

SUMMARY
R-2-hydroxyglutarate (R-2HG), produced at high levels by mutant isocitrate dehydrogenase 1/2 (IDH1/2) enzymes, was reported as an oncometabolite. We show here that R-2HG also exerts a broad anti-leukemic activity in vitro and in vivo by inhibiting leukemia cell proliferation/viability, and promoting cell-cycle arrest and apoptosis. Mechanistically, R-2HG inhibits FTO activity, thereby increasing global N6-methyladenosine (m6A) RNA modification in R-2HG-sensitive leukemia cells, which in turn decreases the stability of MYC/CEBPA transcripts, leading to the suppression of relevant pathways. Ectopically expressed mutant IDH1 and S-2HG recapitulate the effects of R-2HG. High levels of FTO sensitize leukemic cells to R-2HG, whereas hyperactivation of MYC signaling confers resistance that can be reversed by the inhibition of MYC signaling. R-2HG also displays anti-tumor activity in glioma. Collectively, while R-2HG accumulated in IDH1/2-mutant cancers contributes to cancer initiation, our work demonstrates anti-tumor effects of 2HG in inhibiting proliferation/survival of FTO-high cancer cells via targeting FTO/m6A/MYC/CEBPA signaling.
Graphical Abstract
While accumulation of the oncometabolite R-2-hydroxyglutarate (R-2HG) contributes to cancer initiation, it also has anti-tumor effects by increasing global N6-methyladenosine (m6A) RNA modification.
INTRODUCTION
IDH 1 and 2 catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) in an NADP+-dependent manner. Recurrent somatic mutations in IDH1 and IDH2 occur in ~80% of grade II–III gliomas and secondary glioblastoma (GBM), 10%–20% of acute myeloid leukemia (AML) patients, and at a lower rate in other cancers; most of these lesions involve mutations of arginine (R) residues in codon 132 for IDH1 (IDH1R132), and residues 140 and 172 for IDH2 (IDH2R140 and IDH2R172) (Brat et al., 2015; Mardis et al., 2009; Papaemmanuil et al., 2016). IDH mutants induce a neomorphic enzymatic function that catalyzes the conversion of α-KG to the R enantiomer of 2-hydroxyglutarate (R-2HG) (Dang et al., 2009; Ward et al., 2010). R-2HG is structurally close to α-KG and competitively inhibits a series of Fe(II)/α-KG-dependent dioxygenases (Xu et al., 2011). Since IDH mutants block cell differentiation and promote tumor transformation, and inhibition of mutant IDH (IDHi) can reverse this effect, R-2HG (as the major metabolic product of IDH mutants) has been regarded as an oncometabolite (Figueroa et al., 2010; Lu et al., 2012; Rohle et al., 2013; Wang et al., 2013; Xu et al., 2011).
Interestingly, glioma patients with IDH lesions tend to have a better overall survival than those without (Brat et al., 2015; Eckel-Passow et al., 2015), and a similar trend, though still debatable (Marcucci et al., 2010), was reported in AML patients (Chou et al., 2011; Patel et al., 2012). Recent studies further showed that IDH mutations and R-2HG exhibit growth-suppressive activity and glycolysis-inhibitory function in gliomas (Bralten et al., 2011; Fu et al., 2015). Thus, R-2HG appears to exhibit complex effects during initiation, progression, and drug response of different cancers, perhaps through inhibiting different dioxygenases.
Here we show that R-2HG also displays a broad and intrinsic anti-tumor activity in leukemia and glioma, by targeting the fat mass and obesity-associated protein (FTO), a RNA N6-methyladenosine (m6A) demethylase (Jia et al., 2011), and by inducing suppression of MYC/CEBPA-associated pathways through affecting mRNA m6A modification and the fates of target RNAs. Thus, our studies reveal a previously unrecognized link between FTO/m6A-modification/MYC/CEBPA signaling and the function of R-2HG in cancers.
RESULTS
R-2HG Shows Growth-Suppressive Activity in Leukemia
To broadly define the pathological effect of R-2HG in leukemia, we exposed 27 human leukemia cell lines (Table S1), none of which carries common IDH1/2 mutations (Table S2), to a series of concentrations of R-2HG; similar or even higher concentrations of R-2HG have been used in previous studies (Fu et al., 2015; Losman et al., 2013; Lu et al., 2012). Such concentrations should be physiologically relevant as IDH1/2 mutations in AML and glioma patients can result in accumulation of R-2HG up to the millimolar (mM) level. Unexpectedly, R-2HG inhibited cell proliferation and viability in a time- and dose-dependent manner in the majority of these leukemia cell lines (Figures 1A and 1B; Table S3). The inhibition of cell proliferation and viability likely stems from R-2HG-induced cell-cycle arrest and apoptosis (Figures S1A–S1F). The accumulation of intracellular R-2HG is comparable between the sensitive and resistant cells (Figure S1G).
Figure 1. R-2HG Displays Anti-leukemic Activity in vitro and in vivo.

(A) Relative cell viabilities of leukemia cell lines treated with 300 μM cell-permeable R-2HG for the indicated times. The colors represent different time points; the diameter indicates the relative cell viability. H, hour.
(B) Relative cell viabilities of the cell lines at 96 hours post-treatment with different concentrations of R-2HG. The colors represent different R-2HG concentrations; the diameter represents relative cell viability.
(C) Schematic illustration of leukemic mouse models with R-2HG (or PBS) in vivo injection.
(D) Kaplan-Meier curves of leukemic mouse models xeno-transplanted with sensitive (NOMO-1 and MA9.3ITD) or resistant (MA9.3RAS) cells followed by PBS or R-2HG injection. NRGS mice were used for the NOMO-1 and MA9.3RAS models, while NSGS mice were used for the MA9.3ITD model.
(E) Spleen weight of MA9.3ITD- or MA9.3RAS-xenotransplanted mice with PBS or R-2HG injection.
(F) Schematic illustration of leukemic mouse models with IDH1R132H-mediated generation of R-2HG.
(G) Kaplan-Meier curves of NOMO-1_IDH1R132H, MA9.3ITD_IDH1R132H, and NB4_IDH1R132H mouse models with or without doxycycline (Dox) induction. NRGS mice were used for the NOMO-1_IDH1R132H and MA9.3ITD_IDH1R132H models, while NSGS mice were used for the NB4_IDH1R132H model.
(H) Spleen weight of MA9.3ITD_IDH1R132H and NB4_IDH1R132H leukemic mice with or without Dox induction.
(I and J) Engraftment of MA9.3ITD (I) and MA9.3RAS (J) AML cells into PB, BM and spleen upon R-2HG or PBS injection.
(K) Engraftment of MA9.3ITD_IDH1R132H cells into recipient mice with or without Dox induction.
(L) Wright-Giemsa staining of PB from leukemia mouse models. Arrows indicate the immature leukemic cells. Black bar represents 50 μm.
NS, non-significant; *, P<0.05; **, P<0.01; ***, P<0.001; t-test. Error bars, mean ± SEM (n≥3).
For Kaplan-Meier curve, P values were calculated by log-rank test.
See also Figure S1; Tables S1–S5.
It was reported previously that R-2HG promotes cell proliferation and induces leukemic transformation in TF-1 cells under GM-CSF poor conditions (Losman et al., 2013). We repeated the same experiments and found that R-2HG indeed increased cell proliferation under cytokine-poor conditions, but decreased cell growth and viability under standard culture conditions (Figures S1H and S1I). Interestingly, in SKNO-1, another GM-CSF-dependent leukemia cell line, R-2HG notably inhibited cell proliferation and viability under both standard culture conditions and cytokine-poor conditions (Figures S1J and S1K). Moreover, R-2HG also decreased colony-forming activity (Figure S1L) and cell viability (Figure S1M) of human primary AML cells (without IDH1/2 mutations), and showed neither obvious inhibitory nor promoting effects on the viability of human primary AML cells with IDH1/2 mutations (Figure S1N; Table S4). Thus, contrast to the proliferation-promoting effect of R-2HG observed in TF-1 cells under cytokine-poor conditions, R-2HG exhibits a broad growth-suppressive activity in leukemia in general, whereas IDH1/2-mutant leukemia cells can tolerate such an inhibitory activity.
R-2HG Significantly Inhibits Progression of Sensitive AMLs in vivo
We next used “human-in-mouse” xeno-transplantation leukemic models to evaluate the effect of R-2HG on leukemia progression in vivo. Two strategies of R-2HG treatment, including direct R-2HG injection and IDH1R132H-mediated generation of R-2HG, were conducted with immunodeficient NSGS (Wunderlich et al., 2010) and NRGS (Wunderlich et al., 2014) mice. As expected, the in vivo treatment with R-2HG significantly inhibited AML progression and prolonged survival in mice xeno-transplanted with sensitive cells, but not in those with resistant cells (Figures 1C–1D). Further, we transduced doxycycline-inducible IDH1R132H lentiviruses into sensitive (NOMO-1 and MA9.3ITD) or resistant (NB4) AML cells and xeno-transplanted the cells into immunodeficient mice; the mice were then fed a doxycycline (dox) diet or a regular diet (Figure 1F). Similarly, endogenous R-2HG produced by IDH1R132H also significantly delayed AML progression and extended survival in mice with sensitive AML (Figure 1G). Consistently, both exogenous (in vivo injected) and endogenous (IDH1R132H-generated) R-2HG resulted in less aggressive leukemic symptoms in mice with 2HG-sensitive AML cells, including reduced splenomegaly and inhibited engraftments in peripheral blood (PB), bone marrow (BM) and spleen (Figures 1E, and 1H–1K; Table S5). R-2HG treatment led to a substantial decrease of leukemic blasts in PB from both NSGS and NRGS mice with these sensitive cells, but not with the 2HG-resistant AML cells (Figure 1L). Thus, our in vivo results indicate that R-2HG may hold therapeutic potential to treat R-2HG-sensitive AMLs.
Factors Related to R-2HG Sensitivity in Leukemic Cells
To determine the potential factors related to R-2HG sensitivity in leukemic cells, we performed RNA-seq with four (R-2HG-) sensitive and five resistant leukemia cell lines, along with four healthy control samples (Figure 2A). A set of Fe(II)/α-KG-dependent dioxygenases were found to be expressed at significantly higher levels in sensitive AML cells than in resistant AML cells or normal controls (Figure 2A). Our qPCR analysis with an expanded cohort of leukemic and normal control samples confirmed the positive correlation between expression levels of these dioxygenase genes and R-2HG sensitivity across leukemic samples for 7 out of the top 10 dioxygenase genes (Figures 2A, 2B, and S2A). However, only FTO was expressed at a significantly higher level in leukemic samples compared to all three types of normal control cells (Figure 2B).
Figure 2. Identification of Genes and Pathways Related to R-2HG Response.

(A) Identification of potential α-KG-dependent dioxygenases and signaling pathways responsible for the varying sensitivities to R-2HG treatment. Upper panel: the top 10 α-KG-dependent dioxygenases showing a positive correlation with R-2HG sensitivity; Lower panel: the top 3 signaling pathways distinguishing sensitive and resistant leukemia cells.
(B) Expression of FTO in leukemic samples and healthy controls (mononuclear cells (MNCs), CD34+ cells and CD34− cells) and its positive correlation with R-2HG sensitivity. **, P<0.01; unpaired Student’s t-test.
(C) The top 3 signaling pathways suppressed by R-2HG in sensitive (NOMO-1) leukemia cells.
(D) Gene set enrichment analysis (GSEA) of differentially expressed genes in four groups of comparisons.
(E) The normalized enrichment scores of MYC, G2M, and E2F signaling pathways in the four groups of comparisons.
(F) Violin plots summarize the gradient levels MYC, G2M, and E2F signaling cascades in R-2HG resistant, R-2HG sensitive, and healthy control samples.
We next conducted RNA-seq assays with NOMO-1 and MA9.3ITD cells upon R-2HG treatment (Figure 2C). Expression levels of a number of dioxygenase genes were also changed upon R-2HG treatment (Figure S2B; Table S6). Through gene set enrichment analysis (GSEA), we identified 7 gene sets that are strongly correlated with R-2HG sensitivity through the two cohorts of RNA-seq datasets (Figures 2A, 2C, 2D, and S2C). Among these pathways, MYC, G2M, and E2F signaling pathways are hyper-activated in R-2HG-resistant leukemic samples, while being only moderately activated in R-2HG-sensitive samples, relative to normal controls (Figures 2E–2F and S2C–S2E), suggesting their hyper-activation might be responsible for the resistance to R-2HG in resistant leukemic cells. Importantly, the presence of R-2HG suppressed the activities of MYC, G2M, and E2F signaling pathways in R-2HG sensitive AML cells (Figures 2C–2E and S2C–2G). Our qPCR data further confirmed that R-2HG inhibits the expression of the main component genes of MYC signaling in sensitive cells, but not in resistant cells (Figure S2H). The fact that MYC, G2M, and E2F signaling act concordantly to regulate G0/G1, G1/S, and G2/M cell cycle transitions might be a primary mechanism by which R-2HG causes cell cycle arrest and apoptosis in sensitive cells.
R-2HG Targets FTO in Sensitive Leukemic Cells
FTO, the first identified RNA demethylase, is a Fe(II)/α-KG-dependent dioxygenase that catalyzes demethylation of m6A methylation in RNA and plays an oncogenic role in AML (Jia et al., 2011; Li et al., 2017). Based on our previous study on FTO in AML, a recent study suggested that R-2HG may target FTO (Elkashef et al., 2017). Due to the overexpression of FTO and the positive correlation between its expression level and R-2HG sensitivity in leukemia cells (Figure 2B), we sought to determine whether FTO is a critical bona fide target of R-2HG that mediates the effects of R-2HG in sensitive leukemic cells. We found that R-2HG treatment notably increased global m6A abundance in sensitive cells, but not in resistant cells, as detected by both m6A dot blot assay and LC-MS/MS assay with poly(A)+ RNA (Figures 3A–3B). Besides m6A, FTO also displays demethylation activity towards RNA with N6,2′-O-dimethyladenosine (m6Am) in the 5′ cap (Mauer et al., 2017). Our LC-MS/MS assays showed that R-2HG also slightly increases cap m6Am levels in sensitive leukemia cells, but its overall abundance is only ~1/30 of that of m6A in leukemia cells (Figures 3B–3D). Thus, our data suggest that R-2HG likely suppresses FTO activity and thereby elevates m6A RNA modification in the sensitive AML cells.
Figure 3. R-2HG Induces m6A Modification via Direct Inhibiting m6A Demethylation Activity of FTO.

