

Abstract
Src family kinases (SFKs) are a family of protein tyrosine kinases containing nine members: Src, Lyn, Fgr, Hck, Lck, Fyn, Blk, Yes, and Ylk. Although SFK activation is a major immediate signaling event in LPS/Toll-like receptor 4 (TLR4) signaling, its precise role has remained elusive due to various contradictory results obtained from a certain SFK member-deficient mice or cells. The observed inconsistencies may be due to the compensation or redundancy by other SFKs upon a SFK deficiency. The chemical rescuing approach was suggested to induce temporal and precise SFK activation in living cells, thereby limiting the chance of cellular adaption to a SFK-deficient condition.
Using the rescuing approach, we demonstrate that restoring SFK activity not only induces tyrosine phosphorylation of TLR4, but also inhibits LPS-induced NFκB and JNK1/2 activation and consequently suppresses LPS-induced cytokine production. TLR4 normally recruits TIR domain-containing adaptors in response to LPS, however, temporally restored SFK activation disrupts the LPS-induced association of MyD88 and Mal/Tirap with TLR4. Additionally, using kinase-dead SFK-Lyn (Y397/508F) and constitutively active SFK-Lyn (Y508F), we found that the kinase-dead SFK inhibits TLR4 tyrosine phosphorylation with reduced binding affinity to TLR4, while the kinase-active SFK strongly binds to TLR4 and promotes TLR4 tyrosine phosphorylation, suggesting that SFK kinase activity is required for TLR4 tyrosine phosphorylation and TLR4-SFK interaction.
Together, our results demonstrate that SFK activation induces TLR4 tyrosine phosphorylation, consequently dissociating MyD88 and Mal/Tirap from TLR4 and inhibiting LPS-induced inflammatory responses, suggesting a negative feedback loop regulated by SFK-induced tyrosine phosphorylation in TLR4.
Keywords: Chemical rescuing approach, Negative feedback, Sepsis, Src family kinase, Toll like receptor, Tyrosine phosphorylation
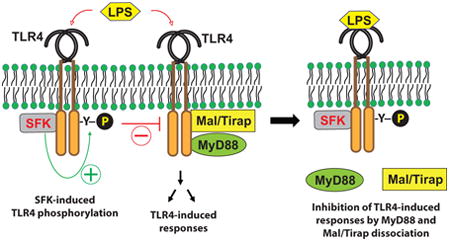
Graphical abstract
1. Introduction
Src family kinases (SFKs) are a family of non-receptor protein tyrosine kinases composed of nine members: Src, Lyn, Fgr, Hck, Lck, Fyn, Blk, Yes, and Ylk. The expression and precise cellular function of each SFK member varies with the cell type and cell stimulatory agent, although SFKs are expressed in the large majority of cells, including immune cells, fibroblasts, and endothelial cells (1). As protein tyrosine kinases, SFKs participate in the regulation of critical cellular functions such as proliferation, migration, differentiation, survival, B- and T-cell immune responses, and microbial sensing by pattern recognition receptors. To regulate these cellular events, SFKs are not only activated by T- and B-cell receptor complexes and microbial pattern recognition receptors, but also by the receptors for growth factors, cytokines, cell adhesion molecules, and G protein-coupled receptors (1-5).
Lipopolysaccharide (LPS), produced by Gram-negative bacteria, is a potent endotoxin capable of inducing the pleiotropic production of inflammatory mediators in many different cell types, resulting in lethal systemic sepsis. In order to induce intracellular signaling, LPS is specifically recognized by Tolllike receptor 4 (TLR4), a type-I transmembrane protein with a leucine-rich repeat (LRR) domain at the extracellular region responsible for binding LPS, and a Toll/IL-1R (TIR) domain at the cytoplasmic region which mediates intracellular signaling. Upon LPS recognition, TLR4 recruits a combination of TIR domain-containing adaptors (MyD88, Mal/Tirap, Tram, and Trif), resulting not only in the downstream activation of the transcription factors NFκB and AP-1 which are the primary contributors to pro-inflammatory gene expression; but also culminating in the activation of IRF3/7, which promote interferon-α/β expression to elicit anti-viral innate immune effector functions.
