

Abstract/summary
An immune response consists of a finely orchestrated interplay between initial recognition of potential microbial threats by the innate immune system and subsequent licensed adaptive immune neutralization. The initial recognition integrates environmental cues derived from pathogen associated molecular patterns (PAMPs) and cell intrinsic damage associated molecular patterns (DAMPs) to contextualize the insult and inform a tailored adaptive response via T and B lymphocytes. While there is much data to support the role of transcriptional responses downstream of pattern recognition receptors (PRRs) in informing the adaptive immune response, markedly less attention has been paid to the role of post-translational responses to PAMP and DAMP recognition by the innate immune system, and how this may influence adaptive immunity. A well characterized post-translational consequence of PRR signaling is the assembly of a multimeric signaling platform, termed the inflammasome, by members of the nucleotide-binding oligomerization domain (Nod), leucine-rich repeat-containing receptors (NLRs) and pyrin and HIN domain (PYHIN) families. Inflammasomes assemble in response to cytosolic perturbations, such as mitochondrial dysfunction and aberrant ion fluxes in the case of the canonical NLRP3 inflammasome or the presence of bacterial lipopolysaccharides in the case of the non-canonical inflammasome. Assembly of the inflammasome allows for the cleavage and activation of inflammatory caspases. These activated inflammatory caspases in turn cleave pro-form inflammatory cytokines into their mature bioactive species and lead to unconventional protein secretion and lytic cell death. In this review, we discuss evidence for inflammasome-mediated instruction and contextualization of infectious and sterile agents to the adaptive immune system.
Graphical Abstract
Introduction
An immune response to an infectious insult begins with sensing of conserved molecular patterns derived from pathogens and host tissue damage. Pathogen associated molecular patterns (PAMPs) consist of conserved microbial ligands, such as cell wall components and nucleic acids, which imbue the host with the ability to detect diverse microbes using a subset of germline-encoded pattern recognition receptors (PRRs) [1,2]. In addition to recognition of microbial products, PRRs can detect structurally related chemical moieties from stressed and damaged host cells. These endogenous PRR ligands are known as damage associated molecular patterns (DAMPs). DAMPs serve as a contextual signal for cellular death and tissue damage [2]. Recognition of PAMPs and DAMPs leads to the induction of pro-inflammatory and anti-microbial programs that activate additional innate immune cells, as well as contextualize the adaptive immune response depending on cytokine milieu [3]. In addition to cytokine and chemokine production, professional antigen presenting cells (APCs), such as dendritic cells (DCs), mature in response to recognition of microbial products [4]. Hallmarks of DC maturation include the upregulation of several activities important for the initiation of T cell-mediated adaptive immune responses [4]. These activities can be grouped into three categories. The first category is upregulation of lysosomal activity, which results in the efficient degradation and loading of microbe-derived peptides onto major histocompatibility complex (MHC) molecules for presentation of antigen to T cells [5,6]. The second category is the increase in cell surface expression of co-stimulatory molecules, such as CD80 and CD86 (originally known as B7-1 and B7-2 respectively), which are required for appropriate T cell activation [7]. The third category is the upregulation and release of cytokines, such as interleukin 12 (IL-12), that can directly act on adaptive immune cells to inform an immune response [8]. These collective activities are necessary to differentiate naïve T cells into their effector and memory cell counterparts [4].
The mechanisms by which PRRs induce cytokine and co-stimulatory molecule expression are best-defined from studies of Toll-like Receptors (TLRs), whose signaling functions have been reviewed expertly in recent years [1,2]. In brief, activation of TLRs upon microbial detection leads to the formation of a large cytoplasmic supramolecular organizing center (SMOC) called the myddosome, whose principal effector function is to promote the activation of pro-inflammatory transcription factors of the NF-κB and AP-1 family [1,9]. These transcription factors function to induce numerous genes important for inflammation and adaptive immunity. Most cytokines that are important for T cell activation are regulated by TLRs, and the expression of these genes is coupled tightly to the secretion of the bioactive cytokine that they encode [2]. However, one family of cytokines differs from this paradigm of coupled cytokine expression and secretion. The IL-1 family is unique in its decoupling of expression and release of bioactive molecules, as many members are transcribed and translated in response to TLR signaling but require additional signals to be effectively released from cells [10,11]. The IL-1 family consists of several cytokines that influence adaptive immunity, with the most-commonly discussed being IL-1α, IL-1β, IL-18 and IL-33 [10,11]. These cytokines differ from most others in that they do not contain an N-terminal secretion signal. Whereas most TLR-dependent chemokines and cytokines are secreted via the biosynthetic pathway immediately upon synthesis, IL-1 family cytokines are produced as latent factors in the intracellular environment of the cell. These latent cytokines are only released after the cell experiences a second stimulus [10,12–14]. Work in recent years has revealed that several families of PRRs induce this second signal that releases IL-1 family cytokines [15–21]. These PRRs include the nucleotide-binding leucine-rich repeat containing (NLR) proteins, the AIM2-like Receptors (ALRs) and the protein Pyrin [15–21]. NLRs, ALRs and Pyrin survey the cytosolic (or nuclear) compartments for microbial products or indications of cellular dysfunction, and upon detection induce the formation of a SMOC called the inflammasome. Inflammasomes, like myddosomes, are not present in resting cells; instead, they assemble downstream of PRR engagement to elicit the activation of specific enzymes that are present within these complexes [1,9]. Whereas myddosomes induce the activation of kinases that promote inflammatory transcription factor activation, inflammasomes induce the activation of the enzyme caspase-1. Caspase-1 plays a critical role in the release of IL-1 family cytokines, as this enzyme cleaves the cytosolic protein gasdermin D (GSDMD) [22–24]. Upon cleavage, the N-terminus of GSDMD oligomerizes into a ring-shaped pore that inserts into the plasma membrane via interactions with acidic phosphoinositides such as a phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) and phosphatidylserine (PS) [25–28]. The GSDMD pore creates ionic imbalances in the cell that ultimately leads to osmotic lysis and the release of IL-1 family cytokines into the extracellular space [25–29]. This form of cell death has been termed pyroptosis [29,30]. Caspase-1 can also cleave members of the IL-1 cytokine family, but the consequences of these cleavage events differ between each family member [10]. IL-1β and IL-18 must be cleaved in order to achieve inflammatory bioactivity [31–34]. IL-1α need not be cleaved to induce bioactivity, and IL-33 cleavage by caspase-1 leads to its inactivation [14,35–38]. The model described above presents an interesting conundrum, as IL-1 family cytokines play important roles in the initiation of adaptive immunity, yet producer cells die in the process of releasing these cytokines. As DCs require hours (and perhaps days) of viability in vivo to effectively stimulate antigen specific T cells [39–42], it is possible that the source of IL-1 important for these events is provided in trans (by cells other than the APCs). An alternative possibility is that mechanisms exist that allow for IL-1 family cytokines to be released from living cells. In this Review, we will discuss both of these possibilities. We discuss examples where inflammasomes and IL-1 family cytokines have been implicated in the generation of adaptive immunity during infection, vaccination, autoimmunity and cancer. We further highlight recent studies that have uncovered a mechanism by which cells may induce the inflammasome-dependent release of IL-1 while retaining viability, and discuss the potential implications of these findings for host defense.
Primer on adaptive immunity
As was alluded to in the prior section, the innate immune system activates the adaptive immune system primarily through the actions of APCs. Professional APCs such as dendritic cells process antigens, thereby degrading proteins into immunostimulatory peptides that are presented on major histocompatibility (MHC) I or II based on the route of antigen acquisition and processing [43]. T cells that detect peptide antigens in the context of MHC are largely grouped into T conventional cells (Tconv) and T regulatory cells (Treg), based on their roles in propagating or regulating immune responses. Antigens that are processed by the proteasome (or immunoproteasome) in the cytosol of nucleated cells can be loaded on MHCI for presentation to a subset of adaptive immune cells known as CD8 T cells [43]. Mature T cells exist in a naïve state prior to antigen exposure [44]. Antigen specific CD8 T cells have major roles in the direct recognition and killing of any infected or transformed cells that present cognate antigen in the context of MHCI [45]. In addition to cytotoxic responses, CD8 T cells can also release instructive cytokines such as interferon-γ (IFN-γ) [45]. Another subset of T cells that utilize alpha beta T cell receptor (TCR) chains that are known to mediate target cell killing and instructive cytokine release are the natural killer T cells [46]. These cells have semi-invariant T cell receptors and primarily detect lipid antigens as opposed to peptide antigens [46]. Antigens that are processed by lysosomal degradation can be loaded on MHC2 for presentation to another major class of adaptive immune cells known as CD4 T cells [43,44]. Naïve CD4 T cells can be influenced by antigen recognition, cytokine milieu, and contextual costimulation by APCs to differentiate naïve T cells into effector T cells of various subtypes including T helper cell 1, T helper cell 2, and T helper cell 17 (Th1, Th2, and Th17 respectively) [44]. These subtypes have defined roles in certain pathogenic contexts, as they are able to release instructive cytokines that shape the subsequent immune response. Th1 cells require the cytokines IL-12 and IFN-γ in addition to TCR engagement and costimulation for efficient differentiation from naïve CD4 T cells [44,47]. These helper cells are important in enhancing the action of innate immune cells such as macrophages through production IFN-γ [44]. This activation of cells such as macrophages is particularly useful in combating immune evasive intracellular bacteria as well as extracellular bacteria. Th2 cells require the cytokine IL-4 in addition to TCR engagement and costimulation for efficient differentiation from naïve CD4 T cells [44,47]. These helper cells are important in the response to parasites including helminths and are implicated in pathological responses such as allergy. Th2 cells produce the instructive cytokines IL-4, IL-5, and IL-13 to skew an immune response towards a type 2 response [44]. Th17 cells require TGF-β and IL-6 with TCR engagement and costimulation for initial differentiation [48]. They also require IL-1 to form stable Th17 cells under certain conditions. These helper cells are associated with the immune response to fungal infections as well as pathology associated with many autoimmune disorders. Th17 cells produce the instructive cytokine IL-17 [48]. Effector T cells can form long-lived memory T cells that are primed for antigen specific re-activation in the event of reinfection [49]. In addition to these effector T cell subsets, CD4 T cells can also form Tregs. These cells repress immune responses in order to maintain tolerance or encourage tissue repair after an immune reaction [50]. These cells require TGF-β and IL-2 for differentiation, in addition to TCR engagement [50]. Gamma delta T cells utilize the gamma and delta chains of the TCR during rearrangement, as opposed to the alpha beta for all the aforementioned T cells. These T cells are present at barrier sites, and can produce instructive cytokines such as IL-17 and IFN-γ [51]. Beyond T cells, the adaptive immune system also comprises other lymphocytes known as B cells. These cells are responsible for the recognition of virtually limitless 3-dimensional epitopes, usually of intact proteins [52]. B cells can undergo selection after somatic hypermutation and class switch recombination to then increase B cell receptor affinity for specific antigens and alter antibody effector function [52]. These highly specific BCRs can then be secreted as antibodies when B cells differentiate into plasma cells [52,53]. These antibodies provide humoral immunity to a host through various effector functions such as neutralization or opsonization of antigen [53]. The adaptive immune system comprises many specialized arms that can unleash subsequent immune programs for specific classes of antigenic insults. All of these responses fundamentally depend on contextualization from innate immune cells.
