCMV tegument protein ppUL35 and SNX5. UL35 proteins negatively regulated SNX5 function, and SNX5 overexpression reduced the virus yield. Virus with mutations in UL35 produced less progeny virus, which could be alleviated by SNX5 knockdown. These changes correlated with an altered subcellular localization of gB.
RESULTS
Identification of SNX5 as interaction partner of UL35.
To identify host cell factors interacting with the UL35 proteins, we performed a yeast (Saccharomyces cerevisiae) two-hybrid screen using full-length UL35 fused to the Gal4 DNA-binding domain as bait. From a human B-cell library we analyzed 73 candidate clones. The most abundantly represented gene was SNX5, which was isolated in five independent clones. To verify the interaction in human cells, we transfected 293T cells with expression plasmids for FLAG-tagged SNX5 or SNX16 and either full-length UL35 or UL35A. After precipitation with the monoclonal antibody M2, directed against the FLAG epitope, coprecipitated ppUL35 proteins were detected with a specific rabbit antiserum. As shown in Fig. 1A, both forms could be coprecipitated with SNX5 but not with SNX16. To detect this interaction in infected cells, we first confirmed that SNX5 is expressed in cells or cell lines susceptible to HCMV. Endogenous SNX5 could be detected in human foreskin fibroblasts (HFF), retina pigment epithelial cells (RPE), and endothelial cells (EA-hy; telomerase-immortalized microvascular endothelial [TIME] cells) (Fig. 1B). Using extracts prepared from HFF cells infected with HCMV strain TB40E-BAC4 (wild-type [wt] strain TB4 [TB4-wt]), we were able to precipitate UL35 with an anti-SNX5 serum also from infected cells (Fig. 1C). In addition, we analyzed the levels of SNX5 during the course of infection. As shown in Fig. 1D, no major changes in SNX5 protein levels were detected; a slight increase of SNX5 at late time points was not consistently observed. In summary, SNX5 was identified as a novel interaction partner for both UL35 protein forms.
FIG 1.

Interaction of UL35 proteins with sorting nexin 5. (A) 293T cells were transfected with expression plasmids for ppUL35, ppUL35A, and FLAG-SNX5 or FLAG-SNX16, as indicated. After 48 h, cell extracts were precipitated with mouse monoclonal antibodies against FLAG (M2). Precipitated proteins were separated by polyacrylamide gel electrophoresis, and coprecipitated UL35 proteins were detected with a specific rabbit antiserum. Expression of the respective proteins in the cell lysates was controlled by immunoblotting (bottom). (B) Cell lines permissive for HCMV were tested for endogenous expression of SNX5. Cell lysates prepared from equal numbers of cells (4 × 104) were subjected to SDS-PAGE and immunoblotting. Endogenous SNX5 was detected by a polyclonal goat anti-SNX5 serum. (C) Coimmunoprecipitation of endogenous proteins from infected cells. HFF were mock treated or infected with TB4-wt at an MOI of 1 and harvested at 120 h postinfection. Cell lysates prepared from 6 × 106 cells were subjected to immunoprecipitation with goat anti-SNX5 serum. Precipitates and lysates were separated by SDS-PAGE and blotted, and proteins were detected with rabbit anti-UL35 serum as the primary antibody. (D) Kinetics of SNX5 expression during infection. HFF cells were mock treated or infected with TB4-wt at an MOI of 1, and cell lysates were prepared at the indicated time points (in hours postinfection). After SDS-PAGE and immunoblotting, proteins were detected with goat anti-SNX5 (top) or anti-β-actin primary antibodies.
Interference of UL35 proteins with SNX5 functions.
SNX5 plays a role in retrograde transport of proteins from early endosomes to the Golgi apparatus (32, 37) and has been shown to be important for macropinocytosis (38, 39). To assess the functional consequences of the interaction between SNX5 and UL35 proteins, we first performed macropinocytosis assays using HeLa cells transfected with eukaryotic expression plasmids coding for FLAG-SNX5 in combination with plasmids expressing UL35 proteins or an empty plasmid. Transfection of SNX5 was necessary to obtain a sufficient basal rate of macropinocytosis (38). After serum starvation, the cells were incubated with epidermal growth factor (EGF) in order to stimulate macropinocytosis and fluorescein isothiocyanate (FITC)-labeled dextran. Uptake of dextran was measured by flow cytometry. As shown in Fig. 2A, expression of SNX5 strongly stimulated the uptake of dextran. Additional expression of both ppUL35 and ppUL35A reproducibly reduced dextran uptake, with ppUL35 having a stronger and statistically significant effect. Similar results but with lower overall uptake rates were obtained in experiments without addition of EGF (data not shown).
FIG 2.
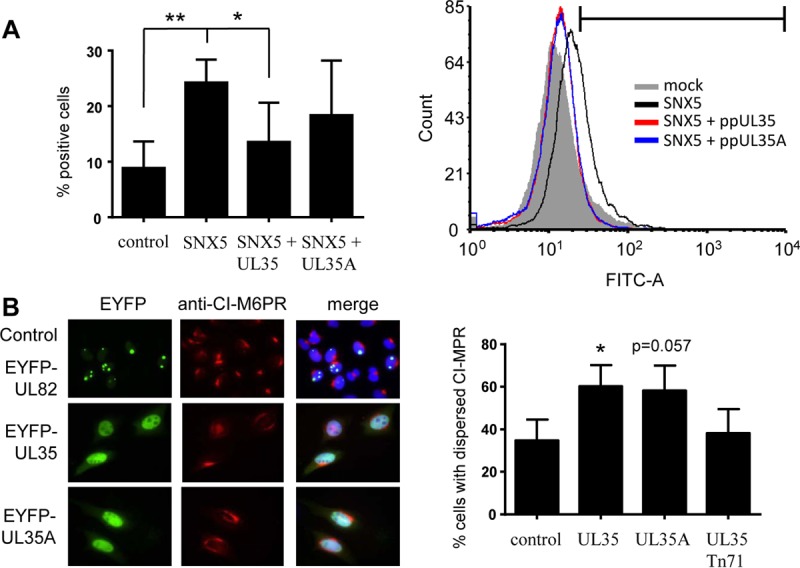
Functional interaction of UL35 proteins with sorting nexin 5. (A) In a macropinocytosis assay, HeLa cells were transfected with an expression plasmid for FLAG-SNX5 and an expression plasmid for ppUL35 or ppUL35A. At 24 h after transfection, the cells were serum starved for 12 h. Cells were incubated for 3 min with EGF (to increase the basal macropinocytosis rate) and FITC-dextran and subsequently detached and fixed with paraformaldehyde. Uptake of FITC-dextran was measured by quantitation of positive cells by flow cytometry. Results are representative of those from at least 3 independent uptake assays; error bars indicate standard deviations. Significance was calculated employing a two-tailed Student's t test with the Welch correction. (B) To analyze the effects of UL35 proteins on the subcellular localization of CI-M6PR, HeLa cells were transfected with expression plasmids for EYFP-UL35 or EYFP-UL35A or for EYFP-UL82 as a control. At 24 h after transfection, cells were fixed with paraformaldehyde and stained with a specific antibody directed against CI-M6PR. At least 100 cells were scored for compact or dispersed localization. Error bars indicate standard deviations. Similar results were obtained in 3 independent experiments. Significance was calculated employing a two-tailed Student's t test with the Welch correction. *, P ≤ 0.05; **, P ≤ 0.01.
Next, we investigated the localization of the cation-independent mannose-6-phosphate receptor (CI-M6PR), which in untreated cells is concentrated in the TGN. Previously, it has been published that small interfering (siRNAs) against SNX5 and SNX6 induced a dispersed localization of CI-M6PR (32, 40). To analyze whether the UL35 proteins modulate CI-M6PR localization, we transfected HeLa cells with plasmids expressing enhanced yellow fluorescent protein (EYFP)-UL35 or EYFP-UL35A fusion constructs or with a control construct expressing EYFP-UL82. As shown in Fig. 2B, both UL35 fusion proteins induced a disperse localization of CI-M6PR, which was not observed for the UL82 fusion protein. Quantification of transfected cells with compact or disperse localization demonstrated a significant increase in the percentage of disperse localization of CI-M6PR in the presence of ppUL35 (P = 0.034) and a clear increase in the presence of ppUL35A (P = 0.057). Altogether, both dextran uptake and CI-M6PR localization experiments suggested that UL35 proteins functionally inactivate SNX5.
Mutagenesis of UL35.