In addition, LPS-TLR4 engagement immediately activates the SFK members Lyn, Fgr, and Hck in macrophages/monocytes and dendritic cells (DCs) (6-9). Although the ability of LPS to activate SFKs has long been known, the question of whether SFKs are a positive or negative regulator in TLR4-mediated signaling pathways remains controversial due to the existence of an array of conflicting results between studies using pharmacologic inhibitors, overexpression of mutated SFKs, RNA interference, and gene-knockout mice.
As a positive regulator in TLR4-induced responses, numerous studies have implicated SFKs. Pharmacologic inhibition of SFKs blocked TLR4-induced responses in several studies (10-12). In another study, SFK-Lyn-/- bone marrow-derived mast cells (BMMCs) had reduced TNFα production in response to LPS compared to wild type BMMCs (13). Overexpression of a constitutively active SFK-Hck upregulated LPS-induced TNFα production in mouse macrophages, while SFK-Hck gene silencing downregulated the cytokine production (14). Together, these observations suggest that SFKs may participate in TLR4-induced responses as a positive regulator.
On the other hand, there exists considerable evidence for a negative regulatory role of SFKs in TLR4-induced responses. One study indicated that overexpression of wild-type or constitutively active SFK-Lyn inhibited LPS-induced NFκB and IRF3 activation, and that silencing SFK-Lyn enhanced TNFα and CCL5/RANTES production in RAW264.7 cells (15). In another study, SFK-Lyn-/- macrophages isolated from SFK-Lyn deficient mice produced more cytokines than wild type macrophages upon LPS stimulation (16). Lamagna et al. recently showed that SFK-Lyn-/- DCs from DC-specific SFK-Lyn knockout mice were hyper-responsive to LPS and IL-1β stimulation (17). These observations suggest that SFKs may play an inhibitory role in TLR4-induced signaling pathways. Nevertheless, given the fact that macrophages from triple SFK-knockout (Lyn/-; Fgr-/-; Hck-/-) mice exhibited normal LPS-induced responses (18), the role of SFKs in LPS signaling still remains controversial.
Given these results, there exist two important questions regarding the involvement of SFKs in LPS signaling: 1) What is the role of SFKs in TLR4-induced responses; and 2) What is the mechanism by which SFKs influence LPS signaling? Despite the considerable observations obtained from gene silencing and knockout approaches, we suspect that such contradictory results might be due to compensation by other SFK members in genetically modified mice or cells with the loss of a certain SFK member (19). To investigate the role of SFKs in LPS-induced responses without the potential compensation of another SFK member, we used the chemical rescue of SFKs in living cells using the mutant SFK member Src (SFK-Src) Arg388 → Ala (R388A), whose enzymatic activity can be temporally and specifically restored by treatment with the small molecule imidazole (20,21).
We found that restoring SFK catalytic activity induced tyrosine phosphorylation in TLR4 and disrupted the association of MyD88 and Mal/Tirap with TLR4, resulting in reduced LPS-induced cytokine production. Therefore, our study provides clear evidence that SFK activation during LPS stimulation plays an inhibitory role in TLR4-mediated signaling pathways, contributing to a negative feedback loop in LPS responses, which may function as an LPS tolerance mechanism.
2. Materialsa and Methods
2.1. SFK-Src R388A and D386N mutant constructs
SFK-Src R388A/Y527F (Src-R/A) and SFK-Src D386N/Y527F (Src-D/N) encoding plasmid constructs were kindly provided by Dr. Philip A. Cole (The Johns Hopkins University, Baltimore) (21). They were subcloned into pcDNA3.1/Hygro (-) vector (Thermo Fisher Scientific, Grand Island, NY) and stably transfected in HEK293 cell or SW480 cells (ATCC, Manassas, VA).