Instruction of adaptive immunity via inflammasome-dependent cytokines
As described above, a major effector function of the inflammasome is the activation of the cysteine protease caspase-1 [15]. Active caspase-1 has clear links to adaptive immunity through the release of IL-1 family cytokines that are important for T cell activation and memory cell formation. In addition to cleavage of IL-1β and IL-18 into their bioactive forms, inflammasomes also facilitate the secretion of the inflammatory proteins HMGB-1 and IL-1α that do not require processing for biological activity [13,54]. Though IL-1α is often secreted in addition to IL-1β after inflammasome-dependent pyroptosis, some stimulations allow for IL-1α secretion independent of the inflammasome [14,35,36]. IL-1α and cleaved IL-1β signal through the heterodimeric IL-1R, which consists of the IL-1R1 and IL-1R accessory protein (IL-1RAcP) [55–57].
IL-1 signaling has numerous activities that are important for adaptive immune responses [11]. IL-1R signaling via IL-1α or cleaved IL-1β can provide a survival signal to naïve T cells, as IL-1 causes a transient release of the survival cytokine IL-2 from T cells [58]. IL-1R signaling also upregulates IL-2R surface expression, potentially providing pro-survival and proliferation signals to stimulated naïve T cells [59]. Furthermore, IL-1R signaling expands naïve and memory T cells and prolongs T cell help to B cells for antibody production [60]. IL-1β signaling allows for a breach in tolerance through expansion of conventional T cells even in the presence of Tregs cells, confirming that IL-1 provides a strong pro-survival and proliferation signal to T cells [61]. IL-1β is also required for the stable differentiation of the T helper cell subset that produces IL-17 (Th17 cells) from naïve T cells [62–65]. As IL-1α also signals through IL-1R, it is likely that IL-1α could substitute for IL-1β in this role. Indeed, in ex vivo cultures naïve T cells treated with IL-1α were able to stably differentiate into Th17 cells [66,67]. Th17 cells are associated with defense against extracellular bacteria and fungal pathogens [68,69]. These cells are also associated with progression of sterile autoimmunity such as multiple sclerosis (MS) [70–72]. IL-1β in conjunction with IL-23 stimulates IL-17 production from a group of γδ T cells that can exacerbate autoimmunity [73].
Not all IL-1 family members signal via the same receptor. IL-33 (first named nuclear factor from high endothelial venules [74]), for example, signals via the receptor ST2, which can be expressed on type-II immunity T helper cell subset (Th2) and T regulatory cells [75–78]. IL-33 also makes use of IL-1RAcP similar to the IL-1R complex [79,80]. A recent study suggests that receptor proximal signaling strategies differ between IL-1R and ST2, despite both using IL-1RAcP to propagate intracellular signaling [81]. IL-33 signaling induces proliferation of Th2 cells and production of allergy-associated cytokines IL-4, -5, -13 [75,76,82,83]. In regulatory T cells, IL-33 signaling via ST2 promotes a tissue repair response through upregulation and release of the anti-inflammatory mediator amphiregulin [84,85]. IL-33 and IL-1α reside in the nucleus of some cells and can be released in a biologically active form after necrotic cell death [74,86–89]. IL-33 was originally assumed to be cleaved by active caspase-1 to release a bioactive fragment similar to the role of caspase-1 in maturation of pro-IL-1β. Instead, it is now appreciated that caspase-1-mediated cleavage inactivates IL-33 [37,38]. Intriguingly, some studies have shown that active caspase-1 and inflammasome components can be released from cells and reside in the extracellular matrix [90,91]. This potentially broadens the scope of influence a pyroptotic cell could have over the local cytokine milieu if extracellular caspase-1 was capable of inactivating IL-33 released from surrounding necrotic cells.
IL-18 is another IL-1 family member that is translated in a pro-form and matured by active caspase-1 after inflammasome activation [34,92]. This cytokine signals through a heterodimeric receptor complex consisting of IL-18Rα, that bears significant homology to IL-1R, and IL-18Rβ [93]. Like IL-1α, pro-IL-18 does not need to be upregulated in many cell types as it is expressed constitutively in macrophages, Kupffer cells, osteoblasts, chondrocytes and synovial fibroblasts [94–97]. IL-18 was first known as interferon γ (IFNγ) inducing factor for its described role in the induction of IFNγ from T cells after infection with Propionibacterium acnes and subsequent LPS challenge [94]. IL-18 in conjunction with other cytokines such as IL-12 serve to increase IFNγ production from type-I immunity T helper subset (Th1) cells and natural killer (NK) cells [98–100]. This synergy likely occurs through IL-12 mediated upregulation of the IL-18R complex on naïve T cells [99,101,102]. IL-18 signaling then results in upregulation of IL-12R on these polarized Th1 cells leading to a feed forward differentiation loop [103].
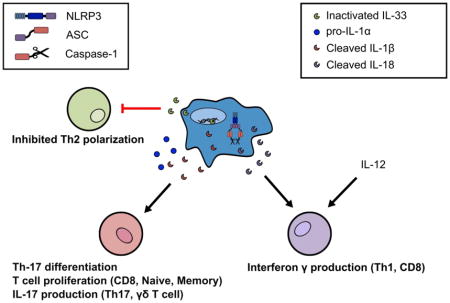
Figure 1 provides a summary of the activities of these IL-1 family members on T cell functions. In subsequent sections, we delve into the roles of inflammasomes and IL-1 family cytokines in driving adaptive immunity to pathogenic insults and sterile inflammation.
Figure 1. Inflammasome-dependent cleavage and release of IL-1 family member cytokines that influence T cell immunity.

Inflammasome activation leads to the cleavage of some IL-1 family members including IL-1β, IL-18, and IL-33. Cleavage of IL-33 by active caspase-1 leads to inactivation of this Th2 skewing cytokine, thus providing a mechanism that inflammasome activity may impinge on Th2 adaptive immune responses. In contrast, cleavage of IL-1β and IL-18 by active caspase-1 results in bioactive cytokines. IL-1β, in addition to pro-IL-1α, can enforce Th-17 stable differentiation and induce T cell proliferation. IL-1β can also cause IL-17 production and secretion from human Th17 cells and murine γδ T cells that may exacerbate autoimmune inflammation. Cleaved IL-18 in conjunction with the cytokine IL-12 enhances IFN-γ from Th1 and CD8 T cells.
Role of inflammasomes in adaptive immune responses to infectious agents
Inflammasomes are well-suited to contextualize pathogen invasion as many of the nucleating receptors are situated within the sterile cytosol compartment of cells. The NLRP3 inflammasome is the best-characterized, with many disparate molecular triggers, such as potassium efflux, lysosomal rupture, reactive oxygen species (ROS) production, and mitochondrial disruption, that initiate assembly of this SMOC [104–109]. Many of these perturbations to homeostasis could indicate pathogenic bacterial invasion. In the case of infections with Streptococcus pneumonia, the pore-forming toxin pneumolysin (PLY) is a major virulence factor that activates the NLRP3 inflammasome [110]. Membrane disruption by PLY causes NLRP3-dependent inflammasome assembly and the consequent caspase-1-dependent cleavage of IL-1β and IL-18 [110–113]. The NLRP3 inflammasome is crucial for the release of IL-1α and mature IL-1β from DCs and macrophages after pneumococcal infection and PLY treatment [110,113]. Treatment of splenocytes with PLY resulted in induction of IL-17A and IFN-γ secretion [110]. PLY is also necessary for IL-17A and IFN-γ secretion in the lungs of mice infected with S. pneumonia [110]. NLRP3 is also important for host survival as NLRP3 KO mice show reduced body weight, increased bacterial burden, and decreased survival compared with wild type mice [113]. The pore-forming adenylate cyclase toxin (CyaA) from Bordetella pertussis also activates the assembly of NLRP3 inflammasomes, and cleaved IL-1β is produced in an NLRP3 and caspase-1-dependent manner following challenge with CyaA [114]. IL-1R KO mice infected with B. pertussis have less Th17 cells and increased bacterial loads [114]. Inflammasomes may also be important for host defense against Mycobacterium tuberculosis as IL-1β knock out (KO) and IL-1R KO mice have increased susceptibility to M. tuberculosis infections [115–117]. Macrophages infected with M. tuberculosis release IL-1β and IL-18 by an NLRP3-dependent process in vitro, although this dependency on NLRP3 is less clear in vivo [115,117,118]. In experiments using heat-killed M. tuberculosis in conjunction with complete Freund’s adjuvant (CFA), caspase-1 and IL-1β are required for generation of Th17 cells [119]. In the context of a live infection with M. tuberculosis, inflammasome-independent IL-1R signaling, likely from release of IL-1α from necrotic cells, can provide differentiation signals to generate Th17 adaptive immunity [115,117,120]. Thus the role of the inflammasome in generating immunity to M. tuberculosis may depend on whether the microbial products are used as an adjuvant or live infection, and this distinction highlights the sufficiency but not necessity of inflammasome signaling to this intracellular pathogen.
The NLRC4 (IPAF) inflammasome is also important in controlling intracellular bacterial infections [16,17,121–124]. This inflammasome is assembled by members of the NAIP family of NLRs, which are direct sensors of bacterial flagellin or subunits of bacterial type III secretion systems [125–128]. Once these bacterial products are detected in the cytosol by their respective receptors, an NLRC4-containing inflammasome is assembled that promotes caspase-1 activation. NLRC4-dependent pyroptosis restricts bacterial infections likely through destruction of a requisite intracellular replication niche [121]. In addition, flagellin derived from Salmonella enterica serovar Typhimurium, Yersinia pseudotuberculosis, and Pseudomonas aeruginosa can promote NLRC4 inflammasome assembly in splenic CD8α DCs resulting in the release of cleaved IL-1β and IL-18 [129]. This pool of IL-18 is capable of stimulating IFNγ secretion from memory CD8 T cells in the spleen [129]. Immune control of Legionella pneumophila infections depends on the flagellin sensing PRR NAIP5 (also known as Birc1e) and NLRC4 [16,122,123]. Using an airway infection model of L. pneumophila, flagellin and NLRC4 were determined to be required for the generation of IL-17 producing T cells [130]. Further characterization illustrated that Th17 differentiation is dependent on caspase-1 and IL-1R after L. pneumophila challenge [130]. Infection with the intracellular pathogen Anaplasma phagocytophilum elicits IL-18 production that is in part due to the NLRC4 inflammasome and is independent of the NLRP3 inflammasome, thereby stimulating the production of IFNγ assayed from serum samples [131]. Moreover, caspase-1 KO and ASC KO mice have reduced Th1 responses that lead to increased susceptibility to A. phagocytophilum infection [131]. Many other bacterial pathogens have been explored for their roles in activating the inflammasome in innate immune cells as reviewed elsewhere [132], but the relative roles of pyroptosis and IL-1 release in generation of adaptive immune responses to these bacteria remains to be defined.