We subsequently set out to identify a UL35 mutant which is unable to interact with SNX5 to functionally analyze the consequences of the interaction between UL35 and SNX5. For this purpose, we generated a UL35 expression plasmid library with pentapeptide insertions in the UL35 protein by transposon mutagenesis. Since both ppUL35 and ppUL35A interacted with SNX5, we concentrated on insertions in the C-terminal region corresponding to the UL35A coding sequence. Out of 100 mutated plasmids, we identified 22 mutants with insertions in this region, which were tested for SNX5 binding by coimmunoprecipitation (co-IP). One insertion mutant, ppUL35-Tn71, that did not precipitate with ppUL35 was identified (Fig. 3A), whereas a mutant with an insertion (Tn72) close by showed no change in binding to SNX5. As another interaction partner of ppUL35, the viral ppUL82, which also binds the C-terminal region of ppUL35 (5), we analyzed the capability of the Tn71 mutant to interact with ppUL82 to verify that the pentapeptide insertion did not disrupt the overall structure of ppUL35. As demonstrated in Fig. 3B, the ppUL35-Tn71 mutant could be precipitated with ppUL82 to the same extent as wild-type ppUL35. Since ppUL35 and ppUL82 cooperatively transactivate the major immediate early enhancer/promoter (MIEP) (4, 5), we analyzed the trans-activating properties of ppUL35 and the ppUL35-Tn71 mutant. After transfection of U373 cells with a luciferase reporter under the control of the major immediate early enhancer/modulator and effector plasmids, we observed a weak activation by both wild-type ppUL35 and mutant ppUL35-Tn71 (Fig. 3C), demonstrating that the trans-activating properties were not changed in the Tn71 mutant. More importantly, both wild-type ppUL35 and mutant ppUL35-Tn71 demonstrated strong cooperative transactivation together with ppUL82 (Fig. 3C), albeit we noted a significant difference between UL35 and UL35-Tn71 in cooperative activation. These results demonstrate that we identified a pentapeptide insertion mutant that is affected in its interaction with SNX5 but not ppUL82 and only slightly impaired in transactivating properties.
FIG 3.

Characterization of a pentapeptide insertion mutant of ppUL35. (A) 293T cells were transfected with expression plasmids for the ppUL35 pentapeptide insertion mutant Tn71 or Tn72 as well as FLAG-SNX5. After 24 h, cell extracts were precipitated with mouse monoclonal antibody against the FLAG (M2) epitope. Precipitated proteins were separated by polyacrylamide gel electrophoresis, and coprecipitated UL35 proteins were detected with a specific rabbit antiserum. Expression of the respective proteins in the cell extracts (lysate controls) was controlled by immunoblotting (bottom). (B) 293T cells were transfected with expression plasmids for wild-type ppUL35 or the Tn71 mutant as well as ppUL82. After 24 h, cell extracts were precipitated with mouse monoclonal antibodies against the ppUL82 (CMV355) or Myc (9E10) epitope. Precipitated proteins were separated by polyacrylamide gel electrophoresis, and coprecipitated UL35 proteins were detected with a specific rabbit antiserum. (C) U373 cells were transfected in triplicate with the HCMV modulator/enhancer luciferase reporter plasmid pHM287 and effector plasmids expressing the indicated genes. All samples contained the same total amount of DNA. After 24 h, luciferase activity was measured. Results are representative of those from 2 or 3 independent experiments. Significance was calculated employing two-tailed Student's t test. *, P ≤ 0.05; ***, P ≤ 0.001; n.s., not significant. (D) HeLa cells were transfected with empty vector as a control or the expression plasmids for wild-type ppUL35 or the ppUL35-Tn71 mutant. At 24 h after transfection, cells were fixed with paraformaldehyde and stained with a rabbit serum directed against ppUL35 and a mouse monoclonal antibody directed against CI-M6PR to assess the subcellular distribution pattern. Two additional experiments yielded comparable results.
Finally, we addressed whether mutant ppUL35-Tn71 was still able to functionally inactivate SNX5. For this purpose, we transfected HeLa cells and subsequently analyzed the localization of CI-M6PR by immunofluorescence. Similarly to control cells, the ppUL35-Tn71-expressing cells showed a compact distribution of CI-M6PR (Fig. 3D), in contrast to the dispersed distribution after transfection of wild-type UL35 proteins (see also Fig. 2B). Quantification of ppUL35-Tn71-expressing cells with compact or disperse CI-M6PR localization showed no difference (P = 0.712) from ppUL82-expressing control cells (Fig. 2C). Therefore, the interaction of UL35 proteins with SNX5 is necessary for the functional inactivation of SNX5.
Mutation of UL35 in the context of the viral genome.
To investigate the role of the ppUL35/SNX5 interaction during viral infection, we constructed several virus mutants in the context of the clinical isolate TB40E using the bacterial artificial chromosome (BAC) TB40E-BAC4 (Fig. 4A). First, we mutated the start codon of UL35 by en passant mutagenesis (41) to obtain TB4-mUL35-M1A. A revertant of this virus was subsequently generated by en passant mutagenesis and named TB4-mUL35-rev. Several further mutants were created by first replacing most of the UL35 open reading frame (ORF) with a kanamycin resistance cassette and a neighboring I-SceI recognition site to generate TB4-UL35-kan-in, which also served as a UL35 knockout mutant. The 50 nucleotides at the start and end of the UL35 gene remained in the genome and served as bridgeheads to reinsert wild-type or modified UL35 genes by recET-based recombination (6). Insertion of the wild-type UL35 gene generated the revertant virus TB4-UL35-kan-in-rev. To construct a virus which lacked expression of ppUL35A, we mutated three methionines (M448, 449, 465A) and obtained TB4-mUL35A. Finally, we transferred the UL35-Tn71 mutant into the virus genome, resulting in TB4-mUL35-Tn71.
FIG 4.

Characterization of HCMV mutants. (A) Generation of mutations in the context of the cloned wild-type genome TB40E-BAC4. The UL35 ORF is shown in gray, with the start codons of ppUL35 and ppUL35A indicated by arrows. Mutation of the UL35 start codon was achieved by en passant mutagenesis (TB4-mUL35-M1A), and a revertant (TB4-mUL35-rev; not shown) was also constructed. Other mutants were generated via marker replacement. First, a marker cassette with kanamycin resistance (black square) and an I-SceI site (white triangle) was inserted into the UL35 ORF (TB4-UL35-kan-in). In a second step, the marker cassette was replaced by wild-type UL35 (TB4-UL35-kan-in-rev; not shown), a triple mutant abolishing UL35A translation (TB4-mUL35A), or the pentapeptide insertion mutant Tn71 (TB4-mUL35-Tn71; the pentapeptide insertion indicated by a black diamond). (B) HFF cells infected with the indicated viruses were harvested at 96 h postinfection, and cell extracts were subjected to immunoblotting. Rabbit antisera directed against ppUL35 or ppUL82 or mouse monoclonal antibodies directed against glycoprotein gB (27-287) or β-actin (13E5) were used to assess protein expression. The numbers on the right are molecular mass markers (in kilodaltons). (C) HFF cells were infected with the indicated viruses at an MOI of 1 (left and right) or an MOI of 0.01 (middle). The supernatant was harvested at the indicated time points and titrated on HFF cells.
All these viruses could be reconstituted after transfection of the respective BACs into HFF; however, reconstitution of TB4-UL35-kan-in and TB4-mUL35A appeared to be much slower than that of the other viruses. We first analyzed the expression of the UL35 proteins in HFF cells infected with the respective viruses. The viruses TB4-mUL35-M1A and TB4-mUL35A lacked expression of ppUL35 and ppUL35A, respectively. All other viruses expressed both forms of the UL35 protein. In addition, expression of several other major structural proteins, such as ppUL82 or gB, could be detected (Fig. 4B).
The replication kinetics of the respective viruses revealed that the mutant virus TB4-mUL35-M1A showed a slightly delayed replication, which was more pronounced when the replication was examined at a low MOI (Fig. 4C, left and middle). In contrast, the deletion mutant virus TB4-UL35-kan-in showed a significantly reduced replication of more than 1 log. When the TB4-mUL35A virus was analyzed, it exhibited a defect similar to that of the knockout virus TB4-UL35-kan-in (Fig. 4C, right). The virus mutant TB4-mUL35-Tn71, which expressed UL35 proteins with the pentapeptide insertion, showed an intermediate phenotype. In summary, virus mutants which lacked expression of ppUL35A showed a significant reduction in virus replication, whereas the pentapeptide insertion virus mutant TB4-mUL35-Tn71 had an intermediate phenotype.
Since UL35 proteins affected intracellular transport processes regulated by SNX5, we suspected that the localization of structural proteins, especially glycoproteins, should be affected in cells infected with the mutant viruses. Therefore, we infected HFF cells with the virus mutants at the same MOI and analyzed the localization of several viral glycoproteins (Fig. 5A). In cells infected with wild-type virus TB4-wt, all glycoproteins investigated showed a focused localization at the assembly center. For the glycoproteins gH, gM, and gN, this focused localization was also detected when cells were infected with the mutant viruses. In contrast, gB displayed a clearly dispersed localization after infection with mutant viruses. When we analyzed the localization of major tegument proteins in cells infected with wild-type virus or virus mutant TB4-mUL35-Tn71, all showed a cytoplasmic localization (Fig. 5B). In summary, when the interaction of ppUL35 and SNX5 was disrupted by UL35 mutagenesis, only the localization of gB of the analyzed viral proteins was significantly affected and showed a dispersed localization.
FIG 5.

