

Abstract
Transcription termination delimits transcription units but also plays important roles in limiting pervasive transcription. We have previously shown that transcription termination occurs when elongating RNA polymerase II (RNAPII) collides with the DNA‐bound general transcription factor Reb1. We demonstrate here that many different DNA‐binding proteins can induce termination by a similar roadblock (RB) mechanism. We generated high‐resolution transcription maps by the direct detection of RNAPII upon nuclear depletion of two essential RB factors or when the canonical termination pathways for coding and non‐coding RNAs are defective. We show that RB termination occurs genomewide and functions independently of (and redundantly with) the main transcription termination pathways. We provide evidence that transcriptional readthrough at canonical terminators is a significant source of pervasive transcription, which is controlled to a large extent by RB termination. Finally, we demonstrate the occurrence of RB termination around centromeres and tRNA genes, which we suggest shields these regions from RNAPII to preserve their functional integrity.
Keywords: pervasive transcription, Rap1, roadblock termination, transcription readthrough, transcription termination mechanism
Subject Categories: RNA Biology, Transcription
Introduction
The compact genome of Saccharomyces cerevisiae is covered by several machineries that need to be temporally and spatially coordinated for allowing the robust reading and perpetuation of the genetic information.
The complexity of the transcriptional landscape is paradigmatic in this regard. Transcription initiation occurs frequently in regions and direction that largely overrun the canonical annotation of genes, a phenomenon known as pervasive transcription. This is due to the inherently loose control imposed on initiation by the structure of chromatin and to the intrinsic bi‐directionality of promoters, which is generally conserved in evolution (Porrua & Libri, 2015). This promiscuity of transcription events is a potential threat to the stability of gene expression programs because many transcription events are susceptible to interfere with each other. Pervasive transcription might also affect other DNA‐related events, such as replication, chromosome segregation, or the expression of RNA polymerase I‐ and III‐dependent genes. The integration of widespread transcription with other cellular processes is a complex process, requiring tools to limit and coordinate concurrent events.
Transcription termination plays essential roles in the control of pervasive transcription. In yeast, two main termination pathways exist. The first depends on the cleavage and polyadenylation factor–cleavage factor (CPF‐CF, referred to as CPF hereafter) and terminates transcription of genes producing mRNAs and some non‐coding RNAs. The CPF complex recognizes signals on the nascent RNA and cleaves the latter, producing a 5′ fragment that is polyadenylated by the Pap1 poly(A) polymerase and exported to the cytoplasm. The 3′ fragment, still associated with the transcribing polymerase, is recognized and degraded by a 5′‐3′ exonuclease, Rat1, which contributes to dismantling the elongation complex by a still elusive mechanism. The CPF is also believed to be directly involved in the polymerase release step of termination by allosterically modifying the properties of the transcription elongation complex (for a recent review, see Porrua et al, 2016).
The second canonical pathway depends on the NNS (Nrd1‐Nab3‐Sen1) complex and was first associated with the production of sn‐ and snoRNAs (Steinmetz et al, 2001). Nrd1 and Nab3 bind the nascent RNA at short motifs containing a well‐conserved 4–5 nucleotides core and are thought to recruit the Sen1 helicase that translocates on the nascent RNA to release the polymerase. Peculiar to this pathway is that the released RNA is polyadenylated by a different poly(A) polymerase, Trf4, functioning within the TRAMP4/5 (Trf4/5‐Air2/1‐Mtr4‐polyadenylation) complex, and trimmed to its mature size in the nucleus by the exosome, a large multisubunit complex that is endowed with 3′–5′ exonuclease activities (Porrua & Libri, 2015).
A large share of the transcripts produced by pervasive transcription do not code for proteins, and to what extent these RNAs have specific functions remains matter of debate. They are sorted in classes, generally defined by the pathways associated with their metabolism. CUTs (cryptic unstable transcripts) have been first described based on their extreme instability (Wyers et al, 2005). These RNAs derive from transcription events terminated by the NNS pathway and are degraded to completion by the TRAMP‐exosome pathway (Wyers et al, 2005; Arigo et al, 2006; Thiebaut et al, 2006). When NNS termination is defective, elongated forms of CUTs are produced that have been recently named NUTs (Nrd1 unterminated transcripts, Schulz et al, 2013). Some of the non‐coding RNAs produced by pervasive transcription are sufficiently stable to be detected in wild‐type cells (SUTs, stable unannotated transcripts, David et al, 2006) or are degraded in the cytoplasm by the nonsense‐mediated decay (NMD) and Xrn1 pathways (XUTs, Xrn1‐sensitive unstable transcripts, van Dijk et al, 2011; Malabat et al, 2015). Finally, some are only detected in particular physiological conditions (MUTs, meiotic unannotated transcripts, Lardenois et al, 2011).
We have recently described an additional pathway of transcription termination that depends on the general regulatory factor (GRF) Reb1. We have shown that the elongating polymerase pauses upstream of DNA‐bound Reb1, which prompts its release by a mechanism that involves its ubiquitylation and presumably degradation (Colin et al, 2014). Insertion of a Reb1 binding site in a region of elongation is sufficient for termination, indicating that this “roadblock” (RB) pathway does not require additional sequence elements. Because, akin to CUTs, the RNAs released are polyadenylated by TRAMP and degraded by the nuclear exosome, these transcripts were dubbed RUTs (Reb1‐dependent unstable transcripts; Colin et al, 2014).
Here we demonstrate that many additional DNA‐binding complexes or factors can elicit RB termination and studied the overall impact of RB termination in the yeast genome. Using an improved crosslinking and cDNA analysis protocol (CRAC, Granneman et al, 2009), we sequenced nascent transcripts to generate the first high‐resolution transcription maps upon depletion of two RB factors, and analyzed the genomewide impact of roadblock termination in wild‐type cells or under conditions defective for NNS‐ or CPF‐dependent termination. We directly demonstrate that RNAPII pausing depends on the roadblock factor and not on sequence elements or other events. We show that many RB events are associated with natural readthrough at canonical CPF or NNS terminators and that RB termination plays a general quality control role in limiting such pervasive transcription events. We studied the mutual relationships between RB termination and the other pathways and conclude that they are functionally independent and act redundantly to provide robust demarcation of adjacent transcription units. Finally, we show that roadblock termination also occurs around centromeres and tRNAs, which we suggest to be protected from the potentially negative interference of surrounding pervasive transcription events.
The faculty of DNA‐associated factors to alter the processivity of elongation complexes, and the diversity of these factors, reveals a major role of RB termination in shaping the transcription landscape. This also underlies a large potential for regulation that likely extends to many organisms.
Results
In vivo selection reveals Rap1‐dependent transcription termination
We have previously described a procedure to select transcription terminators from pools of naïve sequences (Porrua et al, 2012; Colin et al, 2014). Briefly, test sequences are inserted within a transcription unit driven by the tetracycline‐repressible promoter (TETP), roughly 200 nt downstream of the transcription start site. A second promoter, from the GAL1 gene (GAL1 P), is inserted downstream and drives expression of a selectable marker, CUP1, the expression of which is required for yeast growth in copper‐containing medium (Fig 1A). In the absence of a terminator in the test sequence, transcription driven from TETP silences GAL1 P by transcription interference and prevents CUP1 expression, leading to copper sensitivity. When the test sequence induces termination, the CUP1 gene is expressed and yeasts grow on copper‐containing plates. Using this system, we selected terminators from a pool of sequences containing a stretch of 120 random nucleotides. We selected many sequences inducing termination via the NNS pathway and via the Reb1‐dependent roadblock pathway (Porrua et al, 2012; Colin et al, 2014). We also selected sequences that do not belong to either class, some of which contain a motif resembling a Rap1 binding site (Figs 1A and EV1A). Rap1 recognizes its site via a Myb‐like DNA‐binding domain and is involved in many DNA‐associated processes, including telomere maintenance and gene expression (for a review, see Azad & Tomar, 2016). Rap1 is also strongly associated with the positioning and formation of nucleosome‐free regions (NFR; Hartley & Madhani, 2009; Kubik et al, 2015 and references therein).
Figure 1. Analysis of the transcripts produced upon transcription termination induced by Rap1.
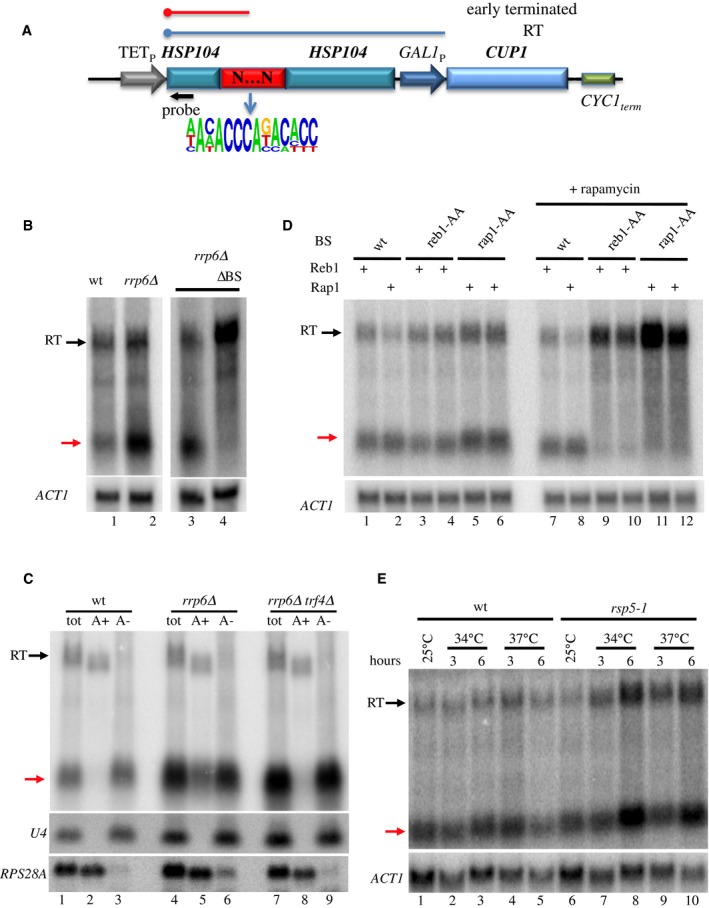
- Scheme of the reporter used for selecting terminators from naïve sequences. TETP: doxycycline‐repressible promoter; GAL1 P: GAL1 promoter. The random sequence (120 nt, red box) was inserted within HSP104 sequences upstream of GAL1 P . The transcripts produced in the presence (red) or absence (blue) of termination signals are indicated. The readthrough (RT) transcript terminates at a cryptic terminator within the GAL1 promoter. A logo (http://weblogo.berkeley.edu/logo.cgi) derived from the putative Rap1 binding sites found in the selected terminators is shown. The approximate position of the oligonucleotide probe used for Northern blot analysis is indicated by a black arrow.
- Northern blot analysis of transcripts produced in the presence of a Rap1‐dependent terminator in wt or rrp6∆ cells as indicated. ∆BS: RNAs derived from a construct containing a precise deletion of the Rap1 binding site (BS). A red arrow indicates the position of the short transcript produced at the Rap1 termination site. RT transcripts are indicated by a black arrow.
- Analysis of the polyadenylation status of transcripts derived from Rap1‐dependent termination. Total, polyadenylated (A+, oligo dT‐selected) and non‐adenylated (A−, oligo dT‐depleted) fractions are analyzed in the strains indicated. Rap1‐terminated and RT transcripts indicated as in (B). U4snRNA and RPS28A RNAs are used as controls for non‐adenylated and adenylated species, respectively.
- Northern blot analysis of strains containing reporters bearing a Rap1‐dependent or a Reb1‐dependent terminator (clone X3, Colin et al, 2014) as indicated. Reb1 (Reb1‐AA) or Rap1 (Rap1‐AA) anchor away strains were used to deplete either protein by the addition of rapamycin (lanes 7–12, two biological replicates). Red and black arrows indicate short and readthrough transcripts as in (B).
- Northern blot analysis of RNAs derived from a reporter containing a Rap1‐dependent terminator in a wild‐type (lanes 1–5) or a thermosensitive rsp5‐1 strain (lanes 6–10) grown at different temperatures as indicated. Note that the short RNA (red arrow) mainly represents nascent RNA associated with the roadblocked polymerase.
Source data are available online for this figure.
Figure EV1. 3′ end mapping of Rap1‐terminated transcripts.