2.2. SFK-Lyn constructs
Wild type murine Lyn cDNA was PCR amplified with primers F1 and R1 and subcloned into Not1 and BamH1 sites of p3×Flag-CMV14 expression vector (Sigma-Aldrich, St. Louis, MO) to obtain 3×Flag-tagged Lyn-WT. The tyrosine at 508 residue was replaced with phenylalanine by PCR with the specific primer set (F2 and R2), and EcoR1/BamH1 fragment of the PCR product replaced the C-terminal region of 3×Flag-Lyn-WT to give rise to 3×Flag-Lyn (Y508F). Although the F2 primer contains a mutation at Tyr397 residue, subcloning of EcoR1/BamH1 fragment results in only an Y508F mutation because the Tyr397 residue is outside of the subcloning sites.
Furthermore, the PCR product with the primer F2 and R2 containing an Y397F mutation was also used as a mega-reverse primer with the forward primer (F1) for PCR. The Xba1/EcoR1 fragment of the PCR product was inserted into the same restriction sites in 3×Flag-Lyn-WT to produce the 3×Flag-Lyn (Y397F) construct that is a kinase-dead mutant. Then, the Xba1/EcoR1 fragment from 3×Flag-Lyn (Y397F) replaced the region in 3×Flag-Lyn (Y508F) to prepare 3×Flag-Lyn (Y397/508F) which is in a kinase-dead open conformation. We confirmed the integrity of the complete cDNA sequence by sequencing all the constructs, and the expression of Lyn constructs was verified by western blotting. All PCR primers used for generating Lyn constructs are the followings: F1, AAA GCG GCC GCA CCC ATG GGA TGT ATT AAA TCA AAA AGG; R1, G ATG AAA GGA TCC CGG TTG CTG CTG ATA CTG CCC; F2, C GAA GAT AAC GAG TTC ACA GCA AGG GAA GG (bold nucleotides indicate a mutated codon of Y397F); R2, G ATG AAT GGA TCC CGG TTG CTG CTG AAA CTG CCC-3″ (bold nucleotides stand for a mutated codon of Y508F);
2.3. Other cDNA expression constructs
We previously described HA-tagged murine TLR4 wild type and P712H mutant cDNA expression constructs and the constitutively active HA-ΔTLR4-WT and dominant negative HA-ΔTLR4(P712H) lacking the extracellular LRR domain (22). We used TLR5-HA (23) and Tram-Flag (24) encoding constructs previously. The NFκB-Luciferase reporter construct and β-galactosidase expression plasmid (HSP70-β-gal) as an internal control were previously described (25).
2.4. Cell culture
Human colonic epithelial cells (NCM460) were cultivated in M3D media from ‘INCELL Corp’ (San Antonio, TX), as previously described (26). Human embryonic kidney (HEK293) and murine macrophages RAW264.7 cells were cultivated as described previously at 37°C in 5 % CO2 air environment (22). Human colon epithelial cells SW480 (ATCC® CCL-228™) were cultivated in Leibovitz's L-15 Medium in ATCC (Manassas, VA) supplemented with fetal bovine serum (FBS) to a final concentration of 10% and Penicillin (50 units/mL)-Streptomycin (50 μg/mL).
2.5. Cell transfection
Cells were transfected with the appropriate plasmid DNA using SuperFect transfection reagent from Qiagen (Valencia, CA). About 18 hours after transfection, cell lysates were prepared in lysis buffer, followed by western blot analysis. To generate stably transfected cells, transfected cells were plated in the proper medium containing selecting antibiotics (zeocin or hygromycin or neomycin) to obtain stably transfected cell pools.
2.6. Antibodies and materials
Anti-phospho-tyrosine antibody (4G10) conjugated with HRP was purchased from EMD Millipore (Billerica, MA). Antibodies of phospho-Src (Y416), Src, phospho-ERK1/2, phospho-JNK1/2, phospho-p105 (NFκB), and phospho-p65 (NFκB) were purchased from Cell Signaling Technology (Danvers, MA). Mal/Tirap and MyD88 antibody (HFL-296) were from Invitrogen and Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), respectively. HA antibody (12CA5) was from Roche Applied Science (Indianapolis, IN). Flag (M2) antibody and imidazole were from Sigma-Aldrich (St. Louis, MO). Ultrapure LPS from E. coli K12 and endotoxin-free water were purchased from InvivoGen (San Diego, CA).
