

Abstract
Ubiquitylation is a fundamental mechanism of signal transduction that regulates immune responses and many other biological processes. Similar to phosphorylation, ubiquitylation is a reversible process that is counter-regulated by ubiquitylating enzymes and deubiquitylating enzymes (DUBs). Despite the identification of a large number of DUBs, our knowledge of the function and activities of this family of enzymes is just starting to accumulate. As described in this Review, recent studies of several DUBs, in particular CYLD and A20, show that deubiquitylation has an important role in the regulation of both innate and adaptive immune responses.
The tagging of ubiquitin molecules to substrate proteins has long been known as a mechanism that targets proteins for degradation by the 26S proteasome. However, it is now clear that ubiquitylation also mediates various non-degradative molecular functions, such as protein trafficking, protein–protein interactions and regulation of signal-transduction events1–3. It has also become apparent that ubiquitylation is a reversible reaction, in which the ubiquitin chains are conjugated and deconjugated by ubiquitylating enzymes and deubiquitylating enzymes (DUBs), respectively (BOX 1). The ubiquitylating enzymes and their role in the immune system have been extensively discussed elsewhere1–3, so this Review focuses on the DUBs, specifically those involved in the regulation of immune responses.
Box 1. Protein ubiquitylation.
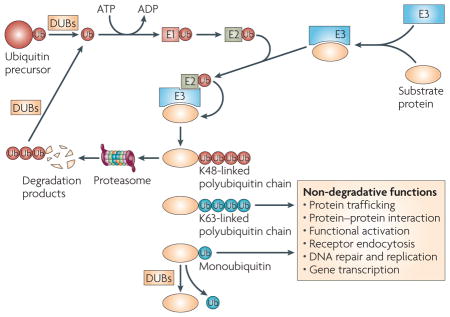
The process of ubiquitylation involves the covalent tagging of a substrate protein with either a single ubiquitin (Ub) molecule (monoubiquitylation) or a chain of ubiquitin molecules linked between their carboxy-terminal glycine residue and an internal lysine residue (polyubiquitylation)2. The two most frequently used internal lysines are lysine 48 and lysine 63, yielding K48- and K63-linked polyubiquitin chains, respectively (see figure). Whereas the K48-linked ubiquitin chains generally mark substrate proteins for proteasomal degradation, the K63-linked ubiquitin chains mediate various non-degradative functions, including protein trafficking, protein–protein interactions and the functional activation of signalling factors1,2,109. Monoubiquitylation also mediates various non-degradative cellular processes, such as receptor endocytosis, DNA repair, DNA replication and gene transcription110. The ubiquitylation reaction is catalysed by the sequential and cooperative actions of three enzymes: the ubiquitin-activating enzyme (E1), the ubiquitin-conjugating enzyme (E2) and the ubiquitin ligase (E3). Deubiquitylating enzymes (DUBs) can deconjugate the ubiquitin chains from ubiquitylated proteins and proteasomal degradation products, thereby reversing the ubiquitylation process and regenerating free ubiquitin molecules.
The substrate specificity of ubiquitylation is determined by the E3 ligases, a large family of proteins that mediate substrate recognition and ubiquitin transfer from E2 enzymes to the substrate2. The E3 ligases typically contain an E2-binding domain, a catalytic domain that mediates ubiquitin transfer from E2 to the substrate proteins, and one or more protein-interaction domains that bind substrate proteins. Some E3 ligases are multi-protein complexes, with the E2- and substrate-binding functions being carried out by separate protein components. The typical E2-binding domains include the really interesting new gene (RING) domain and the homologous to E6-associated protein carboxyl terminus (HECT) domain. Several E3 ligases have been linked to different aspects of immune function, such as T-cell development and activation and the establishment of immunological tolerance3,9.
DUBs are proteases that cleave ubiquitin chains from protein substrates or their degradation remnants, a process that is known as deubiquitylation. The human genome encodes almost 100 DUBs, although the function of most of them remains unknown4. On the basis of their domain structures, DUBs can be divided into five families: the ubiquitin carboxy-terminal hydrolases (UCHs), the ubiquitin-specific proteases (USPs), the ovarian tumour-related proteases (OTUs), the Machado–Joseph disease protein domain proteases (MJDs) and the JAB1/PAB1/MPN-domain-containing metallo-enzymes (JAMMs)4. The UCH family comprises a small number of structurally related DUBs that are known to cleave short ubiquitylated peptides and that have an important role in the recycling of free ubiquitin. The physiological role of UCHs is highlighted by the association of one member, UCHL1, with the development of neurodegenerative diseases5. USPs form the largest family of DUBs, and they are characterized by the presence of two conserved sequence motifs — the cysteine and histidine boxes — in their catalytic domain4. 53 USP genes have been identified in the human genome, although some of these might not encode functional DUBs4. Similarly, there are 54 USP-family members in the mouse genome. As discussed in the following sections, one USP-family member, CYLD, has been extensively studied and shown to regulate various immune functions. OTUs form the second largest mammalian DUB family, being composed of about 24 members in the human genome4,6. The first OTU gene was identified in Drosophila melanogaster, in which it was found to regulate the development of the ovaries; mammalian OTUs were later discovered on the basis of OTU-domain homology6. Several OTU-family members (A20, DUBA, otubain-1 and Cezanne) are involved in the regulation of immune responses, as discussed later. So far, little is known about the MJD and JAMM families of DUBs, and it is unclear whether they have a role in immune regulation.
Similar to the E3 ubiquitin ligases (BOX 1), DUBs have a certain degree of substrate specificity, and this property is contributed by several factors. First, in addition to the catalytic domain, DUBs often contain different protein-interaction domains, which enables them to bind specific target proteins4. Second, some DUBs have a preference for specific ubiquitin branches, such as K48- or K63-linked chains (in which the ubiquitin molecules are linked through lysine 48 or lysine 63, respectively)4,7,8 (BOX 1). So, substrate recognition by DUBs can be determined by the combinatorial specificity for target proteins and for their conjugated ubiquitin branches. Third, the mode of expression and the subcellular localization of DUBs vary extensively, which contribute to their in vivo functional diversity.
This Review focuses on the DUBs that are involved in the regulation of immune responses. I discuss the recent findings regarding the molecular features and signalling function of the immunoregulatory DUBs, as well as their involvement in the regulation of innate and adaptive immune responses.
Immunoregulatory DUBs
It is now well known that ubiquitylation is an important mechanism for regulating immune responses3. Several E3 ligases have been linked to immune function, including the development and activation of lymphocytes, the establishment of immunological tolerance and innate immune functions3,9. In particular, K63-linked ubiquitylation is an important signalling mechanism involved in the activation of nuclear factor-κB (NF-κB), a transcription factor that regulates both innate and adaptive immune responses (BOX 2). Although the role of DUBs in the immune system has been less well studied, significant progress has recently been made in the study of several immunoregulatory DUBs.
Box 2. The NF-κB signalling pathway.
Nuclear factor-κB (NF-κB) comprises a family of transcription factors that regulate numerous genes involved in innate and adaptive immune responses. The mammalian NF-κB family consists of five structurally related members, RELA (also known as p65), RELB, REL (also known as cREL), NF-κB1 (also known as p50) and NF-κB2 (also known as p52), which function as homo- and heterodimers. The NF-κB proteins are normally sequestered in the cytoplasm as inactive complexes by physical association with inhibitors of NF-κB (IκBs) or the precursor proteins of NF-κB1 and NF-κB2, p105 and p100 respectively. Activation of most NF-κB members is mediated by a canonical pathway that involves the proteasomal degradation of IκBs and p105 (Ref. 111), whereas the activation of RELB and NF-κB2 requires a noncanonical pathway that involves processing of p100 (Refs 112,113). The canonical pathway of NF-κB activation is induced by diverse immune receptors, which induce the activation of an IκB kinase (IKK) complex that is composed of two catalytic subunits (IKKα and IKKβ) and the regulatory subunit IKKγ (also known as NEMO)114. The IKK complex phosphorylates IκBs and p105, which triggers K48-linked ubiquitylation and proteasomal degradation of these inhibitors, leading to the nuclear translocation of canonical NF-κB complexes.
IKK activation by Toll-like receptors and cytokine receptors requires members of the TRAF (tumour-necrosis factor receptor-associated factor) family, in particular TRAF2 and TRAF6. These intermediate signalling molecules function as both adaptors and E3 ubiquitin ligases, catalysing K63-linked self-ubiquitylation and the ubiquitylation of other signalling molecules, including receptor-interacting protein 1 (RIP1) and IKKγ. An important function of these ubiquitylation events is to recruit and activate a ubiquitin-dependent IKK-activating kinase, TAK1 (transforming-growth-factor-β-activated kinase 1). TAK1 recognizes the K63-linked ubiquitin chains through its partner protein, TAB2 (TAK1-binding protein 2) or TAB3, which contains a ubiqutin-associated domain. IKKγ also has ubiquitin-binding function and recognizes K63-linked ubiquitin chains29,31. So, the ubiquitin chains conjugated on RIP1 and TRAFs form a platform that recruits TAK1 and IKK complexes, leading to their activation1.