(A) R-2HG treatment (300 μM, 96 hours) increases global m6A levels in sensitive leukemia cells (left panel), but not in resistant cells (right panel). MB, methyl blue.
(B and C) Verification of the m6A abundance (B) and determination of m6Am abundance (C) in poly(A)+ RNA by LC-MS/MS.
(D) Relative m6A and m6Am abundance in leukemia cells.
(E) Identification of the direct binding between R-2HG and FTO via DARTS assays.
(F) CETSAs exhibit the binding affinity of R-2HG to FTO in AML cells.
(G) Proposed oxidative demethylation of m6A to A in RNA by FTO in the presence of Fe(II), α-KG and R-2HG.
(H) Determination of m6A abundance by dot blot in the presence of various R-2HG concentrations and FTO protein in a cell-free system.
(I) Verification of the remaining m6A levels in the presence of FTO and varying R-2HG concentrations by LC-MS/MS. w/o, without; w/, with.
(J) Effects of FTO on cell proliferation/viability in NOMO-1 cells.
(K) Effects of FTO on global m6A modification in NOMO-1 cells. Dot blot assays were conducted with poly(A)+ RNA.
(L) Knockdown of FTO abolishes R-2HG-induced cell proliferation-suppressive effects in NOMO-1 cells.
*, P<0.05; **, P<0.01; ***, P<0.001; t-test. Error bars, mean ± SD (n=3).
See also Figure S3.
R-2HG has also been shown to competitively inhibit the functions of DNA and histone demethylases, such as TET2, JMJD, and KDM, leading to hypermethylated DNA and histones (Chowdhury et al., 2011; Sasaki et al., 2012). Nonetheless, we analyzed the expression levels of genes encoding DNA demethylases (TET1/2/3), m6A RNA demethylases (FTO and ALKBH5), and histone demethylases (including KDM2A, KDM4A, and JMJD6) in leukemic and healthy control samples by qPCR. We found that FTO is the only gene that exhibits a significantly positive correlation with R-2HG sensitivity across the leukemia samples; that is also overexpressed in leukemia samples relative to normal controls (Figures S3A–S3D). Further functional studies showed that R-2HG caused a noticeable decrease in 5-hydroxymethylcytosine (5hmC) modification in resistant cells, but not in sensitive cells (Figures S3E and S3G), suggesting that R-2HG inhibits the activity of TETs in the resistant cells. No consistent, significant increase in histone methylation was observed in either sensitive or resistant leukemic cells upon R-2HG treatment (Figures S3F and S3H). Collectively, our data suggest that FTO might be a main mediator of the effect of R-2HG in 2HG-sensitive AML cells.
To determine if FTO is a direct target of R-2HG, we conducted three types of biochemical experiments. Drug affinity responsive targets stability (DARTS) assays and cellular thermal shift assays (CETSAs) were performed to demonstrate the direct binding between FTO protein and R-2HG in cellulo, while in vitro FTO demethylation activity assays were conducted to verify that R-2HG competitively suppresses FTO enzymatic activity in a cell-free system. Our DARTS data suggest that R-2HG binds to FTO, protecting it from degradation by proteinase in a dose-dependent manner (Figure 3E). The CETSAs detected obvious shifts of the FTO melting curve in the presence of R-2HG, indicating direct binding between FTO and R-2HG inside cells (Figures 3F and S3I). In vitro FTO activity assays confirmed that R-2HG can inhibit FTO demethylase activity (Figures 3G–3I and S3J).
Finally, we also showed that knockdown of endogenous FTO by shRNAs in sensitive cells recapitulates the effects of R-2HG on inhibiting cell viability and increasing global m6A levels of cellular poly(A)+ RNA (Figures 3J–3K and S3K–S3M). Conversely, forced expression of wild-type FTO, but not mutant FTO (carrying two point mutations, H231A and D233A, which disrupt enzymatic activity) (Jia et al., 2011), significantly promotes cell proliferation and decreases total m6A levels (Figures 3J–3K and S3K–S3M). Moreover, knockdown of FTO in R-2HG-sensitive leukemic cells significantly reduced their sensitivity to R-2HG (Figure 3L). Collectively, our data demonstrate that FTO is a direct target of R-2HG and a main mediator of R-2HG-induced growth-suppressive effects in leukemic cells.
The R-2HG⊣FTO⊣m6A Axis Regulates MYC Expression
To identify critical downstream target(s) of the R-2HG⊣FTO⊣m6A axis responsible for growth inhibition, we conducted transcriptome-wide m6A-sequencing (m6A-seq) and RNA-seq with R-2HG- and PBS-treated NOMO-1 cells. The genomic distributions of the m6A peaks and the m6A motifs identified from the two groups (Figures S4A–S4E) are consistent with those reported previously (Wang et al., 2014b). Notably, amongst the 6,024 m6A peaks with significant changes upon R-2HG treatment, the vast majority (5,281; 87.7%) exhibited a notable (p< 0.01) increase in m6A abundance (Figures 4A and 4B). The transcripts with increased m6A levels (m6A-hyper) were also significantly enriched for target genes of MYC, E2F, and G2M signaling pathways (Figure 4C). The m6A-hyper peaks were preferentially located in the CDS at a greater proportion than expected by chance (63.6% vs. 52.8%) (Figures S4C, S4F, and S4G). Approximately 5% of the fold-change-adjusted m6A-hyper peaks are located within the 5′ end of the 5′UTR (150 nt as cut-off) (Figure S4H), which could be attributed to cap m6Am and 5′ UTR m6A peaks, consistent with low cap m6Am in these leukemia cells (Figures 3B–3D). Indeed, only 4.7% of m6A-hyper peaks are located within the first 150 nt of 5′UTR (Figure S4I); the average fold change of these 5′-end m6A-hyper peaks is comparable to that of the other m6A-hyper peaks (Figure S4J). Moreover, although cap m6Am was reported to stabilize target transcripts (Mauer et al., 2017), the 5′-end m6A-hyper peaks identified herein are not preferentially associated with increased mRNA levels upon R-2HG treatment (Figure S4K). In analysis of mRNAs with m6Am, Am, Cm, Gm and Um adjacent to the cap in leukemic cells, we did not observe a bigger fold change in expression of m6Am-initiated mRNAs compared with those of the other mRNA groups upon FTO inhibition or overexpression (Figure S4L), suggesting that cap m6Am has little effects in leukemia cells. Therefore, our data suggest that internal m6A, not cap m6Am, is the relevant substrate of FTO in these 2HG-sensitive leukemic cells.
Figure 4. R-2HG and FTO Regulate MYC Expression via Manipulation of m6A Modification.

(A) The density (line) and frequency (histogram) distributions of m6A peaks in NOMO-1 cells with R-2HG vs. PBS-treatment.
(B) The significantly increased (red) or decreased (blue) m6A peaks (P < 0.05) upon R-2HG treatment in NOMO-1 cells.
(C) GSEA analysis of genes with a significant increase in m6A modification on transcripts after R-2HG treatment.
(D) The m6A abundance in MYC mRNA in R-2HG- or PBS-treated NOMO-1 cells.
(E) Gene-specific m6A qPCR validation of m6A level changes of MYC mRNA in NOMO-1 cells.
(F) Luciferase reporter and mutagenesis assays. HEK-293T cells were co-transfected with MYC-5′UTR or MYC-CDS bearing wild-type or mutant (m6A replaced by T) m6A motifs, together with wild-type FTO, mutant FTO, or control vector.
(G) Luciferase reporter assay-related gene specific m6A qPCR analysis of the m6A levels in the exogenous mRNA transcripts of MYC 5′UTR and CDS.
(H) Relative abundance of MYC 5′UTR (upper panel) and CDS (lower panel) exogenous transcripts with wild-type or mutant m6A site in HEK-293T cells.
(I) Effects of R-2HG on MYC mRNA stability in sensitive or resistant leukemia cells.
(J) Effect of YTHDF2 knockdown on MYC mRNA stability in NOMO-1 and K562 cells.
(K) Schematic illustration of m6A-seq in sensitive cells (MA9.3ITD) with FTO knockdown and in resistant cells (MA9.3RAS) with FTO overexpression.
(L) Verification of the R-2HG levels in each group being subjected to m6A-seq.
(M) Changes of m6A peaks on MYC transcripts in PBS- or R-2HG-treated sensitive cells (MA9.3ITD) upon FTO knockdown, or resistant cells (MA9.3RAS) upon FTO overexpression.
(N) Quantitation of the m6A peaks in Figure 4M.
(O) Response to R-2HG in NOMO-1 cells with MYC knockdown background.
ns, non-significant; *, P<0.05; **P<0.01; ***, P<0.001; t-test. Error bars, mean ± SD (n=3).
See also Figure S4.
MYC targets and E2F targets are the top gene sets repressed by R-2HG in sensitive cells (see Figure 2), and they are believed to be regulated directly or indirectly by MYC (Lin et al., 2012). Notably, our m6A-seq data shows that MYC transcripts are enriched with m6A peaks, which are substantially increased by R-2HG treatment, especially in the 5′UTR and CDS regions (Figure 4D); this was confirmed by gene-specific m6A qPCR assays (Figure 4E). To determine whether MYC is a direct target of FTO, we performed luciferase reporter assays as well as m6A-IP with MYC 5′UTR or CDS containing wild-type or mutant m6A sites (m6A was replaced with T). Forced expression of wild-type FTO, but not mutant FTO, significantly increased luciferase activity of the reporter construct carrying wild-type MYC 5′UTR or CDS, relative to the control; this increase was abrogated when the putative m6A sites were mutated (Figure 4F). Our gene-specific m6A qPCR results showed that the constructs with wild-type m6A motifs contain much higher levels of m6A compared to those with mutant m6A sites, and forced expression of wild-type FTO noticeably reduced m6A abundance (Figure 4G). Moreover, RNAs transcribed from the reporter constructs bearing the (m6)A-T mutant 5′UTR or CDS were more abundant than those from wild-type constructs in cells (Figure 4H), likely because the former were more stable due to the lack of m6A modifications. Overall, our data indicate that FTO can increase the transcript level of MYC through an m6A-dependent mechanism.
In contrast to the effect of FTO, R-2HG notably suppresses MYC expression in sensitive cells, but not in resistant cells (Figure S4M). Furthermore, R-2HG markedly decreased MYC mRNA stability in sensitive cells, but not in resistant cells (Figure 4I), indicating that R-2HG-induced down-regulation of MYC in sensitive cells is likely related to the reduced stability of MYC transcripts. Knockdown of YTHDF2, a major m6A reader that is responsible for the decay of m6A-modified mRNA transcripts (Wang et al., 2014b), and that is known to bind MYC mRNA (Figure S4N), noticeably increased the stability and expression of MYC transcripts in leukemia cells (Figures 4J and S4O). Consistent with R-2HG’s effects, knockdown of FTO also inhibited MYC expression; conversely, ectopically expressed wild-type FTO promoted MYC expression (Figure S4P). Together, our data suggest that the m6A changes caused by FTO or R-2HG affect MYC expression by influencing the stability of MYC transcripts, likely through a YTHDF2-associated mechanism.
We also performed m6A-seq of R-2HG- and PBS-treated sensitive cells (MA9.3ITD) with or without FTO knockdown, and resistant cells (MA9.3RAS) with or without FTO overexpression to further analyze the effects of R-2HG and FTO on m6A modification of MYC transcripts (Figures 4K and 4L). In MA9.3ITD cells, R-2HG increased the abundance of m6A in the 5′UTR and CDS regions of MYC transcripts, and this increase was abrogated by FTO knockdown (Figures 4M and 4N). In contrast, R-2HG showed no obvious effect on m6A abundance on MYC transcripts in parental MA9.3RAS cells, but substantially increased m6A abundance in the 5′UTR and CDS regions of MYC transcripts in FTO-overexpressing MA9.3RAS cells (Figures 4M and 4N). Importantly, knockdown of MYC reduced the response to R-2HG in sensitive cells (Figure 4O), suggesting MYC is an important target of the R-2HG⊣FTO⊣m6A axis. R-2HG treatment also significantly increased expression of RARA and ASB2, two direct targets of FTO (Li et al., 2017), in sensitive cells, but not in the resistant cells (Figures S4Q and S4R). Thus, our data further demonstrate that FTO and its downstream targets (e.g., MYC, ASB2, and RARA) are the major effectors of R-2HG in sensitive leukemia cells.
CEBPA Suppression by the R-2HG⊣FTO⊣m6A Axis Inhibits FTO Transcription
Interestingly, besides inhibition of FTO catalytic activity by R-2HG through direct interaction (see Figures 3E–3I), we also found that extended R-2HG treatment (e.g., 96 hours) can substantially decrease FTO protein levels in sensitive leukemic cells, but not in resistant cells (Figure 5A). Notably, R-2HG treatment for 48 hours shows a minor effect on FTO protein level, but an obvious increase in global m6A abundance in poly(A)+ RNA (Figure 5B), implying that inhibition of FTO expression after extended R-2HG treatment might be a consequence of the increased m6A abundance. As indicated by data shown in Figures S5A–S5H, R-2HG-mediated FTO expression suppression is not related to altered m6A, DNA, or histone modifications. Instead, our nuclear run-on assays suggest that extended R-2HG treatment-mediated down-regulation of FTO is likely due to transcriptional inhibition (Figure 5C). To identify transcription factors (TFs) that may regulate transcription of FTO, we analyzed the correlations in expression between FTO and 92 TF genes which have putative binding sites in the “core promoter” of the FTO gene in four AML datasets (Figure 5D). Among them, CEBPA, MZF1, NFATC3, and RFX3 display a positive correlation (P<0.05), while NFKB2 and TFEB exhibit a negative correlation (P<0.05) in all four datasets (Figures 5E and S5I). Among the 4 positively correlated TFs, only CEBPA demonstrates both increased m6A modification and decreased expression after R-2HG treatment in sensitive cells (Figures 5F and 5G).
Figure 5. R-2HG also Indirectly Modulates FTO Expression at Transcriptional Level.

(A) Extended R-2HG treatment (300 μM, 96 hours) suppresses FTO expression in sensitive, but not in resistant cells.
(B) FTO levels and m6A abundance in NOMO-1 cells treated with PBS or 300 μM R-2HG.
(C) Determination of FTO transcription initiation rate via nuclear run-on assay in sensitive NOMO-1 cells upon R-2HG or PBS treatment for 96 hours.
(D) Pearson correlation analysis between expression of predicted TFs and FTO in four AML datasets.
(E) TFs whose expression shows a positive or negative correlation with FTO across AML datasets.