The role of inflammasomes in antimicrobial defense extends beyond bacterial infections, and includes infections by viral and fungal pathogens as well [133–137]. Direct recognition of viral infection byproducts, such as nucleic acids, and indirect recognition of cellular disruption can activate inflammasomes in response to viruses [134,138–141]. The cytosolic sensor of double stranded DNA, AIM2, has been implicated in the assembly of inflammasomes and the consequent generation of IL-18 after infections with Vaccinia virus and mouse cytomegalovirus [138]. Additionally, the nuclear DNA sensor IFI16 forms an inflammasome in response to viral DNA during Kaposi sarcoma-associated herpes virus infection and mediates pyroptosis in response to abortive human immunodeficiency virus (HIV) infection in human CD4 T cells [139,142,143]. Beyond direct recognition of viral nucleic acids, cells can sense infection with influenza virus after incorporation of a proton selective ion channel into the membrane of host cells. Specifically, insertion of the M2 ion channel into membranes of the Golgi apparatus was required for inflammasome activation during influenza virus infection as shown through in vitro infection studies in bone marrow derived macrophages and dendritic cells [134]. Aberrant ion fluxes from this ion channel much like bacterial pore forming toxins allow for the activation and oligomerization of the NLRP3 inflammasome during influenza infection [134]. Moreover, the inflammasome seems crucial for adaptive responses against influenza as the CD4 and CD8 T cell response as well as influenza specific antibody titers are stunted in caspase-1 and ASC KO mice [133]. IL-1R signaling is important for intact antibody response and T cell recruitment to the lung after influenza infection as seen by impaired IgM production and T cell counts in infected IL-1R KO mice [144]. CD8 T cells are often thought of as the main antiviral effector cells of the adaptive immune system, and signaling through IL-1R enhances CD8 cytolytic programs by upregulating effector proteins, such as granzyme B, as well as promoting antigen-driven proliferation of CD8 T cells [145]. Interestingly, a potential mechanism relating age to influenza susceptibility was identified by the finding that aged mice contain DCs with decreased inflammasome components such as ASC and caspase-1 but increased pro-IL-1β [146]. These mice also show lower IL-1β secretion in response to influenza infection with concomitant deficits in total leukocyte counts in aged mice as compared with their younger counterparts [146]. The addition of an adjuvant that stimulates NLRP3 inflammasome activation increases influenza vaccine efficacy in aged mice [147]. Defects in NLRP3 activation can also be rescued through treatment with the bacterial potassium ionophore nigericin, with rescue of IL-1β production in aged mice [146]. Of note, the inflammasome may also play a harmful role in responses to subsequent bacterial infections that are often associated with influenza infections in the context of aged humans [148]. Using mouse strains deficient in the innate immune proteins MAVS and TLR7 as a model for suppressed immune function, infection with influenza resulted in subsequent mortality that was independent of viral load yet dependent on neutrophil influx, bacterial burden, and caspase-1/-11 [148]. These findings suggest that while inflammasome signaling may aid in adaptive responses to the primary viral infection, inflammasome signaling may also result in tissue damage, with profound effects on organismal survival in response to subsequent bacterial infection. Like studies on adaptive immune responses to M. tuberculosis, the adaptive immune response to influenza infection requires IL-1R signaling in T cells, but the source of the IL-1 can come from cleaved IL-1β or IL-1α release. In addition to direct action on T cells, IL-1R signaling has been implicated in enforcing the maturation and migration of pulmonary DCs to lymphoid tissue to activate influenza specific CD8 T cells [149]. In the natural response to influenza, many studies support the role of the inflammasome in the release of IL-1 in the lung of influenza infected animals and activation of Th1 and CD8 adaptive immune responses [133,141,150]. A study investigating DNA based vaccines through plasmids encoding the hemagglutinin (HA) influenza antigen noted that antibody responses to HA are greatly stunted in AIM2 deficient mice, which do not depend on IL-1β or IL-18 signaling. This study suggests that other consequences of inflammasome signaling, such as pyroptosis and the release of DAMPs, may contribute to formation of adaptive immune responses. In addition to detection of influenza infection, the inflammasome appears to be important in generating an adaptive immune response against the central nervous system (CNS) tropic West Nile Virus (WNV). In vivo infection studies demonstrate that IL-1β is released during WNV infection [151]. In addition, IL-1R deficient mice display stunted antiviral responses from CD8 T cells in the CNS. The source of IL-1 in WNV infected animals likely comes from activation of the inflammasome, as caspase-1 and NLRP3 KO mice are more susceptible to infection with WNV [151].
The adaptive response to fungal infections such as Candidiasis depends on Th17 cells [69]. As the role of IL-1β in the differentiation of Th17 cells is well known, one might expect that the inflammasome instructs the adaptive immune system in fungal infections. Consistent with this idea, hyphae from Candida albicans activates the NLRP3 inflammasome, and NLRP3 KO mice are highly susceptible to disseminated and mucosal C. albicans infection [135–137]. NLRC4 is also involved in the immune response to mucosal C. albicans infection, although the mechanism of NLRC4 activation in this infectious context is unknown [152]. Reduced Th1 and Th17 responses in caspase-1 KO and ASC KO mice provides further evidence for the role of the inflammasome in the adaptive response to C. albicans infection [153]. Lack of these adaptive responses leads to fungal outgrowth and increased morbidity after infection with C. albicans in caspase-1 KO and ASC KO mice [153]. Inflammasome-dependent cytokines are directly implicated in generating adaptive immune responses to C. albicans, as exogenous IL-18 rescues the deficit in Th1 polarization in caspase-1 KO mice [154]. IL-1β treatment of C. albicans specific human Th17 cells elicits both IL-17 and IFNγ secretion ex vivo suggesting that IL-1β enforces a pro-inflammatory cytokine program in these cells beyond its role in Th17 differentiation [155]. In vitro studies have shown that stimulation of yeast derived products such as curdlan and zymosan can activate IL-1β secretion from macrophages and DCs in an NLRP3-dependent manner [156]. Curdlan also induces IgM production and upregulation of CD69 on stimulated B cells that is dependent on NLRP3 but independent of the myddosome forming adaptor protein MyD88 suggesting a role of NLRP3 in formation of humoral immune responses [156]. Additional studies have implicated a signaling pathway mediated by the PRR Dectin-1, which senses fungal PAMPs and results in the assembly of NLRP3 inflammasomes in response to hyphae. This observation illustrates that the inflammasome responsiveness to hyphae can contextualize Th17 responses between colonizing yeast and invasive hyphae. From this discussion of bacterial, viral and fungal infections, a theme has emerged whereby the initiation of adaptive immune responses to microbial encounters depends on the activity of inflammasomes and IL-1 family cytokines. In the next section, we expand the discussion on the influence of the inflammasome on adaptive responses to include non-infectious immune activators.
The inflammasome has also been suggested to affect adaptive immune responses to protozoan parasites. Activation of the NLRP3 inflammasome during infection with Leishmania major can alter the adaptive immune response [157–161]. BALB/c mice that are susceptible to Leishmania major infection lacking the inflammasome components NLRP3, ASC or caspase-1 had lower IL-1β and IL-18 production at infection sites and proved more resistant to infection [157]. Surprisingly, IL-18 was shown to promote Th2 differentiation. Neutralizing IL-18 led to lower Leishmania major titers and signs of infection [157]. C57BL/6 mice infected with the Seidman strain of Leishmania major develop non-healing, chronic lesions and reduced parasite clearance [161]. Mice that were deficient for inflammasome components NLRP3, ASC, or caspase-1/caspase-11 were able to resolve infections and did not develop chronic lesions. Moreover, IL-1β and IL-1R deficient mice also showed increased parasite clearance [161]. Conversely, another study using C57BL/6 mice also notes that NLRP3 inflammasome can activate in response to in vitro infection of macrophages with different Leishmania species, and show a role for inflammasome-dependent IL-1β in promoting nitric oxide synthase (NOS2) production of nitric oxide in vivo [159]. This production of nitric oxide was IL-1R and MyD88 dependent and provided resistance to infection with Leishmania amazonesis, Leishmania braziliensis, and Leishmania infantum chagasi [159]. Leishmania amazonesis was recently shown to activate ROS production and NLRP3 activation in infected macrophages downstream of Dectin-1, Syk and phagocytosis [162]. ROS production and inflammasome activation restricted parasite replication inside macrophages and provided organismal resistance to infection in vivo [159,162]. Moreover, Leishmania can modulate pro-inflammatory cytokine release through cleavage of NLRP3 in infected macrophage through the action of the virulence factor metalloprotease GP63 that is common to all infectious Leishmania species [160]. The role of inflammasome dependent IL-1 family members in modulating the immune response to Leishmania species members may be species and mouse strain specific. Mice deficient for IL-1β and IL-1α are more resistant to infection on the BALB/c background due increased beneficial IFN-γ producing Th1 cells and reduced detrimental IL-4 Th2 cells [163]. Injection of exogenous IL-1β can also increase lesion progression and infection with Leishmania amazonesis on the C57BL/6 background [164]. BALB/c mice deficient in IL-18 and infected with Leishmania mexicana or Leishmania major have delayed lesion growth and lower parasite burden [158,165]. These studies suggest that IL-1 family members can somehow skew the immune response towards a Th2 response that makes BALB/c mice susceptible to Leishmania infection. More intuitively, IL-18 deficient mice on the C57BL/6 background prove more susceptible to Leishmania infection as they have decreased IFN-γ producing T cells [166,167], though another study suggests IL-18 may only be involved in early lesion control [168]. More studies are needed to unify conclusions on the role of IL-1 family members and inflammasome signaling in adaptive responses to Leishmania. Genetic analysis of symptomatic patients from the Amazon infected with Plasmodium vivax revealed associated single nucleotide polymorphisms in the inflammasome receptor NLRP1 [169]. The NLRP3 inflammasome can also activate in response to Plasmodium infection through release of crystalline heme known as hemozoin during the blood growth stage. Hemozoin causes the activation of the NLRP3 inflammasome and release of IL-1 family members in vitro and in vivo [170–172]. In addition to NLPR3 activation, cytosolic DNA can activate the AIM2 inflammasome after Plasmodium infection [173]. The inflammasome might be detrimental to immune response against Plasmodium as mice deficient in inflammasome components NLPR3, ASC, and caspase-1 or IL-1β are resistant to Plasmodium infection [170,171]. Though other studies suggest that inflammasome components and inflammasome-dependent cytokines do not play a significant role in the pathology of Plasmodium infection in vivo [174,175]. Models of parasite and bacterial co-infection demonstrate that infection with Plasmodium falciparum or Plasmodium vivax results in NLRP3 and NLRP12 dependent IL-1β release [176]. Co-infection studies also reveal that inflammasome activation contributes to mortality as mice deficient for NLRP3, NLRP12, ASC, and caspase-1 had higher rates of survival compared to wild type mice [176]. Primary infection with Plasmodium vivax sensitized mice to mortality following either bacterial infection or LPS injection [176].