CYLD
CYLD was originally identified as a tumour suppressor, the mutation of which predisposes patients for the development of tumours of the hair follicles (cylindromas)10. The mutations often occur in the carboxy-terminal portion of CYLD, which contains a DUB domain10, and the DUB function of CYLD was subsequently revealed by functional proteomics11 and linked to signal transduction involved in the activation of NF-κB12–14. So, it is probable that mutations of CYLD disrupt its DUB catalytic activity and cause dysregulated activation of NF-κB, which in turn promotes cell survival and tumorigenesis. A study using Cyld-knockout mice further indicates that CYLD deficiency might promote the transactivation function of NF-κB by increasing the ubiquitylation and nuclear translocation of an NF-κB co-activator protein, BCL-3 (B-cell lymphoma 3)15.
It is now clear that CYLD is a member of the USP family of DUBs that targets multiple signalling molecules, such as members of the TRAF (tumour-necrosis factor (TNF) receptor (TNFR)-associated factor) family, the IKK (inhibitor of NF-κB (IκB) kinase) regulatory subunit IKKγ (also known as NEMO)12–14,16,17, the SRC protein tyrosine kinase LCK18, the NF-κB co-activator protein BCL-3 (Ref. 15), transforming growth factor-β (TGFβ) activated kinase 1 (TAK1)19, receptor-interacting protein 1 (RIP1)20 and transient receptor potential channel A1 (TRPA1)21. Not surprisingly, CYLD regulates diverse biological functions, including host defence against infections22,23, immune-cell development, activation and inflammation18,19,24,25, cell survival12–14, cell proliferation and tumorigenesis15,24, microtubule assembly and cell migration26, mitotic cell entry27, calcium-channel function21, spermatogenesis20 and osteoclastogenesis17.
Consistent with its substrate diversity, CYLD contains several protein-interaction domains, including three cytoskeleton-associated protein glycine-rich (Cap-Gly) domains, two proline-rich motifs and a zinc-finger-like domain10. The Cap-Gly domains typically mediate association with microtubule proteins, whereas the proline-rich motifs interact with proteins through SH3 domains. The first Cap-Gly domain of CYLD mediates its association with microtubules and the regulation of microtubule assembly26. Interestingly, the tertiary structure of the third Cap-Gly domain of CYLD resembles that of an SH3 domain that is known to bind proline-rich sequences28. The possession of both proline-rich motifs and an SH3-like domain could allow CYLD to interact with target proteins containing either SH3 domains or proline-rich motifs. Indeed, the third Cap-Gly domain of CYLD interacts with a proline-rich region in one of its targets, IKKγ (Ref. 28). In addition to these well-defined protein domains, new domains of CYLD have been defined, based on mutational analyses, for the interaction of CYLD with TRAF2, IKKγ and BCL-3 (Refs 13,15).
A prominent feature of the target proteins of CYLD is their conjugation with K63-linked ubiquitin chains, which seem to facilitate protein–protein interactions in the recruitment and activation of downstream signalling molecules by the CYLD target1. In particular, the K63-linked ubiquitin chains of RIP1 form a platform that recruits the IKK complex and its upstream kinase, TAK1, leading to efficient activation of these kinases29–31. Strong biochemical evidence indicates that CYLD is specific for K63-linked ubiquitin chains13,15. Consistent with this, the USP catalytic domain of CYLD has a unique structure that differs from that of other USPs and seems to have specific determinants for mediating the deconjugation of K63-linked ubiquitin chains32. However, it is unclear whether CYLD is exclusively a K63-specific DUB, as CYLD has also been shown to have activity towards K48-linked ubiquitin chains in certain in vitro14 and in vivo18,33 conditions. Results from two studies indicate that CYLD can also deconjugate K48-linked ubiquitin chains to prevent the proteasomal degradation of some target proteins21,33. So, it is possible that the ubiquitin-chain specificity of CYLD is dependent on the target proteins. Moreover, recent evidence indicates that the function of CYLD might also involve adaptor proteins17,34. CYLD binding to one of its targets, TRAF6, requires the adaptor protein p62, which promotes the deubiquitylation of TRAF6 by CYLD17 and probably also modulates the DUB activity of CYLD through induction of CYLD ubiquitylation34.
A20
A20 (also known as TNFAIP3) is a zinc-finger-domain-containing protein that is required for terminating the NF-κB activation signal mediated by innate immune receptors, such as TNFR, Toll-like receptors (TLRs) and an intracellular pattern-recognition molecule, NOD2 (nucleotide-binding oligomerization domain protein 2)35–38. The amino-terminal region of A20 contains an OTU domain, which has DUB activity towards several NF-κB signalling factors, including TRAF6, RIP1, RIP2 and IKKγ (Refs 8,37–40). A unique feature of A20 is its ability to function as both a DUB and an E3 ligase8. A20 catalyses the K48-linked ubiquitylation of RIP1 through its carboxy-terminal zinc-finger domain, an action that targets RIP1 for proteasomal degradation. Concurrently, A20 removes K63-linked ubiquitin chains from RIP1, which not only inactivates the signalling function of RIP1 but might also facilitate its K48-linked ubiquitylation and degradation8. It is important to note, however, that the A20-mediated K48-linked ubiquitylation and degradation of RIP1 might involve a more complex mechanism, as this function of A20 also requires ITCH (itchy homologue E3 ubiquitin protein ligase), a K48-specific E3 ligase that stably associates with A20 (Ref. 41). It is unclear whether both A20 and ITCH ubiquitylate RIP1 or whether ITCH regulates the E3 ligase activity of A20 through ubiquitylation of a regulatory protein.
How the DUB substrate specificity of A20 is regulated is also not completely understood. Although A20 has preferential DUB activity for removing K63-linked ubiquitin chains in vivo8, recombinant A20 or the A20 OTU domain cleaves both K63- and K48-linked ubiquitin chains in vitro with a preference for K48-linked ubiquitin chains42,43. It seems that the substrate specificity of A20 is modulated by the conjugation of ubiquitin chains to specific target proteins. A recent study shows that A20 efficiently removes K63-linked ubiquitin chains when they are conjugated to TRAF6 (Ref. 43). Interestingly, however, A20 does not disassemble the K63-linked ubiquitin chains but instead removes the intact ubiquitin chains by cleaving the linkage between TRAF6 and the ubiquitin chains43. It is therefore probable that the specificity of A20 for K63-linked ubiquitylated targets is not determined by its intrinsic DUB catalytic activity but possibly through its specific interaction with the ubiquitylated proteins. Accumulated evidence indicates that this function of A20 is in turn mediated through adaptor proteins. One such protein is ABIN1 (A20-binding inhibitor of NF-κB 1; also known as TNIP1), which physically associates with A20 and functions as an adaptor to recruit A20 to its target IKKγ (Ref. 40). Small interfering RNA (siRNA)-mediated knockdown of ABIN1 expression prevents A20 from deubiquitylating IKKγ (Ref. 40). Another protein that regulates the DUB function of A20 is TAX1-binding protein 1 (TAX1BP1)44,45, a zinc-finger-domain-containing protein that was originally found to associate with the Tax protein encoded by human T-cell leukaemia virus type 1 (Ref. 46). TAX1BP1 also interacts with A20 (Ref. 47) and mediates the recruitment of A20 to RIP1 and TRAF6. Notably, both ABIN1 and TAX1BP1 contain a ubiquitin-binding domain, which seems to mediate their association with ubiquitylated TRAF6 and RIP1 (Refs 45,48). So, A20 interacts with its targets through ubiquitin-binding adaptor proteins.
Cezanne
Cezanne (cellular zinc finger anti-NF-κB; also known as OTUD7B) was identified on the basis of its sequence homology to A20 (REF. 49). Similar to A20, Cezanne has a DUB catalytic domain that belongs to the OTU family, and it negatively regulates activation of NF-κB in the TNF signalling pathway49,50. In response to TNFR signalling, the expression of Cezanne is induced and it is recruited to the TNFR, where it seems to deubiquitylate RIP1 and thereby negatively regulate the activation of IKK50,51. Cezanne might function, similar to A20, in the feedback control of NF-κB activation, although its functional relationship with A20 is currently unclear.
DUBA
Deubiquitylating enzyme A (DUBA; also known as OTUD5), an OTU-family member, was recently identified through siRNA screening as a negative regulator of type I interferon (IFN) induction7. Similar to A20, DUBA contains an OTU domain and selectively cleaves K63-linked ubiquitin chains in transfected cells. However, unlike A20 and CYLD, DUBA is not required for the negative regulation of NF-κB. Instead, DUBA seems to selectively regulate the activation of IFN-regulatory factor 3 (IRF3) and IRF7, which are transcription factors that regulate IFN expression.