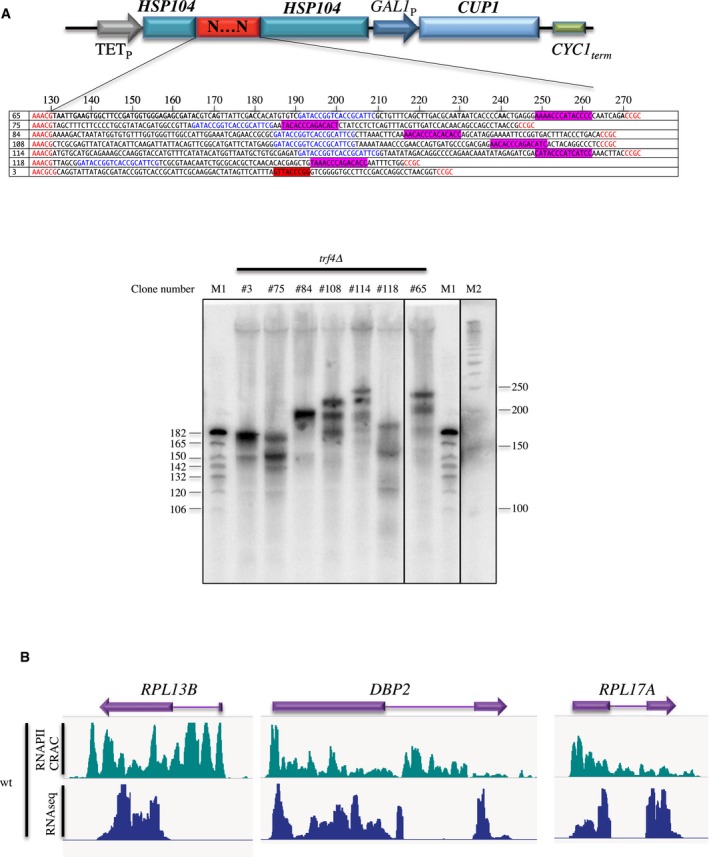
- Top: Schematic drawing of the reporter system used for selecting the RB terminators with the position of the insertion and the sequence of the selected clones containing a Rap1 site (purple). In blue the sequence of a constant linker used for constructing the pool. Flanking HSP104 sequences are indicated in red. Bottom: PAGE‐northern blot analysis of RNAs produced by the different constructs after oligonucleotide‐directed RNaseH cleavage at −130 nt from the start of the insertion. The position from the RNase H cleavage point is indicated above the sequences. The presence of several shorter RNAs might reveal the occurrence of termination at sites of RNAPII piling up. All analyses were done in an trf4∆ strain to detect unstable transcripts. All lanes are derived from the same gel; marker M1 is shown twice for clarity.
- Snapshots showing the RNAPII CRAC and RNAseq signal at intron‐containing genes, illustrating the co‐transcriptional nature of the CRAC signal.
Source data are available online for this figure.
RNA species of a size compatible with termination occurring immediately upstream of the Rap1 site were observed when a selected terminator was present in the reporter construct (Figs 1B, lane 1 and EV1A). These transcripts are only detected when the Rap1 binding site is present (Fig 1B, lanes 3–4) and are strongly sensitive to degradation, as indicated by their marked steady‐state increase in rrp6∆ or rrp6∆trf4∆ mutants of the nuclear 3′–5′ RNA degradation pathway (Fig 1B and C). A major fraction of the transcripts detected in rrp6∆ cells are non‐adenylated (Fig 1C, compare lanes 5 and 6), and a fraction is polyadenylated by Trf4 (compare lanes 5 and 8) and is strongly sensitive to exosomal degradation (Fig 1C, compare lanes 2–3 to 5–6). Non‐adenylated RNAs are also subject to degradation by the exosome (compare lanes 3 and 6), but can be detected in the wild‐type strain (Fig 1C, lanes 1,3), consistent with the notion that they represent the nascent RNAs associated with stalled polymerases.
To demonstrate that Rap1, and not overlapping termination signals, is responsible for releasing RNAPII, we analyzed the RNAs produced upon nuclear depletion of Rap1 with the anchor away methodology (Haruki et al, 2008; Kubik et al, 2015). In the absence of Rap1, we observed the disappearance of the short RNA species, to the profit of a longer species earmarking termination at a downstream site (Fig 1D). As a control, we also show the effect of the nuclear depletion of Reb1 at a site of Reb1‐dependent termination (Colin et al, 2014).
Finally, we have previously shown that release of the roadblocked polymerase from the DNA template occurs following its ubiquitylation that depends on the Rsp5 ubiquitin ligase. The failure to clear the roadblocked RNAPII results in increasing levels of both the nascent transcript (preferentially detected in a wild‐type strain) and the RT transcripts, due to increased opportunity to overcome the RB when the polymerase is not released (Colin et al, 2014). Northern blot analysis confirmed such expected increase when the Rap1‐roadblocked polymerase is less efficiently removed in a thermosensitive rsp5‐1 mutant strain (Fig 1E).
Together, these results demonstrate that Rap1 induces transcription termination by a roadblock mechanism.
Rap1‐dependent transcription termination in the Saccharomyces cerevisiae genome
To assess the natural extent of Rap1‐dependent RB termination, we analyzed the occurrence of RNAPII pausing immediately upstream of Rap1 sites, which is a hallmark of roadblock termination (Colin et al, 2014). We profiled RNAPII occupancy in a wild‐type and a Rap1 anchor away (Rap1‐AA) strain by an improved version of the crosslinking and cDNA analysis protocol (CRAC, Granneman et al, 2009). This approach allows assessing the position of the polymerase at the nucleotide resolution level by sequencing the nascent transcript associated with the largest subunit of the enzyme after in vivo UV crosslinking (RNAPII CRAC, Milligan et al, 2016). The analysis indeed detects nascent transcripts, as demonstrated by the coverage of intronic regions in the RNAPII CRAC dataset but not in the sequencing of mature, total RNAs (Figs 2C and EV1B).
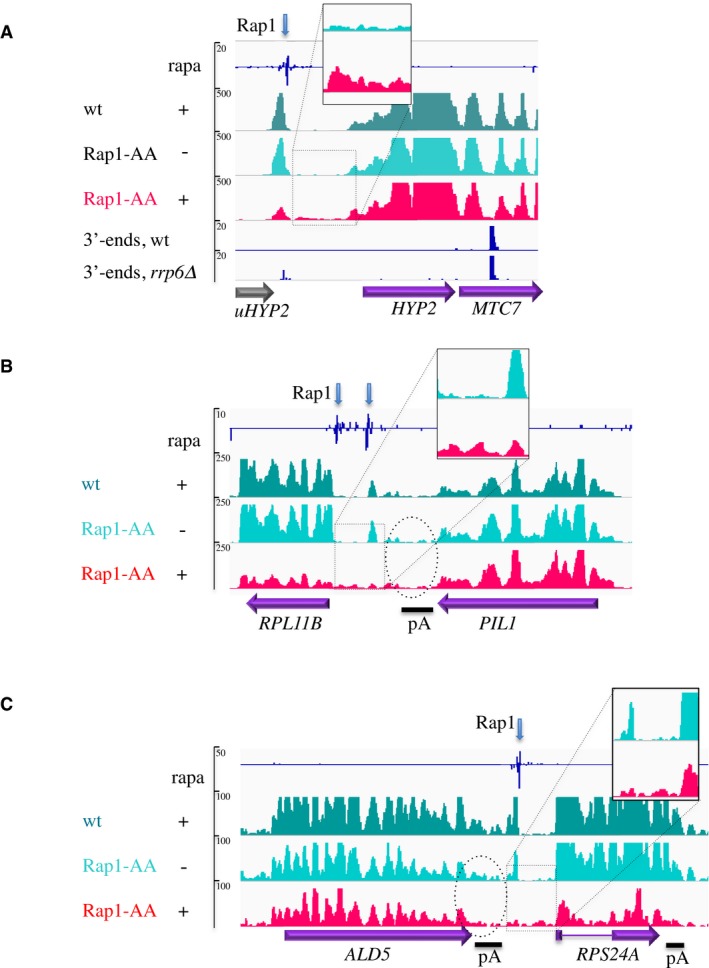
Figure 2. RNAPII occupancy at sites of Rap1 roadblock detected by CRAC analysis.
-
ARNAPII CRAC profile at a site of roadblock upstream of HYP2 (only the signals on the strand of the annotated features are shown). A peak of CRAC RNAPII signal is visible upstream the site of Rap1 occupancy (blue arrow, ChIP exo data, Rhee & Pugh, 2011) in a wild‐type strain in the presence of rapamycin (dark green track) or Rap1‐AA in the absence of the drug (light green track). The roadblock peak is markedly diminished when Rap1 is depleted from the nucleus by the addition of rapamycin to Rap1‐AA cells (red track). Transcription termination at the RB site is accompanied by the production of a non‐annotated cryptic transcript (uHYP2, gray arrow) with a predominant 3′ end located 13 nt upstream of the Rap1 site (data from Roy et al, 2016). The maximum value of the RNAPII peak is 26 nt upstream of the sequence of the Rap1 site. The position of multiple polyadenylation (pA) sites for HYP2 as defined by 3′‐T‐Fill analysis (Wilkening et al, 2013) is indicated. Note the occurrence of transcriptional readthrough after the roadblock when Rap1 is depleted (inset).
-
B, CSame as in (A), with Rap1 sites located between two genes arranged in tandem. The dotted oval underscores the level of polymerase occupancy between the CPF terminator and the roadblock, which is not affected by depletion of the roadblock factor. The maxima of the RNAPII peak are located 33 nt (PIL1) and 15 nt (ALD5) upstream of the first Rap1 binding site.
Notable examples of Rap1‐dependent roadblock sites are shown in Fig 2. CRAC analysis revealed a marked RNAPII peak upstream of sites of Rap1 binding (Rhee & Pugh, 2011; Knight et al, 2014) at the HYP2, RPL11B, and RPS24A loci.
At the HYP2 locus (Fig 2A), a Rap1‐dependent RB terminates transcription of an upstream, non‐annotated transcription unit (dubbed uHYP2), leading to the production of a cryptic transcript as revealed by SAGE analysis (Fig 2A, Neil et al, 2009). At the RPL11B and RPS24A loci (Fig 2B and C), roadblocked polymerases most likely derive from transcription events reading through the upstream terminator (see below). Nuclear depletion of Rap1 by the addition of rapamycin led to the significant decrease of the RNAPII peak and to the spreading of a readthrough signal downstream of the RB site (Fig 2, insets). Rap1‐dependent termination could be confirmed by inserting a short region only containing the two Rap1 sites present at the HYP2 locus in the heterologous context of our reporter system (Appendix Fig S1A).
To extend these findings genomewide, we generated aggregate plots by profiling the average distribution of the RNAPII CRAC signal around aligned sites of Rap1 occupancy (Fig 3). A major peak of average RNAPII occupancy is present downstream of the aligned occupancy sites, due to general presence of genes regulated by Rap1 (Fig 3A, left). Importantly, however, a significant roadblock peak was observed upstream of Rap1 binding, which strongly decreased upon Rap1 depletion (compare the red and blue traces).
Figure 3. Metasite analysis of roadblock termination at Rap1 and Reb1 sites.
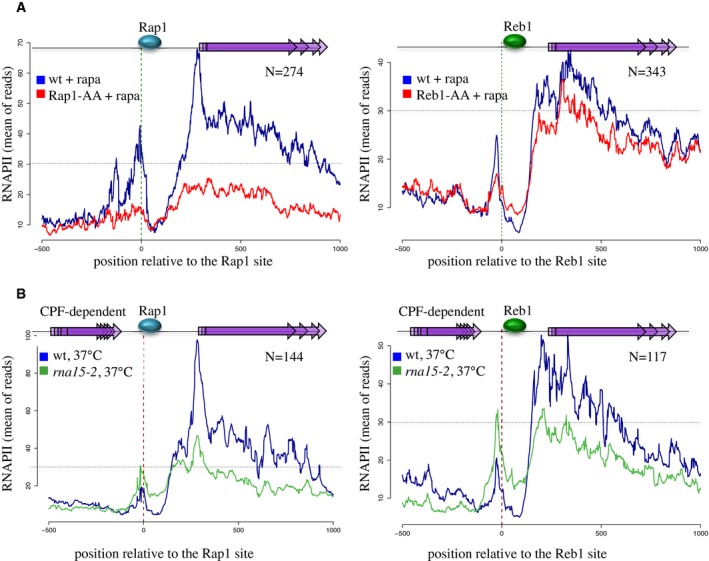
- Average RNAPII CRAC profile at genomic regions aligned on Rap1 (left panel) or Reb1 (right panel) occupancy sites, in the presence (blue) or absence (red) of the roadblock factor. In both cases, the latter was depleted from the nucleus by the addition of rapamycin. Overlapping purple arrows represent features transcribed downstream of Rap1 or Reb1 occupancy sites. Note that the two panels have a different y‐axis scale due to the average higher expression of Rap1‐dependent genes; a dotted horizontal line marks the same average occupancy for comparison.