(F and G) m6A modification and quantitation of m6A abundance on mRNA transcripts (F) and relative expression levels (G) of CEBPA and MZF1 in NOMO-1 cells with R-2HG or PBS treatment for 48 hours.
(H and I) Effects of FTO on regulating CEBPA expression at the protein (H) and RNA (I) levels in NOMO-1 cells.
(J) The effect of R-2HG treatment and YTHDF2 knockdown on the stability of CEBPA mRNA.
(K) The effect of YTHDF2 knockdown on FTO and CEBPA expression.
(L) Expression of CEBPA in AML and healthy control samples according to GSE24006 dataset.
(M) CEBPA enhances transcriptional activity of the FTO promoter as detected by luciferase reporter/mutagenesis assays.
(N) Effects of CEBPA knockdown on cell proliferation/viability in NOMO-1 cells.
(O) Effects of CEBPA on FTO expression in NOMO-1 cells.
(P) Response to R-2HG in NOMO-1 cells with or without CEBPA knockdown.
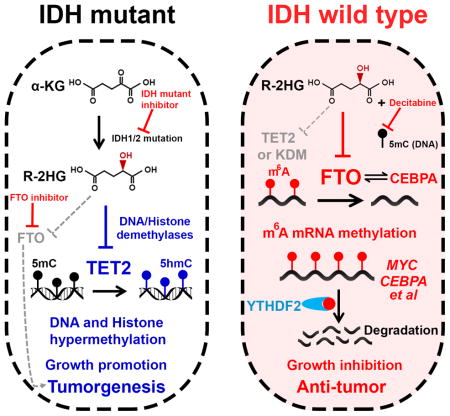
(Q) Model of the anti-leukemic activity of R-2HG through FTO inhibition. In this model, direct interaction between R-2HG and FTO induces global increased m6A modification as well as decreased expression of MYC and CEBPA, which account for the anti-tumor activity of R-2HG; as a feedback mechanism, R-2HG⊣FTO⊣m6A axis-induced down-regulation of CEBPA also decreases FTO expression at transcriptional level.
*P<0.05; **, P<0.01; ***, P<0.001; t-test. Error bars, mean ± SD (n=3).
See also Figure S5.
Consistent with the effects mediated by R-2HG, expression of CEBPA could also be enhanced by forced expression of FTO and inhibited by FTO knockdown (Figures 5H and 5I). The increased m6A modification on CEBPA mRNA upon R-2HG treatment could be recognized by YTHDF2, leading to reduced stability of CEBPA transcripts and reduced expression (Figures 5J and 5K). Moreover, CEBPA was abnormally upregulated in AML patients (Figure 5L), and its forced expression promoted transcriptional activity of the FTO promoter (Figure 5M). Knockdown of CEBPA decreased cell growth, reduced FTO abundance, and largely abolished R-2HG-induced growth-suppressive effects (Figures 5N–5P) in R-2HG-sensitive leukemic cells. Consistent with this, low abundance of endogenous CEBPA in R-2HG-resistant leukemic cells (Figure S5J) likely also contributes to the resistance of the cells to R-2HG. Collectively, suppression of CEBPA by the R-2HG⊣FTO⊣m6A axis could further inhibit FTO transcription as a feedback mechanism (Figure 5Q).
Both Ectopic IDH1 Mutant and S-2HG Exert Similar Effects to R-2HG
To determine whether mutant IDH can recapitulate the phenotypes we observed in R-2HG treated cells, we created leukemic cell lines with inducible expression of mutant IDH. As expected, doxycycline-induced IDH1R132H expression mimicked the phenotypes caused by exogenous R-2HG, such as suppressed FTO and MYC expression, enhanced m6A modification, induced cell cycle arrest, decreased cell proliferation, and increased cell apoptosis in sensitive, but not in resistant cells (Figures 6A–6C and S6A–S6D). Measurement of intracellular R-2HG confirmed the generation of R-2HG by IDH1R132H, to the level that results from 300 μM R-2HG treatment (Figure 6B vs. Figure S1G). Moreover, doxycycline-induced IDH1R132H expression also mimicked the in vivo anti-tumor effects of exogenous R-2HG in mice with sensitive AML (see Figure 1).
Figure 6. Effects of IDH Mutation and S-2HG in Leukemia.

(A) Effect of IDH1R132H on FTO and MYC expression in sensitive cells (NOMO-1 and MA9.3ITD) and resistant (K562) cells.
(B) Confirmation of the intracellular R-2HG accumulation in IDH1R132H infected cells with Dox induction. ***, P<0.001; t-test. Error bars, mean ± SD (n=3).
(C) Effects of IDH1R132H on global m6A modification in poly(A)+ RNA in the sensitive and resistant cells.
(D) The chemical structures of α-KG, R-2HG, and S-2HG as well as their potential inhibition on FTO.
(E) Effects of S-2HG on cell proliferation/viability in multiple R-2HG sensitive and resistant cells. Upper panel, the cells were treated with 300 μM S-2HG for indicated time points; lower panel, the cells were treated for 96 hours with indicated S-2HG concentrations.
(F) m6A dot blot demonstrates that S-2HG also acts as a competitive inhibitor of FTO in a cell-free system.
(G) Analysis of the remaining m6A abundance with presence various S-2HG concentrations and FTO via LC-MS/MS.
See also Figure S6.
S-2HG, the enantiomer of R-2HG, is also structurally and chemically related to α-KG and R-2HG (Figure 6D). We found that S-2HG displayed a growth-suppressive effect in R-2HG-sensitive cells, but not in R-2HG-resistant cells (Figures 6E, S6E, and S6F). S-2HG also directly inhibited the demethylation activity of FTO (Figures 6F, 6G, and S6G). Together, IDH mutation and S-2HG could largely recapitulate effects of R-2HG, including direct inhibition of FTO, increased global m6A modification, and decreased leukemic cell proliferation/viability.
FTO/MYC Homeostasis Controls R-2HG Sensitivity
Since ectopically expressed IDH mutants (e.g., IDH1R132H) display anti-tumor effects, why do they still exist in 10–20% of AML cases? To address this question, we conducted integrative analysis of the TCGA AML microarray dataset (Ley et al., 2013) with our RNA-seq data shown in Figure 2A. We identified 5 core signaling pathways enriched in the IDH-mutant AML samples (relative to IDH-wildtype AML samples, in the whole set or in the normal-karyotype subset), as well as in R-2HG-resistant leukemic cells (relative to R-2HG-sensitive cells) and R-2HG-sensitive cells (compared with healthy controls) (Figures 7A and S7A). Primary AML cells with IDH mutations are also sensitive to JQ1, an inhibitor of MYC signaling (Delmore et al., 2011), often with an IC50 lower than 1 μM, though with a higher IC50 than AML cells with wild-type IDH (Figure 7B; Table S4), likely due to the hyper-activation of MYC signaling in the former. We also confirmed that IDH-mutant AML samples have a higher expression level of MYC and its critical targets (e.g., CDK4 and CDK6), and a lower level of FTO (but not ALKBH5) expression, than IDH wild-type AML samples (Figure 7C; Table S4). Our Western blot assays showed that R-2HG-resistant leukemic cell lines have a much higher level of MYC and a lower level of FTO than the sensitive cell lines; R-2HG treatment caused a substantial decrease in FTO and MYC expression in sensitive cells, but had minimal effects in resistant cells (Figure 7D).
Figure 7. The Abundance of FTO and MYC Control Sensitivity of Leukemic Cells to R-2HG and R-2HG Shows Synergistic Activity with First-line Chemotherapy Drugs.

(A) Venn diagram showing the shared signaling pathways of the 4 indicated groups of comparisons. Information of the sensitive, resistant, and healthy control samples is described in Figure 2A. The other samples listed in the plot are human primary AML samples from the TCGA dataset: IDH mutant, AML samples with mutations in IDH1 and/or IDH2; IDH WT, AML samples with wild-type IDH genes; IDH WT (NK) or IDH mutant (NK), normal-karyotype AML samples with wild-type or mutant IDH genes.
(B) The IC50 values of JQ1 in AML patients with IDH mutations or wild-type IDH genes.
(C) Relative expression levels of MYC, CDK4, CDK6, FTO, and ALKBH5 in primary AML patients with or without IDH mutation as well as in healthy controls (MNCs).
(D) The expression pattern of FTO and MYC in sensitive and resistant cells with or without R-2HG treatment for 96 hours.
(E) Knockdown of MYC increases the sensitivity to R-2HG in resistant K562 cells.
(F) Forced expression of MYC rescues R-2HG-induced (left panel) and IDH1R132H-mediated (middle panel) growth-suppressive effects in sensitive NOMO-1 cells. MYC overexpression was confirmed by Western blot (right panel).
(G) Synergistic therapy of R-2HG in combination with Daunorubicin and Decitabine in vivo. NSGS mice were transplanted with NOMO-1 cells and the combined chemotherapy regimens were started on day 11 post xeno-transplantation as indicated in the upper panel.
(H) Schematic illustration of the relevant abundance of FTO and MYC controlling R-2HG sensitivity.
ns, non-significant; *, P<0.05; **, P<0.01; ***, P<0.001; t-test. Error bars, mean ± SD (n≥3).
For Kaplan-Meier curve, P values were calculated by log-rank test.
Thus, we propose that highly activated MYC signaling diminishes the anti-leukemic effect of R-2HG in most IDH-mutant AML cells. To test this hypothesis, we manipulated MYC levels both in resistant and sensitive cells, and tested their response to R-2HG or IDH mutation. In the resistant cells, both shRNA-induced MYC knockdown and JQ1-mediated MYC suppression could increase their sensitivity to exogenous R-2HG and IDH1R132H (Figures 7E and S7B). In the sensitive cells, forced expression of MYC did render leukemic cells resistant to R-2HG or IDH1R132H, and rescue R-2HG or IDH1R132H-induced proliferation inhibition (Figure 7F).
Furthermore, besides JQ1, we found that R-2HG also exhibits a synergistic effect with a series of first-line chemotherapy drugs such as all-trans retinoic acid (ATRA), Azacitidine (AZA), Decitabine, and Daunorubicin (Figure S7C). This synergistic effect is likely, at least in part, due to their combinational inhibition of MYC expression (Figure S7D). Importantly, the synergistic effect between R-2HG and chemotherapy drugs was also validated in vivo. The combinatorial treatment of R-2HG plus Daunorubicin or Decitabine displayed much better therapeutic effects in the leukemia mouse models xeno-transplanted with R-2HG-sensitive leukemic cells (Figures 7G and S7E).
Together, our data suggest that high abundance of FTO confers R-2HG sensitivity in leukemic cells, whereas hyper-activation of MYC-associated pathways renders leukemic cells (including those with endogenous IDH mutations) resistant to R-2HG (Figure 7H). Moreover, R-2HG shows synergistic anti-tumor effects with first-line chemotherapy drugs, and their combinations hold great therapeutic potential to treat leukemia in the clinic.
As IDH mutations frequently occur in human brain tumors as well, we also investigated the pathological effects of R-2HG in 8 human GBM cell lines (all with wild-type IDH1/2). R-2HG also inhibits cell proliferation and viability of all 8 cell lines (Figures S7F and S7G), and displays synergistic action with a common chemotherapy agent, Temozolomide (TMZ) (data not shown). Thus, our data indicates that R-2HG also exhibits anti-tumor activity in brain tumors.
DISCUSSION
Here we provide compelling in vitro and in vivo evidence demonstrating that exogenous or (IDH mutant-mediated) endogenous R-2HG could exert an intrinsic and broad anti-tumor activity in most leukemia samples we tested (most of which have wild-type IDH1/2).
Mechanistically, R-2HG exerts its anti-tumor effect largely through inhibiting the enzymatic activity of FTO. FTO effectively demethylates internal m6A and the inhibition by 2HG results in increased global m6A in mRNA with only <5% changes attributed to cap m6Am. While the cap m6Am is an effective in vitro substrate of FTO, its level in AML cells is very low. 2HG-mediated FTO inhibition led to accumulation of m6A on MYC transcripts, leading to decreased MYC mRNA stability and down-regulation of MYC signaling. This effect contributes to inhibition of cancer proliferation. We did not observe cap m6Am in MYC transcripts in our sequencing results. If increased m6Am through 2HG-mediated inhibition of FTO could play a role, it should lead to increased transcript stability, which is opposite to the decreased MYC expression level and the cancer inhibition effect induced by 2HG.
R-2HG also causes down-regulation of FTO expression indirectly through epigenetic suppression of CEBPA, an essential hematopoiesis-related transcription factor that is required for leukemogenesis (Ohlsson et al., 2014; Ye et al., 2015), via an m6A-dependent mechanism in which accumulation of internal m6A causes reduced CEBPA transcript levels. Notably, the high FTO (and likely also high CEBPA) abundance and the hyper-activation of MYC signaling confer R-2HG sensitivity and resistance in leukemia cells, respectively.
In contrast to its effect of increasing global m6A levels in sensitive leukemic cells, R-2HG causes a substantial decrease in global 5hmC abundance in resistant leukemic cells, which most likely attributes to R-2HG-mediated inhibition of TET2, a well-recognized tumor suppressor (Moran-Crusio et al., 2011). While the potential anti-leukemic effect of 2HG generated from mutant IDH is abrogated by hyperactivated MYC signaling, naturally occurring IDH mutants induce leukemia initiation and leukemic cell survival by inhibiting TET2. This may explain why IDH mutations still occur in 10%–20% of AML cases (Papaemmanuil et al., 2016), which respond to inhibition of IDH mutants, and are indeed more responsive to hypomethylating agents (Emadi et al., 2015). Since IDH mutations can sufficiently inhibit TET2 function, IDH mutations and TET2 mutations are mutually exclusive in human AMLs (Figueroa et al., 2010).