Otubain-1
Another OTU-family member, otubain-1 (also known as OTUB1), was identified by affinity purification using biotin-conjugated ubiquitin aldehyde (a competitive DUB inhibitor)52. The DUB activity of otubain-1 was shown by its ability to cleave a tetraubiquitin substrate in vitro. In a separate study using a yeast two-hybrid screen, otubain-1 was isolated as a protein that associates with the E3 ligase GRAIL (gene related to anergy in lymphocytes)53. An alternatively spliced isoform of otubain-1, ARF1 (alternative reading frame 1), shares the C-termimal portion with otubain-1 but lacks an intact OTU domain53. Otubain-1 and ARF1 both bind GRAIL, but they have opposite effects in regulating the self-ubiquitylation and degradation of GRAIL. Surprisingly, although otubain-1 is a DUB, it promotes rather than inhibits the K48-linked self-ubiquitylation and proteolysis of GRAIL53. This unexpected function of otubain-1 might be mediated through the inhibition of USP8, a DUB that binds to and deubiquitylates GRAIL; however, it is not known how otubain-1 might inhibit USP8 (Ref. 53). Nevertheless, it seems that the DUB activity of otubain-1 is important, because ARF1, which lacks the OTU domain and therefore the DUB activity of otubain-1, opposes the function of otubain-1 (Ref. 53). It has been proposed that the ratio between otubain-1 and ARF1 might have an important role in regulating the fate and function of GRAIL53.
USP15
A recent study indicates that the USP-family member, USP15, might have a role in the regulation of NF-κB activation54. In contrast to CYLD and A20, which target upstream signalling molecules in the NF-κB pathway, USP15 negatively regulates the K48-linked ubiquitylation of IκBα, a key downstream signalling step that triggers IκBα proteolysis and NF-κB nuclear translocation55. This function of USP15 was discovered on the basis of its physical association with the COP9 signalosome (CSN), a multi-protein complex that has diverse functions, including the regulation of protein ubiquitylation and degradation56. siRNA-mediated knockdown of a CSN subunit, which destabilizes the CSN complex, prolongs the TNF-stimulated ubiquitylation and degradation of IκBα, leading to sustained nuclear translocation of NF-κB54. A similar, although much milder, phenotype was obtained by knockdown of USP15. It is unclear whether CSN associates with additional DUBs or has other mechanisms that interfere with IκBα ubiquitylation.
Cytokine-induced DUBs
The expression of a subfamily of DUBs is induced in lymphocytes by cytokines, such as interleukin-2 (IL-2), IL-3, IL-4, IL-5 and GM-CSF (granulocyte/macrophage colony-stimulating factor)57. These cytokine-induced DUBs, which include mouse DUB1 (also known as USP36), DUB1A, DUB2 and DUB2A, and human DUB3 and USP17, might have a role in regulating the growth and survival of lymphocytes57. For example, overexpression of DUB2 negatively regulates K48-linked ubiquitylation of the common cytokine-receptor γ-chain (γc) by counteracting the ubiquitin-ligase function of CBL58. Consequently, DUB2 stabilizes γc, prolongs IL-2-induced activation of JAK–STAT (Janus kinase–signal transducer and activator of transcription) signalling and promotes the survival of lymphocytes58,59. However, the physiological role of cytokine-induced DUBs in immune regulation has not yet been confirmed by in vivo models.
Deubiquitylation and innate immunity
During the early phase of an infection, pathogen-associated molecular patterns (PAMPs) are detected by host pattern-recognition receptors (PRRs), including TLRs and the cytoplasmic RNA sensors RIG-I (retinoic-acid-inducible gene I) and MDA5 (melanoma differentiation-associated gene 5). Consequently, these PRRs transduce signals that mediate the activation of IKK, which leads to NF-κB activation and the transcription of genes encoding various pro-inflammatory cytokines (FIG. 1). RIG-I, MDA5 and some TLRs also activate the IKK-related kinases, TBK1 (TANK-binding kinase 1) and IKKε, which in turn activate IRF3 and IRF7, resulting in the induction of expression of type I IFNs60. These signalling events, as well as the actions of the cytokines that are produced, form an important part of the innate immune response to infections. This response is subject to tight regulation by negative mechanisms, as the uncontrolled production of pro-inflammatory cytokines and type I IFNs can have severe pathological consequences, such as sepsis, chronic inflammatory diseases and autoimmune disorders61,62. Accumulating evidence indicates that the DUBs have a crucial role in this control of innate immune-receptor signalling.
Figure 1. Regulation of innate immune-receptor signalling by deubiquitylating enzymes (DUBs).

Toll-like receptors (TLRs) stimulate the K63-linked ubiquitylation of tumour-necrosis factor receptor-associated factor 6 (TRAF6) and TRAF3, which leads to the recruitment of downstream signalling molecules. a | Ubiquitylated TRAF6 recruits the IKK (IκB (inhibitor of NF-κB (nuclear factor-κB) kinase) complex (which consists of IKKα, IKKβ and IKKγ) and its activating kinase, transforming growth factor-β-activated kinase 1 (TAK1) — in association with TAK1-binding protein 1 (TAB1) and TAB2 — through the ubiquitin-binding function of IKKγ and TAB2, leading to activation of these kinases. The IKK complex phosphorylates IκB, triggering its K48-linked ubiquitylation and proteasomal degradation. Through TAX1-binding protein 1 (TAX1BP1) and A20-binding inhibitor of NF-κB 1 (ABIN1), A20 binds to and deubiquitylates TRAF6 and IKKγ, respectively, thereby negatively regulating NF-κB signalling. The role of CYLD is less clear (see main text). Deubiquitylation of IκB, which is another mechanism for the negative regulation of NF-κB, involves USP15 (ubiquitin-specific protease 15), a DUB that is associated with the COP9 signalosome (CSN). b | Ubiquitylated TRAF3 recruits the IKK-related kinases, TANK-binding kinase 1 (TBK1) and IKKε, through the adaptor protein TANK (TRAF-family-member-associated NF-κB activator). Similar to IKKγ, TANK is ubiquitylated in the signalling complex, although how the ubiquitylation of TANK contributes to the activation of TBK1 and/or IKKε is unclear. Deubiquitylation of TRAF3 is mediated by DUBA, a crucial and specific negative regulator of type I interferon (IFN) induction. c | The cytoplasmic RNA sensor, retinoic-acid-inducible gene I (RIG-I), undergoes ubiquitylation on binding to viral RNA, which is required for its association with the adaptor, IPS1 (IFNB-promoter stimulator 1), and activation of downstream signalling events. It is currently unclear which DUB regulates the deubiquitylation of RIG-I. IRF3, IFN-regulatory factor 3.
A20 as a global negative regulator of inflammation
The first DUB shown to have a role in innate immune regulation was A20. Genetic deficiency of A20 in mice causes persistent activation of NF-κB by TLRs and TNFR, leading to multiorgan inflammation, cachexia and neonatal lethality36,37. These inflammatory disorders are independent of the adaptive immune system, as they are also detected in mice that lack both A20 and RAG1 (recombination-activating gene 1; which is crucial for both B- and T-cell development)36. It seems that the loss of A20 breaks the tolerance of the innate immune system to the commensal intestinal microflora, leading to aberrant homeostatic TLR signalling and the production of pro-inflammatory mediators63. Indeed, crossing A20-knockout mice with mice that lack the common TLR adaptor MyD88 (myeloid differentiation primary-response gene 88) markedly decreases the severity of inflammation and prevents premature lethality and cachexia of the A20-knockout mice63.
The negative signalling function of A20 involves the deconjugation of K63-linked ubiquitin chains from TRAF6 and RIP1, which are central players of the TLR and TNFR signalling pathways1. Recent evidence indicates that A20 also mediates the deubiquitylation of RIP2 and thereby negatively regulates the activation of NF-κB and the induction of pro-inflammatory cytokines by the intracellular PRR, NOD2 (Ref. 38). In addition, the E3 ligase activity of A20 also contributes to its negative signalling function. A20 catalyses the K48-linked ubiquitylation of RIP1, thereby targeting it for degradation. In the absence of A20, the signal-induced K63-linked ubiquitylated forms of TRAF6 and RIPs can accumulate, leading to prolonged activation of NF-κB and the aberrant expression of pro-inflammatory cytokines. So, A20 is a central player in the control of inflammatory responses (FIG. 1a).
CYLD in host defence against infections
Initial in vitro studies indicated that CYLD has a negative role in the regulation of TLR and TNFR signalling12–14, but how CYLD regulates innate immune responses in vivo is still unclear. Unlike the A20-knockout mice, Cyld-knockout mice do not have severe abnormalities in innate immune-receptor signalling or multiorgan inflammation15,18,23,24. Loss of CYLD in macrophages has moderate or undetectable effects on NF-κB activation by TLR ligands and TNF18,19,24. As A20 is expressed in both a basal and inducible manner by macrophages37, it is possible that CYLD is functionally redundant with A20. The fact that CYLD cannot compensate for A20 in terminating TLR and TNFR signals can probably be attributed to the phosphorylation-mediated inactivation of CYLD in stimulated cells64.