- Same as in (A), using only Rap1 or Reb1 sites located within 300 nt downstream of genes terminated by the CPF pathway. The RNAPII average profile was determined for the wild‐type strain (blue) or an rna15‐2 (green) strain at the non‐permissive temperature of 37°C for the mutant; data from the same cells at permissive temperature have not been plotted, but are available.
Similar RNAPII CRAC analyses were also performed upon depletion of Reb1. Peaks of RNAPII pausing were readily observed at individual sites of Reb1 occupancy that significantly decreased upon Reb1 depletion (Appendix Fig S1B and data not shown). Aggregate plots (Fig 3A, right) show, as for Rap1, a major peak of transcription initiation mainly due to Reb1‐regulated genes, and a prominent RB peak that is Reb1‐dependent.
The detection of a RB at Rap1 sites was not due to crosslinking of Rap1 to the DNA, because it could be observed using techniques that do not rely on crosslinking (NET‐seq, Churchman & Weissman, 2011; Appendix Fig S2A and B) or that rely on the sole crosslinking of the RNA to proteins (PAR‐CLIP, Schaughency et al, 2014; Appendix Fig S2B and data not shown). Finally, the occurrence of transcription termination at Rap1 and Reb1 occupancy sites is consistent with a peak of RNA 3′ ends that coincides with the site of RB and is generally more prominent in a degradation defective rrp6∆ strain (Appendix Fig S2C and D).
Overall, these results demonstrate the widespread occurrence of Rap1‐ and Reb1‐dependent, roadblock transcription termination in S. cerevisiae.
Roadblock termination limits widespread readthrough transcription in the Saccharomyces cerevisiae genome
Many Reb1 and Rap1 sites are located in intergenic regions, frequently downstream of genes. Based on a few model cases, we have previously proposed that roadblock termination might function to limit transcription reading through canonical, CPF‐dependent terminators (Colin et al, 2014), but neither the general validity of this concept, nor the generalized occurrence of readthrough transcription could be demonstrated.
If polymerases fail to terminate at canonical sites with a significant frequency, they are expected to accumulate at sites of Reb1 and Rap1 binding downstream of genes, where they should be easily detected because of the roadblock.
To address this possibility, we restricted our RNAPII CRAC metasite analyses to Reb1 and Rap1 occupancy sites located within 300 nt downstream of mRNA‐coding genes. In these conditions, only polymerases escaping CPF‐dependent termination, if any, should contribute to the metaprofile upstream of the roadblock. As shown in Fig 3B, polymerases accumulate at Rap1 and Reb1 sites downstream of canonical CPF terminators in the wild‐type strain strongly suggesting the existence of a constitutive transcriptional readthrough. To substantiate this notion, we also performed a parallel RNAPII CRAC analysis using a thermosensitive rna15‐2 allele, which impairs CPF termination. In these conditions, we observed a clear increase in the roadblock peak relative to what observed in wt cells, supporting the notion that the flux that aliments roadblocked polymerases originates from upstream transcription units and increases when upstream termination is defective (Fig 3B, compare green and blue traces). As a control, we profiled RNAPII distribution at the same set of CPF‐dependent features upon nuclear depletion of Nrd1 (Schaughency et al, 2014), an essential actor of NNS termination that is not involved in termination of mRNA‐coding genes. In these conditions, we did not observe an increase in the roadblock peak (Fig EV2A and data not shown). Manual inspection of a significant number of these locations ruled out the existence of intergenic transcription initiation based on the recent published repertoire of RNAPII transcripts 5′ ends (data not shown; see also Fig EV2B; Malabat et al, 2015).
Figure EV2. Roadblock termination functions as a fail safe mechanism to limit constitutive readthrough.
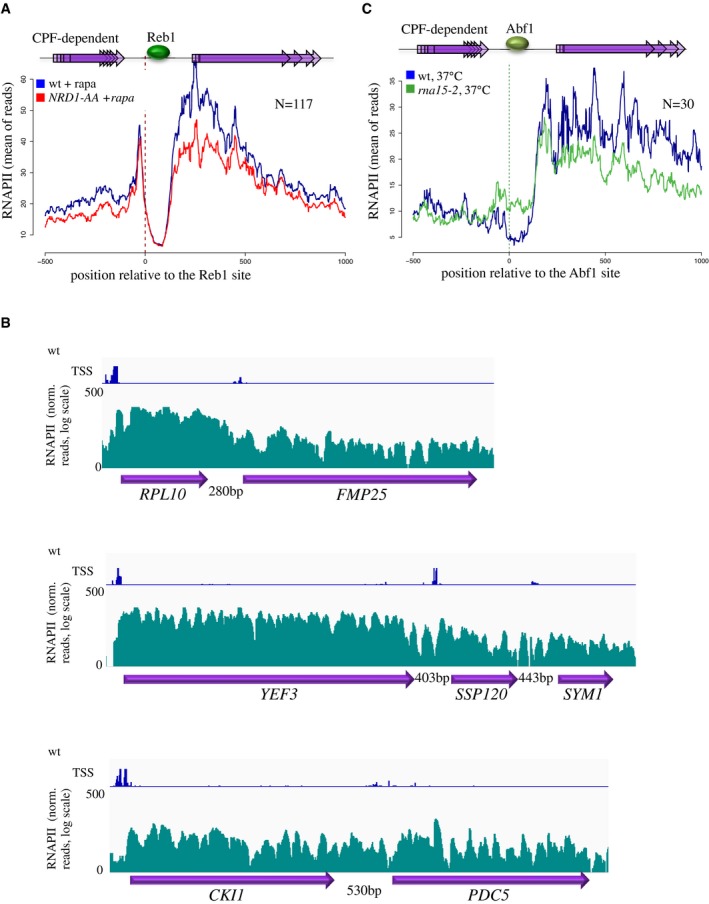
- Aggregate plot showing the average RNAPII CRAC profile at sites of Reb1 occupancy located within 300 nt downstream of genes terminated by the CPF pathway as in Fig 3B. The plot demonstrates that in striking contrast to alteration of the CPF pathway, affecting NNS termination by nuclear depletion of Nrd1 has no significant effects on the accumulation of RNAPII at the site of roadblock. Sites used in these analyses are listed in Dataset EV1.
- Snapshots illustrating the presence of significant levels of intergenic RNAPII CRAC signals at three tandem gene loci. The transcription initiation sites (TSS, Malabat et al, 2015) detected in the regions shown are indicated on top of the RNAPII CRAC tracks. In all these cases, intergenic initiation cannot be detected, indicating that the intergenic RNAPII signal derives from the constitutive readthrough at the CPF‐dependent terminator of the upstream gene. Note that the levels of the readthrough signal in these cases are comparable to the levels of transcription of the downstream gene.
- Aggregate plot showing the average RNAPII CRAC profile at sites of Abf1 occupancy located within 300 nt downstream of genes terminated by the CPF pathway as in Fig 3B. The plots show the average RNAPII occupancy in the wild‐type and in rna15‐1 cells grown for 1.5 hours at the non‐permissive temperature. Sites used in these analyses are listed in Dataset EV1.
Similarly to Reb1 and Rap1, Abf1 belongs to the class of general regulatory factors and contains a myb‐like DNA‐binding domain (Fermi et al, 2017 and references therein). We profiled the RNAPII CRAC signal around sites of Abf1 occupancy downstream of CPF terminators. Although less prominent, a RB peak was observed, which increased, as for Rap1 and Reb1, when termination was impaired in an rna15‐2 mutant (Fig EV2C). A metaprofile analysis using a larger set of Abf1 occupancy sites is shown below in Fig EV3.
Figure EV3. Metasite analyses illustrating the profile of RNAPII CRAC signal at various transcription factor binding sites.
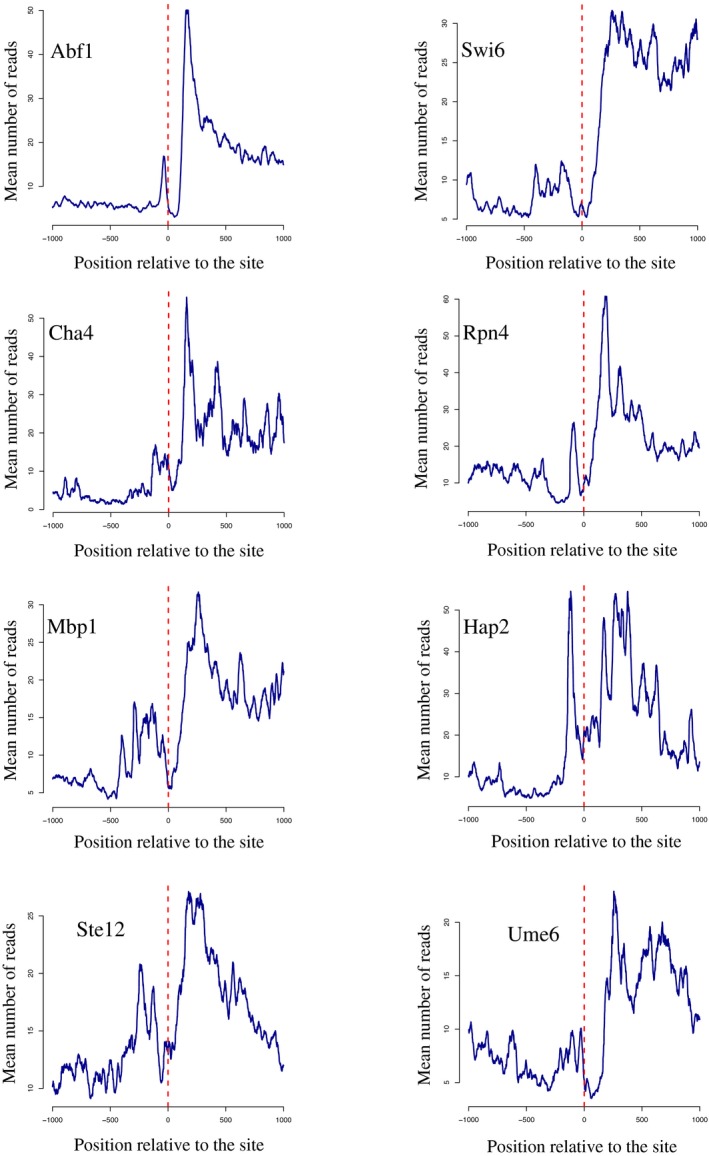
Aggregate plots showing the profile of RNAPII CRAC signal around sites of binding for the transcription factors indicated. The large peak after each binding site corresponds to downstream events of transcription initiation as for the plots in Fig 3A. The roadblock peak precedes the position of aligned binding sites. Sites used in these analyses are listed in Dataset EV1.
Transcriptional readthrough was not restricted to sites containing a downstream Rap1, Reb1, or Abf1 roadblock but could be consistently revealed by the significant detection of intergenic transcription downstream of genes in the absence of dedicated initiation sites. A few representative snapshots are shown in Fig EV2B, in which the levels of readthrough transcription are comparable to the levels of transcription of the downstream gene.
Overall, these results demonstrate the widespread occurrence of transcription readthrough at CPF terminators in strains that are proficient for transcription termination. Such pervasive readthrough events are restricted, to a significant extent, by downstream roadblock termination.