It is very likely that the inhibitory effects of R-2HG or IDH mutations on TET2 and FTO impact different stages or different types of cancer initiation and progression. The TET2 mutations are thought to occur in the early pre-leukemic stage, which may contribute to subsequent cancer initiation (Jan et al., 2012). The inhibition of TET2 affects transcription and hematopoietic stem cell (HSC) self-renewal and differentiation (Moran-Crusio et al., 2011), likely contributing to the initiation of IDH-mutant AMLs. In the 2HG-resistant, IDH-mutant AML cells, TET2 inhibition also contributes to AML proliferation. The inhibition of FTO impacts post-transcriptional regulation, which we show here affects progression and proliferation of a group of 2HG-sensitive cancer cells. Targeting FTO in 2HG-sensitive and wild-type IDH AML cells could be a viable anti-leukemia strategy. The inhibition of FTO by 2HG in certain IDH-mutant AML may explain the benign outcomes observed in some of these cases. Furthermore, R-2HG also suppresses the survival and proliferation of human GBM cells, suggesting that R-2HG has anti-proliferation activity in a broad array of tumors. IDH-mutations occur in around 80% of lower grade (II–III) primary gliomas but in <10% in grade IV primary GBM (Eckel-Passow et al., 2015), implying that the intrinsic inhibition of the oncogenic function of FTO by endogenous R-2HG may prevent rapid progression of glioma precursors toward higher grade glioma and thus limit the incidence of IDH-mutant primary GBM. Importantly, R-2HG may serve as both an oncometabolite to facilitate initiation of cancer, but also a tumor suppressor to suppress further progression of initiated tumors, leading to a beneficial effect in some of these IDH mutant cancers. As a “non-specific” inhibitor, 2HG likely inhibits different demethylases that affect different stages or types of tumor formation and progression. In the case of AML, most IDH-mutant cells are resistant to 2HG inhibition due to upregulated MYC; however, in a portion of IDH-mutant AML cases that exhibit less deleterious outcomes, 2HG may contribute to both cancer initiation and inhibition of further progression resembling those of lower grade IDH-mutant gliomas, in which inhibition of both IDH mutants and FTO could lead to synergistic anti-AML effect.
While IDH mutations in AML or glioma likely contribute to tumorigenesis by suppression of TET2 function, their potential inhibition of FTO function may affect their response to standard chemotherapy. Indeed, consistent with our findings that R-2HG exhibits a synergistic effect with JQ1 and a cohort of first-line therapeutic agents, it was reported that leukemia patients with IDH mutations tend to be more sensitive to treatment with hypomethylating agents (AZA and Decitabine) (Emadi et al., 2015), ATRA (Boutzen et al., 2016), or standard chemotherapy (Daunorubicin and others) (Chou et al., 2011; Patel et al., 2012), than those without. Similarly, our data and previous studies (Wang et al., 2014a) showed that glioma cells carrying IDH mutations are more sensitive to TMZ treatment. This may partially explain why IDH mutations are associated with relatively favorable outcomes in glioma and certain population of AML patients.
Overall, here we report the unexpected intrinsic and broad anti-tumor activity of R-2HG (and likely also S-2HG) in cancer cells without IDH mutations, which appear to involve the suppression of FTO/m6A/MYC/CEBPA signaling. Our studies uncover previously unrecognized activities of R-2HG and the functional importance of FTO, MYC, CEBPA, and reversible m6A RNA modification in 2HG-associated pathways. This work both provides novel insights into the molecular mechanisms underlying tumor pathogenesis and drug response, and paves the way to develop more effective novel therapeutic strategies to treat cancers with or without IDH mutations. Our work suggests that 2HG-type molecules or selective FTO inhibitors, alone and especially in combination with other therapeutic agents (e.g., standard chemotherapeutic drugs), hold great therapeutic potential in treating IDH wild-type cancers with high level expression of FTO (i.e., FTO-high). Our work also suggests that combinational application of both IDH mutant inhibitors and FTO inhibitors could lead to more beneficial outcomes in treating certain IDH-mutant cancers, as FTO inhibitors may block the activity of rebounded expression/function of FTO due to the decreased R-2HG production caused by IDH mutant inhibitors.
STAR METHODS
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
For leukemia cells, U937, THP1, MV4-11, JURKAT, and HEL were obtained from American Type Culture Collection (ATCC) and cultured in endotoxin-free RPMI1640 supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products); TF-1 (ATCC) was maintained in RPMI1640 with 10% FBS and 2ng/ml GM-CSF (PeproTech); K562 (ATCC) was cultured in IMDM with 10% FBS; NOMO-1, ML-2, PL21, ME-1, and NB4 were obtained from DSMZ and kept in RPMI1640 with 10% FBS; SKNO-1 (DSMZ) was maintained in RPMI1640 with 10% FBS and 10ng/ml GM-CSF; KOPN-1, KOCL69, KOCL48, KOCL50, KOCL45 and KOCL51 were maintained in RPMI1640 with 10% FBS; MA9.3 (MLL-AF9-transformed human CD34+ cord blood cell), MA9.3ITD (MLL-AF9 plus FLT3-ITD), MA9.3RAS (MLL-AF9 plus NRasG12D), MA9.6 (MLL-AF9), MA9.6ITD (MLL-AF9 plus FLT3-ITD) and MA9.6RAS (MLL-AF9 plus NRasG12D) were established by Dr. James Mulloy (Wunderlich et al., 2013). MA9.3 and MA9.6 were grown in IMDM supplemented with 20% FBS, 10 ng/ml human cytokines SCF, TPO, FLT3L, IL-3, and IL-6 (PeproTech); while MA9.3ITD, MA9.3RAS, MA9.6ITD and MA9.6RAS were cultured in the same medium without cytokines. The glioblastoma cell lines, including 8MGBA, A172, U87MG, GAMG, T98G, LN229, LN18, and DK-MG, were originally maintained by Dr. David Plas. All of the cells, with exception of DK-MG, were cultured in DMEM with 10% FBS; DK-MG was maintained in RPMI1640 with 10% FBS. BT142 mut/- was kept in NeuroCult NS-A proliferation kit plus 20 ng/ml EGF, 100 ng/ml PDGF-AA, 20 ng/ml bFGF and 2 μg/ml heparin sodium salt. All the cells are not among commonly misidentified cells lines, and were tested for mycoplasma contamination yearly using a PCR Mycoplasma Test Kit (PromoKine). All the cells were treated with cell membrane-permeable version of R-2-Hydroxyglutarate (R-2HG, octyl ester modified) or S-2HG at the indicated concentrations.
Animal Procedures
The NOD/LtSz-scid IL2RG-SGM3 (NSGS) and NRG-SGM3 (NRGS) mice, created by Dr. James Mulloy, were used as the “human in mouse” xeno-transplanation model. For R-2HG injection mouse models, sensitive (NOMO-1 and MA9.3ITD) or resistant (MA9.3RAS) cells were injected into NSGS or NRGS intravenously, and then R-2HG (6mg/kg body weight) or PBS were injected once daily through tail vein for 12 consecutive days starting from day 11 post xeno-transplantation. For IDH1R132H-mediated generation of R-2HG mouse models, sensitive (NOMO-1 and MA9.3ITD) or resistant (NB4) cells were infected with doxycycline-induced IDH1R132H lentivirus and transplanted into NRGS or NSGS mice, and then the mice were fed with doxycycline (dox) diet (to induce expression of IDH1R132H) or with regular diet. 0.1–0.5×106 human leukemia cells were transplanted into each recipient mouse. The engraftment of human leukemic cells into peripheral blood (PB), bone marrow (BM), and spleen of each mouse model was determined by staining with human CD33 via flow cytometry. For the in vivo drug combination assays, R-2HG, daunorubicin and decitabine were reconstituted with PBS, filtered, aliquoted, and stored at −20oC. The chemotherapeutic treatment was started at 11 days post-transplantation. The detailed regimens were as followed, R-2HG (6 mg/kg), i.v., once per day for ten consecutive days; daunorubicin (1.2 mg/kg), i.v., once per day for five consecutive days; decitabine (0.2 mg/kg), i.v., three times per week for two weeks. The mice were checked daily and euthanized by CO2 inhalation once they displayed typical leukemic symptoms, i.e. paralysis, hunched posture, labored breathing, and decreased activity. The PB, BM, spleen, and liver samples were collected for further analyses. All the leukemic animal model studies with NRGS mice were independently performed by Dr. James Mulloy’s lab in the Cincinnati Children’s Hospital Medical Center (CCHMC) animal core facility; all the leukemic animal model studies with NSGS mice were independently conducted by Dr. Jianjun Chen’s lab in the University of Cincinnati Vontz Center animal core facility.
METHODS DETAILS
R-2-Hydroxyglutarate measurement
R-2HG levels were determined with the D-2Hydroxyglutarate Assay Kit (Colorimetric) (K213-100, BioVision). In brief, R-2HG-treated cells and Doxycycline-induced IDH1R132H cells as well as their controls were collected, washed with chilled PBS buffer, and rapidly homogenized with 100 μl ice cold D-2HG buffer via Dounce for 10 min on ice. Samples were centrifuged at 10,000g, 4ºC for 5 min, and the supernatant was collected, mixed with D-2HG substrate and D2HGDH enzyme and incubated for 1 hour at 37ºC. The OD450nm was then measured according to the manufacturer’s instruction.
Leukemic patient and healthy control samples and in vitro colony forming assays
All AML patient samples were obtained at the time of diagnosis or relapse and with informed consent at the University of Chicago Hospital (UCH), City of Hope (COH), or the First Affiliated Hospital of Zhejiang University, and were approved by the institutional review board of the institutes/hospitals. The information about AML patients is exhibited in Supplementary Table 4. The BM mononuclear cells (MNCs) were isolated with NycoPrep 1.077A (Axis-Shield) and stored in liquid nitrogen until used. The healthy PB and BM MNCs were purchased from AllCells; the healthy CD34+ hematopoietic stem/progenitor cells (HSPCs) and CD34− cells were isolated from cord blood samples, which were purchased from CCHMC. For colony forming assay of BM progenitors, 10,000 cells were plated in 24-well plate with 1 mL human methylcellulose complete media (R&D Systems) and the colonies were counted 12 days later.
Cell proliferation/viability, cell cycle and cell apoptosis assays
To study the effects of R-2HG, FTO, or IDH1R132H on cell viability, cells were seeded in 96-well plates at a concentration of 5,000–10,000 cells per well in triplicate and MTT (G4100, Promega) was used to assess cell proliferation and viability following the manufacturer’s instructions. For cell cycle analysis, Propidium iodide (PI) DNA staining was used to assess cells at G0/G1, S, and G2/M phases, while Hoechst 33342 and Pyronin Y were used to determine cells at G0, G1, and S/G2/M stages. For the PI staining, cells were resuspended in Krishan’s reagent (0.05 mg/ml PI, 0.1% trisodium citrate, 0.02 mg/ml ribonuclease A, 0.3% NP-40), incubated at 37°C for 30 minutes and then applied to the flow cytometer. For Hoechst/Pyronin Y staining, the cells were suspended in cell culture medium, incubated at 37°C for 45 minutes with 10ug/mL Hoechst 33342 and further incubated at 37 °C for 15 minutes with Pyronin Y before flow cytometry. Cell apoptosis assays were conducted with FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen) according to the manufacturer’s instructions.
Flow cytometry
All the samples were analyzed on a FACSAria II or LSRFortessa cell analyzer (BD Bioscience). Flow cytometry analyses of mouse BM cells were performed as described previously with some modifications (Li et al., 2017). Data were analyzed with FlowJo software (Tree Star Inc., Ashland, OR). The following reagents were used for staining cells: PE-conjugated anti-human CD33 (eBioscience), FITC-labeled Annexin V (BD Pharmingen), propidium iodide (PI) (BD Pharmingen), Hoechst 33342 (Sigma) and Pyronin Y (Sigma).
Plasmid construction
Wild type FTO-CDS and mutant FTO-CDS (coding region sequence) were amplified from pcDNA3.1_FTO and pcDNA3.1_mutFTO (the two plasmids were created by Dr. Chuan He) by PCR using the following primers: forward 5′-AGAGCTCTAGAACCACCATGGATTACAAAGATGAC-3′ and reverse 5′-CTAAGATTGCGGCCGCCTAGGGTTTTGCTTCCAGAAGC-3′, and then subsequently cloned into lentivector-based pMIRNA1 (SBI). pCDH-puro-MYC was purchased from Addgene (Cambridge, MA). shRNAs against FTO (shFTO-1) and YTHDF2 (shYTHDF2-1 and shYTHDF2-2) were inserted into pLKO.1 vector. The other shRNA against FTO (shFTO-2) were purchased from OriGene. shRNAs specific to MYC and CEBPA were purchased from Dharmacon. The IDH1R132H (provided by Dr. Atsuo Sasaki) was inserted into pTRIPZ lentiviral inducible vector.
RNA extraction, cDNA synthesis, qPCR and m6A dot blot
Total RNA was extracted with miRNeasy Mini Kit (217004, Qiagen) according to the manufacturer’s guidelines and poly(A)+ mRNA was enriched from total RNA with PolyATract mRNA isolation system IV (Promega). For cDNA synthesis, 200 ng total RNA was used for reverse transcription in a 10ul reaction volume with QuantiTect Reverse Transcription Kit (205314, Qiagen) following the manufacturer’s instructions. Then quantitative PCR (qPCR) was performed with SYBR green qPCR Master Mix (K0253, Thermo Fisher) in an AB 7900HT Fast Real-Time PCR system (Applied Biosystem). GAPDH or ACTB was used as endogenous control and each reaction was run in triplicate. m6A dot blots were conducted as previously described with some modifications (Hodge, 1998). Poly(A)+ mRNA samples were denatured at 65 °C for 5 minutes in 3 sample volumes of RNA incubation buffer. An equal volume of chilled 20× SSC buffer (Sigma-Aldrich) was then added before samples were spotted on the Amersham Hybond-N+ membrane (GE Healthcare) with a Bio-Dot Apparatus (Bio-Rad). After UV crosslinking, the membrane was washed with 1 × PBST buffer (Thermo Scientific), blocked with 5% non-fat milk and incubated with anti-m6A antibody (1:2000, Synaptic Systems) overnight at 4°C. Then HRP-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology) was added to the blots for 1 hour at room temperature and the membrane was developed with Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare). The relative signal density of each dot was quantified by Gel-Pro analyzer software.
DNA extraction and 5hmC dot blot
DNA was isolated with a DNeasy Blood and Tissue Kit (69506, Qiagen) according to the manufacturer’s instructions. To assess 5hmC levels, dot blot assays were performed as follows: DNA samples were added to 0.1N NaOH, denatured at 99°C for 5 minutes, neutralized by adding 0.1 volume of 6.6M ammonium acetate, and spotted on Amersham Hybond-N+. After UV crosslinking, the membrane was stained with 0.02% methylene blue (Sigma-Aldrich), washed with 1× PBST buffer, blocked with 5% non-fat milk and incubated with 5hmC antibody (1:5000, Active Motif) overnight at 4°C. Then HRP-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology) was added to the blots for 1 hour at room temperature and the membrane was developed with Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare). The relative signal density of each dot was quantified by Gel-Pro analyzer software.