Recent evidence indicates that CYLD might have a non-redundant role in the regulation of specific aspects of innate immunity. Using an animal model of pneumococcal pneumonia, it has been shown that loss of CYLD protects mice from acute lung injury and lethality induced by Streptococcus pneumoniae23. CYLD seems to negatively regulate S. pneumoniae-induced expression of plasminogen activator inhibitor 1, a factor that protects the host during severe pneumococcal pneumonia by preventing tissue damage and decreasing bacterial translocation to the blood circulation. This function of CYLD is mediated through the negative regulation of the mitogen-activated protein kinase p38 (Ref. 23). In a similar study, CYLD was shown to negatively regulate NF-κB activation and lung inflammation in mice infected with non-typeable Haemophilus influenzae22.
The role of CYLD in regulating the host response to infections is also indicated by the finding that Cyld-knockout mice are more susceptible to the induction of colonic inflammation by dextran sulphate sodium (DSS)24. DSS is known to cause colonic epithelial damage, leading to productive invasion by commensal bacteria and the activation of innate immune cells in the mucosa. The higher sensitivity of Cyld−/− mice to DSS-induced inflammation indicates a role for CYLD in the negative regulation of the mucosal innate immune response to microorganisms.
Regulation of antiviral innate immunity by DUBA
The innate immune response to viral infections involves activation of TBK1 and IKKε, and the downstream transcription factors IRF3 and IRF7 (Ref. 65). A key intermediate molecule, TRAF3, connects TBK1 and IKKε to upstream signalling molecules66,67. Interestingly, DUBA physically interacts with TRAF3 and inhibits the self-ubiquitylation of TRAF3 (Ref. 7) (FIG. 1b). As DUBA also interferes with the binding of TRAF3 to TBK1 (Ref. 7), it is conceivable that the K63-linked ubiquitin chains of TRAF3 might facilitate its binding to the TBK1–IKKε complex. Formation of this intermediate signalling complex also requires the adaptor protein TANK (TRAF-family-member-associated NF-κB activator) 68. Analogous to IKKγ, TANK undergoes K63-linked ubiquitylation69, but how TANK ubiquitylation regulates TBK1–IKKε activation is unclear. A recent study indicates that IKKγ targets TBK1–IKKε through TANK, thereby regulating the induction of type I IFNs70. It remains to be examined whether the ubiquitylation of IKKγ and TANK is required for this newly characterized signalling axis.
Knockdown of DUBA by siRNA has little effect on NF-κB activation, which indicates that DUBA is either not involved or functionally redundant with other DUBs in the regulation of NF-κB signalling. The latter possibility is indicated by the finding that overexpression of DUBA potently inhibits the induction of an NF-κB-driven reporter gene stimulated by constitutively active forms of RIG-I and MDA5 (Ref. 7). As the ubiquitylation of RIG-I is crucial for its signalling function71, it will be important to examine whether DUBA regulates the ubiquitylation of this RNA sensor (FIG. 1c). However, a recent study indicates that CYLD is a DUB that negatively regulates the ubiquitylation of RIG-I and prevents aberrant activation of TBK1–IKKε (Ref. 72). Finally, the in vivo role of DUBA in the regulation of innate immunity awaits studies using animal models.
In vitro studies indicate that A20 also has a role in the negative regulation of antiviral immune responses mediated by TLR3 and RIG-I, in which A20 might target the adaptor protein TRIF (TIR-domain-containing adaptor protein inducing IFNβ) or downstream kinases, TBK1 and IKKε (Refs 73–75). However, a recent study using A20-knockout mice indicates that although A20 negatively regulates TRIF-mediated NF-κB signalling, this DUB is not important for the regulation of TBK1–IKKε activation, as indicated by the normal phosphorylation of the downstream target, IRF3, in A20-deficient macrophages63.
Deubiquitylation in T-cell function
T-cell development
T-cell development in the thymus involves ordered progression through distinct stages of double negative (DN), double positive (DP) and single positive (SP) cells, which are defined based on expression of the T-cell co-receptors CD4 and CD8 (Ref. 76). Signal transduction through the pre-T-cell receptor (pre-TCR) drives the proliferation of DN thymocytes and their progression to the DP stage, whereas progression from the DP to SP stage is tightly regulated by the strength of signals from the rearranged TCR. A TCR signal that is too strong, owing to the recognition of self antigens, leads to negative selection, whereas a signal that is too weak leads to death by neglect. Only those DP thymocytes that receive an adequate TCR signal strength are positively selected and further develop to SP T cells. So, many signalling factors that modulate TCR signalling have a role in T-cell development. However, our knowledge of how ubiquitylation and deubiquitylation regulate T-cell development is scant.
CYLD was the first DUB found to regulate thymocyte development18. Cyld-knockout mice are competent in producing early-stage thymocytes but have a decreased number of SP thymocytes and peripheral T cells. In contrast to its negative role in the regulation of NF-κB, CYLD positively regulates LCK18, a SRC-family protein tyrosine kinase that is required for TCR-proximal signalling and thymocyte development77. CYLD is not required for LCK activation but instead controls the association of activated LCK with its target kinase ZAP70. Therefore, the CYLD-deficient thymocytes have a defect in TCR-stimulated phosphorylation of ZAP70 and downstream signalling events. Notably, CYLD selectively interacts with the active form of LCK. As activation of LCK involves its structural change from a closed to an open conformation78, it is possible that the CYLD-binding domain in LCK is masked in its inactive, closed state.
The ubiquitylation of LCK by CBL (Casitas B-lineage lymphoma) is known to be a negative mechanism that regulates the signalling function of LCK79. However, it is controversial whether this negative regulation of LCK by CBL is through the increased degradation of LCK. One study indicated that loss of CBL expression increases the constitutive level of LCK80, but other studies have not shown this80–82. What seems to be consistent is that CBL negatively regulates phosphorylation of the LCK target, ZAP70 (Refs 81,82). As the opposite phenotype is detected in CYLD-deficient thymocytes, CYLD might oppose some of the E3 ligase functions of CBL18. At least in transfected cells, CYLD inhibits CBL-induced ubiquitylation of LCK18. Notably, CYLD removes both K48- and K63-linked ubiquitin chains from LCK18, which is in contrast to the K63-specific activity of CYLD towards TRAFs and BCL-3 (Refs 13,15). However, as loss of CYLD does not alter the steady-state level of LCK in thymocytes, it is unclear whether CYLD regulates the K48-linked ubiquitylation of LCK in vivo (which targets LCK to the proteasome). Based on studies with both CYLD- and CBL-deficient mice, it seems possible that ubiquitylation of LCK might have a non-degradative effect. One such effect might be to prevent the translocation of LCK to lipid rafts, as LCK is excluded from lipid rafts when it is co-expressed with CBL83. Because lipid rafts provide a microenvironment for concentrating TCR signalling molecules, the translocation of LCK to lipid rafts probably promotes its interaction with ZAP70, which would explain why the LCK–ZAP70 interaction is attenuated in the absence of CYLD18 and promoted in the absence of CBL82.
The finding that CYLD is not required for LCK activation but regulates LCK-mediated ZAP70 activation is consistent with the roles of LCK and ZAP70 in thymocyte development. Whereas LCK is involved in the late and early stages of thymocyte development, ZAP70 specifically regulates the production of SP cells76. Therefore, it is likely that aberrant ubiquitylation of LCK, as a result of loss of CYLD, leads to decreased activation of ZAP70 and attenuated positive selection of SP thymocytes.
T-cell tolerance
The induction of T-cell tolerance occurs at two levels — central tolerance and peripheral tolerance76,84,85. Central tolerance occurs in the thymus and involves negative selection of immature T cells to eliminate those that are self-reactive, as well as the generation of regulatory T (TReg) cells. Peripheral tolerance is mediated by several strategies that render mature T cells non-responsive to self antigens in the periphery. Ubiquitylation is a crucial mechanism that regulates both central and peripheral T-cell tolerance3,9. Several E3 ligases that are involved in T-cell tolerance have been characterized; these include CBL-B, TRAF6, GRAIL, ROQUIN (also known as RC3H1) and ITCH3,9. By contrast, relatively little is known about the DUBs that counteract the function of these E3 ligases.
Otubain-1 was the first DUB to be implicated in the regulation of T-cell anergy53, an important type of peripheral tolerance that can be induced by TCR ligation in the absence of co-stimulatory signals86. As noted already, otubain-1 and its alternatively spliced isoform ARF1 function conversely to control the ubiquitylation and stability of GRAIL53, one of the main E3 ligases to be induced in T cells under anergic conditions9. The E3 ligases are thought to negatively regulate the function or expression of TCR-proximal signalling molecules, such as phospholipase Cγ1 (PLCγ1) and protein kinase C-θ (PKC-θ), thereby attenuating IL-2 production and T-cell proliferation9. GRAIL is also expressed by TReg cells and seems to have a role in the production of inducible regulatory T-cell populations in the periphery87. Consistent with its ability to induce GRAIL degradation through K48-linked polyubiquitylation (see earlier), otubain-1 promotes T-cell activation when overexpressed in mouse T cells, whereas the overexpression of ARF1 inhibits T-cell activation53. It should be noted, however, that a role for otubain-1 in the regulation of immune tolerance has not been examined in vivo.