Roadblock termination and the CPF pathway function independently
RNA polymerase II pausing is generally considered to promote termination by favoring “chasing” of the polymerase by Rat1 at CPF‐dependent genes. It could be conceived that RB pausing functions as part of the CPF pathway for the efficient release of the polymerase. In this perspective, removing the roadblock should significantly affect the overall efficiency of termination. To address this possibility, we assessed whether termination failure could be observed at CPF terminators when the downstream Reb1‐ or Rap1‐dependent roadblocks were removed by nuclear depletion of either factor. Two examples of CPF‐dependent genes with a downstream roadblock are shown in Fig 2. In both cases, transcription termination occurs efficiently at the CPF sites even in the absence of the roadblock as witnessed by the similar decrease in the RNAPII signal at and downstream of the termination region (Fig 2B and C, dotted oval).
To generalize these observations, we compared the RNAPII metaprofile in regions of CPF termination upstream of a Rap1 binding site in the presence and absence of the roadblock factor (Fig 4). The precise location of transcription termination for each gene is not known, but we reproducibly observed a decrease in the RNAPII CRAC signal in the region around the sites of poly(A) addition (Fig 2 and data not shown). This early decrease in the RNAPII signal was to some extent unexpected, but was also observed using NetSeq (data from Harlen et al, 2016; not shown). It might be due to termination at cryptic or earlier sites of poly(A) addition or to the higher speed of the polymerase in this region. Irrespective of its possible components, this signal was clearly different in rna15‐2 cells (see below, Fig 4B), which are termination impaired at the non‐permissive temperature, suggesting that it is linked to the occurrence of termination. We therefore anchored the alignment to the strongest site of poly(A) addition for each gene (Pelechano et al, 2014) and focused our analysis on the region of early termination, to avoid interference with the RNAPII signals at the roadblock. As shown in Fig 4A, a progressive decrease in the average RNAPII signal was observed in wild‐type cells in this region, consistent with the occurrence of termination. As a control, transcription readthrough was clearly observed when termination was impaired in rna15‐2 cells at the non‐permissive temperature (Fig 4B, compare green and blue traces). Importantly, however, upon depletion of Rap1, CPF‐dependent termination occurred efficiently, as witnessed by the identical decline in the RNAPII CRAC signal in the termination region (Fig 4A, compare red and blue traces). Similar results were obtained for the set of CPF‐dependent genes upstream of a Reb1‐dependent roadblock (data not shown). To quantitate these results, we calculated the fractional level of readthrough for each CPF‐dependent gene upstream of a Rap1‐dependent roadblock by dividing the density of reads in the termination region by the density in the gene body (Fig 4C). The distribution of the values obtained is strongly affected by the rna15‐2 mutation, as expected for a bona fide termination defect (P = 10−5), but not by the absence of Rap1 (P = 0.4), demonstrating that removing the roadblock does not significantly impact CPF termination.
Figure 4. Aggregate RNAPII profile at genes followed by a Rap1 site aligned on the poly(A) site.
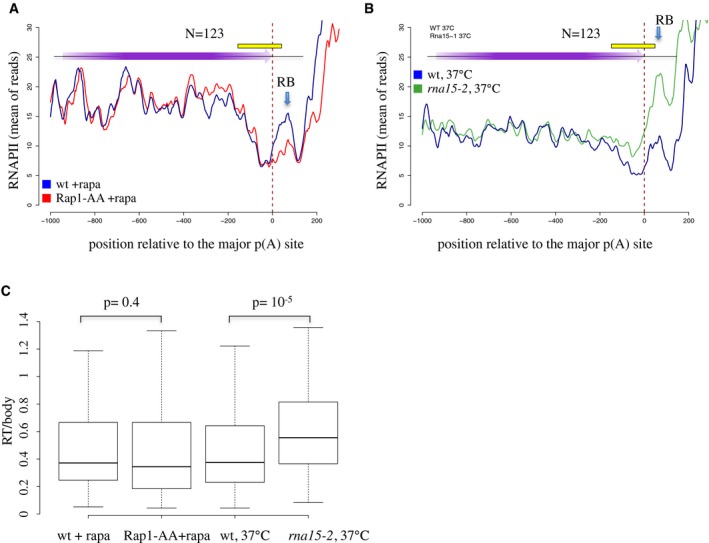
- Genes terminated by the CPF pathway and followed by a Rap1 site were aligned on the major poly(A) site and the average RNAPII CRAC signal was plotted for the wild‐type (blue) or Rap1‐AA (red) strain in the presence of rapamycin to induce the nuclear depletion of Rap1 in Rap1‐AA. The termination region, defined by the region displaying an average decrease of the RNAPII signal, is indicated by a yellow rectangle. Note that the signal in this region is not affected by the depletion of the RB. The roadblock peak (RB), indicated by a blue arrow, is broader and smaller in these plots because genomic regions are not precisely aligned on the RB site.
- As in (A), the average RNAPII profile in wild‐type (blue) and rna15‐2 (green) cells at the non‐permissive temperature was plotted to highlight a bona fide termination defect. To visually appreciate the occurrence of readthrough, we normalized the read counts so that the average RNAPII CRAC signals in gene bodies are comparable in wt and rna15‐2 cells.
- Boxplots representing the distribution of the ratios between the density of reads in the 100 nt immediately preceding the major poly(A) site (RT) and the density of reads in each ORF (body) in the indicated strains and conditions. The plots were generated by the standard R boxplot function. The central line represent the 50th percentile. The top and bottom of the box represent the 75th and 25th percentile respectively. The bottom whisker is the lowest value still within 1.5 Interquartile range (IQR); the top whisker is the highest values still within 1.5 IQR. The number of sites used for the analysis corresponds to the rap1 sites for which an experimentally defined polyadenylation site could be found within 300 bp upstream of the site (123). A two‐sided t‐test has been used to assess statistical significance. Sites used in these analyses are listed in Dataset EV1.
Relationships between RB‐ and NNS‐dependent termination
While this work was in progress, another study (Roy et al, 2016) proposed that roadblock‐ and NNS‐dependent termination are functionally linked, notably suggesting that (i) roadblocked polymerases are released by the NNS pathway and (ii) the roadblock is part of the mechanism of snoRNA termination. We revisited these important questions using our high‐resolution RNAPII CRAC in cells defective for the CPF, NNS, and roadblock pathways.
Roadblock peaks have been shown to increase in strains defective for NNS termination, which was interpreted as evidence that roadblocked polymerases are not efficiently cleared when NNS termination is impaired (Roy et al, 2016). Alternatively, it is possible that this increase in RB peaks is due to the accumulation of polymerases failing to terminate at upstream NNS‐dependent terminators. Consistent with this notion, we observed increased RB peaks only at sites downstream of NNS terminators when the NNS complex is defective (Fig 5 and data not shown). When the RB site follows a CPF terminator, depletion of Nrd1, Sen1, or mutation of Nab3 does not affect the levels of roadblocked polymerases. This is illustrated at the PIL1 and ALD5 loci (Appendix Fig S3A), and more generally in the aggregate RNAPII CRAC profile at Rap1 RB sites downstream of CPF terminators (Fig EV2A). Here, the depletion of Nrd1 poorly affects the signal at the Rap1 roadblock, which is, on the contrary, strongly increased upon impairment of CPF termination in the rna15‐2 mutant (Fig 3B and Appendix Fig S3A). These data demonstrate that the NNS pathway is not generally required for the clearance of roadblocked polymerases.
Figure 5. Analysis of the impact of RNAPII roadblocks in termination of snoRNAs.
-
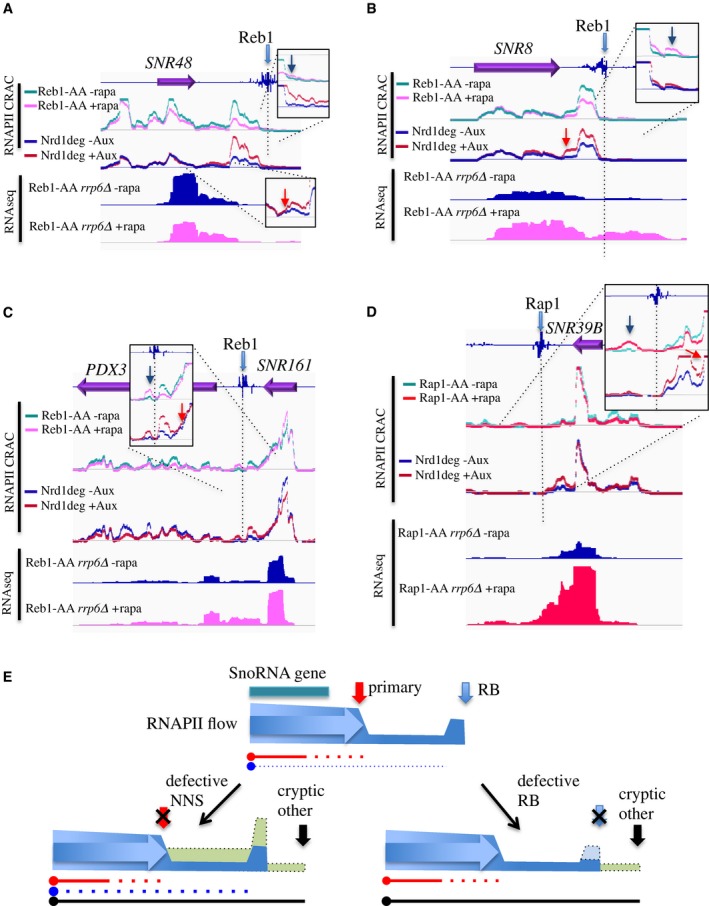
A–DRNAPII CRAC and RNAseq profiles at the indicated genomic loci and conditions (only the signals on the strand of the annotated features are shown). The position of the Reb1 or Rap1 occupancy (Rhee & Pugh, 2011) is indicated (light blue filled arrow). The occupancy profile in the presence or absence of the RB factor or Nrd1 has been overlapped for ease of comparison. The regions of the readthrough after the roadblock (dark blue arrow) or after the NNS terminator (red arrow) have been enlarged in the insets.
-
EModel of primary (NNS) and secondary (RB) termination at snoRNAs that contain a downstream RB site. The flow of RNAPII is indicated in blue, and the internal arrow indicates the direction of transcription. The sites of NNS and RB termination are indicated, respectively, by red and blue arrows; a site of cryptic or alternative (e.g., CPF‐dependent) termination downstream of the RB is indicated by a black arrow. A low level of natural readthrough at the primary site is indicated by a low schematic flow of polymerases (blue) between the NNS and RB sites, which feeds the RB peak. Under defective NNS termination, this readthrough flux increases, together with the RB (left scheme, dotted line, light green). When the RB is affected (right scheme), only the readthrough due to unblocked polymerases (dotted line, light green) downstream of the RB site is observed, terminating at downstream sites (black arrows). The transcripts produced in the different conditions are indicated by plain or dotted lines, which roughly represent the stability and steady‐state levels of the different species. The colors represent the kind of termination (NNS, RB, or cryptic) that leads to the production of a given species.
We also addressed the converse possibility, that is, that the RB pathway could be required for termination of snoRNAs (Roy et al, 2016). We analyzed the polymerase profile around four snoRNAs for which a Reb1 (SNR161, SNR8, SNR48)‐ or Rap1‐dependent (SNR39B) roadblock peak of variable intensity was detected in the termination region (Fig 5). We compared the distribution of polymerases at these NNS‐dependent targets under conditions of defective RB termination by depleting either one of the RB factors. As a control, we generated RNAPII CRAC data upon depletion of Nrd1 with the auxin degron method (Nishimura et al, 2009). Depletion of Nrd1 led to transcription readthrough at the NNS‐dependent terminator as expected, which fed the flow of polymerases accumulating at the downstream roadblock peak (Fig 5A–D, compare red and blue tracks in the insets, red arrows; see also Fig 5E, left scheme). This was clearly visible at the SNR8 and SNR48 loci, where the roadblock is slightly more distal (Fig 5, panels A and B), but also observed at SNR161 and SNR39B where the readthrough signal merges to some extent with the roadblock signal (Fig 5C and D).
Upon depletion of the roadblock factor (Rap1 or Reb1), we did not observe alterations in termination at the primary NNS site, which occurred with similar overall efficiency as in the presence of the RB (Fig 5A–D, note that the RB‐less tracks, pink/red, are always beneath the wt tracks, light blue). A small readthrough was only detected downstream of the RB site (Fig 5, blue arrows), due to the release of polymerases that had accumulated at the roadblock (see Fig 5E, right scheme). These results strongly suggest that the RB is not required for NNS‐dependent termination at these sites.