Protein extraction and Western blotting
For Western blotting, cells were placed on iced and washed twice with chilled PBS. Proteins were extracted with RIPA buffer (Sigma-Aldrich) plus protease inhibitor cocktail and phosphatase inhibitor cocktail (Thermo Fisher Scientific). The protein concentration was determined with the BCA protein assay kit (Thermo Fisher Scientific). An estimated 30–60ug protein was loaded per well on 10% SDS-PAGE gel and transferred onto PVDF membrane (Thermo Fisher Scientific), activated by methanol. Membranes were washed with 1× PBST, blocked with 5% milk and incubated with antibodies against FTO, GAPDH, β-Actin, MYC, CEBPA, FLAG M2, H3K9me3, and H3K36me3. Secondary antibodies (Donkey anti-rabbit IgG-HRP and Goat anti-mouse IgG-HRP) and detection were according to routine laboratory practices.
Drug affinity responsive targets stability (DARTS)
DARTS was conducted to identify the potential target of R-2HG. Briefly, 50×106 cells were lysed in M-PER (78501, Thermo Fisher Scientific) with protease inhibitor cocktail and phosphatase inhibitor cocktail. TNC buffer (50 mM Tris-HCL pH8.0, 50 mM NaCl, and 10 mM CaCl2) was added to the lysate and the protein concentration was determined by BCA assay. Cell lysates were incubated with varying concentration of R-2HG or PBS (vehicle) for 1 hour at room temperature and digested with Pronase (1:3000 for FTO) (10165921001, Roche) for 30 minutes at room temperature. The digestion was stopped by protease inhibitor cocktail and the samples were immediately placed on ice. Subsequently, Western blotting was used to determine whether FTO is a direct target of R-2HG. GAPDH was used as a negative control.
Cellular Thermal Shift Assay (CETSA)
CETSA was performed to determine the direct binding between R-2HG and FTO in cellular. Briefly, . 4×106 NOMO-1, U937 and MA9.3ITD cells were pretreated with 300 μM R-2HG for 12h before being subjected to the CETSA protocol. Cells were chilled on ice, washed with PBS plus protease inhibitor cocktail and then transferred into 200 μl PCR tubes. The cells were heat shocked in the Bio-Rad T100 Thermal Cycler at indicated temperature for 3 min to denature proteins, and immediately cooled down at room temperature for 3 min. Finally, all the samples were subjected to three freeze-thaw cycles with dry ice and Thermal Cycler to lyse cells, and centrifuged at 20,000 g for 20 min at 4°C to clarify and pellet cell debris with aggregated proteins. The supernatant was boiled with 4 × Laemmli Sample Buffer (#1610747, Bio-Rad) for Western blot. The bands were quantified using Gel-Pro analyzer software and plotted with three biological replicates.
Detection of FTO enzymatic activity in cell-free system by dot blot assays
The demethylation assay of FTO was essentially performed as previously described (Jia et al., 2011). Recombinant human FTO protein was purchased from Abcam (ab109039). Single-stranded RNA with m6A modification was synthesized by GE Health with the sequence 5′-AUUGUCA(m6A)CAGCAGC-3′. Demethylation activity assays were conducted in a 20 μl reaction mixture containing the indicated concentration of octyl ester modified R-2HG, 0.1 nmol ssRNA, 200 nM FTO, 283 μM of (NH4)2(SO4)2·6H2O (203505, Sigma-Aldrich), 75 μM of α-KG (K1128, Sigma-Aldrich), 2 mM of L-ascorbic acid (A0278, Sigma-Aldrich), 50 μg/ml of BSA (A2058, Sigma-Aldrich), and 50 mM of HEPES buffer, pH 7.0. The reactions were incubated at 37°C for 3 hours and quenched by the addition of 5 mM EDTA followed by inactivation of FTO enzyme for 5 min at 95ºC. The ssRNAs were precipitated with the addition of one-tenth volumes of 3 M sodium acetate (pH 5.2), glycogen (500 μg/ml, final concentration) and 2.5 volumes of 100% ethanol, and incubated at −80ºC overnight. The RNA pellet was resuspended in 10 μl of RNase-free water and directly subjected to m6A dot blot assay to detect m6A levels.
Detection of FTO demethylation activity in a cell-free system by liquid chromatography tandem-mass spectrometry (LC-MS/MS)
pET28a encoding His-tagged wild type human FTO was transformed into E. coli BL21 Star (DE3) and grown on LB-agar plates containing 30 μg/mL kanamycin. Overnight cultures were used to inoculate 1L of LB with 30 μg/mL kanamycin, which was grown to an OD600 of 1.0 before induction with 0.5 mM IPTG. The culture was then grown overnight at 15ºC prior to harvesting by centrifugation. Cell pellets were resuspended in 50 mM imidazole, 300 mM NaCl, and 50 mM sodium phosphate pH 8.0, and lysed by sonication. The lysate was clarified by centrifugation before purification on a HisTrap Ni-NTA column. Fractions containing His-tagged FTO were pooled and concentrated before further purification on a MonoQ column. Demethylation assays were set up in reaction mixtures containing 0.1 nmol of m6A probe (5′-CGUGG(m6A)CUGGCU-biotin-3′), 0.02 mg/mL FTO, 283 μM (NH4)2Fe(SO4)2·6H2O, 75 μM a-KG, 2 mM L-ascorbic acid, 50 μg/mL BSA, 50 mM HEPES pH 7.5, and varying doses of R-2HG or S-2HG. Reactions were incubated for 3 hours at 37°C, after which 5 mM EDTA was added and reactions were heated at 95°C for 5 mins. The reactions were then precipitated with sodium acetate, glycogen, and ethanol. Resuspended RNA samples were digested with Nuclease P1 (N8630, Sigma, 1 U in 25 mM NaCl, 2.5 mM ZnCl2, 2 hours, 42°C), followed by addition of 100 mM NH4HCO3 and 1 U alkaline phosphatase for another 2 hours digestion at 37°C. Samples were spun through 0.22 μm filters prior to measuring m6A levels by LC-MS/MS on a Sciex Triple Quad 6500 mass spectrometer equipped with an Agilent 1290 Infinity II LC system using a Zorbax Eclipse XDB-C18 column as previously described (Liu et al., 2014).
Quantitation of m6A and m6Am levels by LC-MS/MS
Levels of m6A and m6Am were measured by LC-MS/MS essentially as described previously (Dominissini et al., 2016). Total RNA was isolated from Trizol and then polyadenylated RNA was extracted using a GenElute mRNA miniprep kit (Sigma) or a Dynabeads mRNA DIRECT kit (Thermo Fisher). The mRNA was further purified with the Ribominus Eukaryote System v2 kit (Thermo Fisher). Purified mRNA was then cleaned up with RNA Clean and Concentrator columns (Zymo), using the manufacturer’s protocol that removes RNA species smaller than 200nt. For m6A quantitation, approximately 100–200 ng of RNA was digested with 1 unit of Nuclease P1 from Penicillium citrinum (Sigma, N8630) in 25 mM NaCl and 2.5 mM ZnCl2 for 2 hours at 37°C. 100 mM ammonium bicarbonate and 1 unit of alkaline phosphatase (Sigma, P5931) were then added to the reaction for another 2 hours incubation at 37°C. After digestion, 1 μL of 1.2 M HCl was added to the approximately 40 μL reaction to re-neutralize the pH.
To monitor changes in mRNA cap m6Am, purified mRNA was digested with RNA 5′ pyrophosphohydrolase (RppH; NEB, M0356S) prior to digestion with nuclease P1. mRNA was digested in 1 × NEB buffer 2 with 2 μL of RppH in a 30 μL volume for 2 hours at 37°C. 2 μL of fresh RppH diluted in 10 μL total volume of 1 × NEB buffer 2 was added to each reaction and incubated for another 2 hours at 37°C. After a column cleanup, the reaction was then digested with Nuclease P1 and alkaline phosphatase as described above. The sample was then filtered through a 0.22 μm syringe filter prior to injection on a C18 reverse phase column coupled to an Agilent 6410 triple-quadropole LC mass spectrometer in positive electrospray ionization mode. Quantitation of modifications was based on nucleoside-to-base ion transitions: 268-to-136 for A, 282-150 for m6A, and 296-150 for m6Am. Pure nucleosides were used to generate standard curves, from which the concentrations of A, m6A, and m6Am in the sample were calculated. The level of modification (m6A or m6Am) was then calculated as a percentage of unmodified A to adjust for differences in the amount of digested mRNA injected.
Procedure for synthesis of m6Am standard
The synthesis of m6Am free nucleoside is depicted in the scheme below. Commercially available 2′-O-methylinosine (1) was treated with acetic anhydride in pyridine to protect 3′ and 5′-hydroxyls to give intermediate 2 quantitatively. Treatment of 2 with (chloromethylene) dimethyliminium chloride in chloroform at 40°C for 3 hours converted 2 to 6-chloropurine derivative 3 in quantitative yield. Without further purification, 3 was treated methylamine in ethanol and water overnight at room temperature give product m6Am 4 as a pale solid in 88% yield.

Synthesis scheme of m6Am from 2′-OMe-inosine.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed with EpiTech ChIP OneDay Kit (334471, Qiagen) following the manufacturer’s protocol, with some modifications. Briefly, PBS- and R-2HG-treated NOMO-1 cells (about 5 × 106) were treated with fresh fixing buffer (1% formaldehyde) for 10 min at 37°C to crosslink DNA and proteins. The reaction was terminated by the addition of stop buffer and incubated at room temperature for 5 min. After cell lysis, the cross-linked chromatin was sonicated to an average size of ~5003bp with a Bioruptor Pico Sonication System and was immunoprecipitated with antibodies against 5hmC, H3K9me3, H3K36me3, and IgG. Purified ChIP DNA was amplified by real-time qPCR using specific primers targeting the 5hmC, H3K9me3, and H3K36me3 peaks on FTO.
Lentivirus preparation, precipitation, and infection
Lentivirus particles for pMIRNA1-FTO, pMIRNA1-FTO-Mut, pMIRNA1, pLKO.1-shFTO-1, pLKO.1-shYTHDF2, pLKO.1-shMYC, pLKO.1-shCEBPA, pCDH-MYC, pGFP-shFTO-2, pGFP-shNS (shNS-2), and pLKO.1-shNS, were packaged with pMD2.G, pMDLg/pRRE, and pRSV-Rev (Addgene). Briefly, 0.5 μg pMD2.G, 0.3 μg pMDLg/pRRE, 0.7 μg pRSV-Rev, and 1.5 μg construct for overexpression or knockdown of specific genes were co-transfected into HEK-293T cells in 60 mm cell culture dish with Effectene Transfection Reagent (301427, Qiagen). The pTRIPZ-IDH1R132H was packaged with psPAX2 and pMG2.G. The lentivirus particles were harvested at 48 and 72 hours and concentrated with PEG-it virus precipitation solution (LV810A-1, SBI). For infection, the lentiviruses were directly added into with cells with existence of 4ug/ml polybrene (H9268, Sigma-Aldrich) and then spinoculation was conducted at 32°C, 1000 rpm for 90 min. The infected cells were selected with GFP expression (for FTO, FTO-Mut, and pGFP-shFTO-2) or 1 μg/ml puromycin (for shFTO-1, shYTHDF2, shCEBPA, shMYC, pCDH-MYC, and IDH1R132H) (P8833, Sigma-Aldrich). After selection, 1 μg/ml Doxycycline (D9891, Sigma-Aldrich) was added to induce expression of IDH1R132H.
Nuclear run-on assay
To determine whether suppression of FTO induced by long-term R-2HG treatment is due to transcriptional regulation, we conducted nuclear run-on assay according to the published protocol with some modifications. Nuclei were isolated from NOMO-1 cells upon treatment with PBS or 300 μM R-2HG for 96 hours with Nonidet P-40 lysis buffer (10 mM Tris-Cl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.5% NP-40). The enriched nuclei were suspended in 396 μl nuclear freezing buffer (50 mM Tris-Cl, pH 8.3, 40% glycerol, 5 mM MgCl2, and 0.1 mM EDTA). Then 100 μl of 5× run-on buffer (25 mM Tris-Cl, pH 8.0, 750 mM KCl, 12.5 mM MgCl2, 1.25 mM each of ATP, GTP, and CTP) was added into the nuclei along with 4 μl biotin-16-UTP (11388908910, Sigma-Aldrich) to initiate nuclear run-on reaction. After incubation at 29°C for 30 min, the reaction was quenched by the addition of 0.75 μl 1M CaCl2, and 3 μl RNase-free DNase I following by incubation at 29°C for 10 min. RNA purification was performed with the RNeasy mini kit (74104, Qiagen) according to the manufacturer’s protocol. A small aliquot (5 μl from a total of 50 μl) was saved as “total nuclear RNA” for each sample. Dynabeads M-280 streptavidin (11205D, Thermo Fisher Scientific) were mixed with an equal volume of the isolated RNA samples at 42°C for 20 min and at room temperature for 2 hours. After washing with 2× SSC, the beads were resuspended in 25 μl of nuclease-free water. Reverse transcription and qPCR were performed and ACTB was selected as an internal control. The qPCR products were then applied on a 2% agarose gel analysis and photos were taken by Gel DocTM XR System (BIO-RAD).
RNA-seq and relative data analysis
RNA samples from R-2HG sensitive, resistant cell lines, and healthy controls were extracted with a mirVana miRNA Isolation Kit (Thermo Fisher Scientific) using the total RNA extraction protocol. NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs) was used for polyA RNA purification. Libraries were prepared by PrepX mRNA Library kit (WaferGen) combined Apollo 324 NGS automated library prep system. Libraries at a final concentration of 15 pM were clustered onto a single read (SR) flow cell using Illumina TruSeq SR Cluster kit v3, and sequenced to 50 bp using TruSeq SBS kit on Illumina HiSeq system. Differential gene expression was analyzed by standard Illumina sequence analysis pipeline. The data have been deposited in the GEO repository with the accession number GSE87187. RNA samples from R-2HG- or PBS-treated sensitive leukemia cells were also extracted and purified as described above. Libraries were prepared by NEBNext Ultra Directional RNA Library Prep Kit (New England BioLabs). The libraries were sequenced and analyzed following the same protocol as above. The data have been deposited in the GEO repository with the accession number GSE87189. Gene Set Enrichment Analysis (GSEA) was used to analyze the signal pathway enrichment in different groups of samples.
m6A-seq assays and data analysis
Total RNA was isolated with TRIZOL (15596-018, Life technology). Polyadenylated RNA was extracted using Sigma GenElute mRNA miniprep kit (Sigma). RNA fragmentation Reagents (Ambion) was used to randomly fragment RNA and m6A antibody (Synaptic Systems) was applied for m6A pull down (i.e., m6A IP). Both input and m6A IP samples were prepared for next-generation sequencing (NGS). The library preparation was constructed using the TruSeq Stranded mRNA Sample Prep Kit (Illumina) and was quantified by BioAnalyzer High Sensitivity DNA chip, and then was deeply sequenced on the Illumina HiSeq 2500. The data have been deposited in the GEO repository with the accession number GSE87190.