An X-chromosome-encoded USP-family member, USP9X (also known as FAF), functions as a DUB that regulates the fate of ITCH88, another E3 ligase that is required for the induction of T-cell tolerance89. ITCH deficiency in mice causes the development of a lymphoproliferative disease associated with itching of the skin90. Similar to GRAIL, ITCH undergoes self-ubiquitylation, which triggers its degradation by the proteasome88. USP9X was identified as an ITCH-binding protein by tandem affinity purification and shown to inhibit the self-ubiquitylation and degradation of ITCH. The constitutive level of expression of ITCH is increased by overexpression of USP9X and decreased by RNAi-mediated USP9X knockdown88. Whether USP9X regulates T-cell tolerance has not been investigated.
T-cell activation
Peripheral T-cell activation involves the formation of an intermediate signalling complex, composed of CARMA1 (caspase recruitment domain (CARD) membrane-associated guanylate kinase 1), BCL-10 and MALT1 (mucosa-associated-lymphoid-tissue lymphoma-translocation gene 1)91 (FIG. 2). On activation by the TCR, CARMA1 functions as a scaffold protein that recruits BCL-10 and MALT1 into the lipid raft, which in turn triggers the assembly of a ubiquitin-dependent signalosome containing the K63-specific E2 dimer, UBC13 (ubiquitin-conjugating enzyme 13)–UEV 1A (ubiquitin-conjugating enzyme E2 variant 1A), and probably also the E3 ligase TRAF6 (Refs 92,93). Within this signalosome, MALT1 is conjugated with K63-linked ubiquitin chains, which might function to recruit and activate TAK1 (Ref. 94), a kinase that mediates activation of the IKK complex and JNK in T cells95–97. Activation of IKK by the CARMA1–BCL-10–MALT1 complex also requires K63-linked ubiquitylation of IKKγ, although how IKKγ ubiquitylation triggers the activity of IKKα and IKKβ is unclear92,93. In addition, recent studies indicate that the catalytic activation of TAK1 requires its conjugation with polyubiquitin chains98. Consistent with these biochemical results, genetic deficiency of Ubc13 results in the attenuation of TCR-mediated activation of TAK1 and downstream kinases99. What is unclear, however, is which E3 ligase(s) mediates these K63-linked ubiquitylation events. MALT1 was shown to have E3 ligase activity and to catalyse K63-linked ubiquitylation of IKKγ (Ref. 93). By contrast, another study indicates that MALT1 induces IKKγ ubiquitylation by associating with and activating the E3 ligase TRAF6 (Ref. 92). Additional biochemical studies also support the role of TRAF6 as an E3 ligase in the TCR signalling pathway94; however, this idea is challenged by the finding that T-cell-specific ablation of TRAF6 does not affect TCR-stimulated NF-κB activation, but rather promotes TCR signalling100. It remains to be examined whether TRAF6 is functionally redundant with other TRAF-family members, such as TRAF2, in mediating TCR signalling.
Figure 2. Regulation of TCR signalling by CYL D.

Stimulation of the T-cell receptor (TCR) and CD28 induces the assembly of an intermediate signalling complex composed of CARMA1 (caspase recruitment domain (CARD) membrane-associated guanylate kinase 1), BCL-10 (B-cell lymphoma 10) and MALT1 (mucosa-associated-lymphoid-tissue lymphoma-translocation gene 1). This core complex is also associated with the ubiquitylating enzymes UBC13 (ubiquitin-conjugating enzyme 13), UEV1A (ubiqutin-conjugating enzyme E2 variant 1A) and TRAF6 (tumour-necrosis factor receptor-associated factor 6), which catalyse the K63-linked ubiquitylation of MALT1, triggering the recruitment of IKK (IκB (inhibitor of nuclear factor-κB) kinase) and TAK1 (transforming growth factor-β-activated kinase 1) complexes. Within the CARMA1–BCL-10–MALT1 signalosome, both TAK1 and IKKγ become ubiquitylated, and these ubiquitylation events are important for triggering the catalytic activity of TAK1 and the IKK complex, respectively. CYLD is an essential deubiquitylating enzyme (DUB) that prevents the spontaneous ubiquitylation and activation of TAK1. CYLD deficiency causes constitutive activation of TAK1 and its downstream IKK and JNK (JUN N-terminal kinase) signalling pathways. CYLD positively regulates the TCR-proximal kinase LCK in thymocytes. Whether this function of CYLD is retained in peripheral T cells is not clear. AP1, activator protein 1; TAB, TAK1-binding protein.
Given the dynamic role of ubiquitylation in mediating the signalling function of the CARMA1–BCL-10–MALT1 complex, it is highly predictable that TCR signalling is subject to negative regulation by DUBs. So far, the role of A20 in the regulation of TCR signalling is unclear. Nevertheless, a recent study has shown that TCR and CD28 signals induce A20 cleavage in T cells, which is mediated by the paracaspase activity of MALT1 (Ref. 101). It is therefore possible that A20 has a negative role in regulating TCR signalling and that this function of A20 can be temporarily over-ridden during T-cell activation.
An important function of CYLD in controlling TCR signalling has been shown using Cyld−/− mice19,24 (FIG. 2). The loss of CYLD in T cells causes constitutive activation of TAK1 and its downstream signalling molecules, IKK and JNK (JUN N-terminal kinase). Although it is not yet clear precisely how CYLD regulates the function of the CARMA1–BCL-10–MALT1 signalosome, CYLD deubiquitylates TAK1 and thereby suppresses its catalytic activity19. The fact that loss of CYLD causes spontaneous activation of TAK1 implies that TAK1 ubiquitylation is a dynamic event that occurs even in resting T cells. However, it is unclear whether the activation of TAK1 in CYLD-deficient T cells bypasses the requirement for TCR-proximal signalling molecules.
In keeping with its crucial role in TAK1 regulation, CYLD is indispensable for maintaining normal T-cell activation. CYLD-deficient T cells are hyper-responsive to TCR and CD28 co-stimulation in vitro and have abnormalities in vivo19,24. The Cyld-knockout mice were shown to either be predisposed to or spontaneously develop intestinal inflammation that was similar to human inflammatory bowel disease (IBD)19,24, an autoimmune inflammatory disease caused by inappropriate response of the mucosal immune system to the intestinal microflora102. T cells might have an important role in this pathology, as the adoptive transfer of CYLD-deficient T cells to Rag1-knockout mice is sufficient to induce intestinal inflammation19. Notably, the CYLD gene lies adjacent to the NOD2 gene in both human and mouse genomes. NOD2 is known to regulate IBD by modulating the innate immune response102. As CYLD regulates T-cell responses, it is conceivable that NOD2 and CYLD might function cooperatively to prevent chronic mucosal immune responses to the intestinal microflora.
Deubiquitylation in B-cell function
After a series of developmental events in the bone marrow, immature B cells migrate to the spleen, where they undergo further maturation to become mature B cells. The peripheral maturation and homeostasis of B cells require two NF-κB signalling pathways, the canonical NF-κB pathway and the non-canonical NF-κB pathway, which function cooperatively to regulate genes involved in survival and proliferation103. The canonical NF-κB pathway is stimulated by the B-cell receptor (BCR) and TLRs, whereas the non-canonical NF-κB pathway is regulated by a TNFR-family member, B-cell-activating-factor receptor (BAFFR)104,105.
Although little is known about how ubiquitylation and deubiquitylation regulate B-cell maturation and homeostasis, recent gene-targeting studies have established that CYLD is a crucial B-cell regulator. CYLD deficiency results in constitutive activation of the canonical NF-κB pathway in B cells, which is associated with increased expression of several NF-κB target genes, including those encoding B-cell activation markers (CD21, CD23, CD80 and CD86) and NF-κB members (NF-κB2 and RELB)106. Although the B-cell target of CYLD is unknown, it is probable that CYLD regulates a signalling molecule upstream of IKK, as IKK is constitutively activated in CYLD-deficient B cells106. In keeping with the chronic NF-κB activation, CYLD-deficient B cells have abnormalities in both maturation and homeostasis, characterized by the hyper-production of marginal-zone B cells and B-cell hyperplasia in peripheral lymphoid organs. The crucial role of CYLD in regulating B-cell activation is also indicated by the association of CYLD genetic mutations with human multiple myeloma, a late-stage B-cell malignancy107,108. The Cyld-knockout mice have enlarged mesenteric lymph nodes, which indicates that they produce abnormal mucosal immune responses to the intestinal microflora. Indeed, although the other lymphoid organs are mainly normal in naive Cyld-knockout mice, they undergo abnormal enlargement after immunization. These findings are consistent with the hyper-responsive phenotype of CYLD-deficient B cells to in vitro stimulation.