The small readthrough at the RB in the absence of Rap1 or Reb1 leads to the production of longer transcripts that might have diverse fates and stability, depending on many factors including their sequence and the nature of downstream termination. We analyzed the levels of these RNAs by RNAseq in the presence or absence of the RB. To visualize the primary product of termination that is trimmed to the mature snoRNA by the nuclear exosome, we performed this analysis in an rrp6∆ strain. As shown in Fig 5A–D, variable levels of readthrough transcripts accumulated at three of the four snoRNAs studied in the absence of the RB. The strongest accumulation was observed at the SNR39B site and intermediate levels at SNR161 and SNR8, but in all cases the transcript levels hardly mirrored the levels of polymerases reading through the site of RB detected by CRAC. This indicates that the abundance of these RNAs is mainly dictated by their stability and not by the levels of readthrough transcription.
Together with the results shown in the previous section, our data strongly support the notion that the roadblock pathway functions as a fail‐safe mechanism to neutralize natural readthrough transcription at both the CPF‐ and NNS‐dependent canonical terminators.
Functional importance of fail‐safe transcription termination
As shown in Fig 2, depletion of Rap1 strongly affects transcription of RPL11B and RPS24A. These genes might be downregulated either because the absence of the roadblock exposes their promoters to transcriptional interference or because they require Rap1 for transcriptional activation. To distinguish between these non‐exclusive possibilities, we investigated whether the RB alone could be sufficient to restore, at least partially, their expression. To this end, we depleted Rap1 in cells expressing the well‐characterized DNA‐binding domain of Rap1 (Rap1‐DBD, aa. 358–601), which is not expected to activate transcription, but supports roadblock termination, as verified by RT–qPCR upstream of HYP2 (Appendix Fig S3B). As a control, we used strains containing the wild‐type Rap1 or an empty plasmid and sequenced the RNAs produced in these cells. Because expression of Rap1‐DBD alone affects growth in a dominant‐negative manner, we could not perform reliable CRAC experiments in these conditions, but sequenced the transcriptome at two different time points after Rap1 depletion. Consistent with the RNAPII CRAC data, expression of RPL11B and RPS24A RNAs was markedly affected by the depletion of endogenous Rap1 and restored by the concomitant expression of wt Rap1 (Fig 6A and B, compare red and blue tracks). Importantly, expression of the DNA‐binding domain alone of Rap1 is sufficient to restore RPL11B and RPS24A RNAs to wild‐type levels (Fig 6A and B, purple tracks). This is not due to Rap1‐DBD retaining a general activation function as demonstrated by the failure of the latter to restore expression of genuine Rap1 targets sites such as RPS0A (Fig 6C), RPL29, or MF(ALPHA)1 (data not shown).
Figure 6. Expression of the Rap1 DNA‐binding domain restores expression of genes containing an upstream roadblock.

-
A–CRNAseq profiles at two tandem features containing an upstream Rap1 roadblock site (RPS24A and RPL11B, A and B). A feature (RPS0A) that depends on Rap1 for transcription activation but for which no upstream RB can be detected is shown in (C) as a control. Only the signals on the strand of the annotated features are shown. Wild‐type Rap1, the Rap1 DNA‐binding domain (Rap1‐DBD), or an empty plasmid was expressed in the Rap1‐AA strain and the endogenous protein was depleted from the nucleus upon addition of rapamycin for the times indicated. RNAseq tracks are shown for the target genes and for neighboring genes as a control. Adjacent loci have been separated by vertical lines when the two features have very different expression levels and different scales have been used.
Together, these results support the notion that the constitutive readthrough at CPF (and possibly NNS) terminators can be sufficient for silencing downstream genes, underscoring the importance of the protective action of roadblock factors.
Extent of roadblock termination in the Saccharomyces cerevisiae genome
In light of the results shown here on Rap1 and Reb1, we assessed more generally the occurrence of roadblock termination at sites of occupancy for DNA‐binding proteins or complexes. We first used published data on the genomewide distribution of transcription factors (Harbison et al, 2004) and profiled RNAPII occupancy at genomic regions aligned on sites of binding as defined by MacIsaac et al (2006). We found evidence for RB termination at many such sites, some of which are shown in Fig EV3. These profiles are indicative of roadblock occurring at a variable distance upstream of the protein‐binding site, likely reflecting the topology of the collision between RNAPII and the DNA‐bound factor or complex of factors.
We also observed prominent levels of RNAPII roadblocks at centromeres and tRNA genes. In S. cerevisiae, centromeres are defined by a set of short, well‐conserved sequence elements located in a 125‐nt region. These sequences, CDEI, CDEII, and CDEIII (Fig 7), are specifically occupied by DNA‐binding factors that are part of the kinetochore (for a review, see Westermann et al, 2007; Biggins, 2013). The 8‐bp CDEI is directly recognized by Cbf1, a DNA‐binding factor that is also a transcriptional activator. CDEIII (26 nt) is instead recognized by CBF3, a complex of four proteins, while the CDEII sequence (78–86 nt) is wrapped around a specific centromeric nucleosome (CENP‐A), containing a histone H3 variant, Cse4. We analyzed the distribution of polymerase around centromeres using both PAR‐CLIP (Schaughency et al, 2014) and our RNAPII CRAC data. A marked peak of localized RNAPII pausing was clearly observed at individual centromeres (Figs 7B and EV4A and B), the average position of which was roughly 25 nt upstream of CDEI as shown in the aggregate plot (Fig 7A), strongly suggesting that Cbf1 induces roadblock termination, at least in the context of centromeres.
Figure 7. RNAPII roadblock occurs at centromeres and RNAPIII genes.
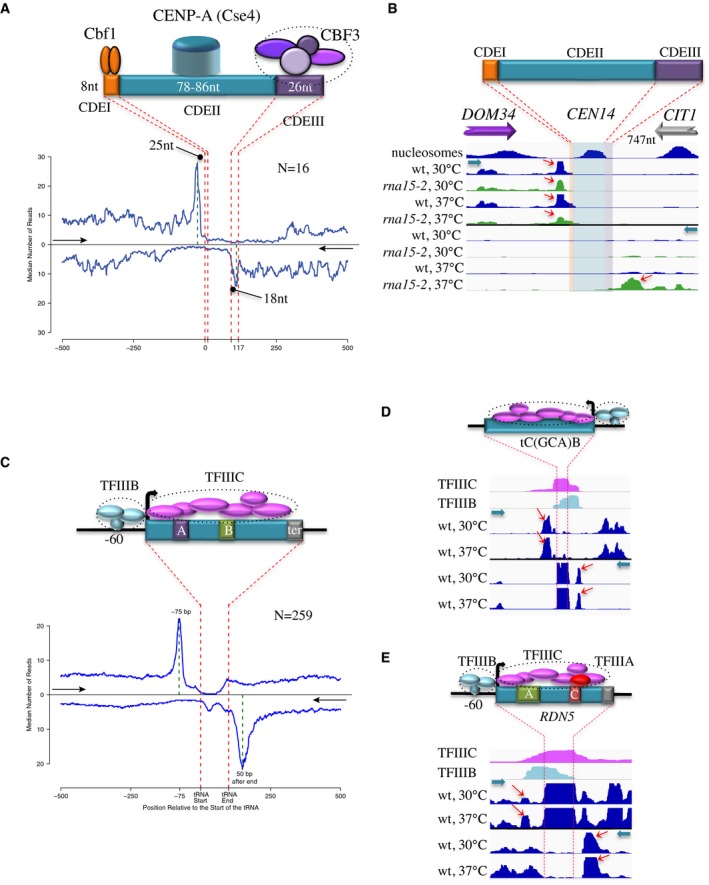
- Aggregate plot of RNAPII occupancy (median reads count, PAR‐CLIP data) around centromeres. Centromeres have been aligned on the beginning of the CDEI (top plot) or the CDEIII sequence (bottom plot) and a virtual centromere has been reconstituted by aligning the two plots based on the average length of the centromere. The 5′–3′ direction is indicated by a black arrow for each plot. The structure of the centromere and the interacting factors are schematically shown on the top.
- Snapshot showing the distribution of polymerases around CEN14. RNAPII CRAC distribution is shown for both wild‐type and rna15‐2 cells at the permissive and non‐permissive temperature for the mutant. A roadblock peak (red arrow) is observed upstream of CDEI in all conditions. Detection of the roadblock upstream of the CDEIII sequence requires increasing readthrough transcription at the upstream gene (CIT1) with the rna15‐2 mutation (bottom track). Cyan arrows indicate the direction of transcription.
- Metaprofile analysis of RNAPII distribution (median reads count, PAR‐CLIP data) around tRNA genes. Genomic regions were aligned on the transcription start sites (top) or the transcription termination site (bottom) and the plots combined as for centromeres. A scheme of tRNA genes and associated factors is shown on the top of the plots. The 5′–3′ direction is indicated by a black arrow for each plot. Note that reads in the body of tRNAs have been removed because representing contamination from mature tRNAs (Schaughency et al, 2014).
- Snapshot of RNAPII distribution around tC(GCA)B. The profiles in wt cells grown at 30°C and 37°C have been shown as duplicates, as only minor differences are observed. Roadblock peaks are indicated by red arrows. The footprints of TFIIIB and TFIIIC (Nagarajavel et al, 2013) are shown for comparison. The direction of transcription is indicated by cyan arrows. Reads in the body of tRNAs are from contaminating tRNAs and should not be considered as bona fide RNAPII CRAC signals.
- RNAPII CRAC profile around the gene coding for 5S rRNA (RDN5) in wt cells at 30°C and 37°C as in (D). Roadblock peaks are indicated by red arrows. The footprints of TFIIIB and TFIIIC are shown. A scheme of the gene and the factors bound is shown in the top of the figure. As for tRNAs, the strong signal in the body of the gene should not be considered as a bona fide RNAPII signal, but contaminating 5S RNA.
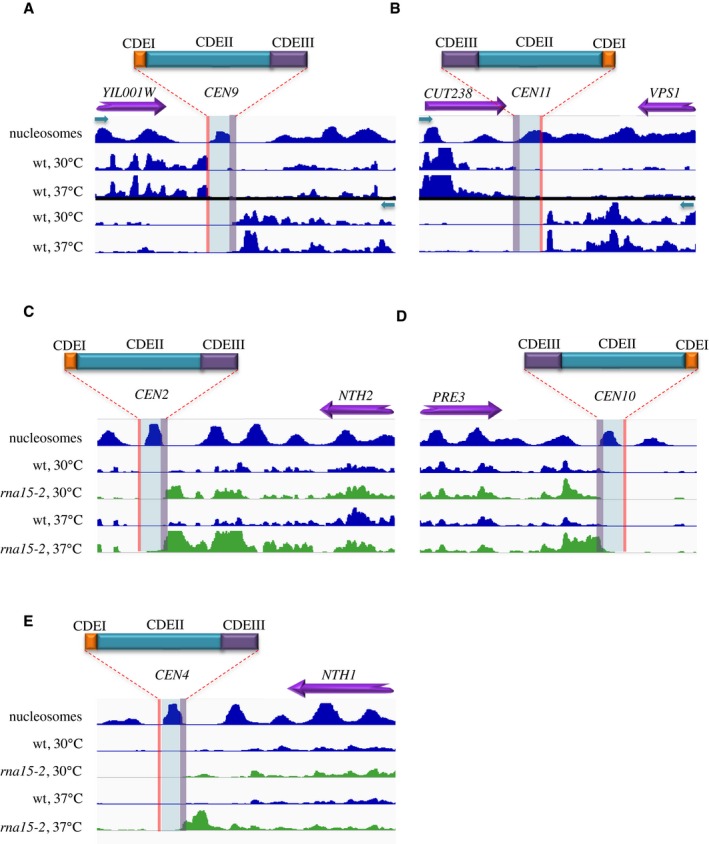
Figure EV4. Snapshots showing the RNAPII CRAC signal around representative centromeres.
-
A–EThe structure of each centromere is schematically shown on top and reported, scaled, on the tracks. In (A, B), both strands are shown and the direction of transcription is indicated by a green arrow. In (C–E), only one strand is shown as no significant levels of transcription could be detected on the other strand. RNAPII CRAC tracks derived from rna15‐1 cells are shown at the permissive and non‐permissive temperature to illustrate the effect of increased readthrough transcription at convergent genes, which leads to the accumulation of RNAPII signal at the roadblock.