Localization of glycoprotein B in infected fibroblasts. HFF cells were infected with wild-type or mutant HCMV, as indicated. Cells were fixed with paraformaldehyde at 96 h postinfection and stained (red) for glycoproteins gB (monoclonal 27-287), gH (monoclonal 14-4b), gM (monoclonal IMP91-3/1), and gN (monoclonal 14-16A) (A) or tegument proteins ppUL32 (rabbit serum XP1), ppUL83 (monoclonal 28-77), and ppUL82 (monoclonal CMV355) and suitable secondary antibodies (B). Nuclear counterstaining was achieved using DAPI (blue).
Effect of SNX5 overexpression and depletion.
Since ppUL35 functionally counteracted SNX5, we reasoned that overexpression of SNX5 should impede virus replication. Therefore, we transfected HFF cells with SNX5 or control plasmid and infected them with HCMV strain TB4-wt after 24 h. At 7 days postinfection (dpi), the virus titer was significantly reduced by more than 50% in SNX5-transfected cells (Fig. 6A). In a converse experiment, we depleted the SNX5 protein amount by siRNA treatment (Fig. 6B). In TB4-wt-infected cells, differences between SNX5 and control siRNA-transfected cells were not observed (Fig. 6C), as expected if the role of ppUL35 was the functional inactivation of SNX5. When infections were performed with the TB4-mUL35-Tn71 mutant, we detected a reduction of virus titers by 1 log compared to that of wild-type virus in control siRNA-transfected cells. In contrast, the titer returned to wild-type levels when this virus was grown on SNX5 siRNA-transfected cells (Fig. 6C). A reduction of almost 2 logs in the titers of TB4-UL35-kan-in and TB4-mUL35a in control siRNA-transfected cells indicated additional replication defects in these mutants. In SNX5-depleted cells, the titer of both mutants increased significantly but did not reach wild-type levels. This clearly demonstrates that expression of ppUL35 during HCMV replication counteracts SNX5 function.
FIG 6.
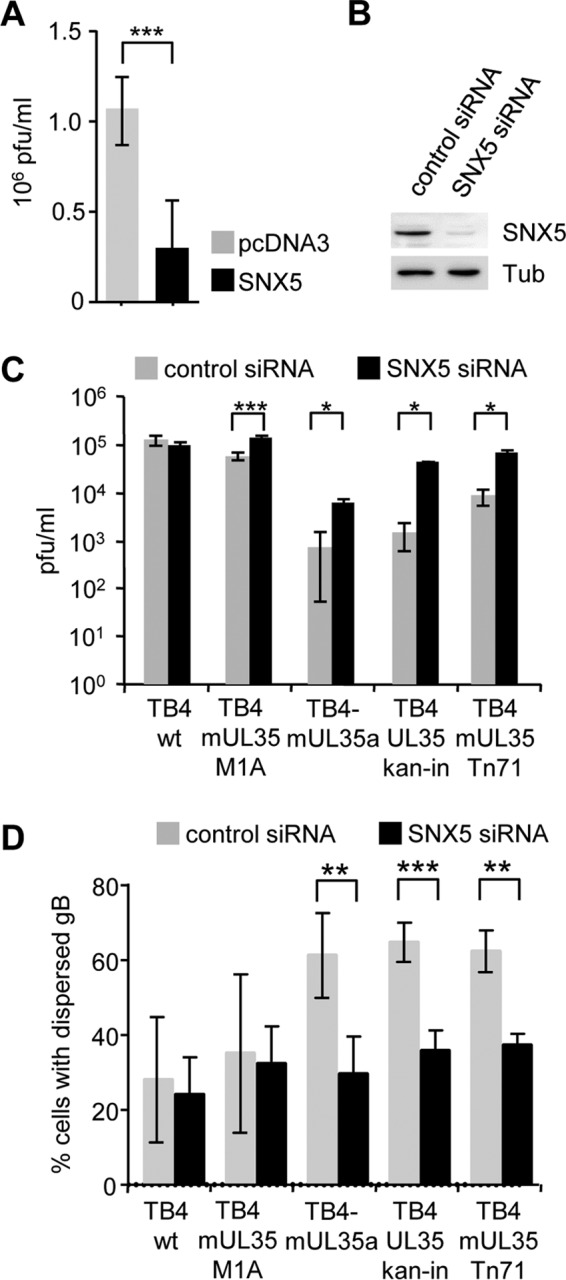
Effect of SNX5 overexpression and depletion. (A) HFF cells were transfected with an expression plasmid for FLAG-SNX5 or control plasmid and infected with TB40E-BAC4 24 h later. At 7 dpi, supernatants were harvested and the virus titers were determined. Three independent experiments were performed, and significance was calculated employing a two-tailed Student's t test. (B) HFF cells were transfected with control or SNX5 siRNA. Cell lysates were analyzed by immunoblotting with staining with polyclonal rabbit anti-SNX5 serum. Immunoblotting with antitubulin served as a loading control. (C) HFF cells transfected with control or SNX5 siRNA were infected with the indicated HCMV wild-type and mutant viruses 24 h later. Supernatants were harvested at 7 dpi, and the virus titer was determined. Results of a representative experiment (from two independent experiments) performed in triplicate are shown. (D) HFF cells were transfected with SNX5 or control siRNA and infected with the indicated wild-type and mutant HCMV 24 h later. At 96 h postinfection, cells were fixed with paraformaldehyde and stained for glycoprotein B. At least 50 cells were scored for compact or dispersed localization. Four to five independent experiments were performed, and significance was calculated employing a two-tailed Student's t test with the Welch correction. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Finally, we analyzed if SNX5 depletion also affects the localization of gB in the context of infection with wild-type and mutant viruses. As shown in Fig. 6D, only a few cells showed a dispersed localization in TB4-wt-infected cultures. This was irrespective of the status of SNX5 depletion, since UL35 proteins were active in these cells. In control siRNA-transfected cells infected with mutant viruses, we observed a much higher level of dispersed localization for gB, due to a lack of full UL35 activity. Depletion of SNX5 reestablished the lower levels of dispersed localization that were seen in wild-type virus-infected cells. Therefore, UL35 proteins functionally inactivate cellular SNX5 to allow a proper localization of gB.
DISCUSSION
Hijacking and tight regulation of host cell transport functions are vital for most viruses to ensure proper localization of virus components during virus particle assembly. HCMV regulates a complex sequence of transport steps during virion assembly. Studies investigating HCMV particle maturation revealed that endosomal and secretory transport pathways are of eminent importance for virus assembly. The HCMV assembly compartment consists of concentric rearranged vesicles derived from the endosome and the TGN (9, 10) and located close to the nucleus. Marker proteins of different compartments and vesicle types colocalized in the same vesicles; however, it is not yet clear how these rearrangements occur and what functional consequences arise from these findings (10). Several reports revealed an important role of components of the endocytosis and retrograde transport machinery, such as the ESCRT complex (16), FIP4 (42), and dynein (43), for HCMV maturation. Experimental evidence also indicated the importance of secretory pathway components like Rab27a (14), Bicaudal D1 (15), Rab6 (17), Syntaxin3 (44), and SNAP-23 (45) for virus maturation. Depletion of these proteins led to reduced virus titers and in most cases to a disturbed localization of viral proteins during the late phase of infection. Major HCMV proteins directing host cell factors and viral protein transport are ppUL32 (46), pUL96 (46), pUL103 (21), pUL71 (20, 47, 48), and pUL94 (49). Previously, we showed significant mislocalization of several viral proteins during virion maturation in the late phase upon infection with recombinant HCMV in which the UL35 gene had been deleted (6). In the present study, we identified SNX5 to be a binding partner of UL35 proteins and demonstrate the importance of this interaction for virus replication and localization of glycoprotein B.
SNX5 is a part of the retromer multiprotein complex which recognizes cargo proteins in the early endosome and directs their retrograde transport to the TGN. This transport pathway is of particular interest because herpesvirus envelope proteins, such as gB, are transported from the endosome to the TGN before they are integrated into the virus particle. In transient-transfection assays, expression of ppUL35 or ppUL35A led to a dispersed localization of CI-M6PR, a well-studied cargo of the retromer complex (50). This mislocalization was very similar to the relocalization of CI-M6PR from the TGN to endosomes after siRNA-induced knockdown of SNX5 expression (40). Therefore, we suspected that UL35 proteins might functionally inactivate SNX5. By pentapeptide-scanning mutagenesis, we identified a mutant with Tn71 in the C terminus of ppUL35, which was unable to bind SNX5 and to relocalize CI-M6PR. Other functions residing in the C terminus of ppUL35, such as binding to and functional interaction with ppUL82, were not affected by this mutation. This demonstrated that mutant ppUL35-Tn71 is specifically deficient in physical and functional interaction with SNX5. Therefore, the direct binding of UL35 proteins to SNX5 is necessary to affect the normal function of SNX5.