B-cell abnormalities were also observed in mice that express a shorter isoform of CYLD (sCYLD), which is encoded by a natural splicing variant of the Cyld gene that lacks exons 7 and 8 (Cyldex7/8). sCYLD retains DUB activity but lacks the domain that mediates binding to TRAF2 and IKKγ (Ref. 25). Interestingly, the pathological phenotype of the Cyldex7/8 mice was similar to, but more prominent than, that of the Cyld-knockout mice25. For example, although both the Cyldex7/8 mice and the Cyld-knockout mice have enlarged lymphoid organs, this abnormality occurs spontaneously in the former mice but is mostly induced by immunization in the Cyld-knockout mice25,106. The phenotype of the Cyldex7/8 mice probably reflects both loss of the negative signalling function of wild-type CYLD and gain of some positive signalling functions of sCYLD. As sCYLD retains its DUB activity but is defective in binding to some, but not all, of its targets25, it is tempting to propose that CYLD might have dual signalling functions, with the negative one being lost in sCYLD (FIG. 3). This hypothesis is also based on studies of CYLD in other cell types. Specifically, CYLD positively regulates LCK in thymocytes and negatively regulates TAK1 in peripheral T cells18,19. It is currently unknown whether CYLD has DUB activity towards the LCK homologues SYK and LYN in B cells.
Figure 3. Dual signalling functions of CYL D in B cells as indicated by studies of a natural variant, sCYL D.

a | B-cell receptor (BCR) signalling activates the inhibitor of nuclear factor-κB (NF-κB) kinase (IKK)–NF-κB signalling pathway through the CARMA1–BCL-10–MALT1 signalosome. Although the precise targets of CYLD in B cells remain unclear, CYLD negatively regulates the IKK complex and NF-κB. Based on recent work on a short isoform of CYLD (sCYLD), it is conceivable that CYLD might also have a positive signalling target, possibly one of the SRC kinases (LYN or SYK) as their T-cell homologue, LCK, is positively regulated by CYLD. b | sCYLD is encoded by a natural splicing variant of the CYLD gene that lacks exons 7 and 8. sCYLD does not contain the TRAF2- and IKKγ-binding domains that are required for negative regulation of NF-κB, but it retains the deubiquitylating enzyme (DUB) activity. One implication is that sCYLD is defective in binding to its negative targets (such as IKKγ) but remains effective towards its positive targets, which would explain the B-cell hyper-activation in mice expressing sCYLD. BCL-10, B-cell lymphoma 10; CARMA1, caspase recruitment domain (CARD) membrane-associated guanylate kinase 1; Cap-Gly, cytoskeleton-associated protein glycine-rich domain; MALT1, mucosa-associated lymphoid-tissue lymphoma-translocation gene 1.
Concluding remarks
Recent studies have provided important information about how deubiquitylation regulates immune responses. Several DUBs have been shown to regulate signal transduction in different compartments of the immune system, ranging from innate immune responses to the development and activation of lymphocytes. The work on CYLD and A20 provides in vivo evidence for the requirement of DUBs in maintaining normal immune functions. Given the involvement of a large number of E3 ligases in immune regulation, it is probable that more immunoregulatory DUBs will be discovered. With the availability of the sequence information of various human and mouse DUB genes, functional screening using RNAi libraries will probably lead to the characterization of new DUBs that regulate specific pathways of immune-receptor signalling. However, an important challenge in future studies will be to identify the natural substrates of the immunoregulatory DUBs. So far, most of the studies are based on binding and deubiquitylation assays using in vitro or overexpression systems. An obvious caveat of such experimental systems is that the DUBs might nonspecifically deubiquitylate substrates that are not targeted under physiological conditions.
The studies of CYLD and A20 emphasize the importance of in vivo model systems to elucidate the physiological functions of DUBs. Despite their similar signalling functions in cell lines, CYLD and A20 have marked differences in the regulation of immune responses in vivo. In addition to the specificity issue mentioned above, an important factor that determines the in vivo functional specificity of DUBs is their expression pattern, a factor that is often neglected in transfection studies. Moreover, the function of some DUBs, such as A20, requires accessory proteins that might also have specific modes of expression and subcellular localization in vivo. Another lesson learnt from the gene-targeting studies is that the in vivo functions of DUBs might be far more complex than expected. This is particularly evident for CYLD, which has been linked to several immune and non-immune functions. Notably, CYLD deficiency can lead to either harmful or beneficial consequences for the host immune system depending on the nature of the response. So, manipulation of the function of CYLD, and other immunoregulatory DUBs, might be exploited as a new therapeutic approach for treating immunological diseases.
Acknowledgments
Work in my laboratory is supported by the University of Texas MD Anderson Cancer Center and the National Institutes of Health, USA, Grants R01 AI064639, R01 AI057555, R01 CA94922. I would like to thank the members of my laboratory for discussions and input, and I apologize to those authors whose work was not cited because of space limitations.
Glossary
- 26S proteasome
A giant multicatalytic protease that resides in the cytosol and nucleus. The 20S core, which contains three distinct catalytic subunits, can be appended at either end by a 19S cap or an 11S cap. The binding of two 19S caps to the 20S core forms the 26S proteasome, which degrades polyubiquitylated proteins.
- E3 ubiquitin ligases
Enzymes that are required to attach the molecular tag ubiquitin to proteins. Depending on the position and number of the ubiquitin molecules that are attached, the ubiquitin tag can target proteins for degradation in the proteasomal complex, sort them to specific subcellular compartments or modify their biological activity.
- SH3 domains
Protein-interaction domains that are commonly found in signal-transduction molecules. They specifically interact with certain proline-containing peptides (containing either (R/K)XXPXXP or PXXPXR motifs, where X denotes any amino acid) to facilitate protein–protein interactions that are required for protein function or subcellular localization.
- Small interfering RNA (siRNA)-mediated knockdown
Double-stranded RNAs (dsRNAs) with sequences that precisely match a given gene are able to ‘knock down’ the expression of that gene by directing RNA-degrading enzymes to destroy the encoded mRNA transcript. The two most common forms of dsRNAs used for gene silencing are short — usually 21-bp long — siRNAs or the plasmid-delivered short hairpin RNAs (shRNAs).
- Yeast two-hybrid screen
A screening system for protein–protein interactions that result in the transcription of a reporter gene when a bait protein attached to a DNAbinding domain comes into contact with a prey protein bound to a transcriptional activator.
- COP9 signalosome
(CSN). A multi-protein complex conserved in eukaryotic organisms that contains eight subunits, which have structural homology with the lid subunits of the 19S regulatory particle of the 26S proteasome. A prominent function of the COP9 signalosome is to regulate the activity of cullin-based E3 ubiquitin ligases, thereby modulating protein ubiquitylation and degradation.
- Common cytokine-receptor γ-chain
(γc). A type I cytokine receptor chain that is shared by the receptors for interleukin-2 (IL-2), IL-4, IL-7, IL-9, IL-15 and IL-21.
- Pathogen-associated molecular patterns
(PAMPs). Molecular patterns that are found in pathogens but not mammalian cells. Examples include terminally mannosylated and polymannosylated compounds, which bind the mannose receptor, and various microbial products, such as bacterial lipopolysaccharides, hypomethylated DNA, flagellin and double-stranded RNA, which bind Toll-like receptors.
- Pattern-recognition receptors
(PPRs). Host receptors (such as Toll-like receptors) that can sense pathogen-associated molecular patterns and initiate signalling cascades (involving activation of nuclear factor-κB) that lead to an innate immune response.
- Anergy
A state of T cells that have been stimulated through their T-cell receptors in the absence of the ligation of CD28. On restimulation, these T cells are unable to produce interleukin-2 or to proliferate, even in the presence of costimulatory signals.
- Canonical NF-κB pathway
A typical pathway of NF-κB activation that involves phosphorylation and degradation of the prototypical NF-κB inhibitor, IκBα.
- Non-canonical NF-κB pathway
A pathway of NF-κB activation that does not involve IκBα degradation but relies on the processing of an NF-κB precursor protein, p100, leading to nuclear translocation of the p52–RELB NF-κB heterodimer.