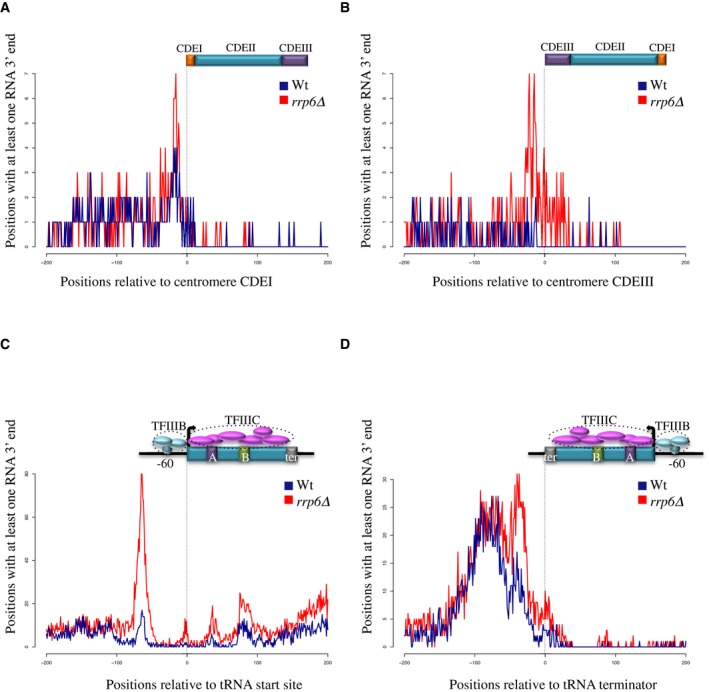
A RB peak was also observed around CDEIII and upstream of CDEII, as shown in the RNAPII CRAC metaprofile and at individual centromeres (Figs 7B and EV4). In many cases, the RB was more prominently observed when incoming transcription was increased by affecting termination of convergent genes in rna15‐2 cells (Figs 7B and EV4C–E, rna15‐2 tracks). Termination by the RB pathway occurred in these regions, as witnessed by the presence of RNA 3′ ends peaks that overlap the peaks of pausing and that often represent unstable transcripts (compare the wt and the rrp6∆ profile, Fig EV5A and B). Interestingly, two peaks of termination can be observed around CDEIII (Fig EV5B), one within and another upstream of it, indicating heterogeneity in the position of the RB around (Fig 7A).
Figure EV5. Metasite analysis of RNA 3′ ends around centromeres and tRNAs.
-
A–DAggregate plots showing the distribution of RNA 3′ ends around centromeres and tRNAs in wt and rrp6∆ cells to detect unstable RNAs. The orientation of centromeres and tRNA relative to transcription is shown by schematic drawings. Note the presence of 3′ ends peaks indicating the occurrence of transcription termination at sites of RB.
In apparent contrast with published data (Ohkuni & Kitagawa, 2011, 2012), no evidence of transcription within CDEII was observed, which we find to be virtually free of polymerases. Note that this is unlikely due to failure from mapping A‐T‐rich reads to this region, because reads with virtually identical sequences (i.e., containing one or two mismatches) could be efficiently mapped to other regions in the genome (data not shown).
Transcription of tRNA genes depends on internal promoters, which harbor sequences (A and B boxes) that are bound by the TFIIIC hexameric complex (Arimbasseri & Maraia, 2016). TFIIIC covers the whole tRNA sequence and interacts with TFIIIB, composed of three subunits, which binds at position −60 relative to the transcription start site (Nagarajavel et al, 2013). The aggregate profile of RNAPII distribution around tRNA genes is presented in Fig 7C and one representative example in Fig 7D, together with the mapped “bootprints” of TFIIIB and TFIIIC (Nagarajavel et al, 2013). A prominent peak of RNAPII accumulation was observed at position −75 relative to the start site consistent with a roadblock induced by TFIIIB bound at position −60. Interestingly, a major roadblock was also observed for RNAPII transcription running antisense to tRNA genes, peaking roughly 50 nt upstream of the aligned U‐rich tract that defines the RNAPIII termination signal. Evidence for the production of unstable transcripts for termination occurring upstream of tRNA start sites and for antisense transcription downstream of tRNA terminators is provided by the distribution of stable and unstable RNA 3′ ends around these features (Fig EV5C and D).
Evidence for RNAPII roadblocks was also observed around the gene coding for the ribosomal 5S RNA subunit, which is an RNAPIII gene with a structure similar to that of tRNA genes (Fig 7E).
TFIIIC was also found to bind at locations distinct from tRNA genes, in the absence of TFIIIB (ETC, extra TFIIIC sites, Roberts et al, 2003; Moqtaderi & Struhl, 2004; Nagarajavel et al, 2013). We could not find evidence of transcriptional roadblock at ETC sites, suggesting that the sole binding of TFIIIC is not sufficient to prevent RNAPII elongation (data not shown). Together, these data illustrate the genomewide extension of roadblock termination and underscore its large potential for modeling the yeast transcriptional landscape.
Discussion
In a previous study, we have demonstrated that transcription termination occurs when the RNAPII encounters the factor Reb1 bound to the DNA. Here we generated high‐resolution genomewide RNAPII transcription maps data under conditions of defective RB, CPF, or NNS termination to study the overall impact of RB termination on the yeast genome, and the functional relationships with the other pathways.
We provide evidence that natural readthrough at canonical CPF and NNS terminators constitutes an additional and functionally significant source of pervasive transcription in S. cerevisiae. We demonstrate that the canonical pathways and RB termination function independently from each other but act redundantly at the end of transcription units, limiting pervasive readthrough and favoring insulation of transcription events. Finally, we extend the repertoire of roadblocking factors, which we propose to play major roles in determining the distribution of transcription events.
Rap1 is a roadblock termination factor
We demonstrated that Rap1, a DNA‐binding factor that has roles in transcription activation, gene silencing, and telomere homeostasis (Azad & Tomar, 2016), is also a roadblock termination factor. An earlier study showed that the fortuitous introduction of a Rap1 site in a Ty1 retrotransposon leads to RNAPII stalling and repression of gene expression (Yarrington et al, 2012). Based on the analysis of the RNA produced, which was reported to be non‐adenylated and insensitive to exosome degradation, it was concluded that termination of transcription does not occur in this system. In contrast to this early study, we show that roadblock termination occurs at Rap1 binding sites, leading to the production of RNAs that can be polyadenylated by Trf4 and are degraded for a large part by the nuclear exosome. Importantly, nuclear depletion of Rap1 prevents termination, indicating that the protein—and not the presence of termination signals that might overlap its binding site—is essential for the release of the polymerase. Failure to detect the nuclear degradation of the adenylated and non‐adenylated RNAs for technical reasons in the study by Yarrington et al (2012) might account for the discrepancies; alternatively, release of the polymerase might not occur in the Ty1 retrotransposon model for unknown reasons.
The mechanism of roadblock termination
Similar to what previously shown for Reb1 (Colin et al, 2014), release of the polymerase paused upstream of the roadblock depends on its ubiquitylation by Rsp5 and possibly its degradation. Thus, this pathway is not restricted to Reb1‐dependent termination but presumably extends to all cases of roadblock, and possibly of most RNAPII pausing that cannot be resolved in a more “conservative” manner (Wilson et al, 2013). We generally observed very sharp peaks of stalling at the roadblock sites, which is compatible with one or two polymerases on average roadblocked at a time and indicates that, at steady state, the clearance due to the Rsp5 pathway must be as efficient as the feeding of the peak by incoming polymerases.
It has been recently proposed, mostly based on the analysis of RNA 3′ ends in several mutant conditions, that the NNS and the RB termination pathways are functionally interconnected, in that the NNS is required for releasing roadblocked polymerases and, conversely, that the presence of a RB is necessary for NNS termination (Roy et al, 2016). The analyses presented here based on the direct and high‐resolution detection of RNAPII transcription in conditions defective for RB, CPF, or NNS termination do not generally support this model. We observed that RB termination is largely insensitive to depletion of Nrd1 (e.g., see Fig EV2 and Appendix Fig S3), or mutation of Nab3 (data not shown), which is also consistent with the findings that the insertion of the sole Reb1 (Colin et al, 2014) or Rap1 sites (Fig 1 and Appendix Fig S1A) in the heterologous context of the HSP104 gene is sufficient for efficient, NNS‐independent termination. Similarly to what reported by Roy et al (2016), we detected increased RNAPII occupancy at some roadblock sites upon impairment of NNS function (e.g., Fig 5), but we show that this is due to the accumulation of polymerases that fail to terminate at NNS terminators upstream of the RB rather than to the general defective clearance of stalled elongation complexes.
We favor a model according to which polymerases are not recycled for further steps of transcription when encountering a RB (as if it were released by the NNS complex) but degraded, together with the RNA that is produced. This might look uneconomical, but the energetic balance might still be favorable in light of the evolutionary cost of developing highly efficient, error‐proof termination processes. In this respect, the genomewide analyses reported here suggest that roadblock termination is likely devoted to controlling a relatively low fraction of polymerases that might nevertheless significantly affect the efficiency or robustness of neighboring processes.
Relationships between RB and the main pathways of termination in Saccharomyces cerevisiae
Many studies support the notion that pausing is a prerequisite for transcription termination (for a recent review, see Porrua et al, 2016). Slowing down the speed of the polymerase has been shown to promote earlier termination at NNS targets (Hazelbaker et al, 2012), and RNAPII pausing sites are preferential sites of Sen1‐dependent termination in vitro (Porrua & Libri, 2013). This is thought to be due to a kinetic competition between RNAPII elongation and translocation on the RNA by Sen1, a concept that might also apply to CPF termination whereby “pursuing” of the polymerase is operated by the Rat1/XRN2 exonuclease (Fong et al, 2015). Whether pausing is induced by intrinsic components of the NNS or the CPF pathway, or by extrinsic factors, remains a poorly understood and important facet of termination.
We considered the possibility that roadblock pausing could be required for upstream termination, both CPF‐ and NNS‐dependent. However, we did not detect the termination defects predicted by this model upon depletion of Rap1 or Reb1. In these conditions, readthrough was only observed downstream of the RB site, due to the release of a low fraction of polymerases that had accumulated at the RB. Transcription beyond the site of RB in the absence of Rap1 or Reb1 might produce RNAs that are more stable than the ones derived from RB‐ or NNS‐dependent termination, as we suggest occurring at the SNR8, SNR161, and SNR39B sites. In the absence of the RB, the levels of these transcripts do not match the actual levels of transcriptional readthrough, which might lead to overestimating the impact of the RB on termination, possibly explaining the discrepancies with the model proposed by Roy et al (2016).
Although it remains possible that in the absence of Reb1 or Rap1, other roadblock events take over to slow down the RNAPII and promote termination, we favor the notion that pausing is induced by components of the NNS or CPF complexes, or depends on specific sequences, the nature of which remains elusive.
Widespread transcriptional readthrough at gene terminators in Saccharomyces cerevisiae
It is generally accepted that pervasive transcription is mainly generated by the leaky control imposed on transcription initiation by the structure of chromatin. Many studies have shown that altering the positioning or the modification status of nucleosomes (see for instance Churchman & Weissman, 2011; Marquardt et al, 2014; Venkatesh et al, 2016) significantly impacts the relative extent of initiation at divergent or cryptic promoters, generating pervasive transcription events. Our data strongly suggest that in addition to the leaky control on initiation, leaky termination generates pervasive transcription events, which might significantly impact gene expression and other cellular events. Extensive occurrence of transcriptional readthrough has been observed in the B. subtilis (Nicolas et al, 2012) and E. coli transcriptome (Stringer et al, 2014). In S. pombe, antisense transcripts derived from readthrough transcription are produced in wild‐type cells and degraded by the exosome (Zofall et al, 2009) while in human cells readthrough transcripts induced by osmotic stress have been shown to represent a considerable fraction of pervasive transcription (Vilborg et al, 2015). However, the direct demonstration of widespread and constitutive transcriptional readthrough in wild‐type cells was not attempted in these studies. Detection of these events in the present study is facilitated by the sensitivity and resolution of RNAPII CRAC and by the analysis of RB sites, where polymerases escaping termination accumulate. Because the genes used for the metasite analyses in Fig 3B were solely selected based on Reb1 or Rap1 downstream binding, they can be reasonably considered as a random sampling for their efficiency of termination. The average detection of transcriptional readthrough at these terminators therefore strongly suggests that leaky termination occurs genomewide, widening the repertoire and the potential impact of pervasive transcription. This conclusion is supported by the direct detection of significant readthrough signals in intergenic regions downstream of many genes, which do not reflect the occurrence of independent intergenic initiation (see for instance Appendix Fig S2C).