HCMV replication was significantly affected in cells overexpressing SNX5, whereas siRNA depletion of SNX5 had no effect on wild-type virus replication. The decrease is consistent with a role of ppUL35 in functionally interfering with the SNX5 function. When the ppUL35 function was abolished by deletion of UL35, strong replication defects were observed, and these were partially restored by siRNA depletion of SNX5. More specifically, when the SNX5-binding function of ppUL35 was abolished by transferring mutant UL35-Tn71 into the context of the viral genome, a suppression of virus replication of 1 log was observed, a magnitude also observed for other factors in the secretory pathway which affect HCMV maturation (14, 15, 17, 44). This also demonstrates that the 50% decrease in virus titer observed after transfection of cells with SNX5 is likely an underestimate since not all HFF cells were transfected. Importantly, depletion of SNX5 was able to fully revert replication of this mutant virus to wild-type levels. This clearly demonstrated the importance for the viral life cycle of ppUL35/ppUL35A targeting SNX5.
Previously, we had described a strong effect of ppUL35 on the localization of major tegument proteins of HCMV (6). Genomic UL35 deletion led to a nuclear accumulation of both ppUL82 and ppUL83 in the late phase, where both proteins would normally localize to the assembly center. Since both proteins were localized in the cytoplasm of cells infected with TB4-mUL35-Tn71, we could rule out a role of SNX5 in this process. However, upon closer examination, we noted a more dispersed cytoplasmic distribution of gB in cells infected with the full UL35 deletion mutant of AD169 (6). When we analyzed the localization of major viral glycoproteins, we observed the same dispersed cytoplasmic distribution in cells infected with UL35 mutant viruses, instead of a clear punctual localization in wild-type HCMV-infected cells. Notably, this diffuse localization was also observed in TB4-mUL35-Tn71-infected cells. The condensed assembly center localization of gB seen in wild-type-infected cells could be restored by siRNA depletion of SNX5, clearly demonstrating the importance of UL35 proteins in proper gB localization. No altered localization was seen for glycoproteins, such as gH, gM, or gN, but we cannot rule out the possibility that other glycoproteins might be affected in the same way as gB. These observations demonstrate that UL35 proteins have multiple functions and that interaction with SNX5 specifically affects localization of gB.
Components of herpesvirus virions likely assemble along several maturation pathways, which converge in the assembly center (11, 51, 52). Glycoprotein B is a major glycoprotein in the HCMV envelope, is highly conserved in other herpesviruses, and has a role in virus entry and cell fusion. Similarly to the gB of other herpesviruses, the HCMV gB could be detected on the cell surface, and it has been proposed that gB has to be retrieved from the cell membrane via endocytosis before assembly into virions (53–55). HCMV gB harbors a cluster of acidic amino acids and two dileucine motifs in the cytosolic tail (53, 54) which serve as sorting signals for the retrograde transport of gB. The requirement of endocytosis of gB and other envelope proteins for incorporation into the particle is a subject of a controversial discussion. Several reports revealed that endocytosis of herpesvirus envelope proteins is not necessary for incorporation into the virus particle (26, 56, 57). A report using dominant negative dynamin mutants suggested that endocytosis of HCMV gB (26) is not critical for particle maturation. On the other hand, expression of endocytosis-deficient herpes simplex virus 1 gB (58) or varicella-zoster virus gE (59) had a negative impact on virus replication. Endocytosis of gpUL132 from the plasma membrane is required for integration into the HCMV particle and for optimal replication (60). As discussed by Kropff and colleagues (60), intracellular transport and endocytosis are redundant and can compensate for the loss of a single component or a whole pathway. Endocytosis can be executed by dynamin- and clathrin-independent uptake mechanisms. Thus, it has to be considered that gB and other herpesvirus envelope proteins bypass endocytosis by direct transport from the TGN to the endosome (27). This might even render endocytosis not essential for all herpesvirus envelope proteins.
As a potential model for the SNX5-ppUL35/ppUL35A interaction, we suggest three possible functional roles. (i) Through the interaction of ppUL35 with SNX5, the complete retromer is inhibited (inhibition). (ii) The interaction of ppUL35 with SNX5 leads to the inhibition of SNX5-dependent recruitment and transport processes (exclusion of targets). (iii) Instead of complete inhibition of SNX5, ppUL35/35A selectively alters recruitment and transport processes: certain proteins are preferentially transported to the TGN (accumulation). Models (i) and (ii) have a similar effect and are based on the complete or partial inhibition of the retromer. However, complete inhibition of the retromer by inhibition of SNX5 might be bypassed by redundant SNXs. Nonetheless, the possibility of compensation of the inhibited transport of excluded proteins by other pathways also has to be taken into account. It should also be noted that the exclusion of certain cargoes from one pathway and their transport by another pathway could be a desired outcome of infection and a contribution to the formation of cVAC, yet it is not clear whether different transport pathways yield different localizations in the TGN or the assembly complex and whether this is favorable for the maturation of HCMV particles. With the functional roles of SNX5 still being unraveled, the cell biological role of SNX5 in gB transport pathways will clearly need further investigation.
Reports on vesicular stomatitis virus (VSV) and human papillomavirus (HPV) underlined the significance of sorting nexins for viral replication. The release of VSV nucleocapsids from late endosomes is regulated by SNX16, an SNX without additional protein domains. SNX16 overexpression resulted in reduced VSV replication (61, 62). Studies with HPV-11 and HPV-16 identified SNX17 to be a binding partner of capsid protein L2 (63). SNX17 is essential for the release of viral DNA from the endosome during the first steps of viral infection. Gene silencing of SNX17 by siRNA led to a reduced virus titer of HPV-11 and HPV-16. Both studies revealed roles for SNX during uptake and uncoating of virus particles. Since our experiments were performed in human fibroblasts, where HCMV uptake appears at the plasma membrane, an involvement of SNX5 during virus entry is unlikely. In contrast, our study reports for the first time a role for a SNX during the phase of particle assembly.
In summary, we show that ppUL35 and ppUL35A interact with SNX5, thereby impairing its function. Inactivation of the interaction with SNX5 by mutation led to reduced virus replication and interfered with the correct localization of gB during virion maturation. Depletion of SNX5 restored both the replication and localization of gB. We extended previous reports on UL35 interactions and functions, clearly defining them as multifunctional proteins.
MATERIALS AND METHODS
Oligonucleotides.
The following oligonucleotides were synthesized by Sigma (Taufkirchen, Germany) or Biomers (Ulm, Germany): ETH85 (5′-CTGAACGTCGACGCCCAAGGATCGCGAGCCCCATCG-3′), ETH86 (5′-GAAGCTTGGCGGCCGCGAGATGCCGTAGGTTTTCGGCC-3′), ul35alaala (5′-CTCCAGCGCATCTATAGCGCTGCGATCGAGGGCGCCTCTCGG-3′), UL35 M465A (5′-CTGACACCCAAGCGCTTCGCGGAGCTCCTCGACAGAGCGCCT-3′), epm_fwd_UL35_dATG (5′-TCCGCTCTGGTTTTGGTTTCGTTTTCAAAGGGAGCCCCATCGCGGCCCAAGGATCGCGAGCTAGGGATAACAGGGTAATCGATTT-3′), epm_bwd_UL35_dATG (5′-CGGGCAGTGGCGGGCCCGATGGGGCTCGCGATCCTTGGGCCGCGATGGGGCTCCCTTTGAAAACGGCCAGTGTTACAACCAATTAACC-3′), epm_fwd_UL35revATG (5′-GGTTTTGGTTTCGTTTTCAAAGGGAGCCCCATCATGGCCCAAGGATCGCGAGCTAGGGATAACAGGGTAATCGATTT-3′), epm_bwd_UL35revATG (5′-GGCGGGCCCGATGGGGCTCGCGATCCTTGGGCCATGATGGGGCTCCCTTTGAAAACGGCCAGTGTTACAACCAATTAACC-3′), epm_delUL35_fwd (5′-ATGGCCCAAGGATCGCGAGCCCCATCGGGCCCGCCACTGCCCGTTCTCCTAGGGATAACAGGGTAATCGATTT-3′), epm_delUL35_bwd (5′-GAGATTCCGTAGGTTTTCGGCCAGATCGTCCCGCGGGTCTTCGACGTCGGCCAGTGTTACAACCAATTAACC-3′), hSNX5-5Sal (5′-CCCGTCGACACCATGGCCGCGGTTCCCGAGTTG-3′), pCAGGS-3′ (5′-CAGAAGTCAGATGCTCAAGGG-3′), hSNX16-5X (5′-CGCTCGAGATGGCAACTCCTTATGTCCCAG-3′), and hSNX16-3N (5′-GGGCGGCCGCTTAGTCTTCTTCAGCATCATATGC-3′).
Plasmids and siRNA reagents.
The plasmids pcDNA-UL82 and pCMV71 expressing ppUL82, pcDNA3-UL35 expressing ppUL35, and the HCMV enhancer/modulator luciferase reporter plasmid pHM287 were described previously (5, 64). For prokaryotic expression, UL82 was subcloned from pUC18-UL82 (5) into pMAL-c2x (New England BioLabs, Frankfurt, Germany) using BamHI and SalI, resulting in pMal-UL82. Expression plasmid pCMV-3×FLAG-SNX5, coding for FLAG-tagged SNX5, was kindly provided by Rohan Teasdale (65). Plasmid pCAGGS-FLAG-SNX5 was generated by amplification of SNX5 from pCAGGS-mseCFP-SNX5 (40) using primers hSNX5-5Sal/pCAGGS-3′, digestion with SalI and NotI, and ligation into the pCAGGS-FLAG backbone cut with XhoI and NotI. Sorting nexin 16 (SNX16) was amplified using primers hSNX16-5X/hSNX16-3N and cDNAs from A549 and 293T cells, digested with XhoI and NotI, and cloned into the pCAGGS-FLAG backbone to give plasmids pCAGGS-FLAG-SNX16 and pCAGGS-FLAG-SNX16-X1, respectively.