Genetic analyses of a North American patient cohort revealed NLRP1 SNPs associated with congenital toxoplasmosis [177]. Human monocytes infected with Toxoplasma gondii release IL-1β [177,178]. Knock down of caspase-1 and ASC inflammasome components in human monocytes demonstrate that inflammasomes form after infection with Toxoplasma gondii [177]. Toxoplasma gondii infection can also activate the NLRP3 inflammasome [179]. Mice deficient in NLRP1, NLRP3, ASC, or caspase-1 are more susceptible to Toxoplasma gondii suggesting that inflammasome signaling in vivo is protective for the host [179,180]. IL-18 is detectable in the serum of mice infected with Toxoplasma gondii though both IL-1 and IL-18 are protective as mice deficient for either IL-18 or IL-1R are more susceptible to Toxoplasma gondii infection [179,180]. Though there is evidence that inflammasome signaling and inflammasome dependent cytokines are important for host defense to Toxoplasma gondii, further studies are needed to establish how the inflammasome affects adaptive immune responses in the context of protozoan parasitic infections with Toxoplasma gondii.
Additional studies suggest that the NLPR3 inflammasome can form in response to infection with other protozoan parasites such as Trypanosoma cruzi and Entamoeba histolytica [181–184]. Though these studies have not established a role for the inflammasome signaling in informing the adaptive immune response. Further studies are required to determine how inflammasome activation in vivo can affect immune responses against Trypanosoma cruzi and Entamoeba histolytica. A recent study also suggests that host-protozoan interactions with Tritrichomonas musculis can activate the inflammasome with the consequence of providing adaptive immune protection from bacterial infections [185].
Role of the inflammasome in autoimmunity
As we have discussed for infectious diseases, IL-1β and IL-18 likely contribute to differentiation of naïve and primed T cells into effector Th1 and Th17 cells that mediate autoimmune disease. A major candidate autoimmune disease that is potentially instructed by inflammasome activities is multiple sclerosis (MS). Evidence supporting this statement is ample, and includes patient samples that display increased expression of IL-1β and IL-18 as well as caspase-1 [186–188]. In addition, serum levels of IL-18 are increased in MS patients [189,190]. The mouse model for MS known as experimental autoimmune encephalomyelitis (EAE) utilizes CFA to cause an immune response against major CNS antigens, such as myelin oligodendrocyte glycoprotein (MOG). In vivo studies that immunize against MOG have shown dependency on caspase-1 for disease progression [191]. As these studies utilize CFA, which contains heat killed M. tuberculosis, the adjuvant may be the inflammasome-stimulating component in EAE. M. tuberculosis is not unique in its ability to promote inflammasome activity in EAE, as studies using pertussis toxin to prompt EAE have identified the pyrin inflammasome as the inducer of cleaved IL-1β release [192]. Interestingly, in contrast to caspase-1, which is often implicated in inflammasome activities during infections, a recent study implicates IL-1β processing in T cells by a caspase-8-dependent mechanism in propagating a Th17 response in EAE, although the canonical inflammasome adaptor protein ASC is still required [193]. The cytokines IL-1β and IL-18 derived from DCs have been shown to be important for generation of Th17 and γδ T cell responses in EAE [66,73,194].
Beyond MS, genomic studies have implicated polymorphisms in the inflammasome associated receptors NLRP1 and NLRP3 with susceptibility to rheumatoid arthritis (RA) [195,196]. Mechanistic insight into this association are lacking, although one study suggests that inflammasome signaling and basal priming in mice lacking A20, a negative regulator of TLR signaling, can exacerbate arthritis in mouse models [197]. Systemic lupus erythematous (SLE) is another autoimmune disease that has potential involvement of inflammasome activation in perpetuating systemic inflammation. Immune complexes containing double stranded DNA (dsDNA) activate the NLRP3 and AIM2 inflammasomes [198,199]. The generation of antinuclear antibodies by auto reactive B cells depends on IL-1β, potentially offering a feed-forward loop of inflammation [200]. Unsurprisingly, supernatants from cells stimulated with dsDNA immune complexes that activate the NLRP3 inflammasome are able to stimulate IL-17 secretion from memory CD4 T cells [198]. Like with RA, NLRP1 has been genetically associated with SLE in patients [201]. Caspase-1 activation is associated with vascular damage in a mouse model of SLE [202]. Consistent with these results, macrophages from SLE patients contain active NLRP3 inflammasomes [203]. Furthermore, expression of the cryopyrin associated periodic syndromes (CAPS)-associated mutant NLRP3 R258W leads to severe organ damage in a mouse model of lupus [204]. Like with MS, inflammasome dependent cytokines IL-1β and IL-18 show elevated levels in the serum of lupus patients [205–207]. Studies suggest that increased IL-18 and localization of plasmacytoid DCs to the kidneys might activate tissue resident T cells and mediate kidney damage in SLE, which is a major consequence of lupus in patients [206,207]. More studies are required to determine if inflammasome activation is a requisite step in the progression of autoimmunity. Of particular interest would be parsing out the relative roles of pyroptosis from the release of T cell stimulating cytokines such as IL-1β and IL-18 after inflammasome activation as has been investigated in auto-inflammatory disorders [208].
Inflammasomes and vaccines
As has been discussed above, ample and diverse evidence supports the role of IL-1 family cytokines in T cell mediated immunity. As IL-1 release is classically associated with inflammasome-mediated cell death, a conundrum emerges. How can a T cell response depend on IL-1 (released from dead cells) and also depend on antigen presentation by a living cell? While it is possible that the IL-1 necessary for T cell activation and differentiation is provided in trans, from dying cells, another possibility is that mechanisms of IL-1 release from living APCs exist. Recent work from our group suggests that DCs can indeed be induced to released inflammasome-dependent IL-1 while retaining viability [209]. When murine DCs are treated with TLR ligands, such as bacterial lipopolysaccharide (LPS), these cells will upregulate MHC and costimulatory molecule expression and release of inflammatory cytokines that contain N-terminal signal sequences [3,4]. However, they will not release IL-1 family cytokines that are present in their intracellular compartment as previously discussed. When DCs are exposed to TLR ligands and a DAMP, such as oxidized phosphorylcholine derivatives, these cells assemble inflammasomes and induce the release of IL-1 family cytokines while retaining viability [209]. The addition of IL-1 release coupled with the repertoire of TLR-dependent cytokines that normally occur during TLR activation renders these cells highly immunostimulatory, and have therefore been described as hyperactive DCs [209]. Interestingly, the ability of living DCs to release IL-1 and TLR-dependent cytokines allows these hyperactive cells to induce more robust antigen-specific T cell responses than are normally elicited by traditionally activated DCs [209]. At least two other studies have also recently reported stimulations that cause inflammasome-dependent IL-1β release in the absence of pyroptosis. These stimulations include responses to LPS in human and porcine monocytes and peptidoglycan in mouse macrophages [210,211]. In fact, much like our group has demonstrated that using oxidized phospholipids as an adjuvant can induce stronger antigen specific T cell responses in vivo, a recent study revealed that peptidoglycan from a mutant strain of Staphylococcus aureus that is highly sensitive to degradation by the enzyme lysozyme can cause protective immunity against subsequent Staphylococcus aureus infection through the generation of a memory T cell response [212]. These studies raise an important question regarding the potential role of the inflammasome in licensing adaptive immunity, and the potential applications of inflammasome activation towards next generation vaccine development. In addition to stimulation-specific resistance to pyroptosis, several studies also note cell type-specific responses downstream of inflammasome activation. Neutrophils treated with ATP activate the NLRP3 inflammasome or infected with Salmonella activate the NLRC4 inflammasome resulting in release IL-1 family members without signs of lysis such as lactate dehydrogenase release [213,214]. In a majority of papers that link inflammasome activation to adaptive immunity the link stems from release of IL-1β and IL-18. The role of cell lysis in secretion of these cytokines is intimately connected to inflammasome activation where secretion of these cytokines occurs secondary to cell lysis [12,29,215]. It is interesting to note that while membrane permeability seems to be a requisite step for IL-1 release, pyroptosis can be prevented by preventing osmotic lysis with glycine buffering with marginal effect on IL-1 release [29,216–218]. With the knowledge that membrane permeability seems to be required for IL-1β release and cleavage of GSDMD by inflammatory caspases-1/-11 causes large pore formation in the plasma membrane, one could speculate that GSDMD pores may facilitate IL-1 release from living cells [25–28,217]. GSDMD-independent mechanisms of IL-1 release likely exist, as strongly pyroptotic stimuli do not cause IL-1 release at acute time points in GSDMD KO macrophages, though these macrophages can undergo secondary necrosis after longer stimulations [23]. These recent studies change the paradigm of cell lysis concomitant with IL-1β and IL-18 secretion, and may reveal the relative roles cellular lysis and pro-inflammatory cytokines play in informing a subsequent immune response.
While the role of hyperactive APCs in adaptive immunity needs further exploration, it is worth considering whether the activities of current adjuvants may depend on this novel cell activation state. For example, a major adjuvant in human vaccines is aluminum hydroxide (alum), a particulate that activates the NLRP3 inflammasome in phagocytes through rupture of lysosomal compartments [105,219–221]. Consequently, alum causes the release of cleaved IL-1β and IL-18 from macrophages by a caspase-1-dependent process [222]. During alum-dependent responses in vivo, DAMPs, such as oxidized lipids described above, are likely released from dying cells. Indeed, DAMPs have already been implicated in alum adjuvanticity, as alum-dependent responses depend on more than just release of pro-inflammatory cytokines. Alum-mediated release of DNA during cell death as well as permeabilization of lysosomes that release cathepsins within cells influence protective immunity [223,224]. These released DAMPs may act with any available PAMPs that might also be present to hyperactivate DCs and influence the development of T cell mediated immunity. Alternatively, it is possible that adjuvants can be engineered to specifically introduce oxidized lipids that supplement the adjuvants already present in these vaccines, as a means of promoting more effective T cell responses.
Further studies are needed to characterize the relative roles of lytic cell death and release of cellular antigens and DAMPs from the release of pro-inflammatory cytokines in the context of adjuvants.