Extensive occurrence of transcriptional readthrough might confer additional evolutionary advantages over the generation of ex novo genes from pervasive initiation (Carvunis et al, 2012; Wu & Sharp, 2013). Readthrough transcripts might evolve to generate new functions from existing modules, leading for instance to the fusion of contiguous ORFs or the generation of protein extensions.
The non‐quantitative feature of termination also brings about a large potential for regulation, allowing anticorrelated expression of tandem genes and possibly its modulation by alterations in the efficiency of termination.
Roadblock occurs at many genomic sites
We show here that although the elongation complex is armed to progress relatively efficiently through nucleosomes in vivo, it is significantly affected by the presence of many other factors bound to the DNA. Two notable examples are centromeres and tRNAs.
Roadblock peaks for RNAPII were observed upstream of both centromere edges, where they are roadblocked presumably by Cbf1 and the CBF3 complex, binding, respectively, the CDEI site and the CDEIII sequence, which is consistent with early observations (Doheny et al, 1993). Very little, if any, RNAPII CRAC signal can be detected within the centromeric DNA in general and particularly in CDEII, suggesting that centromeres use intrinsic roadblock barriers to prevent trans‐centromere transcription. This might underlie a requirement for maintaining the identity of the centromeric nucleosome, containing a specific variant of histone H3, Cse4, the occupancy of which might be affected by through transcription, and is consistent with the notion that directing strong transcription toward a centromere generally inactivates it (Hill & Bloom, 1987; Doheny et al, 1993). In contrast to these conclusions, earlier studies have proposed that a relatively moderate level of transcription through centromeres is actually required for function, which was supported by the detection of trans‐centromeric transcripts by RT‐qPCR, and by genetic experiments (Ohkuni & Kitagawa, 2011). Although we might have failed to detect RNAPII CRAC signals corresponding to very low levels of these trans‐centromeric transcripts, we do not fully understand the basis of this discrepancy, and future work is required to elucidate the role of transcription at the point centromeres of S. cerevisiae.
We also show that RNAPII is roadblocked at the 5′ and 3′ ends of tRNA genes. The existence of a 5′ end roadblock is consistent with earlier studies on the tV(UAC)D locus, proposing a role for TFIIIB in preventing upstream intergenic transcription from entering the tRNA gene body (Korde et al, 2014; see also Roy et al, 2016). Here we extend this finding to a genomewide perspective, and additionally demonstrate the existence of an additional roadblock barrier that prevents antisense RNAPII transcription from crossing tRNA genes. The 3′ RB is aligned to the tRNA terminators, to which, in turn, is also aligned the trailing edge of TFIIIC footprint (Nagarajavel et al, 2013). However, the binding of TFIIIC alone at ETC sites is not sufficient for inducing a RB, which might indicate that additional factors (presumably TFIIIB) must be present to stabilize the interaction of TFIIIC with the DNA and induce a RB. Alternatively, it is possible that the specific topology of these transcription units, which are believed to be circularized for a more efficient transcription re‐initiation (Dieci et al, 2013), or the general high persistence of RNAPIII at these sites might underlie the formation of the 3′ end RB.
In conclusion, our results suggest that the transcriptional landscape is modeled to a large extent by non‐histone proteins bound to the DNA, which has a considerable impact in the partitioning of DNA‐linked activities and in the control of pervasive transcription events. Aside from operating a quality control mechanism on the efficiency of termination at canonical sites, roadblock pausing (and termination) of polymerases has a large potential for shaping and regulating the transcriptome, in yeast and most likely many other organisms.
Materials and Methods
Yeast strains and plasmids
Yeast strains used in this study are listed in Appendix Table S1. Plasmids are listed in Appendix Table S3. The reporter construct used for selecting the Rap1‐dependent terminators was previously described (Porrua et al, 2012). The full sequences of the selected terminators are available upon request. Rap1 constructs were expressed from the RAP1 gene promoter to avoid growth defects due to the overexpression of Rap1 derivatives. The whole Rap1 coding sequence and 598 nt of the upstream region were cloned in a pCM185 backbone (Garí et al, 1997) in which the TRP1 marker was replaced with the S. pombe HIS5 gene. The Tet promoter and hybrid transactivator of pCM185 were deleted. The Rap1‐DBD construct was obtained by replacing the Rap1 coding sequence with a fragment of the gene coding for amino acids 358–601.
RNA analyses
Northern blot analyses were performed as previously described (Colin et al, 2014). RT–qPCR was performed with standard procedures, using the primers listed in Appendix Table S2. Amplification efficiencies were calculated for every primer pair in each amplification reaction.
Growth conditions and preparation of cells for CRAC
Two liters of yeast cells expressing Rpb1‐HTP tag (Granneman et al, 2009) were grown at 30°C to OD600 = 0.6 in CSM‐Trp medium. For nuclear depletion of Reb1 and Rap1, rapamycin was added to anchor away strains or control untagged wild type for two hours to a final concentration of 1 μg/ml. rna15‐2 or wild‐type cells were grown at 30°C to OD600 = 0.6; 1 volume of media preheated at 30 or 37°C was added and cultures were incubated at 30 or 37°C for 1.5 h. Nrd1 was depleted with the auxin degron system (Nishimura et al, 2009) by adding IAA (indole‐3‐acetic acid, Sigma) 100 μM to Nrd1‐AID cells for 60 min before crosslinking.
Cells were submitted to UV crosslink using a W5 UV crosslinking unit (UVO3 Ltd) for 50 s, harvested by centrifugation, washed in cold PBS, and resuspended in TN150 buffer (50 mM Tris pH 7.8, 150 mM NaCl, 0.1% NP‐40, and 5 mM beta‐mercaptoethanol, 2.4 ml/g of cells) supplemented with protease inhibitors (Complete™, EDTA‐free Protease Inhibitor Cocktail). The suspension was flash frozen in droplets, and cells were mechanically broken with a Mixer Mill MM 400 (5 cycles of 3 min at 20 Hz).
CRAC
The CRAC protocol used in this study is derived from Granneman et al (2009) with a few modifications. Cell powders were thawed and the resulting extracts were treated for one hour at 25°C with DNase I (165 U/g of cells) to solubilize chromatin and then clarified by centrifugation (20 min at 20,000 × g at 4°C).
IgG purification was performed with M‐280 tosylactivated dynabeads coupled with rabbit IgG (15 mg of beads per sample). The complexes were eluted with TEV protease and treated with 0.2 U of RNase cocktail (RNace‐IT, Agilent) to reduce the size of the nascent RNA. High salt washes for both purification steps were done at 1 M NaCl for increased stringency. The dephosphorylation step required for cleaving the 2′–5′ cyclic phosphate left by RNase treatment (Granneman et al, 2009) was omitted to enrich for nascent transcripts, the 3′ end of which being protected from RNase treatment.
After overnight binding on Ni‐NTA column (Qiagen, 100 μl of slurry per sample), sequencing adaptors were added on the RNA as described in the original procedure. Adaptors were modified for sequencing from the 3′ end. The 3′ ligation was realized with T4 rnl 2 truncated K227Q enzyme (NEB) instead of classical T4 RNA ligase.
RNA–protein complexes were eluted in 400 μl of elution buffer (50 mM Tris pH 7.8, 50 mM NaCl, 150 mM imidazole, 0.1% NP‐40, 5 mM beta‐mercaptoethanol). Eluates were concentrated with Vivacon® ultrafiltration spin columns 30‐kDa MWCO to a final volume of 120 μl. The protein fractionation step was performed with a Gel Elution Liquid Fraction Entrapment Electrophoresis (GelFree) system (Expedeon). Rpb1‐containing fractions were treated with 100 μg of proteinase K in a buffer containing 0.5 % SDS. RNAs were purified and reverse‐transcribed using reverse transcriptase Superscript IV (Invitrogen).
The absolute concentration of cDNAs in the reaction was estimated by quantitative PCR using a standard of known concentration. Amplifications were performed separately in 25 μl reactions containing each 2 μl of cDNA for typically 7–9 PCR cycles (LaTaq, Takara). The PCRs from all the samples were pooled and treated for 1 h at 37°C with 200 U/ml of Exonuclease I (NEB). The DNA was purified using NucleoSpin® Gel and PCR Clean‐up (Macherey‐Nagel) and sequenced using Illumina technology.
Dataset processing
CRAC samples were demultiplexed using the pyBarcodeFilter script from the pyCRACutility suite (Webb et al, 2014). Subsequently, the 5′ adaptor (Appendix Table S2, read at the 3′ end of reads) was clipped with Cutadapt ({m 10}, Martin, 2011) and the resulting insert quality‐trimmed from the 3′ end using Trimmomatic rolling mean clipping (Bolger et al, 2014) (window size = 5, minimum quality = 25). At this stage, the pyCRAC script pyFastqDuplicateRemover was used to collapse PCR duplicates using a 6‐nucleotide random tag included in the 3′ adaptor (Appendix Table S2, read at the 5′ end of reads). During demultiplexing, pyBarcodeFilter retains this information in the header of each sequence. This information is used at this stage to better discern between identical inserts and PCR duplicates of the same insert. The resulting sequences are reverse complemented with Fastx reverse complement (part of the fastx toolkit, http://hannonlab.cshl.edu/fastx_toolkit/) and mapped to the R64 genome (Cherry et al, 2012) with bowtie2 (using “‐N 1” option) (Langmead & Salzberg, 2012).
RNAseq samples were demultiplexed by the sequencing platform with bcl2fastq2 v2.15.0; adaptor trimming of standard Illumina TruSeq adaptors was performed with cutadapt 1.9.1. Samples were subsequently quality‐trimmed with trimmomatic (see above) and mapped to the R64 genome with bowtie2 (default options).
Metagene analyses and boxplots
For each feature included in the analysis, we extracted the polymerase occupancy values at every position around the feature and plotted the mean or median over all the values for that position in the final aggregate plot. To limit the influence of outliers on the final plot when using the mean to summarize the data, we excluded from the analysis every value at each site that was above the mean + 5 standard deviations.
To assess the occurrence of a termination defect at genes upstream of Rap1 RB sites upon Rap1 depletion or in rna15‐2 cells, we selected a subset of genes whose termination region (operationally defined as the region around the strongest site of polyadenylation) was within 300 bp upstream of a Rap1 site. To avoid interference with the signal at the RB site, we then calculated the average polymerase occupancy in the early segment of the termination region, that is, in a window 100 nucleotides immediately before the major site of polyadenylation. This value was then divided by the average polymerase occupancy signal across the whole body of the gene. A decline in the RNAPII signal in wt cell and a significantly higher signal in rna15‐2 cells in this region confirmed that loss of RNAPII indeed starts occurring in this region, presumably associated with multiple sites of 3′ end processing. The overall distribution of these ratios for several datasets was represented with boxplots and the statistical significance assessed with paired t‐tests in order to account for different termination efficiencies at each polyadenylation site.
Dataset availability
All datasets used in this study are available under GEO numbers GSE97913 and GSE97915.
Author contributions
All authors designed and performed experimental work. DL and JC supervised the work. DL wrote the paper. JC, DC, TC, and OP reviewed and edited the draft. TC curated the data and generated software for analysis. DL was responsible for funding acquisition.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Acknowledgments
We would like to thank F. Feuerbach, T. H. Jensen, and T. Villa for critical reading of the manuscript. D. Shore and R. Morse for the kind gift of the Rap1‐AA and the rap1‐2 strains, respectively. We also thank D. Tollervey and S. Granneman for help with the CRAC protocol. This work was supported by the Centre National de la Recherche Scientifique (C.N.R.S.), the Fondation pour la Recherche Medicale (F.R.M., programme équipe 2013), l'Agence Nationale pour la Recherche (A.N.R., grants ANR‐08‐Blan‐0038‐01, ANR‐12‐BSV8‐0014‐01, and ANR‐16‐CE12‐0022‐01), and the Fondation Bettencourt (prix Coup d'Elan 2009) and the LABEX Who am I. J.B.B., T.C., and D.C. have been supported by fellowships from the French Ministry of Research. This work has benefited from the facilities and expertise of the high‐throughput sequencing core facility of I2BC (Centre de Recherche de Gif—http://www.i2bc-saclay.fr/).