For data analysis, the following pipeline was used to identify m6A peaks. The reads from input and m6A IP samples were aligned to human reference genome GRCh38 using Tophat. Both the MACS2 (Zhang et al., 2008) callpeak function with parameter extsize 85, and exomePeak (Meng et al., 2014) with default settings, were used to call m6A peaks based on the .bam files generated by Tophat. To achieve high specificity, only the m6A peaks called by both MACS2 and exomePeak were retained for the further analysis. The m6A peaks were annotated using an ad hoc perl script. Sequence motifs enriched in m6A peak regions compared to control regions were identified using HOMER. The differentially methylated m6A peaks were also identified by MACS2 bdgdiff function and exomePeak, the peaks called by both MACS2 and exomePeak were retained. Circos and Integrative Genomics Viewer (IGV) were used to visualize the distributions of the m6A peaks. The RNA-seq reads were normalized using Cufflinks. Cuffdiff was used to calculate differentially expressed genes.
Gene-specific m6A qPCR
To assess the relative abundance of specific mRNA transcripts in m6A IP and input groups, qPCR was performed. m6A RNA immunoprecipitation (MeRIP) was performed with Magna MeRIP m6A kit (17-10499, Millipore) according to the manufacturer’s instructions. Reverse transcription and qPCR were performed with Qiagen’s RT kit and 2× SYBR green qPCR Master Mix. Cycle threshold (Ct) values were used to determine the relative enrichment of mRNA.
RNA stability assay
Actinomycin D (A9415, Sigma-Aldrich) was added to leukemia cells at 5 μg/ml to assess RNA stability. After incubation for indicated time points, the cells were collected and RNA samples were extracted for reverse transcription and qPCR. The mRNA degradation rate was estimated according to published protocols (Liu et al., 2014). mRNA transcription was inhibited with Actinomycin D and the degradation rate of RNA (Kdecay) was estimated by following equation:
C0 is the concentration of mRNA at time 0, t is the transcription inhibition time, and C is the mRNA concentration at the time t. Thus the Kdecay can be derived by the exponential decay fitting of C/C0 versus time t. The half-time (t1/2), which means C/C0=50%/100%=1/2, can be calculated by the following equation:
Rearrangement of the above equation leads to the mRNA half-life time value, t1/2= ln2/Kdecay.
Dual-luciferase reporter and mutagenesis assays and related gene-specific m6A qPCR
To determine whether FTO-induced expression of MYC is dependent on m6A modification and whether forced expression of CEBPA could promote transcriptional activity of FTO promoter, we performed dual-luciferase reporter and mutagenesis assays with pMIR-REPORT-MYC-CDS-WT (wild type CDS of MYC), pMIR-REPORT-MYC-CDS-Mut (mutant CDS of MYC, m6A was replaced by T in the m6A motifs), pGL3-Basic-MYC-5′UTR-WT (wild type 5′UTR of MYC), pGL3-Basic-MYC-5′UTR-Mut (mutant 5′UTR of MYC, m6A was replaced by T in the m6A motifs), pGL3-Basic-FTO promoter WT and pGL3-Basic-FTO promoter Mut (the CEBPA binding site was mutant). All the fragments inserted into pMIR-REPORT and pGL3 basic vectors were directly synthesized from IDT. All the plasmids, extracted with QIAGEN Plasmid Mini Kit (12125, Qiagen), were transfected into HEK-293T cells with pRL-TK (a control reporter vector) and pMIRNA1-FTO, or pMIRNA1-FTO-Mut or pCMV6-CEBPA. The relative luciferase activities were assessed with Dual-luciferase reporter assay system (E1910, Promega) at 48 hours. Each group was repeated in triplicate. Meanwhile, we also collected poly(A)+ mRNAs from these groups, conducted MeRIP with Magna MeRIP m6A kit (17-10499, Millipore), and performed gene-specific m6A qPCR to determine the m6A abundance on the transcripts.
Software and AML datasets
JASPAR software was used to predict the potential binding of transcription factors (TFs) on the FTO “core promoter” region, from −201 to −1 relative to the transcription start site (TSS). The four AML patient datasets, including TCGA set, GSE37642, GSE14468 set, and an in-house set (Li et al., 2013), were used to determine the correlation between the predicted TFs and FTO in expression. The TCGA set (n=177), GSE14468 set (n=518) and in-house set (n=87) were generated by RNA-seq, Affymetrix U133Plus2.0 arrays, and Affymetrix human exon arrays, respectively. GSE37642 set (n=562) were generated by Affymetrix U133Plus2.0 (n=140) and U133A and B (U133A+B) (n=422) arrays.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed with GrapPad Prism 6 and were presented as mean ± SEM or mean ± SD as indicated. Two-tailed Student’s t test was used to compare means between groups as indicated; p < 0.05 was considered significant. For Figures 1D, 1G, and 7G, the Kaplan-Meier survival curves were charted with GraphPad Prism 6 and the P values were calculated using the log rank test. For Western blot results, representative figures from three biological replicates were shown. Densitometry analysis of the bands from Western blot or dots from dot blot were performed with Gel-Pro analyzer and normalized to the loading controls.
DATA AND SOFTWARE AVAILABILITY
The RNA-Seq data obtained in this study has been uploaded to NCBI GEO datasets, under accession number GSE87187 and GSE87189. The m6A-Seq data reported in this paper also has been uploaded to NCBI GEO datasets under the accession number GSE87190.
Supplementary Material
Table S2. IDH1/2 mutation status in the 27 leukemia cell lines, Related to Figure 1
Table S4. Information of the AML patients used in the functional studies and/or qPCR assays, Related to Figures 1 and 7
(A and B) Analysis of the effect of R-2HG on the cell cycle via PI-staining in NOMO-1 (A, B), U937, and NB4 (B) cells.
(C and D) Analysis of the effect of R-2HG (300 μM) on the cell cycle via Hoechst/Pyronin Y staining in NOMO-1 (C, D), U937, and NB4 (D) cells.
(E and F) Effects of R-2HG (300 μM) on cell apoptosis in NOMO-1 (E, F), U937, and NB4 (F) cells. L, living cells; EA, early apoptotic cells; LA, late apoptotic cells.
(G) Analysis of intracellular R-2HG levels after treatment with PBS or 300 μM of R-2HG.
(H and I) Effects of R-2HG on cell proliferation (upper panel; cell density detected by MTT assays), viability (middle panel; detected by MTT assays) and growth (lower level; detected by cell number counts) of TF-1 cells cultured under standard culture condition (with 2 ng/mL GM-CSF) (H) or GM-CSF-poor conditions (0.1 ng/mL) (I).
(J and K) Functions of R-2HG on cell proliferation (upper panel), viability (middle panel) and growth (lower level) of SKNO-1 cells cultured under standard culture condition (with 10 ng/mL GM-CSF) (J) or GM-CSF-poor conditions (0.1 ng/mL) (K).
(L and M) Effects of R-2HG on colony-forming capacity (L) and cell viability (M) of leukemic blast cells isolated from primary AML patients.
(N) Effects of R-2HG (300 μM) on cell proliferation/viability in human primary AML samples with or without naturally occurring IDH1/2 mutations.
*, P<0.05; **, P<0.01; ***P<0.001; t-test. Error bars, mean ± SD (n=3). h, hour(s); d, day(s).
(A) qPCR analysis of expression patterns of the top 10 α-KG dependent enzymes (see Figure 2A) in 30 human leukemic samples (including the 27 cell lines shown in Figures 1A and 1B, and 3 primary leukemic samples), and in an expanded cohort of healthy control samples, including 16 normal mononuclear cell (MNC) samples isolated from peripheral blood or bone marrow of healthy donors, and 10 CD34+ and 12 CD34− MNC samples isolated from cord blood samples. Pearson correlation analysis of the correlation between expression levels of the individual α-KG dependent enzyme genes and the sensitivity of the 27 leukemia cell line samples to R-2HG treatment is also shown. The relative expression levels amongst different normal control sample groups and the leukemic sample group (upper panels), and Pearson correlation analysis (lower panels) of P4HB, LCP1, PHF2, PKM, PHF8, PLOD3, KDM2B, PHYH and ALKBH7 are shown. The result for FTO is displayed in Figure 2B.
(B) The expression changes of all the α-KG dependent/related dioxygenases (with expression values in all twelve samples) after 48 hour treatment with 300 μM R-2HG in NOMO-1 and MA9.3ITD cells.
(C) The core signaling pathways identified by RNA-seq. Based on the RNA-seq data from the samples shown in Figure 2A and in Figure 2C, GSEA identified 7 core enriched gene sets (or signaling pathways) from the following four groups of comparisons: resistant leukemia cells vs. sensitive leukemia cells; sensitive leukemia cells vs. healthy control cells; PBS-treated NOMO-1 vs. R-2HG-treated NOMO-1; and PBS-treated MA9.3ITD vs. R-2HG-treated MA9.3ITD. Among the 7 gene sets, MYC targets V1, MYC targets V2, G2M checkpoint and E2F targets were consistently enriched in resistant cells compared with sensitive cells. They were also enriched in sensitive cells compared with healthy controls, and notably suppressed by R-2HG treatment in both NOMO-1 and MA9.3ITD cells, whereas the other three genes sets including cholesterol homeostasis, inflammatory response, and TNFA signaling via NF-kB largely show the opposite pattern. ES, enrichment score. P < 0.001 and FDR < 0.05 were used as cut-off for statistic significance.
(D) Venn diagram displaying the core genes enriched amongst the four gene sets including ‘MYC targets V1’, ‘MYC targets V2’, ‘G2M checkpoint’ and ‘E2F targets’ shared by both ‘resistant vs. sensitive’ and ‘sensitive vs. healthy control’ comparisons.
(E) Heat map of the 146 shared, core enriched genes. They showed the highest abundance in R-2HG-resistant leukemia cells and the lowest abundance in healthy controls, with an intermediate level of abundance in R-2HG-sensitive leukemic cells.
(F) Venn diagram showing the core genes enriched amongst the aforementioned four gene sets shared by both ‘PBS-treated NOMO-1 vs. R-2HG-treated NOMO-1’ and ‘PBS-treated MA9.3ITD vs. R-2HG-treated MA9.3ITD’ comparisons.
(G) Heat map of the 185 shared core genes enriched, which were consistently and significantly suppressed by R-2HG both in NOMO-1 and MA9.3ITD cells.
(H) Relative expression of major component genes (including CCNA2, CDK2, CDK4, CDK6, DDX21, MCM2 and MCM7) of the MYC pathways in sensitive (NOMO-1) cells (left panel) or resistant (K562) cells (right panel) with or without R-2HG treatment, as detected by qPCR.
*, P<0.05; **, P<0.01; t-test. Error bars, mean ± SD (n≥3).
(A) Schematic illustration of enzymes mediating demethylation of DNA, RNA, and protein. All of these enzymes are Fe (II)/α-KG-dependent dioxygenases. TET family members (TET1/2/3) mediate demethylation of DNA by converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). ALKBH5 and FTO, members of the AlkB family, are responsible for the removal of m6A modification on mRNA. KDM (histone lysine demethylase) and JMJD (Jumonji Domain-containing histone demethylase) catalyze the demethylation of histones.
(B–D) Relative expression of the DNA demethylase genes (TET1/2/3; B), RNA demethylase genes (ALKBH5 and FTO; C), and histone demethylase genes (KDM2A, KDM4A, and JMJD6; D) in 30 leukemia cells and 38 healthy controls (upper panels), and Pearson correlation analysis of their expression and sensitivities to R-2HG in the 27 leukemia cell line samples (lower panels). See the legend of Figure S2A for sample information.
(E–H) The 5hmC levels (E,G) and histone methylation status (F,H) in R-2HG sensitive or resistant leukemic cells with or without R-2HG treatment. 5hmC levels were determined by dot blot. Methylene blue (MB) is shown as a loading control. β-actin was used as loading control.
(I) The Western blot results for FTO from CETSA with PBS- and R-2HG-treated AML cells.
(J) LC-MS/MS quantification of remaining m6A abundance in the presence of varying R-2HG concentrations.
(K) Confirmation of FTO overexpression and knockdown efficacy in NOMO-1 cells.
(L) Quantification of m6A levels in poly(A)+ RNA isolated from sensitive NOMO-1 cells upon FTO overexpression and knockdown.
(M) Effect of FTO overexpression or knockdown on cell proliferation in sensitive MA9.3ITD cells.
* P<0.05; **P<0.01; ***, P<0.001; t-test. Error bars, mean ± SEM (n≥3).
(A) Identification of m6A peaks by two algorithms. The layers from outer to inner represent the m6A peaks identified by MACS2, exomePeak, and the overlap from both algorithms, respectively.
(B) The density distribution of m6A peaks across the length of mRNA transcripts. Each region of 5′ untranslated region (5′UTR), coding region (CDS), and 3′ untranslated region (3′UTR) was split into 100 segments, and the percentage of m6A peaks that fall within each segment was determined.
(C) The proportion (upper panel) and enrichment (lower panel) of the m6A peak distribution in the 5′UTR, start codon, CDS, stop codon or 3′UTR region across the mRNA transcripts. The enrichment was calculated by the number of m6A peaks normalized by the length of the region.
(D) Distribution of the m6A motifs in exonic or intronic regions of protein-coding and non-coding genes, and in other regions.
(E) The m6A motifs detected by HOMER include the predominant consensus motif DRACH ([G/A/U] [G/A] m6AC [U/A/C]).
(F) Distribution of the m6A peaks with increased abundance (termed m6A-hyper peaks) upon R-2HG treatment across RNA regions. The abundance was calculated by the reads from m6A-seq.
(G) The pie chart shows the percentage of nucleotides mapped to the m6A peaks with increased (Hyper) or decreased (Hypo) abundance, upon R-2HG treatment, in 5′UTR, start codon, CDS, stop codon, 3′UTR, intron and non-coding RNA (ncRNA).
(H) The distribution and fold-change-adjusted density of the m6A-hyper peaks across different RNA regions. The density of m6A-hyper peaks was adjusted by (i.e., multiplied by) the increased fold changes in R-2HG-treated cells relative to the PBS-treated cells. 150 nt of 5′UTR was selected as a “cut-off” to represent the reads covering transcriptional start sites in mRNA, where the m6Am modification is potentially existing.
(I) The proportion of the m6A-hyper peaks in the 5′ end of the 5′ UTR (using 150 nt as cut-off) and the other regions, including the 5′UTR (behind the first 150 nt), CDS and 3′UTR.