Inflammasomes and cancer
Inflammasome signaling may also contribute to immune responses in the context of cancer. A clear role of inflammasome protection from cancer has been observed in the gut, as inflammasomes are necessary components for maintenance of an intact barrier [225–228]. Mice deficient for components of the inflammasome such as ASC, NLRP3, and NLRP6 are more susceptible to dextran sulfate sodium (DSS) induced colitis, highlighting that inflammasomes can limit chronic inflammation in the gut through production of IL-18 [225,229]. NLRP6 in hematopoietic cells prevents increased susceptibility to DSS-induced colitis and tumorigenesis [227]. NLRP3 has also been confirmed to play a role in preventing colitis-induced tumorigenesis [228]. An additional study links the NLRC4 inflammasome to control of tumorigenesis through prevention of chronic inflammation in the colon [230]. The link of inflammasomes to adaptive immune control of tumorigenesis could stem from the production of IL-18 in response to invading gut bacteria. One study has shown that reduction of IL-18 through loss of NLRP3 leads to a reduction of IFNγ production in the gut [226]. IL-1β may serve to be pro-tumoral depending on the context as IL-1β has been shown to promote tumor invasiveness [231,232]. In addition, the release of angiogenic factors downstream of IL-1R signaling may serve to promote early tumor formation [232,233]. In a mouse model of breast cancer, blocking IL-1R signaling through administration of the IL-1Ra prevents tumor-associated macrophage (TAM) and myeloid-derived suppressor cell (MDSC) accumulation in tumors as well as preventing metastases. Adding to the context-specific efficacy of inflammasome signaling on adaptive immune responses to tumors, studies have implicated NLRP3 activation in tumors with accumulation of MDSCs that can suppress adaptive immune responses to tumors [234,235]. Anti-tumor effects of inflammasome signaling may be important for the adaptive immune response to late stage tumors after cancer treatment. After chemotherapy, DCs activate the NLRP3 inflammasome and prime tumor specific CD8 T cells leading to an anti-tumor adaptive immune response [236]. Engineered tumor cells that express bacterial flagellin cause a robust CD8 T cell response against the tumors that depends on synergistic activation of TLR5 signaling and the NLRC4 inflammasome [237]. The role of inflammasome signaling in generating an adaptive immune response against tumors deserves additional studies, as the current literature would suggest that inflammation downstream of the inflammasome could be anti-tumoral or pro-tumoral depending on the particular tumor and intervening therapy.
Adaptive immune control of inflammasome signaling
A majority of the inflammasome biology literature centers around inflammasome activation in traditional innate phagocytes, such as macrophages and DCs. Recent studies have sought to identify if inflammasomes can be active in other cell types. In response to radiation, one study has found inflammasome assembly to occur in adaptive immune cells, such as T and B cells. Inflammasome assembly in these cells proceeds independently of NLRP3 [238]. A recent study also using radiation identifies AIM2 to be the responding receptor to DNA damage in the gut of irradiated mice in epithelial and bone marrow derived cells [239]. This study also highlights the role of cell death in tissue inflammation, in contrast to the role of inflammasome-mediated cytokines such as IL-1β and IL-18 [239]. One group has identified that HIV infection can result in inflammasome assembly and pyroptosis in CD4 T cells [142,143]. This group determined that abortive infections with HIV causes assembly of inflammasomes in lymphoid, but not blood, CD4 T cells that leads to eventual cell lysis [142,143,240]. These studies suggest that T cells are able to form functional inflammasomes in response to viral infections, and that pyroptosis may contribute to CD4 T cell decline in the context of HIV infection [142,143]. It is interesting to note that CD4 T cells from humans seem to constitutively express pro-IL-1β, whereas many myeloid cells require a NF-κB activating stimulus to express this caspase-1 substrate [143]. Another recent study has also identified T cell-intrinsic inflammasome assembly downstream of T cell receptor (TCR) engagement in coordination with complement receptor signaling [241]. T cell intrinsic ASC is required for Th17 mediated pathology in EAE. This study suggests that T cells with active inflammasomes can release active IL-1β downstream of caspase-8. This IL-1β may serve as an autocrine survival signal to these pathogenic T cells [193]. NLRP3 is also required for the production of Th2 cytokines, though this appears to be inflammasome-independent [242]. These recent findings suggest that inflammasomes and their components can operate within adaptive immune cells such as T cells.
Adaptive immune cells also can influence inflammasome assembly in other cell types. Innate-like natural killer (NK) T cells also coordinate inflammasome activation in traditional phagocytes. Activated human NK T cells trigger IL-1β processing and secretion from monocytes in the absence of significant lysis in a caspase-8 dependent mechanism [243]. Co-culture of iNK T cells with monocytes was sufficient to prompt release of IL-1β, and this phenotype was not recapitulated with transwell experiments suggesting that NK T cell and monocytes must contact each other [243]. Moreover, NK T cells positively regulate priming of the NLRP3 inflammasome through release of TNF-α that acts to upregulate IL-1β and NLRP3 in macrophages in response to α-galactosylceramide treatment [244]. As IL-1β can activate proliferation and effector functions from different T cell subsets, the concept of adaptive immune induction of IL-1β secretion from either T cells or innate immune cells is intriguing. Further studies are required to determine the molecular events that determine T cell initiated inflammasomes in cell intrinsic and extrinsic situations. The adaptive immune compartment has also been implicated in negative regulation of inflammasome signaling. IFNγ can negatively regulate NLRP3 inflammasome activation [245], though IFNγ has also been reported to increase caspase-11 expression leading to non-canonical inflammasome signaling in the context of bacterial infection [246]. In addition, adaptive immune cells such Tr1 cells can produce suppressive cytokines, such as IL-10, that can negatively regulate NLRP3 inflammasome signaling [247]. Interestingly, another study shows direct contact from effector and memory CD4 T cells can abolish NLRP1 and NLRP3 inflammasome activation in macrophages [248]. These negative regulatory mechanisms downstream of adaptive immune activation could serve as a way to diminish innate immune responses after the adaptive immune system has started responding to a threat. Figure 2 summarizes how T cells can regulate the assembly of inflammasomes in cell intrinsic contexts as well as in other cell types. Further studies should investigate whether T cells can survive inflammasome activation like has been recently described for traditional phagocytes as this could serve as an autocrine survival factor particularly for CD8, Th17, and certain γδ T cells.
Figure 2. T cell regulation of cell-intrinsic and extrinsic inflammasomes.

A. DNA sensing pathways activate after abortive HIV infection and radiation therapy, which causes assembly of the IFI16 and AIM2 inflammasomes respectively in T cells. Abortive HIV infection proceeds to pyroptosis in human lymphoid derived CD4 T cells, but not in blood CD4 T cells. These cells have high levels of pro-IL-1β prior to infection, thus are able to release cleaved IL-1β after inflammasome assembly. Radiation therapy induces DNA damage that results in AIM2 inflammasome assembly and pyroptotic cell death in many cell types including T cells in mice. B. TCR signaling likely upregulates pro-IL-1β and NLRP3, and intracellular complement receptor signaling results in ROS production and inflammasome assembly. T cell intrinsic ASC is required for Th17-mediated pathology in EAE. These T cells might release cleaved IL-1β downstream of ATP sensing from other necrotic cells, and this IL-1β may provide autocrine signaling promoting pathogenic Th17 cell survival. C. T cells capable of secreting IFN-γ may inhibit NLRP3 inflammasome signaling. Tr1 cells secrete IL-10 and other suppressive cytokines. These cytokines have been shown to inhibit the assembly of the NLRP3 inflammasome. NK T cells can induce IL-1β release from monocytes by an ill-defined mechanism. NK T cells can also release TNF-α that can serve as a priming signal for the NLRP3 inflammasome.
Concluding remarks
The role of inflammasome-associated IL-1 family cytokines in shaping adaptive immune responses is now well established in regards to differentiation of Th17 cells and promoting effector functions of Th1 cells and CD8 T cells. The additional role that cell lysis may play after inflammasome activation through the release of additional DAMPs and self-antigens deserves further study especially in the context of driving autoimmunity. The separation of pyroptosis and unconventional protein secretion downstream of inflammasome activation also remains to be elucidated in terms of molecular determinants that contribute to this cell fate decision. The role of the adaptive immune system in regulation of inflammasome activation requires further study to determine the extent to which adaptive immune cells contribute to inflammasome signaling in infectious and sterile contexts.
Highlights.
Interleukin-1 (IL-1) family cytokines play varying and essential roles in T cell mediated responses during infection, vaccination, autoimmunity and cancer.
Inflammasomes are important regulators of IL-1 family cytokine activity and release.
Most stimulators of inflammasome activity cause IL-1 release from pyroptotic cells, but recent studies identified inflammasomes that permit IL-1 release from living cells.
Cells that release IL-1 while maintaining viability have been identified as “hyperactive”, and are highly potent activators of adaptive immunity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 3.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 5.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 6.Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Mirza A, et al. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 1997;388:787–792. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- 7.Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I, et al. Activation of human dendritic cells through CD40 cross-linking. Journal of Experimental Medicine. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, et al. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. Journal of Experimental Medicine. 1997;186:1819–1829. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nature Publishing Group. 2014;14:821–826. doi: 10.1038/nri3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev. 2008;223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x. [DOI] [PubMed] [Google Scholar]
- 11.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brough D, Rothwell NJ. Caspase-1-dependent processing of pro-interleukin-1beta is cytosolic and precedes cell death. J Cell Sci. 2007;120:772–781. doi: 10.1242/jcs.03377. [DOI] [PubMed] [Google Scholar]
- 13.Keller M, Rüegg A, Werner S, Beer H-D. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 14.Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 15.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 16.Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol. 2006;7:318–325. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- 17.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 18.Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 21.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 22.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 23.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 24.He W-T, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 27.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USa. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. Embo J. 2016;35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 30.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infection and Immunity. 2005;73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, Hillman AG, et al. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proceedings of the National Academy of Sciences. 1989;86:5227–5231. doi: 10.1073/pnas.86.14.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howard AD, Kostura MJ, Thornberry N, Ding GJ, Limjuco G, Weidner J, et al. IL-1-converting enzyme requires aspartic acid residues for processing of the IL-1 beta precursor at two distinct sites and does not cleave 31-kDa IL-1 alpha. The Journal of Immunology. 1991;147:2964–2969. [PubMed] [Google Scholar]