Plasmid pEP-kan-S2 was a kind gift from Karsten Tischer (66). Plasmid pcDNA3-UL35Ad3ATG was generated by site-directed mutagenesis with oligonucleotides ul35alaala and UL35 M465A using the method of Sawano and Miyawaki (67). To obtain a helper plasmid for en passant mutagenesis, pKD46 (68) was modified by insertion of the gene for the homing endonuclease I-SceI from plasmid pST98-AS (69). The resulting plasmid, pKD46S, expresses the Red recombinases γ, β, and exo of phage lambda under the control of an l-arabinose-inducible promoter and I-SceI under the control of a tetracycline-inducible promoter. All plasmid constructs were verified by DNA sequencing (Markus Schilhabel, Institute for Clinical Molecular Biology sequencing facility, Kiel, Germany).
siRNA directed against SNX5 (Ambion Silencer Select) and control siRNA (Ambion Silencer Select negative control) were purchased from Life Technologies and used at 2 pmol/well (on a 48-well plate for replication analysis) or 20 pmol/well (on a 24-well plate for immunofluorescence).
Cell culture and transfection.
Primary human foreskin fibroblasts (HFF) and HeLa, 293T, A549, EA-hy, and U373 cells were maintained in Dulbecco's modified Eagle medium (DMEM). RPE1 cells (70) were maintained in a 1:1 mixture of DMEM and Ham's F-12 medium. All media were supplemented with 10% fetal calf serum (FCS), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell culture supplements were purchased from PAA Cell Culture Company (Cölbe, Germany). Telomerase-immortalized microvascular endothelial (TIME) cells (71) were maintained in EBM-2 basal medium with an EGM-2 MV SingleQuot kit (Lonza, Basel, Switzerland). Transfection of plasmids into HeLa cells was performed using the Lipofectamine LTX reagent (Life Technologies, Darmstadt, Germany). The Lipofectamine RNAimax reagent (Life Technologies, Darmstadt, Germany) was used to transfect siRNA. Electroporation of plasmids into HFF cells was performed using an amaXa Nucleofector device. Briefly, 5 × 105 cells were resuspended in 100 μl phosphate-buffered saline (PBS), mixed with 3 μg plasmid DNA, and subjected to electroporation using the program T-030. When transfected with an enhanced green fluorescent protein-expressing plasmid, we routinely achieved transfection efficiencies of 60 to 80% in HFF.
Ethics statement.
Primary human fibroblasts were cultivated from dissociated foreskin samples from anonymous donors on the basis of a statement of the Ethikkommission der Medizinischen Fakultät der Christian-Albrechts-Universität zu Kiel, reference number D467/13: “This is to certify that according to the Professional Code for Physicians in Germany (‘Berufsordnung für Ärzte, BO § 15′) the use of human fibroblast cultures of foreskin samples does not require the approval of an Ethics Committee, since these foreskin samples originate from indicated surgical procedures and are considered clinical waste. Given the ‘waste status’ of the obtained samples these were collected anonymously.”
Antibodies.
Polyclonal rabbit antiserum against ppUL35 was a kind gift of Bonita Biegalke (4). To generate a rabbit antiserum against ppUL82, a maltose-binding protein–ppUL82 fusion protein was expressed in Escherichia coli BL21(DE3). Purified protein was sent to Pineda (Berlin, Germany) to generate a rabbit antiserum. A rabbit monoclonal antibody against β-actin (13E5) was purchased from Cell Signaling Technologies (Frankfurt, Germany), polyclonal rabbit anti-SNX5 antibody (H-40) and polyclonal goat anti-SNX5 were from Santa Cruz (Heidelberg, Germany), and mouse monoclonal antibody directed against CI-M6PR was from Novus Biologicals (Littleton, USA). Murine monoclonal antibody M2 against the FLAG epitope was from Sigma-Aldrich (Taufkirchen, Germany). Monoclonal antibodies against gB (27-287) (72), ppUL83 (28-77) (73), ppUL82 (CMV355) (74), gH (14-4b) (75), gM (IMP91-3/1) (76), gN (14-16A) (77), IE1 (p63-27) (78), and the Myc epitope (9E10) (79) were used as cell culture supernatants. Horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from Dianova (Hamburg, Germany). Fluorophore-conjugated secondary antibodies were from Life Technologies (Darmstadt, Germany).
Yeast two-hybrid screening.
For yeast two-hybrid screening, full-length UL35 was amplified from cosmid pCM1017 (80) using primers ETH85/ETH86 and cloned into plasmid pPC97mT by use of the restriction enzymes SalI and NotI. Bait vector pPC97mT was derived from pPC97 (81) by replacing the polylinker as described in reference 5 and replacing the LEU2 gene with TRP1 containing the ApaI/SpeI fragment from pPC86. Bait plasmid pPC97mT-UL35 was transformed into Saccharomyces cerevisiae strain Y153 using the lithium acetate method and stably maintained by selection for tryptophan prototrophy. Subsequently, yeast cells were transformed with a pACT-based cDNA library derived from Epstein-Barr virus-transformed human peripheral lymphocytes (82), and more than 106 primary transformants were selected on His-Leu-Trp-dropout plates containing 20 mM 3-aminotriazole. His-positive colonies were then analyzed for β-galactosidase activity by filter lift experiments. Out of a total of 99 candidate clones positive in both assays, 73 clones could be recovered in E. coli and sequenced.
BAC mutagenesis.
UL35 mutagenesis was carried out in the HCMV bacterial artificial chromosome (BAC) TB40E-BAC4, which is derived from a clinical isolate (83). E. coli DH10B containing TB40E-BAC4 and pKD46S was cultivated overnight in 5 ml LB medium with chloramphenicol for maintaining the BAC and ampicillin for maintaining pKD46S. Samples of 2.5 ml of the overnight culture were transferred to 250 ml fresh LB medium and cultivated at 32°C until the culture reached an optical density (600 nm) of 0.1. Addition of 2.5 ml 10% l-arabinose and propagation for an additional 1 h at 32°C induced the expression of the Red recombinase system from plasmid pKD46S. Finally, the bacteria were cooled on ice for 20 min, washed three times with ice-cold sterile water, and washed once with 10 ml ice-cold 10% glycerol. After decanting the supernatant, the cells were resuspended in the remaining glycerol. Recombination-competent DH10B cells were either stored at −80°C or used for electroporation.
For deletion of ppUL35 expression, the start codon (ATG) was mutated into a codon for alanine (CTG), and this mutant was named TB4-mUL35-M1A. Mutation of this codon was performed by markerless en passant recombination (66). First, the kanamycin resistance cassette from plasmid pEP-kan-S2 was amplified by PCR with oligonucleotides epm_fwd_UL35_dATG and epm_bwd_UL35_dATG containing 50 nucleotides homologous to the region upstream and downstream of the start codon of UL35 and a CTG instead of ATG of the start codon. The resulting amplification product contained sequences homologous to UL35 flanking the kanamycin resistance cassette and the 18-bp recognition site of the I-SceI endonuclease upstream of the kanamycin resistance cassette.
The PCR product was purified from the agarose gel and electroporated (0.1-cm-gap-width cuvette, 2.5 kV, 25 μF, 200 Ω) into recombination-competent E. coli DH10B containing TB40E-BAC. After electroporation, the bacteria were incubated in 1 ml LB medium at 32°C for 2 h and finally plated on LB agar plates with chloramphenicol and kanamycin. Insertion of the PCR product into the UL35 ORF was facilitated by Red recombination. After 2 days, colonies were screened with colony PCR and primers for UL35 to verify insertion of the PCR product into the UL35 ORF. For deletion of the kanamycin resistance cassette, positive colonies were cultivated overnight in 5 ml LB medium with chloramphenicol and kanamycin. Samples (100 μl) of the overnight culture were transferred into 3 ml fresh LB medium with chloramphenicol, kanamycin, 0.1% l-arabinose, and anhydrotetracycline for 5 h and afterwards plated on LB agar plates with chloramphenicol, ampicillin, and anhydrotetracycline. Anhydrotetracycline induced the expression of I-SceI endonuclease, which created a double-strand DNA break for the second Red recombination. After incubation of the plates at 32°C for 2 days, colonies were screened for the absence of the kanamycin resistance cassette. The UL35 region of positive clones was amplified by PCR and sequenced to detect the introduced point mutation.
In order to generate a revertant virus, we used the same procedure starting with TB4-mUL35-M1A and primers epm_fwd_UL35revATG and epm_bwd_UL35revATG with an ATG instead of the CTG. To remove pKD46S from the resulting bacterial strains, cultures were subjected to growth at 42°C overnight, which inhibited pKD46S replication due to a temperature-sensitive origin of replication.