(J) Fold change of the m6A-hyper peaks upon R-2HG treatment in different RNA regions as indicated. The values are log(2) transformed.
(K) Correlation analysis between changed m6A reads in the 5′ end of 5′UTR (150 nt as cut-off) and corresponding gene expression. The results show that increased m6A reads in the 5′ end of 5′UTR are not positively correlated with gene expression, indicating that our transcriptome-wide mapping of m6A peaks likely contains few misannotated m6Am modifications.
(L) Fold changes of mRNA with m6Am, Am, Cm, Gm, and Um adjacent to the cap in leukemia cells upon R-2HG-induced FTO inhibition or FTO overexpression. Each box shows the first quartile, median and third quartile; while whiskers represent 5–95 percentile. For “R-2HG vs PBS” NOMO-1, n=1,542 (m6Am); 1,247 (Am); 2,475 (Cm); 1,798 (Gm); and 2,383 (Um); For “R-2HG vs PBS” MA9.3ITD, n=1,528 (m6Am); 1,178 (Am); 2,365 (Cm); 1,700 (Gm); and 2,276 (Um); For “FTO vs Ctrl” MA9.3RAS, n=1,477 (m6Am); 939 (Am); 1,826(Cm); 1,342 (Gm); and 1,875 (Um). ns, non-significant; *P<0.05; **P<0.01; ***P<0.001, unpaired Student’s t-test.
(M) Effect of R-2HG on MYC expression in sensitive and resistant cells.
(N) CLIP-seq data (GSE49339) demonstrates binding of YTHDF2 on MYC mRNA.
(O) Confirmation of YTHDF2 knockdown efficacy and its effect on MYC expression in sensitive NOMO-1 and resistant K562 cells.
(P) Effect of FTO overexpression or knockdown on MYC expression. Forced expression of wild-type FTO increased MYC expression compared with mutant FTO or control group, and FTO knockdown decreased MYC expression in sensitive (MA9.3ITD) leukemia cells.
(Q and R) R-2HG treatment increases ASB2 (Q) and RARA (R) expression in sensitive cells, but not in resistant cells.
**, P<0.01; ***, P<0.001; t-test. Error bars, mean ± SD (n=3).
(A) m6A abundance on FTO mRNA as measured by m6A-seq in NOMO-1 cells.
(B) Effects of R-2HG treatment or YTHDF2 knockdown on FTO mRNA stability.
(C–E) Genome browser views of the potential 5hmC (C), H3K9me3 (D) and H3K36me3 (E) peaks across the FTO genomic locus in human AML samples.
(F–H) Verification of the 5hmC (F), H3K9me3 (G), and H3K36me3 (H) modification via ChIP-qPCR with PBS- and R-2HG-treated NOMO-1 sensitive cells. For each group, we designed three different primers near the relative peaks shown in the upper panels (i.e., Figures S5C–S5E).
(I) Correlation analysis of the transcription factor genes and FTO in expression in four different human AML datasets. CEBPA, MZF1, NFATC3, and RFX3 display significant positive correlation with FTO expression (P<0.05) in all the four datasets, while NFKB2 and TFEB exhibit negative correlation (P<0.05) in all the four datasets.
(J) Relative expression levels of CEBPA in R-2HG sensitive and resistant leukemic cells. Its expression level in HEL cells was set to 1.
(A) Effects of doxycycline (Dox)-induced IDH1R132H on FTO and MYC levels in sensitive (NOMO-1 and MA9.3ITD) and resistant (K562) cells. The results are derived from quantification of Western blot data in three independent experiments.
(B–D) The effects of Dox-induced IDH1R132H on cell cycle (B), cell proliferation (C) and apoptosis (D) in sensitive NOMO-1 and MA9.3ITD cells, as well as in resistant K562 cells.
(E and F) S-2HG displays similar functions on cell proliferation (E) and viability (F) to R-2HG in leukemia cell lines. Consistent with the methods to determine R-2HG’s effects on cell proliferation/viability (see Table S3), several R-2HG sensitive cells (NOMO-1, U937, MA9.3ITD, ML-2, and MONOMAC-6) and R-2HG resistant cells (SKNO-1, KASUMI-1, and K562) were chosen and exposed to 20, 100, and 300 μM cell-permeable S-2HG for indicated time points..
(G) Determination of remaining m6A levels in the presence of various S-2HG concentrations via LC-MS/MS.
ns, non-significant; *P<0.05; **P<0.01; ***P<0.001; unpaired Student’s t-test; Error bars, mean ± SD (n=3).
(A) The 5 core gene sets shared by four group comparisons, including ‘IDH mutant vs. IDH WT’ (i.e., AML patients with IDH mutations vs. AML patients without IDH mutations), ‘IDH mutant (NK) vs. IDH WT (NK)’ (i.e., normal karyotype (NK) AML patients with IDH mutations vs. NK-AML patients without IDH mutations), ‘resistant vs. sensitive’ (i.e., R-2HG resistant leukemic cell lines vs. R-2HG sensitive leukemic cell lines; see the samples in Figure 2A), and ‘sensitive vs. healthy control’ (i.e., R-2HG-sensitive leukemic cell lines vs. healthy control samples; see the samples in Figure 2A). MYC targets V1, MYC targets V2, G2M check point, and E2F targets are modestly activated in AML patients without IDH mutations and R-2HG sensitive cells, but hyper-activated both in AML patients with IDH mutations and R-2HG resistant cells, which may result in the tolerance to R-2HG-induced inhibition of cell proliferation.
(B) Effects of JQ1-induced inhibition of MYC signaling on altering sensitivity to R-2HG or IDH1 mutation in resistant K562 and NB4 cells.
(C) The synergistic effects between R-2HG and clinical chemotherapeutic drugs, including ATRA, Daunorubicin, AZA, and Decitabine, on inhibiting leukemic cell viability (using MONOMAC-6 as a representative). The cell viability was determined 48 hours post-treatment.
(D) R-2HG treatment or chemotherapeutic treatment, especially their combinations, decrease MYC levels in leukemic cells (using MONOMAC-6 as a representative).
(E) Wright-Giemsa staining of peripheral blood smears from NOMO-1 mouse models with the six different chemotherapeutic regimens, including only with PBS, PBS + daunorubicin (PBS+Dauno), PBS + decitabine (PBS+DAC), R-2HG + PBS (R-2HG), R-2HG + daunorubicin (R-2HG+Dauno) and R-2HG + decitabine (R-2HG+DAC). Arrows indicate the immature leukemic cells in each group. Black bar represents 20 μm.
(F) Effects of R-2HG on cell proliferation/growth in brain tumor (GBM) cells without IDH1/2 mutations. Cells were seeded in a 96-well plate at 5,000–10,000 cells/well and proliferation was assessed by MTT assay.
(G) R-2HG inhibits the cell viability of the GBM cells in a dose-dependent manner. The relative cell viabilities of the 8 glioma cell lines were detected by MTT 96 hours post-treatment with 20 μM, 100 μM, or 300 μM R-2HG.
*, P<0.05; **, P<0.01; ***, P<0.001; t-test. Error bars, mean ± SD (n=3).
HIGHLIGHTS.
R-2HG exhibits broad and variable anti-proliferation effects in leukemia and glioma
R-2HG increases global m6A RNA modification in the sensitive cells via targeting FTO
The R-2HG⊣FTO⊣m6A axis regulates MYC/CEBPA expression and downstream pathways
The FTO/MYC homeostasis controls the response/sensitivity of leukemia cells to R-2HG
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH) R01 Grants CA214965 (J.C.), CA211614 (J.C.), CA178454 (J.C.), CA182528 (J.C.), RM1 HG008935 (C.H.), GM071440 (C.H.), R50 CA211404 (M.W.), and Foundation of Innovation Team for Basic and Clinical Research of Zhejiang Province (Grant 2011R50015) (J.J.). J.C. is a Leukemia & Lymphoma Society (LLS) Scholar. C.H. is an investigator of the Howard Hughes Medical Institute (HHMI). S.N. is an HHMI Fellow of the Damon Runyon Cancer Research Foundation (DRG-2215-15). A.T.S. is supported by two NIH grants (R01NS089815 and R21NS100077). We (J.C. and R. S.) have a patent application filed based on the discoveries presented. C.H. is a scientific founder of the Accent Therapeutics, Inc.
Footnotes
Supplemental Information includes seven figures and seven tables and can be found with this article online.
AUTHOR CONTRIBUTIONS
Conceptualization, R.S. and J.C.; Supervision, R.S., C.H., and J.C.; Formal Analysis, L.D.; Investigation, R.S., C.L., S.N., M.W., Y.Q., X.D., X.W., Y.W., C.H., M.Y., J.S., Z.J.,Q.D., D.Z., T.W., K.Y., H.W., H.H., K.F., X.Q., and J.C.; Resources, B.Z., J.Q., M.W., A.T.S.,D.P., J.E.B., M.W., G.M., X.J., and J.M.; Funding Acquisition, S.N., M.W., A.T.S., J.J., C.H., and J.C.; Writing-Original Draft, Writing-Review & Editing, R.S., L.D., S.N., C.H., and J.C.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boutzen H, Saland E, Larrue C, de Toni F, Gales L, Castelli FA, Cathebas M, Zaghdoudi S, Stuani L, Kaoma T, et al. Isocitrate dehydrogenase 1 mutations prime the all-trans retinoic acid myeloid differentiation pathway in acute myeloid leukemia. The Journal of experimental medicine. 2016;213:483–497. doi: 10.1084/jem.20150736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bralten LB, Kloosterhof NK, Balvers R, Sacchetti A, Lapre L, Lamfers M, Leenstra S, de Jonge H, Kros JM, Jansen EE, et al. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Annals of neurology. 2011;69:455–463. doi: 10.1002/ana.22390. [DOI] [PubMed] [Google Scholar]
- Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WC, Lei WC, Ko BS, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Wu SJ, Huang SY, et al. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia. 2011;25:246–253. doi: 10.1038/leu.2010.267. [DOI] [PubMed] [Google Scholar]
- Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO reports. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530:441–446. doi: 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med. 2015;372:2499–2508. doi: 10.1056/NEJMoa1407279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkashef SM, Lin AP, Myers J, Sill H, Jiang D, Dahia PLM, Aguiar RCT. IDH Mutation, Competitive Inhibition of FTO, and RNA Methylation. Cancer cell. 2017;31:619–620. doi: 10.1016/j.ccell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emadi A, Faramand R, Carter-Cooper B, Tolu S, Ford LA, Lapidus RG, Wetzler M, Wang ES, Etemadi A, Griffiths EA. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am J Hematol. 2015;90:E77–79. doi: 10.1002/ajh.23965. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Chin RM, Vergnes L, Hwang H, Deng G, Xing Y, Pai MY, Li S, Ta L, Fazlollahi F, et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 2015;22:508–515. doi: 10.1016/j.cmet.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge R. Preparation of RNA dot-blots. Methods in molecular biology. 1998;86:73–75. doi: 10.1385/0-89603-494-1:73. [DOI] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science translational medicine. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ, Laird PW, Baty JD, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Herold T, He C, Valk PJ, Chen P, Jurinovic V, Mansmann U, Radmacher MD, Maharry KS, Sun M, et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:1172–1181. doi: 10.1200/JCO.2012.44.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31:127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. The New England journal of medicine. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature. 2017;541:371–375. doi: 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Lu Z, Liu H, Zhang L, Zhang S, Chen Y, Rao MK, Huang Y. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods. 2014;69:274–281. doi: 10.1016/j.ymeth.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson E, Hasemann MS, Willer A, Lauridsen FK, Rapin N, Jendholm J, Porse BT. Initiation of MLL-rearranged AML is dependent on C/EBPalpha. J Exp Med. 2014;211:5–13. doi: 10.1084/jem.20130932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. The New England journal of medicine. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Wang JB, Dong DF, Wang MD, Gao K. IDH1 overexpression induced chemotherapy resistance and IDH1 mutation enhanced chemotherapy sensitivity in Glioma cells in vitro and in vivo. Asian Pac J Cancer Prev. 2014a;15:427–432. doi: 10.7314/apjcp.2014.15.1.427. [DOI] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014b;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M, Brooks RA, Panchal R, Rhyasen GW, Danet-Desnoyers G, Mulloy JC. OKT3 prevents xenogeneic GVHD and allows reliable xenograft initiation from unfractionated human hematopoietic tissues. Blood. 2014;123:e134–144. doi: 10.1182/blood-2014-02-556340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, Mulloy JC. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24:1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M, Mizukawa B, Chou FS, Sexton C, Shrestha M, Saunthararajah Y, Mulloy JC. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121:e90–97. doi: 10.1182/blood-2012-10-464677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Zhang H, Yang H, Koche R, Staber PB, Cusan M, Levantini E, Welner RS, Bach CS, Zhang J, et al. Hematopoietic Differentiation Is Required for Initiation of Acute Myeloid Leukemia. Cell Stem Cell. 2015;17:611–623. doi: 10.1016/j.stem.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. IDH1/2 mutation status in the 27 leukemia cell lines, Related to Figure 1
Table S4. Information of the AML patients used in the functional studies and/or qPCR assays, Related to Figures 1 and 7
(A and B) Analysis of the effect of R-2HG on the cell cycle via PI-staining in NOMO-1 (A, B), U937, and NB4 (B) cells.
(C and D) Analysis of the effect of R-2HG (300 μM) on the cell cycle via Hoechst/Pyronin Y staining in NOMO-1 (C, D), U937, and NB4 (D) cells.
(E and F) Effects of R-2HG (300 μM) on cell apoptosis in NOMO-1 (E, F), U937, and NB4 (F) cells. L, living cells; EA, early apoptotic cells; LA, late apoptotic cells.
(G) Analysis of intracellular R-2HG levels after treatment with PBS or 300 μM of R-2HG.
(H and I) Effects of R-2HG on cell proliferation (upper panel; cell density detected by MTT assays), viability (middle panel; detected by MTT assays) and growth (lower level; detected by cell number counts) of TF-1 cells cultured under standard culture condition (with 2 ng/mL GM-CSF) (H) or GM-CSF-poor conditions (0.1 ng/mL) (I).
(J and K) Functions of R-2HG on cell proliferation (upper panel), viability (middle panel) and growth (lower level) of SKNO-1 cells cultured under standard culture condition (with 10 ng/mL GM-CSF) (J) or GM-CSF-poor conditions (0.1 ng/mL) (K).
(L and M) Effects of R-2HG on colony-forming capacity (L) and cell viability (M) of leukemic blast cells isolated from primary AML patients.