- 33.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 34.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 35.Chen C-J, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 36.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, et al. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–4843. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- 37.Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. Journal of Biological Chemistry. 2009;284:19420–19426. doi: 10.1074/jbc.M901744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cayrol C, Girard J-P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USa. 2009;106:9021–9026. doi: 10.1073/pnas.0812690106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ingulli E, Mondino A, Khoruts A, Jenkins MK. In vivo detection of dendritic cell antigen presentation to CD4(+) T cells. Journal of Experimental Medicine. 1997;185:2133–2141. doi: 10.1084/jem.185.12.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol. 2002;3:265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- 41.Hommel M, Kyewski B. Dynamic changes during the immune response in T cell-antigen-presenting cell clusters isolated from lymph nodes. Journal of Experimental Medicine. 2003;197:269–280. doi: 10.1084/jem.20021512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mempel TR, Henrickson SE, von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 43.Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 47.Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol Rev. 2013;252:12–23. doi: 10.1111/imr.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood. 2013;121:2402–2414. doi: 10.1182/blood-2012-09-378653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nature Publishing Group. 2016;16:102–111. doi: 10.1038/nri.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nature Publishing Group. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol. 2010;11:681–688. doi: 10.1038/ni.1900. [DOI] [PubMed] [Google Scholar]
- 53.Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nature Publishing Group. 2015;15:160–171. doi: 10.1038/nri3795. [DOI] [PubMed] [Google Scholar]
- 54.Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–4392. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greenfeder SA, Nunes P, Kwee L, Labow M, Chizzonite RA, Ju G. Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. Journal of Biological Chemistry. 1995;270:13757–13765. doi: 10.1074/jbc.270.23.13757. [DOI] [PubMed] [Google Scholar]
- 56.Wesche H, Korherr C, Kracht M, Falk W, Resch K, Martin MU. The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1-induced activation of interleukin-1 receptor-associated kinase (IRAK) and stress-activated protein kinases (SAP kinases) Journal of Biological Chemistry. 1997;272:7727–7731. doi: 10.1074/jbc.272.12.7727. [DOI] [PubMed] [Google Scholar]
- 57.Vigers GP, Anderson LJ, Caffes P, Brandhuber BJ. Crystal structure of the type-I interleukin-1 receptor complexed with interleukin-1beta. Nature. 1997;386:190–194. doi: 10.1038/386190a0. [DOI] [PubMed] [Google Scholar]
- 58.Plaetinck G, Combe MC, Corthésy P, Sperisen P, Kanamori H, Honjo T, et al. Control of IL-2 receptor-alpha expression by IL-1, tumor necrosis factor, and IL-2. Complex regulation via elements in the 5′ flanking region. The Journal of Immunology. 1990;145:3340–3347. [PubMed] [Google Scholar]
- 59.Plaetinck G, Combe MC, Corthésy P, Sperisen P, Kanamori H, Honjo T, et al. Control of IL-2 receptor-alpha expression by IL-1, tumor necrosis factor, and IL-2. Complex regulation via elements in the 5′ flanking region. The Journal of Immunology. 1990;145:3340–3347. [PubMed] [Google Scholar]
- 60.Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci USa. 2009;106:7119–7124. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, et al. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. The Journal of Immunology. 2006;176:7278–7287. doi: 10.4049/jimmunol.176.12.7278. [DOI] [PubMed] [Google Scholar]
- 62.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 63.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 64.Kryczek I, Wei S, Vatan L, Escara-Wilke J, Szeliga W, Keller ET, et al. Cutting edge: opposite effects of IL-1 and IL-2 on the regulation of IL-17+ T cell pool IL-1 subverts IL-2-mediated suppression. The Journal of Immunology. 2007;179:1423–1426. doi: 10.4049/jimmunol.179.3.1423. [DOI] [PubMed] [Google Scholar]
- 65.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 66.Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. Journal of Experimental Medicine. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 69.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 70.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 71.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 74.Baekkevold ES, Roussigné M, Yamanaka T, Johansen F-E, Jahnsen FL, Amalric F, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Ajpa. 2003;163:69–79. doi: 10.1016/S0002-9440(10)63631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu D, Chan WL, Leung BP, Huang FP, Wheeler R, Piedrafita D, et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. Journal of Experimental Medicine. 1998;187:787–794. doi: 10.1084/jem.187.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meisel C, Bonhagen K, Löhning M, Coyle AJ, Gutierrez-Ramos JC, Radbruch A, et al. Regulation and function of T1/ST2 expression on CD4+ T cells: induction of type 2 cytokine production by T1/ST2 cross-linking. The Journal of Immunology. 2001;166:3143–3150. doi: 10.4049/jimmunol.166.5.3143. [DOI] [PubMed] [Google Scholar]
- 77.Trajkovic V, Sweet MJ, Xu D. T1/ST2--an IL-1 receptor-like modulator of immune responses. Cytokine Growth Factor Rev. 2004;15:87–95. doi: 10.1016/j.cytogfr.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 78.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an Interleukin-1-like Cytokine that Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 79.Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci USa. 2007;104:18660–18665. doi: 10.1073/pnas.0705939104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. The Journal of Immunology. 2007;179:2551–2555. doi: 10.4049/jimmunol.179.4.2551. [DOI] [PubMed] [Google Scholar]
- 81.Günther S, Deredge D, Bowers AL, Luchini A, Bonsor DA, Beadenkopf R, et al. IL-1 Family Cytokines Use Distinct Molecular Mechanisms to Signal through Their Shared Co-receptor. Immunity. 2017;47:510–523.e4. doi: 10.1016/j.immuni.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Löhning M, Stroehmann A, Coyle AJ, Grogan JL, Lin S, Gutierrez-Ramos JC, et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proceedings of the National Academy of Sciences. 1998;95:6930–6935. doi: 10.1073/pnas.95.12.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an Interleukin-1-like Cytokine that Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 84.Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. 2015;162:1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, et al. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity. 2016;44:355–367. doi: 10.1016/j.immuni.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Buryskova M, Pospisek M, Grothey A, Simmet T, Burysek L. Intracellular interleukin-1alpha functionally interacts with histone acetyltransferase complexes. Journal of Biological Chemistry. 2004;279:4017–4026. doi: 10.1074/jbc.M306342200. [DOI] [PubMed] [Google Scholar]
- 87.Werman A, Werman-Venkert R, White R, Lee J-K, Werman B, Krelin Y, et al. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proceedings of the National Academy of Sciences. 2004;101:2434–2439. doi: 10.1073/pnas.0308705101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, et al. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci USa. 2010;107:2574–2579. doi: 10.1073/pnas.0915018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moussion C, Ortega N, Girard J-P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS ONE. 2008;3:e3331. doi: 10.1371/journal.pone.0003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and “prionoid” activities that propagate inflammation. Nat Immunol. 2014;15:727–737. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–748. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 92.Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science. 1997;275:206–209. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- 93.Thomassen E, Bird TA, Renshaw BR, Kennedy MK, Sims JE. Binding of interleukin-18 to the interleukin-1 receptor homologous receptor IL-1Rrp1 leads to activation of signaling pathways similar to those used by interleukin-1. Journal of Interferon & Cytokine Research. 1998;18:1077–1088. doi: 10.1089/jir.1998.18.1077. [DOI] [PubMed] [Google Scholar]
- 94.Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 95.Udagawa N, Horwood NJ, Elliott J, Mackay A, Owens J, Okamura H, et al. Interleukin-18 (interferon-gamma-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-gamma to inhibit osteoclast formation. Journal of Experimental Medicine. 1997;185:1005–1012. doi: 10.1084/jem.185.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Olee T, Hashimoto S, Quach J, Lotz M. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. The Journal of Immunology. 1999;162:1096–1100. [PubMed] [Google Scholar]
- 97.Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest. 1999;104:1393–1401. doi: 10.1172/JCI7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ahn HJ, Maruo S, Tomura M, Mu J, Hamaoka T, Nakanishi K, et al. A mechanism underlying synergy between IL-12 and IFN-gamma-inducing factor in enhanced production of IFN-gamma. The Journal of Immunology. 1997;159:2125–2131. [PubMed] [Google Scholar]
- 99.Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, et al. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. The Journal of Immunology. 1998;161:3400–3407. [PubMed] [Google Scholar]
- 100.Xu D, Chan WL, Leung BP, Hunter D, Schulz K, Carter RW, et al. Selective expression and functions of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells. Journal of Experimental Medicine. 1998;188:1485–1492. doi: 10.1084/jem.188.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci USa. 2009;106:13463–13468. doi: 10.1073/pnas.0906988106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Blom L, Poulsen LK. IL-1 family members IL-18 and IL-33 upregulate the inflammatory potential of differentiated human Th1 and Th2 cultures. J Immunol. 2012;189:4331–4337. doi: 10.4049/jimmunol.1103685. [DOI] [PubMed] [Google Scholar]
- 103.Chang JT, Segal BM, Nakanishi K, Okamura H, Shevach EM. The costimulatory effect of IL-18 on the induction of antigen-specific IFN-gamma production by resting T cells is IL-12 dependent and is mediated by up-regulation of the IL-12 receptor beta2 subunit. Eur J Immunol. 2000;30:1113–1119. doi: 10.1002/(SICI)1521-4141(200004)30:4<1113::AID-IMMU1113>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 104.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 107.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Koedel U, Winkler F, Angele B, Fontana A, Flavell RA, Pfister H-W. Role of Caspase-1 in experimental pneumococcal meningitis: Evidence from pharmacologic Caspase inhibition and Caspase-1-deficient mice. Ann Neurol. 2002;51:319–329. doi: 10.1002/ana.10103. [DOI] [PubMed] [Google Scholar]
- 112.Shoma S, Tsuchiya K, Kawamura I, Nomura T, Hara H, Uchiyama R, et al. Critical involvement of pneumolysin in production of interleukin-1alpha and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae in vitro: a novel function of pneumolysin in caspase-1 activation. Infection and Immunity. 2008;76:1547–1557. doi: 10.1128/IAI.01269-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. 2011;187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- 114.Dunne A, Ross PJ, Pospisilova E, Masin J, Meaney A, Sutton CE, et al. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J Immunol. 2010;185:1711–1719. doi: 10.4049/jimmunol.1000105. [DOI] [PubMed] [Google Scholar]