The EMBO Journal (2018) 37: e97490 29351914
Contributor Information
Jessie Colin, Email: jessie.colin@uvsq.fr.
Domenico Libri, Email: domenico.libri@ijm.fr.
References
- Arigo JT, Eyler DE, Carroll KL, Corden JL (2006) Termination of cryptic unstable transcripts is directed by yeast RNA‐binding proteins Nrd1 and Nab3. Mol Cell 23: 841–851 [DOI] [PubMed] [Google Scholar]
- Arimbasseri AG, Maraia RJ (2016) RNA polymerase III advances: structural and trna functional views. Trends Biochem Sci 41: 546–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad GK, Tomar RS (2016) The multifunctional transcription factor Rap1: a regulator of yeast physiology. Front Biosci (Landmark Ed) 21: 918–930 [DOI] [PubMed] [Google Scholar]
- Biggins S (2013) The composition, functions, and regulation of the budding yeast kinetochore. Genetics 194: 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvunis A‐R, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, Simonis N, Charloteaux B, Hidalgo CA, Barbette J, Santhanam B, Brar GA, Weissman JS, Regev A, Thierry‐Mieg N, Cusick ME, Vidal M (2012) Proto‐genes and de novo gene birth. Nature 487: 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, Fisk DG, Hirschman JE, Hitz BC, Karra K, Krieger CJ, Miyasato SR, Nash RS, Park J, Skrzypek MS, Simison M et al (2012) Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res 40: D700–D705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS (2011) Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469: 368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin J, Candelli T, Porrua O, Boulay J, Zhu C, Lacroute F, Steinmetz LM, Libri D (2014) Roadblock termination by reb1p restricts cryptic and readthrough transcription. Mol Cell 56: 667–680 [DOI] [PubMed] [Google Scholar]
- David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM (2006) A high‐resolution map of transcription in the yeast genome. Proc Natl Acad Sci USA 103: 5320–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieci G, Bosio MC, Fermi B, Ferrari R (2013) Transcription reinitiation by RNA polymerase III. Biochim Biophys Acta 1829: 331–341 [DOI] [PubMed] [Google Scholar]
- van Dijk EL, Chen CL, d'Aubenton‐Carafa Y, Gourvennec S, Kwapisz M, Roche V, Bertrand C, Silvain M, Legoix‐Né P, Loeillet S, Nicolas A, Thermes C, Morillon A (2011) XUTs are a class of Xrn1‐sensitive antisense regulatory non‐coding RNA in yeast. Nature 475: 114–117 [DOI] [PubMed] [Google Scholar]
- Doheny KF, Sorger PK, Hyman AA, Tugendreich S, Spencer F, Hieter P (1993) Identification of essential components of the S. cerevisiae kinetochore. Cell 73: 761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermi B, Bosio MC, Dieci G (2017) Multiple roles of the general regulatory factor Abf1 in yeast ribosome biogenesis. Curr Genet 63: 65–68 [DOI] [PubMed] [Google Scholar]
- Fong N, Brannan K, Erickson B, Kim H, Cortazar MA, Sheridan RM, Nguyen T, Karp S, Bentley DL (2015) Effects of transcription elongation rate and Xrn2 exonuclease activity on RNA polymerase II termination suggest widespread kinetic competition. Mol Cell 60: 256–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garí E, Piedrafita L, Aldea M, Herrero E (1997) A set of vectors with a tetracycline‐regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae . Yeast 13: 837–848 [DOI] [PubMed] [Google Scholar]
- Granneman S, Kudla G, Petfalski E, Tollervey D (2009) Identification of protein binding sites on U3 snoRNA and pre‐rRNA by UV cross‐linking and high‐throughput analysis of cDNAs. Proc Natl Acad Sci USA 106: 9613–9618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe PA, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA (2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, Trotta KL, Smith EE, Mosaheb MM, Fuchs SM, Churchman LS (2016) Comprehensive RNA polymerase II interactomes reveal distinct and varied roles for each phospho‐CTD residue. Cell Rep 15: 2147–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD (2009) Mechanisms that specify promoter nucleosome location and identity. Cell 137: 445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK (2008) The anchor‐away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31: 925–932 [DOI] [PubMed] [Google Scholar]
- Hazelbaker DZ, Marquardt S, Wlotzka W, Buratowski S (2012) Kinetic competition between RNA Polymerase II and Sen1‐dependent transcription termination. Mol Cell 49: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A, Bloom K (1987) Genetic manipulation of centromere function. Mol Cell Biol 7: 2397–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight B, Kubik S, Ghosh B, Bruzzone MJ, Geertz M, Martin V, Dénervaud N, Jacquet P, Ozkan B, Rougemont J, Maerkl SJ, Naef F, Shore D (2014) Two distinct promoter architectures centered on dynamic nucleosomes control ribosomal protein gene transcription. Genes Dev 28: 1695–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korde A, Rosselot JM, Donze D (2014) Intergenic transcriptional interference is blocked by RNA polymerase III transcription factor TFIIIB in Saccharomyces cerevisiae . Genetics 196: 427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Jacquet P, Falcone J‐L, Rougemont J, Shore D (2015) Nucleosome stability distinguishes two different promoter types at All protein‐coding genes in yeast. Mol Cell 60: 422–434 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardenois A, Liu Y, Walther T, Chalmel F, Evrard B, Granovskaia M, Chu A, Davis RW, Steinmetz LM, Primig M (2011) Execution of the meiotic noncoding RNA expression program and the onset of gametogenesis in yeast require the conserved exosome subunit Rrp6. Proc Natl Acad Sci USA 108: 1058–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E (2006) An improved map of conserved regulatory sites for Saccharomyces cerevisiae . BMC Bioinformatics 7: 113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabat C, Feuerbach F, Ma L, Saveanu C, Jacquier A (2015) Quality control of transcription start site selection by nonsense‐mediated‐mRNA decay. eLife 4: e06722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt S, Escalante‐Chong R, Pho N, Wang J, Churchman LS, Springer M, Buratowski S (2014) A chromatin‐based mechanism for limiting divergent noncoding transcription. Cell 157: 1712–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 17: 10 [Google Scholar]
- Milligan L, Huynh‐Thu VA, Delan‐Forino C, Tuck A, Petfalski E, Lombraña R, Sanguinetti G, Kudla G, Tollervey D (2016) Strand‐specific, high‐resolution mapping of modified RNA polymerase II. Mol Syst Biol 12: 874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqtaderi Z, Struhl K (2004) Genome‐wide occupancy profile of the RNA polymerase III machinery in Saccharomyces cerevisiae reveals loci with incomplete transcription complexes. Mol Cell Biol 24: 4118–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajavel V, Iben JR, Howard BH, Maraia RJ, Clark DJ (2013) Global ‘bootprinting’ reveals the elastic architecture of the yeast TFIIIB‐TFIIIC transcription complex in vivo . Nucleic Acids Res 41: 8135–8143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil H, Malabat C, d'Aubenton‐Carafa Y, Xu Z, Steinmetz LM, Jacquier A (2009) Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457: 1038–1042 [DOI] [PubMed] [Google Scholar]
- Nicolas P, Mäder U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S, Becher D, Bisicchia P, Botella E, Delumeau O, Doherty G, Denham EL, Fogg MJ, Fromion V, Goelzer A, Hansen A et al (2012) Condition‐dependent transcriptome reveals high‐level regulatory architecture in Bacillus subtilis . Science 335: 1103–1106 [DOI] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M (2009) An auxin‐based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6: 917–922 [DOI] [PubMed] [Google Scholar]
- Ohkuni K, Kitagawa K (2011) Endogenous transcription at the centromere facilitates centromere activity in budding yeast. Curr Biol 21: 1695–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuni K, Kitagawa K (2012) Role of transcription at centromeres in budding yeast. Transcription 3: 193–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Jakob P, Steinmetz LM (2014) Genome‐wide identification of transcript start and end sites by transcript isoform sequencing. Nat Protoc 9: 1740–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, Hobor F, Boulay J, Kubicek K, D'Aubenton‐Carafa Y, Gudipati RK, Stefl R, Libri D (2012) In vivo SELEX reveals novel sequence and structural determinants of Nrd1‐Nab3‐Sen1‐dependent transcription termination. EMBO J 31: 3935–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, Libri D (2013) A bacterial‐like mechanism for transcription termination by the Sen1p helicase in budding yeast. Nat Struct Mol Biol 20: 884–891 [DOI] [PubMed] [Google Scholar]
- Porrua O, Libri D (2015) Transcription termination and the control of the transcriptome: why, where and how to stop. Nat Rev Mol Cell Biol 16: 190–202 [DOI] [PubMed] [Google Scholar]
- Porrua O, Boudvillain M, Libri D (2016) Transcription termination: variations on common themes. Trends Genet 32: 508–522 [DOI] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF (2011) Comprehensive genome‐wide protein‐DNA interactions detected at single‐nucleotide resolution. Cell 147: 1408–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DN, Stewart AJ, Huff JT, Cairns BR (2003) The RNA polymerase III transcriptome revealed by genome‐wide localization and activity‐occupancy relationships. Proc Natl Acad Sci USA 100: 14695–14700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy K, Gabunilas J, Gillespie A, Ngo D, Chanfreau GF (2016) Common genomic elements promote transcriptional and DNA replication roadblocks. Genome Res 26: 1363–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaughency P, Merran J, Corden JL (2014) Genome‐wide mapping of yeast RNA polymerase II termination. PLoS Genet 10: e1004632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Schwalb B, Kiesel A, Baejen C, Torkler P, Gagneur J, Soeding J, Cramer P (2013) Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell 155: 1075–1087 [DOI] [PubMed] [Google Scholar]
- Steinmetz EJ, Conrad NK, Brow DA, Corden JL (2001) RNA‐binding protein Nrd1 directs poly(A)‐independent 3′‐end formation of RNA polymerase II transcripts. Nature 413: 327–331 [DOI] [PubMed] [Google Scholar]
- Stringer AM, Currenti S, Bonocora RP, Baranowski C, Petrone BL, Palumbo MJ, Reilly AA, Zhang Z, Erill I, Wade JT (2014) Genome‐scale analyses of Escherichia coli and Salmonella enterica AraC reveal noncanonical targets and an expanded core regulon. J Bacteriol 196: 660–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiebaut M, Kisseleva‐Romanova E, Rougemaille M, Boulay J, Libri D (2006) Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the nrd1‐nab3 pathway in genome surveillance. Mol Cell 23: 853–864 [DOI] [PubMed] [Google Scholar]
- Venkatesh S, Li H, Gogol MM, Workman JL (2016) Selective suppression of antisense transcription by Set2‐mediated H3K36 methylation. Nat Commun 7: 13610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, Passarelli MC, Yario TA, Tycowski KT, Steitz JA (2015) Widespread inducible transcription downstream of human genes. Mol Cell 59: 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb S, Hector RD, Kudla G, Granneman S (2014) PAR‐CLIP data indicate that Nrd1‐Nab3‐dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biol 15: R8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann S, Drubin DG, Barnes G (2007) Structures and functions of yeast kinetochore complexes. Annu Rev Biochem 76: 563–591 [DOI] [PubMed] [Google Scholar]
- Wilkening S, Pelechano V, Järvelin AI, Tekkedil MM, Anders S, Benes V, Steinmetz LM (2013) An efficient method for genome‐wide polyadenylation site mapping and RNA quantification. Nucleic Acids Res 41: e65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Harreman M, Svejstrup JQ (2013) Ubiquitylation and degradation of elongating RNA polymerase II: the last resort. Biochim Biophys Acta 1829: 151–157 [DOI] [PubMed] [Google Scholar]
- Wu X, Sharp PA (2013) Divergent transcription: a driving force for new gene origination? Cell 155: 990–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyers F, Rougemaille M, Badis G, Rousselle J‐C, Dufour M‐E, Boulay J, Régnault B, Devaux F, Namane A, Séraphin B, Libri D, Jacquier A (2005) Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121: 725–737 [DOI] [PubMed] [Google Scholar]
- Yarrington RM, Richardson SM, Lisa Huang CR, Boeke JD (2012) Novel transcript truncating function of Rap1p revealed by synthetic codon‐optimized Ty1 retrotransposon. Genetics 190: 523–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Fischer T, Zhang K, Zhou M, Cui B, Veenstra TD, Grewal SIS (2009) Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 461: 419–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1