Footnotes
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
A20 | ABIN1 | Cezanne | CYLD | DUB1 | DUB1A | DUB2 | DUB2A | DUB3 | DUBA | otubain-1 | TAX1BP1 | UCHL1 | USP15 | USP17
FURTHER INFORMATION
Shao-Cong Sun’s homepage: http://www.mdanderson.org/departments/immunology
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26:3214–3226. doi: 10.1038/sj.onc.1210413. [DOI] [PubMed] [Google Scholar]
- 2.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 3.Liu YC, Penninger J, Karin M. Immunity by ubiquitylation: a reversible process of modification. Nature Rev Immunol. 2005;5:941–952. doi: 10.1038/nri1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nijman SM, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Gong B, Leznik E. The role of ubiquitin C-terminal hydrolase L1 in neurodegenerative disorders. Drug News Perspect. 2007;20:365–370. doi: 10.1358/dnp.2007.20.6.1138160. [DOI] [PubMed] [Google Scholar]
- 6.Makarova KS, Aravind L, Koonin EV. A novel superfamily of predicted cysteine proteases from eukaryotes, viruses and Chlamydia pneumoniae. Trends Biochem Sci. 2000;25:50–52. doi: 10.1016/s0968-0004(99)01530-3. [DOI] [PubMed] [Google Scholar]
- 7.Kayagaki N, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. This paper identifies DUBA as a key regulator of antiviral innate immunity. [DOI] [PubMed] [Google Scholar]
- 8.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. This paper shows that A20 has both DUB and E3-ligase functions. [DOI] [PubMed] [Google Scholar]
- 9.Lin AE, Mak TW. The role of E3 ligases in autoimmunity and the regulation of autoreactive T cells. Curr Opin Immunol. 2007;19:665–673. doi: 10.1016/j.coi.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Bignell GR, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nature Genet. 2000;25:160–165. doi: 10.1038/76006. This paper reports the identification of CYLD as a tumour suppressor. [DOI] [PubMed] [Google Scholar]
- 11.Borodovsky A, et al. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol. 2002;9:1149–1159. doi: 10.1016/s1074-5521(02)00248-x. [DOI] [PubMed] [Google Scholar]
- 12.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 13.Kovalenko A, et al. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 14.Trompouki E, et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. References 12–14 are key reports of the signalling function of CYLD. [DOI] [PubMed] [Google Scholar]
- 15.Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking bcl-3-dependent NF-κB signaling. Cell. 2006;125:665–677. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida H, Jono H, Kai H, Li JD. The tumor suppressor CYLD acts as a negative regulator for Toll-like receptor 2 signaling via negative cross-talk with TRAF6 and TRAF7. J Biol Chem. 2005;280:41111–41121. doi: 10.1074/jbc.M509526200. [DOI] [PubMed] [Google Scholar]
- 17.Jin W, et al. Deubiquitinating enzyme CYLD regulates RANK signaling and osteoclastogenesis. J Clinic Invest. doi: 10.1172/JCI34257. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reiley WW, et al. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nature Immunol. 2006;7:411–417. doi: 10.1038/ni1315. This paper was the first to show the in vivo role of CYLD in regulating immune functions. [DOI] [PubMed] [Google Scholar]
- 19.Reiley WW, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med. 2007;204:1475–1485. doi: 10.1084/jem.20062694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright A, et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell. 2007;13:705–716. doi: 10.1016/j.devcel.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Stokes A, et al. TRPA1 is a substrate for deubiquitination by the tumor suppressor CYLD. Cell Signal. 2006;18:1584–1594. doi: 10.1016/j.cellsig.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Lim JH, et al. Tumor suppressor CYLD acts as a negative regulator for non-typeable Haemophilus influenza-induced inflammation in the middle ear and lung of mice. PLoS ONE. 2007:e1032. doi: 10.1371/journal.pone.0001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim JH, et al. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity. 2007;27:349–360. doi: 10.1016/j.immuni.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, et al. Impaired regulation of NF-κB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest. 2006;116:3042–3049. doi: 10.1172/JCI28746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hövelmeyer N, et al. Regulation of B cell homeostasis and activation by the tumor suppressor gene CYLD. J Exp Med. 2007;204:2615–2627. doi: 10.1084/jem.20070318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao J, et al. The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J Biol Chem. 2008;283:8802–8809. doi: 10.1074/jbc.M708470200. [DOI] [PubMed] [Google Scholar]
- 27.Stegmeier F, et al. The tumor suppressor CYLD regulates entry into mitosis. Proc Natl Acad Sci USA. 2007;104:8869–8874. doi: 10.1073/pnas.0703268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saito K, et al. The CAP-Gly domain of CYLD associates with the proline-rich sequence in NEMO/IKKγ. Structure. 2004;12:1719–1728. doi: 10.1016/j.str.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 29.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor α-induced NF-κB activation. J Biol Chem. 2006;281:13636–13643. doi: 10.1074/jbc.M600620200. [DOI] [PubMed] [Google Scholar]
- 31.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation. Nature Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 32.Komander D, et al. The structure of the CYLD USP domain explains its specificity for Lys63-linked polyubiquitin and reveals a B Box module. Mol Cell. 2008;29:451–464. doi: 10.1016/j.molcel.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 33.Xue L, et al. Tumor suppressor CYLD regulates JNKinduced cell death in Drosophila. Dev Cell. 2007;13:446–454. doi: 10.1016/j.devcel.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 34.Wooten MW, et al. Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J Biol Chem. 2008;283:6783–6789. doi: 10.1074/jbc.M709496200. [DOI] [PubMed] [Google Scholar]
- 35.Beyaert R, Heyninck K, Van Huffel S. A20 and A20-binding proteins as cellular inhibitors of nuclear factor-κB-dependent gene expression and apoptosis. Biochem Pharmacol. 2000;60:1143–1151. doi: 10.1016/s0006-2952(00)00404-4. [DOI] [PubMed] [Google Scholar]
- 36.Lee EG, et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. This study was the first to report the in vivo role of A20 in regulating inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nature Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 38.Hitotsumatsu O, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28:381–390. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evans PC, et al. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J. 2004;378:727–734. doi: 10.1042/BJ20031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mauro C, et al. ABIN-1 binds to NEMO/IKKγ and co-operates with A20 in inhibiting NF-κB. J Biol Chem. 2006;281:18482–18488. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 41.Shembade N, et al. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nature Immunol. 2008;9:254–262. doi: 10.1038/ni1563. [DOI] [PubMed] [Google Scholar]
- 42.Komander D, Barford D. Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem J. 2008;409:77–85. doi: 10.1042/BJ20071399. [DOI] [PubMed] [Google Scholar]
- 43.Lin SC, et al. Molecular basis for the unique deubiquitinating activity of the NF-κB inhibitor A20. J Mol Biol. 2008;376:526–540. doi: 10.1016/j.jmb.2007.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shembade N, Harhaj NS, Liebl DJ, Harhaj EW. Essential role for TAX1BP1 in the termination of TNF-α-, IL-1- and LPS-mediated NF-κB and JNK signaling. EMBO J. 2007;26:3910–3922. doi: 10.1038/sj.emboj.7601823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iha H, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-κB activation. EMBO J. 2008;27:629–641. doi: 10.1038/emboj.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin DY, et al. A human suppressor of c-Jun N-terminal kinase 1 activation by tumor necrosis factor α. J Biol Chem. 1997;272:25816–25823. doi: 10.1074/jbc.272.41.25816. [DOI] [PubMed] [Google Scholar]
- 47.De Valck D, et al. The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene. 1999;18:4182–4190. doi: 10.1038/sj.onc.1202787. [DOI] [PubMed] [Google Scholar]
- 48.Wagner S, et al. Ubiquitin binding mediates the NF-κB inhibitory potential of ABINs. Oncogene. 2008 Jan 21; doi: 10.1038/sj.onc.1211042. [DOI] [PubMed] [Google Scholar]
- 49.Evans PC, et al. Isolation and characterization of two novel A20-like proteins. Biochem J. 2001;357:617–623. doi: 10.1042/0264-6021:3570617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans PC, et al. A novel type of deubiquitinating enzyme. J Biol Chem. 2003;278:23180–23186. doi: 10.1074/jbc.M301863200. [DOI] [PubMed] [Google Scholar]
- 51.Enesa K, et al. NF-κB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J Biol Chem. 2008;283:7036–7045. doi: 10.1074/jbc.M708690200. [DOI] [PubMed] [Google Scholar]
- 52.Balakirev MY, Tcherniuk SO, Jaquinod M, Chroboczek J. Otubains: a new family of cysteine proteases in the ubiquitin pathway. EMBO Rep. 2003;4:517–522. doi: 10.1038/sj.embor.embor824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soares L, et al. Two isoforms of otubain 1 regulate T cell anergy via GRAIL. Nature Immunol. 2004;5:45–54. doi: 10.1038/ni1017. [DOI] [PubMed] [Google Scholar]