(N) Effects of R-2HG (300 μM) on cell proliferation/viability in human primary AML samples with or without naturally occurring IDH1/2 mutations.
*, P<0.05; **, P<0.01; ***P<0.001; t-test. Error bars, mean ± SD (n=3). h, hour(s); d, day(s).
(A) qPCR analysis of expression patterns of the top 10 α-KG dependent enzymes (see Figure 2A) in 30 human leukemic samples (including the 27 cell lines shown in Figures 1A and 1B, and 3 primary leukemic samples), and in an expanded cohort of healthy control samples, including 16 normal mononuclear cell (MNC) samples isolated from peripheral blood or bone marrow of healthy donors, and 10 CD34+ and 12 CD34− MNC samples isolated from cord blood samples. Pearson correlation analysis of the correlation between expression levels of the individual α-KG dependent enzyme genes and the sensitivity of the 27 leukemia cell line samples to R-2HG treatment is also shown. The relative expression levels amongst different normal control sample groups and the leukemic sample group (upper panels), and Pearson correlation analysis (lower panels) of P4HB, LCP1, PHF2, PKM, PHF8, PLOD3, KDM2B, PHYH and ALKBH7 are shown. The result for FTO is displayed in Figure 2B.
(B) The expression changes of all the α-KG dependent/related dioxygenases (with expression values in all twelve samples) after 48 hour treatment with 300 μM R-2HG in NOMO-1 and MA9.3ITD cells.
(C) The core signaling pathways identified by RNA-seq. Based on the RNA-seq data from the samples shown in Figure 2A and in Figure 2C, GSEA identified 7 core enriched gene sets (or signaling pathways) from the following four groups of comparisons: resistant leukemia cells vs. sensitive leukemia cells; sensitive leukemia cells vs. healthy control cells; PBS-treated NOMO-1 vs. R-2HG-treated NOMO-1; and PBS-treated MA9.3ITD vs. R-2HG-treated MA9.3ITD. Among the 7 gene sets, MYC targets V1, MYC targets V2, G2M checkpoint and E2F targets were consistently enriched in resistant cells compared with sensitive cells. They were also enriched in sensitive cells compared with healthy controls, and notably suppressed by R-2HG treatment in both NOMO-1 and MA9.3ITD cells, whereas the other three genes sets including cholesterol homeostasis, inflammatory response, and TNFA signaling via NF-kB largely show the opposite pattern. ES, enrichment score. P < 0.001 and FDR < 0.05 were used as cut-off for statistic significance.
(D) Venn diagram displaying the core genes enriched amongst the four gene sets including ‘MYC targets V1’, ‘MYC targets V2’, ‘G2M checkpoint’ and ‘E2F targets’ shared by both ‘resistant vs. sensitive’ and ‘sensitive vs. healthy control’ comparisons.
(E) Heat map of the 146 shared, core enriched genes. They showed the highest abundance in R-2HG-resistant leukemia cells and the lowest abundance in healthy controls, with an intermediate level of abundance in R-2HG-sensitive leukemic cells.
(F) Venn diagram showing the core genes enriched amongst the aforementioned four gene sets shared by both ‘PBS-treated NOMO-1 vs. R-2HG-treated NOMO-1’ and ‘PBS-treated MA9.3ITD vs. R-2HG-treated MA9.3ITD’ comparisons.
(G) Heat map of the 185 shared core genes enriched, which were consistently and significantly suppressed by R-2HG both in NOMO-1 and MA9.3ITD cells.
(H) Relative expression of major component genes (including CCNA2, CDK2, CDK4, CDK6, DDX21, MCM2 and MCM7) of the MYC pathways in sensitive (NOMO-1) cells (left panel) or resistant (K562) cells (right panel) with or without R-2HG treatment, as detected by qPCR.
*, P<0.05; **, P<0.01; t-test. Error bars, mean ± SD (n≥3).
(A) Schematic illustration of enzymes mediating demethylation of DNA, RNA, and protein. All of these enzymes are Fe (II)/α-KG-dependent dioxygenases. TET family members (TET1/2/3) mediate demethylation of DNA by converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). ALKBH5 and FTO, members of the AlkB family, are responsible for the removal of m6A modification on mRNA. KDM (histone lysine demethylase) and JMJD (Jumonji Domain-containing histone demethylase) catalyze the demethylation of histones.
(B–D) Relative expression of the DNA demethylase genes (TET1/2/3; B), RNA demethylase genes (ALKBH5 and FTO; C), and histone demethylase genes (KDM2A, KDM4A, and JMJD6; D) in 30 leukemia cells and 38 healthy controls (upper panels), and Pearson correlation analysis of their expression and sensitivities to R-2HG in the 27 leukemia cell line samples (lower panels). See the legend of Figure S2A for sample information.
(E–H) The 5hmC levels (E,G) and histone methylation status (F,H) in R-2HG sensitive or resistant leukemic cells with or without R-2HG treatment. 5hmC levels were determined by dot blot. Methylene blue (MB) is shown as a loading control. β-actin was used as loading control.
(I) The Western blot results for FTO from CETSA with PBS- and R-2HG-treated AML cells.
(J) LC-MS/MS quantification of remaining m6A abundance in the presence of varying R-2HG concentrations.
(K) Confirmation of FTO overexpression and knockdown efficacy in NOMO-1 cells.
(L) Quantification of m6A levels in poly(A)+ RNA isolated from sensitive NOMO-1 cells upon FTO overexpression and knockdown.
(M) Effect of FTO overexpression or knockdown on cell proliferation in sensitive MA9.3ITD cells.
* P<0.05; **P<0.01; ***, P<0.001; t-test. Error bars, mean ± SEM (n≥3).
(A) Identification of m6A peaks by two algorithms. The layers from outer to inner represent the m6A peaks identified by MACS2, exomePeak, and the overlap from both algorithms, respectively.
(B) The density distribution of m6A peaks across the length of mRNA transcripts. Each region of 5′ untranslated region (5′UTR), coding region (CDS), and 3′ untranslated region (3′UTR) was split into 100 segments, and the percentage of m6A peaks that fall within each segment was determined.
(C) The proportion (upper panel) and enrichment (lower panel) of the m6A peak distribution in the 5′UTR, start codon, CDS, stop codon or 3′UTR region across the mRNA transcripts. The enrichment was calculated by the number of m6A peaks normalized by the length of the region.
(D) Distribution of the m6A motifs in exonic or intronic regions of protein-coding and non-coding genes, and in other regions.
(E) The m6A motifs detected by HOMER include the predominant consensus motif DRACH ([G/A/U] [G/A] m6AC [U/A/C]).
(F) Distribution of the m6A peaks with increased abundance (termed m6A-hyper peaks) upon R-2HG treatment across RNA regions. The abundance was calculated by the reads from m6A-seq.
(G) The pie chart shows the percentage of nucleotides mapped to the m6A peaks with increased (Hyper) or decreased (Hypo) abundance, upon R-2HG treatment, in 5′UTR, start codon, CDS, stop codon, 3′UTR, intron and non-coding RNA (ncRNA).
(H) The distribution and fold-change-adjusted density of the m6A-hyper peaks across different RNA regions. The density of m6A-hyper peaks was adjusted by (i.e., multiplied by) the increased fold changes in R-2HG-treated cells relative to the PBS-treated cells. 150 nt of 5′UTR was selected as a “cut-off” to represent the reads covering transcriptional start sites in mRNA, where the m6Am modification is potentially existing.
(I) The proportion of the m6A-hyper peaks in the 5′ end of the 5′ UTR (using 150 nt as cut-off) and the other regions, including the 5′UTR (behind the first 150 nt), CDS and 3′UTR.
(J) Fold change of the m6A-hyper peaks upon R-2HG treatment in different RNA regions as indicated. The values are log(2) transformed.
(K) Correlation analysis between changed m6A reads in the 5′ end of 5′UTR (150 nt as cut-off) and corresponding gene expression. The results show that increased m6A reads in the 5′ end of 5′UTR are not positively correlated with gene expression, indicating that our transcriptome-wide mapping of m6A peaks likely contains few misannotated m6Am modifications.
(L) Fold changes of mRNA with m6Am, Am, Cm, Gm, and Um adjacent to the cap in leukemia cells upon R-2HG-induced FTO inhibition or FTO overexpression. Each box shows the first quartile, median and third quartile; while whiskers represent 5–95 percentile. For “R-2HG vs PBS” NOMO-1, n=1,542 (m6Am); 1,247 (Am); 2,475 (Cm); 1,798 (Gm); and 2,383 (Um); For “R-2HG vs PBS” MA9.3ITD, n=1,528 (m6Am); 1,178 (Am); 2,365 (Cm); 1,700 (Gm); and 2,276 (Um); For “FTO vs Ctrl” MA9.3RAS, n=1,477 (m6Am); 939 (Am); 1,826(Cm); 1,342 (Gm); and 1,875 (Um). ns, non-significant; *P<0.05; **P<0.01; ***P<0.001, unpaired Student’s t-test.
(M) Effect of R-2HG on MYC expression in sensitive and resistant cells.
(N) CLIP-seq data (GSE49339) demonstrates binding of YTHDF2 on MYC mRNA.
(O) Confirmation of YTHDF2 knockdown efficacy and its effect on MYC expression in sensitive NOMO-1 and resistant K562 cells.
(P) Effect of FTO overexpression or knockdown on MYC expression. Forced expression of wild-type FTO increased MYC expression compared with mutant FTO or control group, and FTO knockdown decreased MYC expression in sensitive (MA9.3ITD) leukemia cells.
(Q and R) R-2HG treatment increases ASB2 (Q) and RARA (R) expression in sensitive cells, but not in resistant cells.
**, P<0.01; ***, P<0.001; t-test. Error bars, mean ± SD (n=3).
(A) m6A abundance on FTO mRNA as measured by m6A-seq in NOMO-1 cells.
(B) Effects of R-2HG treatment or YTHDF2 knockdown on FTO mRNA stability.
(C–E) Genome browser views of the potential 5hmC (C), H3K9me3 (D) and H3K36me3 (E) peaks across the FTO genomic locus in human AML samples.
(F–H) Verification of the 5hmC (F), H3K9me3 (G), and H3K36me3 (H) modification via ChIP-qPCR with PBS- and R-2HG-treated NOMO-1 sensitive cells. For each group, we designed three different primers near the relative peaks shown in the upper panels (i.e., Figures S5C–S5E).
(I) Correlation analysis of the transcription factor genes and FTO in expression in four different human AML datasets. CEBPA, MZF1, NFATC3, and RFX3 display significant positive correlation with FTO expression (P<0.05) in all the four datasets, while NFKB2 and TFEB exhibit negative correlation (P<0.05) in all the four datasets.
(J) Relative expression levels of CEBPA in R-2HG sensitive and resistant leukemic cells. Its expression level in HEL cells was set to 1.
(A) Effects of doxycycline (Dox)-induced IDH1R132H on FTO and MYC levels in sensitive (NOMO-1 and MA9.3ITD) and resistant (K562) cells. The results are derived from quantification of Western blot data in three independent experiments.
(B–D) The effects of Dox-induced IDH1R132H on cell cycle (B), cell proliferation (C) and apoptosis (D) in sensitive NOMO-1 and MA9.3ITD cells, as well as in resistant K562 cells.
(E and F) S-2HG displays similar functions on cell proliferation (E) and viability (F) to R-2HG in leukemia cell lines. Consistent with the methods to determine R-2HG’s effects on cell proliferation/viability (see Table S3), several R-2HG sensitive cells (NOMO-1, U937, MA9.3ITD, ML-2, and MONOMAC-6) and R-2HG resistant cells (SKNO-1, KASUMI-1, and K562) were chosen and exposed to 20, 100, and 300 μM cell-permeable S-2HG for indicated time points..
(G) Determination of remaining m6A levels in the presence of various S-2HG concentrations via LC-MS/MS.
ns, non-significant; *P<0.05; **P<0.01; ***P<0.001; unpaired Student’s t-test; Error bars, mean ± SD (n=3).
(A) The 5 core gene sets shared by four group comparisons, including ‘IDH mutant vs. IDH WT’ (i.e., AML patients with IDH mutations vs. AML patients without IDH mutations), ‘IDH mutant (NK) vs. IDH WT (NK)’ (i.e., normal karyotype (NK) AML patients with IDH mutations vs. NK-AML patients without IDH mutations), ‘resistant vs. sensitive’ (i.e., R-2HG resistant leukemic cell lines vs. R-2HG sensitive leukemic cell lines; see the samples in Figure 2A), and ‘sensitive vs. healthy control’ (i.e., R-2HG-sensitive leukemic cell lines vs. healthy control samples; see the samples in Figure 2A). MYC targets V1, MYC targets V2, G2M check point, and E2F targets are modestly activated in AML patients without IDH mutations and R-2HG sensitive cells, but hyper-activated both in AML patients with IDH mutations and R-2HG resistant cells, which may result in the tolerance to R-2HG-induced inhibition of cell proliferation.
(B) Effects of JQ1-induced inhibition of MYC signaling on altering sensitivity to R-2HG or IDH1 mutation in resistant K562 and NB4 cells.
(C) The synergistic effects between R-2HG and clinical chemotherapeutic drugs, including ATRA, Daunorubicin, AZA, and Decitabine, on inhibiting leukemic cell viability (using MONOMAC-6 as a representative). The cell viability was determined 48 hours post-treatment.
(D) R-2HG treatment or chemotherapeutic treatment, especially their combinations, decrease MYC levels in leukemic cells (using MONOMAC-6 as a representative).
(E) Wright-Giemsa staining of peripheral blood smears from NOMO-1 mouse models with the six different chemotherapeutic regimens, including only with PBS, PBS + daunorubicin (PBS+Dauno), PBS + decitabine (PBS+DAC), R-2HG + PBS (R-2HG), R-2HG + daunorubicin (R-2HG+Dauno) and R-2HG + decitabine (R-2HG+DAC). Arrows indicate the immature leukemic cells in each group. Black bar represents 20 μm.
(F) Effects of R-2HG on cell proliferation/growth in brain tumor (GBM) cells without IDH1/2 mutations. Cells were seeded in a 96-well plate at 5,000–10,000 cells/well and proliferation was assessed by MTT assay.
(G) R-2HG inhibits the cell viability of the GBM cells in a dose-dependent manner. The relative cell viabilities of the 8 glioma cell lines were detected by MTT 96 hours post-treatment with 20 μM, 100 μM, or 300 μM R-2HG.
*, P<0.05; **, P<0.01; ***, P<0.001; t-test. Error bars, mean ± SD (n=3).