- 115.Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, et al. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol. 2010;184:3326–3330. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. 2011;35:1023–1034. doi: 10.1016/j.immuni.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McElvania-TeKippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, et al. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS ONE. 2010;5:e12320. doi: 10.1371/journal.pone.0012320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1β production in human macrophages. J Immunol. 2011;187:2540–2547. doi: 10.4049/jimmunol.1100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shenderov K, Barber DL, Mayer-Barber KD, Gurcha SS, Jankovic D, Feng CG, et al. Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through mincle/CARD9 signaling and the inflammasome. J Immunol. 2013;190:5722–5730. doi: 10.4049/jimmunol.1203343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Walter K, Hölscher C, Tschopp J, Ehlers S. NALP3 is not necessary for early protection against experimental tuberculosis. Immunobiology. 2010;215:804–811. doi: 10.1016/j.imbio.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 121.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, et al. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. Journal of Experimental Medicine. 2006;203:1093–1104. doi: 10.1084/jem.20051659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Amer A, Franchi L, Kanneganti T-D, Body-Malapel M, Ozören N, Brady G, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. Journal of Biological Chemistry. 2006;281:35217–35223. doi: 10.1074/jbc.M604933200. [DOI] [PubMed] [Google Scholar]
- 124.Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2006;2:e18. doi: 10.1371/journal.ppat.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 127.Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USa. 2013;110:14408–14413. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA. Cutting edge: Mouse NAIP1 detects the type III secretion system needle protein. J Immunol. 2013;191:3986–3989. doi: 10.4049/jimmunol.1301549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kupz A, Guarda G, Gebhardt T, Sander LE, Short KR, Diavatopoulos DA, et al. NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8+ T cells. Nat Immunol. 2012;13:162–169. doi: 10.1038/ni.2195. [DOI] [PubMed] [Google Scholar]
- 130.Trunk G, Oxenius A. Innate instruction of CD4+ T cell immunity in respiratory bacterial infection. J Immunol. 2012;189:616–628. doi: 10.4049/jimmunol.1200924. [DOI] [PubMed] [Google Scholar]
- 131.Pedra JHF, Sutterwala FS, Sukumaran B, Ogura Y, Qian F, Montgomery RR, et al. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-gamma axis during Anaplasma phagocytophilum infection. The Journal of Immunology. 2007;179:4783–4791. doi: 10.4049/jimmunol.179.7.4783. [DOI] [PubMed] [Google Scholar]
- 132.von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013;31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- 133.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11:404–410. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Joly S, Ma N, Sadler JJ, Soll DR, Cassel SL, Sutterwala FS. Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J Immunol. 2009;183:3578–3581. doi: 10.4049/jimmunol.0901323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host and Microbe. 2009;5:487–497. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gross O, Poeck H, Bscheider M, Dostert C, Hannesschläger N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 138.Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell Host and Microbe. 2011;9:363–375. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kanneganti T-D, Body-Malapel M, Amer A, Park J-H, Whitfield J, Franchi L, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. Journal of Biological Chemistry. 2006;281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 141.Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, et al. IFI16 DNA Sensor Is Required for Death of Lymphoid CD4 T Cells Abortively Infected with HIV. Science. 2014;343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Doitsh G, Galloway NLK, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol. 2005;79:6441–6448. doi: 10.1128/JVI.79.10.6441-6448.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med. 2013;210:491–502. doi: 10.1084/jem.20122006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stout-Delgado HW, Vaughan SE, Shirali AC, Jaramillo RJ, Harrod KS. Impaired NLRP3 inflammasome function in elderly mice during influenza infection is rescued by treatment with nigericin. J Immunol. 2012;188:2815–2824. doi: 10.4049/jimmunol.1103051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schneider-Ohrum K, Giles BM, Weirback HK, Williams BL, DeAlmeida DR, Ross TM. Adjuvants that stimulate TLR3 or NLPR3 pathways enhance the efficiency of influenza virus-like particle vaccines in aged mice. Vaccine. 2011;29:9081–9092. doi: 10.1016/j.vaccine.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, et al. M×1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. 2016;352:463–466. doi: 10.1126/science.aaf3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8+ T cell responses to influenza A virus. Nat Immunol. 2013;14:246–253. doi: 10.1038/ni.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thomas PG, Dash P, Aldridge JR, Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, et al. IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 2012;8:e1003039. doi: 10.1371/journal.ppat.1003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tomalka J, Ganesan S, Azodi E, Patel K, Majmudar P, Hall BA, et al. A novel role for the NLRC4 inflammasome in mucosal defenses against the fungal pathogen Candida albicans. PLoS Pathog. 2011;7:e1002379. doi: 10.1371/journal.ppat.1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.van de Veerdonk FL, Joosten LAB, Shaw PJ, Smeekens SP, Malireddi RKS, Van der Meer JWM, et al. The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Eur J Immunol. 2011;41:2260–2268. doi: 10.1002/eji.201041226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mencacci A, Bacci A, Cenci E, Montagnoli C, Fiorucci S, Casagrande A, et al. Interleukin 18 restores defective Th1 immunity to Candida albicans in caspase 1-deficient mice. Infection and Immunity. 2000;68:5126–5131. doi: 10.1128/iai.68.9.5126-5131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature. 2012;484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
- 156.Kumar H, Kumagai Y, Tsuchida T, Koenig PA, Satoh T, Guo Z, et al. Involvement of the NLRP3 inflammasome in innate and humoral adaptive immune responses to fungal beta-glucan. J Immunol. 2009;183:8061–8067. doi: 10.4049/jimmunol.0902477. [DOI] [PubMed] [Google Scholar]
- 157.Gurung P, Karki R, Vogel P, Watanabe M, Bix M, Lamkanfi M, et al. An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. J Clin Invest. 2015;125:1329–1338. doi: 10.1172/JCI79526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bryson KJ, Wei X-Q, Alexander J. Interleukin-18 enhances a Th2 biased response and susceptibility to Leishmania mexicana in BALB/c mice. Microbes Infect. 2008;10:834–839. doi: 10.1016/j.micinf.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 159.Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva ALN, Mineo TWP, et al. Inflammasome-derived IL-1β production induces nitric oxide-mediated resistance to Leishmania. Nat Med. 2013;19:909–915. doi: 10.1038/nm.3221. [DOI] [PubMed] [Google Scholar]
- 160.Shio MT, Christian JG, Jung JY, Chang K-P, Olivier M. PKC/ROS-Mediated NLRP3 Inflammasome Activation Is Attenuated by Leishmania Zinc-Metalloprotease during Infection. PLoS Negl Trop Dis. 2015;9:e0003868. doi: 10.1371/journal.pntd.0003868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Charmoy M, Hurrell BP, Romano A, Lee SH, Ribeiro-Gomes F, Riteau N, et al. The Nlrp3 inflammasome, IL-1β, and neutrophil recruitment are required for susceptibility to a nonhealing strain of Leishmania major in C57BL/6 mice. Eur J Immunol. 2016;46:897–911. doi: 10.1002/eji.201546015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lima-Junior DS, Mineo TWP, Calich VLG, Zamboni DS. Dectin-1 Activation during Leishmania amazonensis Phagocytosis Prompts Syk-Dependent Reactive Oxygen Species Production To Trigger Inflammasome Assembly and Restriction of Parasite Replication. J Immunol. 2017;199:2055–2068. doi: 10.4049/jimmunol.1700258. [DOI] [PubMed] [Google Scholar]
- 163.Voronov E, Dotan S, Gayvoronsky L, White RM, Cohen I, Krelin Y, et al. IL-1-induced inflammation promotes development of leishmaniasis in susceptible BALB/c mice. International Immunology. 2010;22:245–257. doi: 10.1093/intimm/dxq006. [DOI] [PubMed] [Google Scholar]
- 164.Xin L, Li Y, Soong L. Role of interleukin-1beta in activating the CD11c(high) CD45RB- dendritic cell subset and priming Leishmania amazonensis-specific CD4+ T cells in vitro and in vivo. Infection and Immunity. 2007;75:5018–5026. doi: 10.1128/IAI.00499-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wei X-Q, Niedbala W, Xu D, Luo Z-X, Pollock KGJ, Brewer JM. Host genetic background determines whether IL-18 deficiency results in increased susceptibility or resistance to murine Leishmania major infection. Immunol Lett. 2004;94:35–37. doi: 10.1016/j.imlet.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 166.Wei XQ, Leung BP, Niedbala W, Piedrafita D, Feng GJ, Sweet M, et al. Altered immune responses and susceptibility to Leishmania major and Staphylococcus aureus infection in IL-18-deficient mice. The Journal of Immunology. 1999;163:2821–2828. [PubMed] [Google Scholar]
- 167.Ohkusu K, Yoshimoto T, Takeda K, Ogura T, Kashiwamura S, Iwakura Y, et al. Potentiality of interleukin-18 as a useful reagent for treatment and prevention of Leishmania major infection. Infection and Immunity. 2000;68:2449–2456. doi: 10.1128/iai.68.5.2449-2456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Monteforte GM, Takeda K, Rodriguez-Sosa M, Akira S, David JR, Satoskar AR. Genetically resistant mice lacking IL-18 gene develop Th1 response and control cutaneous Leishmania major infection. The Journal of Immunology. 2000;164:5890–5893. doi: 10.4049/jimmunol.164.11.5890. [DOI] [PubMed] [Google Scholar]
- 169.Santos MLS, Reis EC, Bricher PN, Sousa TN, Brito CFA, Lacerda MVG, et al. Contribution of inflammasome genetics in Plasmodium vivax malaria. Infect Genet Evol. 2016;40:162–166. doi: 10.1016/j.meegid.2016.02.038. [DOI] [PubMed] [Google Scholar]
- 170.Shio MT, Tiemi Shio M, Eisenbarth SC, Savaria M, Vinet AF, Bellemare M-J, et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. 2009;5:e1000559. doi: 10.1371/journal.ppat.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE. 2009;4:e6510. doi: 10.1371/journal.pone.0006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Griffith JW, Sun T, McIntosh MT, Bucala R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol. 2009;183:5208–5220. doi: 10.4049/jimmunol.0713552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, et al. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. CellReports. 2014;6:196–210. doi: 10.1016/j.celrep.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Reimer T, Shaw MH, Franchi L, Coban C, Ishii KJ, Akira S, et al. Experimental cerebral malaria progresses independently of the Nlrp3 inflammasome. Eur J Immunol. 2010;40:764–769. doi: 10.1002/eji.200939996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kordes M, Matuschewski K, Hafalla JCR. Caspase-1 activation of interleukin-1β (IL-1β) and IL-18 is dispensable for induction of experimental cerebral malaria. Infection and Immunity. 2011;79:3633–3641. doi: 10.1128/IAI.05459-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza MDC, Franklin BS, et al. Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog. 2014;10:e1003885. doi: 10.1371/journal.ppat.1003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Witola WH, Mui E, Hargrave A, Liu S, Hypolite M, Montpetit A, et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infection and Immunity. 2011;79:756–766. doi: 10.1128/IAI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gov L, Karimzadeh A, Ueno N, Lodoen MB. Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. MBio. 2013;4:e00255–13. doi: 10.1128/mBio.00255-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio. 2014;5:e01117–13. doi: 10.1128/mBio.01117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ewald SE, Chavarria-Smith J, Boothroyd JC. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infection and Immunity. 2014;82:460–468. doi: 10.1128/IAI.01170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mortimer L, Moreau F, Cornick S, Chadee K. Gal-lectin-dependent contact activates the inflammasome by invasive Entamoeba histolytica. Mucosal Immunol. 2014;7:829–841. doi: 10.1038/mi.2013.100. [DOI] [PubMed] [Google Scholar]
- 182.Mortimer L, Moreau F, Cornick S, Chadee K. The NLRP3 Inflammasome Is a Pathogen Sensor for Invasive Entamoeba histolytica via Activation of α5β1 Integrin at the Macrophage-Amebae Intercellular Junction. PLoS Pathog. 2015;11:e1004887. doi: 10.1371/journal.ppat.1004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gonçalves VM, Matteucci KC, Buzzo CL, Miollo BH, Ferrante D, Torrecilhas AC, et al. NLRP3 controls Trypanosoma cruzi infection through a caspase-1-dependent IL-1R-independent NO production. PLoS Negl Trop Dis. 2013;7:e2469. doi: 10.1371/journal.pntd.0002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Silva GK, Costa RS, Silveira TN, Caetano BC, Horta CV, Gutierrez FRS, et al. Apoptosis-associated speck-like protein containing a caspase recruitment domain inflammasomes mediate IL-1β response and host resistance to Trypanosoma cruzi infection. J Immunol. 2013;191:3373–3383. doi: 10.4049/jimmunol.1203293. [DOI] [PubMed] [Google Scholar]