- 54.Schweitzer K, Bozko PM, Dubiel W, Naumann M. CSN controls NF-κB by deubiquitinylation of IκBα. EMBO J. 2007;26:1532–1541. doi: 10.1038/sj.emboj.7601600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scherer DC, Brockman JA, Chen A, Maniatis T, Ballard DW. Signal-induced degradation of IκBα requires site-specific ubiquitination. Proc Natl Acad Sci USA. 1995;92:11259–11263. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwechheimer C, Deng XW. COP9 signalosome revisited: a novel mediator of protein degradation. Trends Cell Biol. 2001;11:420–426. doi: 10.1016/s0962-8924(01)02091-8. [DOI] [PubMed] [Google Scholar]
- 57.Baek KH. Cytokine-regulated protein degradation by the ubiquitination system. Curr Protein Pept Sci. 2006;7:171–177. doi: 10.2174/138920306776359740. [DOI] [PubMed] [Google Scholar]
- 58.Gesbert F, Malardé V, Dautry-Varsat A. Ubiquitination of the common cytokine receptor gammac and regulation of expression by an ubiquitination/deubiquitination machinery. Biochem Biophys Res Commun. 2005;334:474–480. doi: 10.1016/j.bbrc.2005.06.121. [DOI] [PubMed] [Google Scholar]
- 59.Migone TS, et al. The deubiquitinating enzyme DUB-2 prolongs cytokine-induced signal transducers and activators of transcription activation and suppresses apoptosis following cytokine withdrawal. Blood. 2001;98:1935–1941. doi: 10.1182/blood.v98.6.1935. [DOI] [PubMed] [Google Scholar]
- 60.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 61.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of Toll-like receptor-mediated immune responses. Nature Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 62.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nature Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 63.Turer EE, et al. Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J Exp Med. 2008;205:451–464. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reiley W, Zhang M, Wu X, Graner E, Sun SC. Regulation of the deubiquitinating enzyme CYLD by IκB kinase γ-dependent phosphorylation. Mol Cell Biol. 2005;25:3886–3895. doi: 10.1128/MCB.25.10.3886-3895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hiscott J. Convergence of the NF-κB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 2007;18:483–490. doi: 10.1016/j.cytogfr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 66.Häcker H, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 67.Oganesyan G, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 68.Guo B, Cheng G. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem. 2007;282:11817–11826. doi: 10.1074/jbc.M700017200. [DOI] [PubMed] [Google Scholar]
- 69.Gatot JS, et al. Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKε-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J Biol Chem. 2007;282:31131–31146. doi: 10.1074/jbc.M701690200. [DOI] [PubMed] [Google Scholar]
- 70.Zhao T, et al. The NEMO adaptor bridges the nuclear factor-κB and interferon regulatory factor signaling pathways. Nature Immunol. 2007;8:592–600. doi: 10.1038/ni1465. [DOI] [PubMed] [Google Scholar]
- 71.Gack MU, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. This paper reports the regulation of RIG-I signalling function by K63-linked ubiquitylation. [DOI] [PubMed] [Google Scholar]
- 72.Zhang M, et al. Regulation of IKK-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem. 2008 doi: 10.1074/jbc.M801451200. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang YY, Li L, Han KJ, Zhai Z, Shu HB. A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-κB and ISRE and IFN-β promoter. FEBS J. 2004;576:86–90. doi: 10.1016/j.febslet.2004.08.071. [DOI] [PubMed] [Google Scholar]
- 74.Saitoh T, et al. A20 is a negative regulator of IFN regulatory factor 3 signaling. J Immunol. 2005;174:1507–1512. doi: 10.4049/jimmunol.174.3.1507. [DOI] [PubMed] [Google Scholar]
- 75.Lin R, et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem. 2006;281:2095–2103. doi: 10.1074/jbc.M510326200. [DOI] [PubMed] [Google Scholar]
- 76.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 77.Molina TJ, et al. Profound block in thymocyte development in mice lacking p56lck. Nature. 1992;357:161–164. doi: 10.1038/357161a0. [DOI] [PubMed] [Google Scholar]
- 78.Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- 79.Rao N, et al. Negative regulation of Lck by Cbl ubiquitin ligase. Proc Natl Acad Sci USA. 2002;99:3794–3799. doi: 10.1073/pnas.062055999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thien CB, Bowtell DD, Langdon WY. Perturbed regulation of ZAP-70 and sustained tyrosine phosphorylation of LAT and SLP-76 in c-Cbl-deficient thymocytes. J Immunol. 1999;162:7133–7139. [PubMed] [Google Scholar]
- 81.Naramura M, Kole HK, Hu RJ, Gu H. Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc Natl Acad Sci USA. 1998;95:15547–15552. doi: 10.1073/pnas.95.26.15547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murphy MA, et al. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol. 1998;18:4872–4882. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hawash IY, Kesavan KP, Magee AI, Geahlen RL, Harrison ML. The Lck SH3 domain negatively regulates localization to lipid rafts through an interaction with c-Cbl. J Biol Chem. 2002;277:5683–5691. doi: 10.1074/jbc.M110002200. [DOI] [PubMed] [Google Scholar]
- 84.Kronenberg M, Rudensky A. Regulation of immunity by self-reactive T cells. Nature. 2005;435:598–604. doi: 10.1038/nature03725. [DOI] [PubMed] [Google Scholar]
- 85.Kyewski B, Klein L. A central role for central tolerance. Annu Rev Immunol. 2006;24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601. [DOI] [PubMed] [Google Scholar]
- 86.Choi S, Schwartz RH. Molecular mechanisms for adaptive tolerance and other T cell anergy models. Semin Immunol. 2007;19:140–152. doi: 10.1016/j.smim.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.MacKenzie DA, et al. GRAIL is up-regulated in CD4+ CD25+ T regulatory cells and is sufficient for conversion of T cells to a regulatory phenotype. J Biol Chem. 2007;282:9696–9702. doi: 10.1074/jbc.M604192200. [DOI] [PubMed] [Google Scholar]
- 88.Mouchantaf R, et al. The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J Biol Chem. 2006;281:38738–38747. doi: 10.1074/jbc.M605959200. [DOI] [PubMed] [Google Scholar]
- 89.Liu YC. The E3 ubiquitin ligase Itch in T cell activation, differentiation, and tolerance. Semin Immunol. 2007;19:197–205. doi: 10.1016/j.smim.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perry WL, et al. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nature Genet. 1998;18:143–146. doi: 10.1038/ng0298-143. [DOI] [PubMed] [Google Scholar]
- 91.Rawlings DJ, Sommer K, Moreno-García ME. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nature Rev Immunol. 2006;6:799–812. doi: 10.1038/nri1944. [DOI] [PubMed] [Google Scholar]
- 92.Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 93.Zhou H, et al. Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature. 2004;427:167–171. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]
- 94.Oeckinghaus A, et al. Malt1 ubiquitination triggers NF-κB signaling upon T-cell activation. EMBO J. 2007;26:4634–4645. doi: 10.1038/sj.emboj.7601897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu HH, Xie M, Schneider MD, Chen ZJ. Essential role of TAK1 in thymocyte development and activation. Proc Natl Acad Sci USA. 2006;103:11677–11682. doi: 10.1073/pnas.0603089103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sato S, et al. TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T cells. Int Immunol. 2006;18:1405–1411. doi: 10.1093/intimm/dxl082. [DOI] [PubMed] [Google Scholar]
- 97.Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nature Immunol. 2006;7:851–858. doi: 10.1038/ni1355. [DOI] [PubMed] [Google Scholar]
- 98.Thiefes A, et al. The Yersinia enterocolitica effector YopP inhibits host cell signalling by inactivating the protein kinase TAK1 in the IL-1 signalling pathway. EMBO Rep. 2006;7:838–844. doi: 10.1038/sj.embor.7400754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yamamoto M, et al. Cutting Edge: pivotal function of Ubc13 in thymocyte TCR signaling. J Immunol. 2006;177:7520–7524. doi: 10.4049/jimmunol.177.11.7520. [DOI] [PubMed] [Google Scholar]
- 100.King CG, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nature Med. 2006;12:1088–1092. doi: 10.1038/nm1449. [DOI] [PubMed] [Google Scholar]
- 101.Coornaert B, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-κB inhibitor A20. Nature Immunol. 2008;9:263–271. doi: 10.1038/ni1561. [DOI] [PubMed] [Google Scholar]
- 102.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 103.Sen R. Control of B lymphocyte apoptosis by the transcription factor NF-κB. Immunity. 2006;25:871–883. doi: 10.1016/j.immuni.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 104.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nature Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- 105.Kayagaki N, et al. BAFF/BLyS receptor 3 binds the B-cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity. 2002;17:515–524. doi: 10.1016/s1074-7613(02)00425-9. [DOI] [PubMed] [Google Scholar]
- 106.Jin W, et al. Deubiquitinating enzyme CYLD regulates the peripheral development and naive phenotype maintenance of B cells. J Biol Chem. 2007;282:15884–15893. doi: 10.1074/jbc.M609952200. [DOI] [PubMed] [Google Scholar]
- 107.Annunziata CM, et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Keats JJ, et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Haglund K, Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sigismund S, Polo S, Di Fiore PP. Signaling through monoubiquitination. Curr Top Microbiol Immunol. 2004;286:149–185. doi: 10.1007/978-3-540-69494-6_6. [DOI] [PubMed] [Google Scholar]
- 111.Beinke S, Ley SC. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J. 2004;382:393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xiao G, Harhaj EW, Sun SC. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 113.Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 114.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;357:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]