Deletion of the full UL35 ORF was achieved by insertion of the kanamycin resistance cassette and leaving only 50 bp of the original UL35 ORF flanking the kanamycin resistance cassette. For this purpose, mutant primers epm_delUL35_fwd and epm_delUL35_bwd were used. The resulting BAC was called TB4-UL35-kan-in. To produce BACs with deleted ppUL35A expression, a UL35 expression cassette with mutated UL35A start codons (M448, 449, 465A) was cut out from the pcDNA3-UL35Ad3ATG plasmid with restriction enzymes, purified from an agarose gel, and transformed into recombination-competent E. coli containing TB4-UL35-kan-in. Red recombination exchanged the kanamycin resistance cassette against the mutated UL35 ORF. Colonies were screened with colony PCR, and mutations were validated via cycle sequencing (Institute for Clinical Molecular Biology, Kiel, Germany). The same protocol was used to insert the UL35 gene which was mutated by transposon insertion (UL35-Tn71). Plasmid pKD46S was removed by growth at 42°C. The resulting BACs were tested for integrity by restriction enzyme digestion. The UL35 ORF and its flanking sequences were sequenced to verify the introduced mutations.
Virus reconstitution, titration, and replication curves.
For virus reconstitution, BAC DNA was isolated from E. coli using a Nucleobond PC-100 kit (Macherey-Nagel, Düren, Germany) and dissolved in a final volume of 100 μl. HFF cells were detached, washed two times with medium, and resuspended in 400 μl cell culture medium. The cell suspension was mixed with 50 μl BAC DNA and 10 μg of pCMV71. Electroporation was performed with a Bio-Rad Gene Pulser device at 280 V and 1,500 μF. The cells were immediately supplemented with fresh medium and transferred to 75-cm2 tissue culture bottles. On the next day, the cells were washed and maintained in fresh medium. When a cytopathic effect was visible for the majority of the cells, they were detached with trypsin, mixed with uninfected HFF, and transferred into 175-cm2 tissue culture bottles. Virus supernatant was harvested after approximately 10 days, when a confluent cytopathic effect was evident. Viral particles were concentrated by ultracentrifugation in an SW28 rotor (Beckman Coulter, Krefeld, Germany) at 22,000 rpm for 1 h at 4°C. Pellets were resuspended in a 1:1 mixture of medium and 0.2 M sucrose phosphate (84). Aliquots were stored at −80°C. Virus titers were determined by limiting dilution assays by counting infected HFF cells stained with ppUL83 monoclonal antibody 65-33 at 3 dpi. In order to prepare one-step replication curves, HFF cells were seeded into a 48-well plate with a density of 2.5 × 103 cells per well and infected with recombinant HCMV. Supernatants were harvested at the time points indicated above, and the virus titer was determined as described above. The results are presented as the number of PFU per milliliter.
Transposon mutagenesis.
For fine mapping of ppUL35, we generated UL35 mutants by pentapeptide-scanning mutagenesis, by which 15 bp encoding 5 additional amino acids was inserted into the UL35 ORF in a transposon-mediated process. We used a GPS-linker scanning kit (New England BioLabs, Frankfurt, Germany) or a mutation generation system (Finnzymes, Vantaa, Finland) for in vitro mutagenesis of plasmid pcDNA-UL35 according to the instructions of the manufacturers. The mutated plasmids were transformed into E. coli DH10B, and the resulting colonies were screened for transposon insertion into the UL35 ORF by colony PCR. Plasmid DNA of positive clones was isolated and digested with PmeI (with the New England BioLabs kit) or NotI (with the Finnzymes kit), religated, and transformed into E. coli. Removal of the transposon was controlled by colony PCR. The pentapeptide insertion sites were determined by DNA sequencing.
Coimmunoprecipitation.
293T cells in 6-well plates were transfected with plasmids expressing ppUL35 or ppUL35A and FLAG-SNX5. After 48 h, cells were harvested and lysed in 500 μl coimmunoprecipitation (co-IP) lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 5 mM EDTA, 0.5% NP-40). Centrifugation at 33,000 rpm for 30 min at 4°C cleared the lysate from the cell debris and large protein complexes. For the precipitation of SNX5, protein G Sepharose (GE Healthcare) was blocked with 5% gelatin from cold water fish skin (Sigma) in co-IP lysis buffer for 1 h and incubated with the antibody indicated above for 1 h. After each incubation step, protein G Sepharose beads were centrifuged at 2,000 rpm for 2 min and washed with co-IP lysis buffer two times. Prebound protein G Sepharose was added to the cell lysate and incubated for 10 min in a tube rotator at room temperature. The beads were washed eight times with co-IP lysis buffer and eluted with 10 μl SDS-PAGE sample buffer.
Immunoblotting.
Samples from cell extracts or coimmunoprecipitates were separated on 10 or 12% polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes with a Transblot SD apparatus (Bio-Rad). The membranes were blocked for 1 h in blocking buffer (5% skim milk powder, PBS, 0.1% Tween 20). For protein detection, filters were incubated with specific primary antibody at room temperature for 1 h. Rabbit anti-UL35 serum was diluted 1:5,000, rabbit anti-ppUL82 serum was diluted 1:1,000 in blocking buffer, and anti-β-actin rabbit monoclonal antibody 13E5 was diluted 1:1,000. Hybridoma supernatants were used undiluted. After primary antibody incubation, membranes were rinsed once and washed three times in PBS. Thereafter, the blots were incubated for 1 h with HRP-conjugated secondary antibodies (Dianova) diluted 1:10,000 in blocking buffer. Finally, the membranes were again rinsed once and washed three times with PBS. Bound antibodies were detected using the Super Signal West Pico chemiluminescent substrate (Thermo) supplemented 1:10 with the Super Signal West Femto substrate (Thermo) and the digital documentation instrument LAS3000 (Fujifilm, Düsseldorf, Germany).
Macropinocytosis assay.
The macropinocytosis assay was performed as described by Lim et al. (38), except that the final analysis was done by flow cytometry. Briefly, HeLa cells seeded in 6-well plates were transfected with 1 μg of the plasmids indicated above per well and grown for 24 h. Cells were serum starved for 12 h and then treated with EGF (100 ng/ml; Peprotech) and FITC-coupled dextran (average molecular mass, 70 kDa; 100 μg/ml; Sigma) for 3 min. After detachment with trypsin-EDTA at 4°C, cells were fixed in 2% paraformaldehyde (final concentration) in PBS, and the uptake of FITC-coupled dextran was measured by flow cytometry. Results are presented as percent positive cells.
Indirect immunofluorescence microscopy.
For immunofluorescence microscopy, cells were seeded onto glass coverslips in 24-well plates at 4 × 104 cells per well the day before transfection or infection. For fixation, the cells were washed three times with PBS, incubated in 4% paraformaldehyde in PBS for 10 min, and washed three times with PBS. Fixed cells were permeabilized with PBS–0.2% Triton X-100 for 10 min and washed two times with PBS. To block nonspecific binding, the coverslips were incubated in 10% FCS in PBS for 30 min and subsequently covered with primary antibodies for 1 h. Purchased antibodies were diluted according to the instructions of the manufacturers, and hybridoma supernatants were used undiluted. After having been washed three times with PBS, the coverslips were incubated for 1 h with secondary antibody coupled with Alexa Fluor 488 or Alexa Fluor 567 (Life Technologies), diluted 1:2,000 in blocking buffer. The coverslips were washed three times with PBS and mounted on slides with Vectashield mounting medium with DAPI (4′,6-diamidino-2-phenylindole; Vector, Burlingame, CA). Images were analyzed on an IX81 fluorescence microscope with an MT10 light source (Olympus, Hamburg, Germany). For quantification of the CI-M6PR distribution in transfection experiments, at least 100 cells were analyzed; in infection experiments, at least 50 cells were analyzed.
Luciferase reporter assay.
For luciferase assays, 3 × 105 cells per well of U373-MG cells were seeded into 6-well dishes the day before transfection. The cells were transfected with Lipofectamine LTX with 1 μg pcDNA3, pcDNA-UL35, or pcDNA-UL35-Tn71 and 0.2 μg pHM287 according to the instructions of the manufacturer. After 48 h, cells were lysed with 500 μl of passive lysis buffer (Promega, Mannheim, Germany). Insoluble cell debris was pelleted by centrifugation, and the supernatant was collected. Luciferase activity was measured in a Plate Chameleon V microplate luminometer (Hidex, Turku, Finland) using a commercial kit (Promega, Mannheim, Germany).
ACKNOWLEDGMENTS
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Wi 1725/2-3) and by the Excellence Center Inflammation at Interfaces, Kiel, Germany.
We thank U. Koszinowski, G. Hahn, R. Teasdale, E. Kiyokawa, and B. K. Tischer for plasmids and the TB40E-BAC4 construct and T. Stamminger, M. Mach, and B. Biegalke for murine monoclonal antibodies and rabbit antiserum against pUL35. We thank S. Pöhlmann for helpful comments and generous support and O. Braum for critically reading the manuscript.