- 185.Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A, et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell. 2016;167:444–456.e14. doi: 10.1016/j.cell.2016.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Furlan R, Filippi M, Bergami A, Rocca MA, Martinelli V, Poliani PL, et al. Peripheral levels of caspase-1 mRNA correlate with disease activity in patients with multiple sclerosis; a preliminary study. J Neurol Neurosurg Psychiatr. 1999;67:785–788. doi: 10.1136/jnnp.67.6.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ming X, Li W, Maeda Y, Blumberg B, Raval S, Cook SD, et al. Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J Neurol Sci. 2002;197:9–18. doi: 10.1016/s0022-510x(02)00030-8. [DOI] [PubMed] [Google Scholar]
- 188.Huang W-X, Huang P, Hillert J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler. 2004;10:482–487. doi: 10.1191/1352458504ms1071oa. [DOI] [PubMed] [Google Scholar]
- 189.Nicoletti F, Di Marco R, Mangano K, Patti F, Reggio E, Nicoletti A, et al. Increased serum levels of interleukin-18 in patients with multiple sclerosis. Neurology. 2001;57:342–344. doi: 10.1212/wnl.57.2.342. [DOI] [PubMed] [Google Scholar]
- 190.Chen Y-C, Chen S-D, Miao L, Liu Z-G, Li W, Zhao Z-X, et al. Serum levels of interleukin (IL)-18, IL-23 and IL-17 in Chinese patients with multiple sclerosis. J Neuroimmunol. 2012;243:56–60. doi: 10.1016/j.jneuroim.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 191.Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, et al. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. The Journal of Immunology. 1999;163:2403–2409. [PubMed] [Google Scholar]
- 192.Dumas A, Amiable N, de Rivero Vaccari JP, Chae JJ, Keane RW, Lacroix S, et al. The inflammasome pyrin contributes to pertussis toxin-induced IL-1β synthesis, neutrophil intravascular crawling and autoimmune encephalomyelitis. PLoS Pathog. 2014;10:e1004150. doi: 10.1371/journal.ppat.1004150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Martin BN, Wang C, Zhang C-J, Kang Z, Gulen MF, Zepp JA, et al. T cell-intrinsic ASC critically promotes TH17-mediated experimental autoimmune encephalomyelitis. Nat Immunol. 2016;17:583–592. doi: 10.1038/ni.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KHG. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol. 2011;186:5738–5748. doi: 10.4049/jimmunol.1003597. [DOI] [PubMed] [Google Scholar]
- 195.Sui J, Li H, Fang Y, Liu Y, Li M, Zhong B, et al. NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in han chinese. Arthritis Rheum. 2012;64:647–654. doi: 10.1002/art.33370. [DOI] [PubMed] [Google Scholar]
- 196.Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, et al. Biologics in Rheumatoid Arthritis Genetics, Genomics Study Syndicate (BRAGGSS) Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis. 2014;73:1202–1210. doi: 10.1136/annrheumdis-2013-203276. [DOI] [PubMed] [Google Scholar]
- 197.Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512:69–73. doi: 10.1038/nature13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shin MS, Kang Y, Lee N, Wahl ER, Kim SH, Kang KS, et al. Self double-stranded (ds)DNA induces IL-1β production from human monocytes by activating NLRP3 inflammasome in the presence of anti-dsDNA antibodies. J Immunol. 2013;190:1407–1415. doi: 10.4049/jimmunol.1201195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhang W, Cai Y, Xu W, Yin Z, Gao X, Xiong S. AIM2 facilitates the apoptotic DNA-induced systemic lupus erythematosus via arbitrating macrophage functional maturation. J Clin Immunol. 2013;33:925–937. doi: 10.1007/s10875-013-9881-6. [DOI] [PubMed] [Google Scholar]
- 200.Voronov E, Dayan M, Zinger H, Gayvoronsky L, Lin J-P, Iwakura Y, et al. IL-1 beta-deficient mice are resistant to induction of experimental SLE. Eur Cytokine Netw. 2006;17:109–116. [PubMed] [Google Scholar]
- 201.Pontillo A, Girardelli M, Kamada AJ, Pancotto JAT, Donadi EA, Crovella S, et al. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity. 2012;45:271–278. doi: 10.3109/08916934.2011.637532. [DOI] [PubMed] [Google Scholar]
- 202.Kahlenberg JM, Yalavarthi S, Zhao W, Hodgin JB, Reed TJ, Tsuji NM, et al. An essential role of caspase 1 in the induction of murine lupus and its associated vascular damage. Arthritis Rheumatol. 2014;66:152–162. doi: 10.1002/art.38225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yang C-A, Huang S-T, Chiang B-L. Sex-dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatology (Oxford) 2015;54:324–331. doi: 10.1093/rheumatology/keu318. [DOI] [PubMed] [Google Scholar]
- 204.Lu A, Li H, Niu J, Wu S, Xue G, Yao X, et al. Hyperactivation of the NLRP3 Inflammasome in Myeloid Cells Leads to Severe Organ Damage in Experimental Lupus. J Immunol. 2017;198:1119–1129. doi: 10.4049/jimmunol.1600659. [DOI] [PubMed] [Google Scholar]
- 205.Dellalibera-Joviliano R, Dos Reis ML, de Cunha FQ, Donadi EA. Kinins and cytokines in plasma and cerebrospinal fluid of patients with neuropsychiatric lupus. J Rheumatol. 2003;30:485–492. [PubMed] [Google Scholar]
- 206.Calvani N, Richards HB, Tucci M, Pannarale G, Silvestris F. Up-regulation of IL-18 and predominance of a Th1 immune response is a hallmark of lupus nephritis. Clin Exp Immunol. 2004;138:171–178. doi: 10.1111/j.1365-2249.2004.02588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tucci M, Quatraro C, Lombardi L, Pellegrino C, Dammacco F, Silvestris F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum. 2008;58:251–262. doi: 10.1002/art.23186. [DOI] [PubMed] [Google Scholar]
- 208.Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest. 2013;123:4695–4705. doi: 10.1172/JCI71543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352:1232–1236. doi: 10.1126/science.aaf3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016;44:833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 211.Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell. 2016;166:624–636. doi: 10.1016/j.cell.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sanchez M, Kolar SL, Müller S, Reyes CN, Wolf AJ, Ogawa C, et al. O-Acetylation of Peptidoglycan Limits Helper T Cell Priming and Permits Staphylococcus aureus Reinfection. Cell Host and Microbe. 2017 doi: 10.1016/j.chom.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E. Neutrophil P2×7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nature Communications. 2016;7:10555. doi: 10.1038/ncomms10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Chen KW, Groβ CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. CellReports. 2014;8:570–582. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 215.Cullen SP, Kearney CJ, Clancy DM, Martin SJ. Diverse Activators of the NLRP3 Inflammasome Promote IL-1β Secretion by Triggering Necrosis. CellReports. 2015;11:1535–1548. doi: 10.1016/j.celrep.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 216.Edgeworth JD, Spencer J, Phalipon A, Griffin GE, Sansonetti PJ. Cytotoxicity, interleukin-1beta processing following Shigella flexneri infection of human monocyte-derived dendritic cells. Eur J Immunol. 2002;32:1464–1471. doi: 10.1002/1521-4141(200205)32:5<1464::AID-IMMU1464>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 217.Martín-Sánchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016;23:1219–1231. doi: 10.1038/cdd.2015.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000;38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- 219.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Li H, Willingham SB, Ting JP-Y, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. The Journal of Immunology. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kool M, Pétrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–3759. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 222.Li H, Nookala S, Re F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. The Journal of Immunology. 2007;178:5271–5276. doi: 10.4049/jimmunol.178.8.5271. [DOI] [PubMed] [Google Scholar]
- 223.Marichal T, Ohata K, Bedoret D, Mesnil C, Sabatel C, Kobiyama K, et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med. 2011;17:996–1002. doi: 10.1038/nm.2403. [DOI] [PubMed] [Google Scholar]
- 224.Jacobson LS, Lima H, Goldberg MF, Gocheva V, Tsiperson V, Sutterwala FS, et al. Cathepsin-mediated necrosis controls the adaptive immune response by Th2 (T helper type 2)-associated adjuvants. J Biol Chem. 2013;288:7481–7491. doi: 10.1074/jbc.M112.400655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti T-D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010;185:4912–4920. doi: 10.4049/jimmunol.1002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Chen GY, Liu M, Wang F, Bertin J, Núñez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol. 2011;186:7187–7194. doi: 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Allen IC, TeKippe EM, Woodford R-MT, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–1056. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti T-D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci USa. 2010;107:21635–21640. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proceedings of the National Academy of Sciences. 2003;100:2645–2650. doi: 10.1073/pnas.0437939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007;67:1062–1071. doi: 10.1158/0008-5472.CAN-06-2956. [DOI] [PubMed] [Google Scholar]
- 233.Saijo Y, Tanaka M, Miki M, Usui K, Suzuki T, Maemondo M, et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. The Journal of Immunology. 2002;169:469–475. doi: 10.4049/jimmunol.169.1.469. [DOI] [PubMed] [Google Scholar]
- 234.van Deventer HW, Burgents JE, Wu QP, Woodford R-MT, Brickey WJ, Allen IC, et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. 2010;70:10161–10169. doi: 10.1158/0008-5472.CAN-10-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bruchard M, Mignot G, Derangère V, Chalmin F, Chevriaux A, Végran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19:57–64. doi: 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- 236.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 237.Garaude J, Kent A, van Rooijen N, Blander JM. Simultaneous targeting of toll- and nod-like receptors induces effective tumor-specific immune responses. Science Translational Medicine. 2012;4:120ra16–120ra16. doi: 10.1126/scitranslmed.3002868. [DOI] [PubMed] [Google Scholar]
- 238.Stoecklein VM, Osuka A, Ishikawa S, Lederer MR, Wanke-Jellinek L, Lederer JA. Radiation exposure induces inflammasome pathway activation in immune cells. J Immunol. 2015;194:1178–1189. doi: 10.4049/jimmunol.1303051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Hu B, Jin C, Li H-B, Tong J, Ouyang X, Cetinbas NM, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science. 2016;354:765–768. doi: 10.1126/science.aaf7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Muñoz-Arias I, Doitsh G, Yang Z, Sowinski S, Ruelas D, Greene WC. Blood-Derived CD4 T Cells Naturally Resist Pyroptosis during Abortive HIV-1 Infection. Cell Host and Microbe. 2015;18:463–470. doi: 10.1016/j.chom.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. 2016;352:aad1210. doi: 10.1126/science.aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bruchard M, Rebé C, Derangère V, Togbé D, Ryffel B, Boidot R, et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol. 2015:1–15. doi: 10.1038/ni.3202. [DOI] [PubMed] [Google Scholar]
- 243.Felley LE, Sharma A, Theisen E, Romero-Masters JC, Sauer J-D, Gumperz JE. Human Invariant NKT Cells Induce IL-1β Secretion by Peripheral Blood Monocytes via a P2×7-Independent Pathway. J Immunol. 2016;197:2455–2464. doi: 10.4049/jimmunol.1600790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chow MT, Duret H, Andrews DM, Faveeuw C, Möller A, Smyth MJ, et al. Type I NKT-cell-mediated TNF-α is a positive regulator of NLRP3 inflammasome priming. Eur J Immunol. 2014;44:2111–2120. doi: 10.1002/eji.201344329. [DOI] [PubMed] [Google Scholar]
- 245.Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol. 2013;14:52–60. doi: 10.1038/ni.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JP-Y, Coers J, et al. Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host and Microbe. 2015;18:320–332. doi: 10.1016/j.chom.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Yao Y, Vent-Schmidt J, McGeough MD, Wong M, Hoffman HM, Steiner TS, et al. Tr1 Cells, but Not Foxp3+ Regulatory T Cells, Suppress NLRP3 Inflammasome Activation via an IL-10-Dependent Mechanism. J Immunol. 2015;195:488–497. doi: 10.4049/jimmunol.1403225. [DOI] [PubMed] [Google Scholar]
- 248.Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, et al. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009;460:269–273. doi: 10.1038/nature08100. [DOI] [PubMed] [Google Scholar]