REFERENCES
- 1.Davison AJ, Eberle R, Ehlers B, Hayward GS, McGeoch DJ, Minson AC, Pellett PE, Roizman B, Studdert MJ, Thiry E. 2009. The order Herpesvirales. Arch Virol 154:171–177. doi: 10.1007/s00705-008-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalejta RF. 2008. Tegument proteins of human cytomegalovirus. Microbiol Mol Biol Rev 72:249–265. doi: 10.1128/MMBR.00040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winkler M. 2003. Interactions and functions of human cytomegalovirus tegument proteins, p 113–121. In Pr̈osch S, Cinatl J, Scholz M (ed), New aspects of CMV-related immunopathology. Karger, Basel, Switzerland. [Google Scholar]
- 4.Liu Y, Biegalke BJ. 2002. The human cytomegalovirus UL35 gene encodes two proteins with different functions. J Virol 76:2460–2468. doi: 10.1128/jvi.76.5.2460-2468.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schierling K, Stamminger T, Mertens T, Winkler M. 2004. Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J Virol 78:9512–9523. doi: 10.1128/JVI.78.17.9512-9523.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schierling K, Buser C, Mertens T, Winkler M. 2005. Human cytomegalovirus tegument protein ppUL35 is important for viral replication and particle formation. J Virol 79:3084–3096. doi: 10.1128/JVI.79.5.3084-3096.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salsman J, Wang X, Frappier L. 2011. Nuclear body formation and PML body remodeling by the human cytomegalovirus protein UL35. Virology 414:119–129. doi: 10.1016/j.virol.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 8.Salsman J, Jagannathan M, Paladino P, Chan PK, Dellaire G, Raught B, Frappier L. 2012. Proteomic profiling of the human cytomegalovirus UL35 gene products reveals a role for UL35 in the DNA repair response. J Virol 86:806–820. doi: 10.1128/JVI.05442-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das S, Pellett PE. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J Virol 85:5864–5879. doi: 10.1128/JVI.00155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res 143:222–234. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 12.Challacombe JF, Rechtsteiner A, Gottardo R, Rocha LM, Browne EP, Shenk T, Altherr MR, Brettin TS. 2004. Evaluation of the host transcriptional response to human cytomegalovirus infection. Physiol Genomics 18:51–62. doi: 10.1152/physiolgenomics.00155.2003. [DOI] [PubMed] [Google Scholar]
- 13.Hertel L, Mocarski ES. 2004. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of pseudomitosis independent of US28 function. J Virol 78:11988–12011. doi: 10.1128/JVI.78.21.11988-12011.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fraile-Ramos A, Cepeda V, Elstak E, van der Sluijs P. 2010. Rab27a is required for human cytomegalovirus assembly. PLoS One 5:e15318. doi: 10.1371/journal.pone.0015318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Indran SV, Ballestas ME, Britt WJ. 2010. Bicaudal D1-dependent trafficking of human cytomegalovirus tegument protein pp150 in virus-infected cells. J Virol 84:3162–3177. doi: 10.1128/JVI.01776-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tandon R, AuCoin DP, Mocarski ES. 2009. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J Virol 83:10797–10807. doi: 10.1128/JVI.01093-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Indran SV, Britt WJ. 2011. A role for the small GTPase Rab6 in assembly of human cytomegalovirus. J Virol 85:5213–5219. doi: 10.1128/JVI.02605-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das S, Ortiz DA, Gurczynski SJ, Khan F, Pellett PE. 2014. Identification of human cytomegalovirus genes important for biogenesis of the cytoplasmic virion assembly complex. J Virol 88:9086–9099. doi: 10.1128/JVI.01141-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rebmann GM, Grabski R, Sanchez V, Britt WJ. 2016. Phosphorylation of Golgi peripheral membrane protein Grasp65 is an integral step in the formation of the human cytomegalovirus cytoplasmic assembly compartment. mBio 7:e01554-. doi: 10.1128/mBio.01554-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Womack A, Shenk T. 2010. Human cytomegalovirus tegument protein pUL71 is required for efficient virion egress. mBio 1:e00282-. doi: 10.1128/mBio.00282-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahlqvist J, Mocarski E. 2011. Cytomegalovirus UL103 controls virion and dense body egress. J Virol 85:5125–5135. doi: 10.1128/JVI.01682-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eickmann M, Gicklhorn D, Radsak K. 2006. Glycoproteins trafficking in virion morphogenesis, p 245–264. In Reddehase MJ. (ed), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 23.Mocarski ES, Shenk T, Pass R. 2007. Cytomegaloviruses, p 2701–2772. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 24.Britt WJ, Vugler LG. 1992. Oligomerization of the human cytomegalovirus major envelope glycoprotein complex gB (gp55-116). J Virol 66:6747–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Britt WJ, Auger D. 1986. Synthesis and processing of the envelope gp55-116 complex of human cytomegalovirus. J Virol 58:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jarvis MA, Fish KN, Soderberg-Naucler C, Streblow DN, Meyers HL, Thomas G, Nelson JA. 2002. Retrieval of human cytomegalovirus glycoprotein B from cell surface is not required for virus envelopment in astrocytoma cells. J Virol 76:5147–5155. doi: 10.1128/JVI.76.10.5147-5155.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crump CM, Hung CH, Thomas L, Wan L, Thomas G. 2003. Role of PACS-1 in trafficking of human cytomegalovirus glycoprotein B and virus production. J Virol 77:11105–11113. doi: 10.1128/JVI.77.20.11105-11113.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cullen PJ, Korswagen HC. 2012. Sorting nexins provide diversity for retromer-dependent trafficking events. Nat Cell Biol 14:29–37. doi: 10.1038/ncb2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cullen PJ. 2008. Endosomal sorting and signalling: an emerging role for sorting nexins. Nat Rev Mol Cell Biol 9:574–582. doi: 10.1038/nrm2427. [DOI] [PubMed] [Google Scholar]
- 30.Mim C, Unger VM. 2012. Membrane curvature and its generation by BAR proteins. Trends Biochem Sci 37:526–533. doi: 10.1016/j.tibs.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffin CT, Trejo J, Magnuson T. 2005. Genetic evidence for a mammalian retromer complex containing sorting nexins 1 and 2. Proc Natl Acad Sci USA 102:15173–15177. doi: 10.1073/pnas.0409558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wassmer T, Attar N, Bujny MV, Oakley J, Traer CJ, Cullen PJ. 2007. A loss-of-function screen reveals SNX5 and SNX6 as potential components of the mammalian retromer. J Cell Sci 120:45–54. doi: 10.1242/jcs.03302. [DOI] [PubMed] [Google Scholar]
- 33.Carlton J, Bujny M, Peter BJ, Oorschot VM, Rutherford A, Mellor H, Klumperman J, McMahon HT, Cullen PJ. 2004. Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high-curvature membranes and 3-phosphoinositides. Curr Biol 14:1791–1800. doi: 10.1016/j.cub.2004.09.077. [DOI] [PubMed] [Google Scholar]
- 34.Haft CR, de la Luz SM, Bafford R, Lesniak MA, Barr VA, Taylor SI. 2000. Human orthologs of yeast vacuolar protein sorting proteins Vps26, 29, and 35: assembly into multimeric complexes. Mol Biol Cell 11:4105–4116. doi: 10.1091/mbc.11.12.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bugarcic A, Zhe Y, Kerr MC, Griffin J, Collins BM, Teasdale RD. 2011. Vps26A and Vps26B subunits define distinct retromer complexes. Traffic 12:1759–1773. doi: 10.1111/j.1600-0854.2011.01284.x. [DOI] [PubMed] [Google Scholar]
- 36.Kerr MC, Bennetts JS, Simpson F, Thomas EC, Flegg C, Gleeson PA, Wicking C, Teasdale RD. 2005. A novel mammalian retromer component, Vps26B. Traffic 6:991–1001. doi: 10.1111/j.1600-0854.2005.00328.x. [DOI] [PubMed] [Google Scholar]
- 37.Wassmer T, Attar N, Harterink M, van Weering JR, Traer CJ, Oakley J, Goud B, Stephens DJ, Verkade P, Korswagen HC, Cullen PJ. 2009. The retromer coat complex coordinates endosomal sorting and dynein-mediated transport, with carrier recognition by the trans-Golgi network. Dev Cell 17:110–122. doi: 10.1016/j.devcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim JP, Wang JT, Kerr MC, Teasdale RD, Gleeson PA. 2008. A role for SNX5 in the regulation of macropinocytosis. BMC Cell Biol 9:58. doi: 10.1186/1471-2121-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang JT, Kerr MC, Karunaratne S, Jeanes A, Yap AS, Teasdale RD. 2010. The SNX-PX-BAR family in macropinocytosis: the regulation of macropinosome formation by SNX-PX-BAR proteins. PLoS One 5:e13763. doi: 10.1371/journal.pone.0013763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hara S, Kiyokawa E, Iemura S, Natsume T, Wassmer T, Cullen PJ, Hiai H, Matsuda M. 2008. The DHR1 domain of DOCK180 binds to SNX5 and regulates cation-independent mannose 6-phosphate receptor transport. Mol Biol Cell 19:3823–3835. doi: 10.1091/mbc.E08-03-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless Red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 42.Krzyzaniak MA, Mach M, Britt WJ. 2009. HCMV-encoded glycoprotein M (UL100) interacts with Rab11 effector protein FIP4. Traffic 10:1439–1457. doi: 10.1111/j.1600-0854.2009.00967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchkovich NJ, Maguire TG, Alwine JC. 2010. Role of the endoplasmic reticulum chaperone BiP, SUN domain proteins, and dynein in altering nuclear morphology during human cytomegalovirus infection. J Virol 84:7005–7017. doi: 10.1128/JVI.00719-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cepeda V, Fraile-Ramos A. 2011. A role for the SNARE protein syntaxin 3 in human cytomegalovirus morphogenesis. Cell Microbiol 13:846–858. doi: 10.1111/j.1462-5822.2011.01583.x. [DOI] [PubMed] [Google Scholar]
- 45.Liu ST, Sharon-Friling R, Ivanova P, Milne SB, Myers DS, Rabinowitz JD, Brown HA, Shenk T. 2011. Synaptic vesicle-like lipidome of human cytomegalovirus virions reveals a role for SNARE machinery in virion egress. Proc Natl Acad Sci USA 108:12869–12874. doi: 10.1073/pnas.1109796108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tandon R, Mocarski ES. 2011. Cytomegalovirus pUL96 is critical for the stability of pp150-associated nucleocapsids. J Virol 85:7129–7141. doi: 10.1128/JVI.02549-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schauflinger M, Fischer D, Schreiber A, Chevillotte M, Walther P, Mertens T, von Einem J. 2011. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J Virol 85:3821–3832. doi: 10.1128/JVI.01540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meissner CS, Suffner S, Schauflinger M, von Einem J, Bogner E. 2012. A leucine zipper motif of a tegument protein triggers final envelopment of human cytomegalovirus. J Virol 86:3370–3382. doi: 10.1128/JVI.06556-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phillips SL, Bresnahan WA. 2012. The human cytomegalovirus (HCMV) tegument protein UL94 is essential for secondary envelopment of HCMV virions. J Virol 86:2523–2532. doi: 10.1128/JVI.06548-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seaman MN. 2005. Recycle your receptors with retromer. Trends Cell Biol 15:68–75. doi: 10.1016/j.tcb.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 51.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. doi: 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moorman NJ, Sharon-Friling R, Shenk T, Cristea IM. 2010. A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol Cell Proteomics 9:851–860. doi: 10.1074/mcp.M900485-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tugizov S, Wang Y, Qadri I, Navarro D, Maidji E, Pereira L. 1995. Mutated forms of human cytomegalovirus glycoprotein B are impaired in inducing syncytium formation. Virology 209:580–591. doi: 10.1006/viro.1995.1290. [DOI] [PubMed] [Google Scholar]
- 54.Tugizov S, Maidji E, Xiao J, Pereira L. 1999. An acidic cluster in the cytosolic domain of human cytomegalovirus glycoprotein B is a signal for endocytosis from the plasma membrane. J Virol 73:8677–8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Radsak K, Eickmann M, Mockenhaupt T, Bogner E, Kern H, Eis-Hubinger A, Reschke M. 1996. Retrieval of human cytomegalovirus glycoprotein B from the infected cell surface for virus envelopment. Arch Virol 141:557–572. doi: 10.1007/BF01718317. [DOI] [PubMed] [Google Scholar]
- 56.Tirabassi RS, Enquist LW. 1999. Mutation of the YXXL endocytosis motif in the cytoplasmic tail of pseudorabies virus gE. J Virol 73:2717–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nixdorf R, Klupp BG, Karger A, Mettenleiter TC. 2000. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J Virol 74:7137–7145. doi: 10.1128/JVI.74.15.7137-7145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beitia Ortiz de Zarate I, Cantero-Aguilar L, Longo M, Berlioz-Torrent C, Rozenberg F. 2007. Contribution of endocytic motifs in the cytoplasmic tail of herpes simplex virus type 1 glycoprotein B to virus replication and cell-cell fusion. J Virol 81:13889–13903. doi: 10.1128/JVI.01231-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moffat J, Mo C, Cheng JJ, Sommer M, Zerboni L, Stamatis S, Arvin AM. 2004. Functions of the C-terminal domain of varicella-zoster virus glycoprotein E in viral replication in vitro and skin and T-cell tropism in vivo. J Virol 78:12406–12415. doi: 10.1128/JVI.78.22.12406-12415.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kropff B, Koedel Y, Britt W, Mach M. 2010. Optimal replication of human cytomegalovirus correlates with endocytosis of glycoprotein gpUL132. J Virol 84:7039–7052. doi: 10.1128/JVI.01644-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Le Blanc I, Luyet PP, Pons V, Ferguson C, Emans N, Petiot A, Mayran N, Demaurex N, Faure J, Sadoul R, Parton RG, Gruenberg J. 2005. Endosome-to-cytosol transport of viral nucleocapsids. Nat Cell Biol 7:653–664. doi: 10.1038/ncb1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brankatschk B, Pons V, Parton RG, Gruenberg J. 2011. Role of SNX16 in the dynamics of tubulo-cisternal membrane domains of late endosomes. PLoS One 6:e21771. doi: 10.1371/journal.pone.0021771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bergant Marušič M, Ozbun MA, Campos SK, Myers MP, Banks L. 2012. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 13:455–467. doi: 10.1111/j.1600-0854.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu B, Stinski MF. 1992. Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis-acting elements. J Virol 66:4434–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teasdale RD, Loci D, Houghton F, Karlsson L, Gleeson PA. 2001. A large family of endosome-localized proteins related to sorting nexin 1. Biochem J 358:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 67.Sawano A, Miyawaki A. 2000. Directed evolution of green fluorescent protein by a new versatile PCR strategy for site-directed and semi-random mutagenesis. Nucleic Acids Res 28:E78. doi: 10.1093/nar/28.16.e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Posfai G, Kolisnychenko V, Bereczki Z, Blattner FR. 1999. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res 27:4409–4415. doi: 10.1093/nar/27.22.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 71.Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D. 2002. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J Virol 76:2440–2448. doi: 10.1128/jvi.76.5.2440-2448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Utz U, Britt W, Vugler L, Mach M. 1989. Identification of a neutralizing epitope on glycoprotein gp58 of human cytomegalovirus. J Virol 63:1995–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Waldo FB, Britt WJ, Tomana M, Julian BA, Mestecky J. 1989. Non-specific mesangial staining with antibodies against cytomegalovirus in immunoglobulin-A nephropathy. Lancet i:129–131. [DOI] [PubMed] [Google Scholar]
- 74.Nowak B, Gmeiner A, Sarnow P, Levine AJ, Fleckenstein B. 1984. Physical mapping of human cytomegalovirus genes: identification of DNA sequences coding for a virion phosphoprotein of 71 kDa and a viral 65-kDa polypeptide. Virology 134:91–102. doi: 10.1016/0042-6822(84)90275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simpson JA, Chow JC, Baker J, Avdalovic N, Yuan S, Au D, Co MS, Vasquez M, Britt WJ, Coelingh KL. 1993. Neutralizing monoclonal antibodies that distinguish three antigenic sites on human cytomegalovirus glycoprotein H have conformationally distinct binding sites. J Virol 67:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mach M, Kropff B, Dal MP, Britt W. 2000. Complex formation by human cytomegalovirus glycoproteins M (gpUL100) and N (gpUL73). J Virol 74:11881–11892. doi: 10.1128/JVI.74.24.11881-11892.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Britt WJ, Auger D. 1985. Identification of a 65 000 dalton virion envelope protein of human cytomegalovirus. Virus Res 4:31–36. doi: 10.1016/0168-1702(85)90018-8. [DOI] [PubMed] [Google Scholar]
- 78.Plachter B, Britt W, Vornhagen R, Stamminger T, Jahn G. 1993. Analysis of proteins encoded by IE regions 1 and 2 of human cytomegalovirus using monoclonal antibodies generated against recombinant antigens. Virology 193:642–652. doi: 10.1006/viro.1993.1172. [DOI] [PubMed] [Google Scholar]
- 79.Evan GI, Lewis GK, Ramsay G, Bishop JM. 1985. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol 5:3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fleckenstein B, Muller I, Collins J. 1982. Cloning of the complete human cytomegalovirus genome in cosmids. Gene 18:39–46. doi: 10.1016/0378-1119(82)90054-3. [DOI] [PubMed] [Google Scholar]
- 81.Vidal M, Brachmann RK, Fattaey A, Harlow E, Boeke JD. 1996. Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc Natl Acad Sci USA 93:10315–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Durfee T, Becherer K, Chen PL, Yeh SH, Yang Y, Kilburn AE, Lee WH, Elledge SJ. 1993. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev 7:555–569. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- 83.Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368. doi: 10.1099/vir.0.83286-0. [DOI] [PubMed] [Google Scholar]
- 84.Howell CL, Miller MJ. 1983. Effect of sucrose phosphate and sorbitol on infectivity of enveloped viruses during storage. J Clin Microbiol 18:658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]