2.7. Western blot (immunoblot) analysis
Cell lysates were prepared in lysis buffer [150 mM NaCl, 50 mM Tris-Cl (pH 8.0), 5 mM EDTA, 1% Nonidet P-40] containing a protease inhibitor cocktail (Roche, Indianapolis, IN) and a phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). The equal amount of total protein was subjected to SDS-PAGE analysis, and immunoblotting with the appropriate antibodies was performed as previously described (25). For immunoblotting with anti-phospho-tyrosine antibody (4G10), transferred PVDF blot was incubated with HRP-conjugated 4G10 antibody (1:2,500) in 3% IgG-free BSA-containing TBS mixed with Tween 20 (0.05%) for 1 hour with shaking. After washing three times, the antigen-antibody complexes were visualized by enhanced chemiluminescence (ECL).
2.8. Immunoprecipitation assay
Total cell lysates were prepared in lysis buffer as described above. Equal amounts of total protein were mixed with monoclonal anti-HA–agarose antibody and incubated at 4 °C for 4-5 hours. The precipitants were washed three times with lysis buffer and fractionated in SDS-PAGE, followed by the immunoblotting procedure described above.
2.9. Measuring IL-6 and IL-8 production
An enzyme-linked immunosorbent assay (ELISA) was performed to measure the level of the cytokine expression using the appropriate kits from R&D Systems (Minneapolis, MN), following the manufacturer's instructions. All assays were performed in triplicate, and data were shown as mean + SEM.
2.10. Luciferase reporter assays
RAW264.7 cells were plated in six-well plates (0.5 × 106 cells/well) and co-transfected with NFκB-luciferase reporter and Src-R/A constructs, including a β-galactosidase expression plasmid (HSP70-β-gal) as an internal control, using SuperFect transfection reagent (Qiagen, Valencia, CA), according to the manufacturer's instructions. One day after transfection, cells were stimulated with LPS and imidazole for 6 hours, and the relative luciferase activity was determined by the normalization with β-galactosidase activity as previously described (25). The total amount of plasmid DNA was kept consistent by adding the empty vector for each transfection. All assays were performed in triplicate. Data were shown as mean + SEM.
3. Results
3.1. Chemical rescue of SFK activity induces tyrosine phosphorylation of TLR4, but not TLR5
It has been suggested that a protein tyrosine kinase containing active-site mutations can be restored by the functional substitution of the missing side chain with a small molecule like imidazole. The small molecule may effectively stand in for the side chain of a mutated amino acid if it contains similar electronic or steric features (20,21). Arg388, found in SFK-Src, is a conserved residue in protein tyrosine kinases. Qiao et al. showed that when the Arg388 residue in SFK-Src was mutated to Ala (Src R388A), the catalytic activity could be effectively rescued by imidazole (21). In this way, the chemical rescue approach allows highly specific and precise temporal activation of SFKs in living cells (20,21). In studying the role of SFKs, the chemical rescue approach has a great advantage over gene silencing or gene knockout systems, in which surviving cells or genetically modified mice might have become adapted to a SFK gene-deficient environment, and consequently adopted a compensatory or redundancy mechanism using other SFK members to overcome the gene deficiency (19).
The tyrosine phosphorylation at the C-terminal regulatory tail of SFKs by C-terminal Src kinase (Csk) results in a closed conformation, leading to an inactive form of the enzyme (Fig. 1A). In contrast, dephosphorylation at the tyrosine residue leads to an open conformation, giving rise to a constitutively active SFK (Fig. 1B) (27,28). However, the presence of the C-terminal Y527F mutation in SFK-Src prevents inhibitory phosphorylation by Csk. When this mutation is present, the catalytic activity of the SFK-Src R388A/Y527F (Src-R/A) mutant can be rescued by imidazole treatment, while preventing inhibitory phosphorylation by Csk. On the other hand, the SFK-Src Asp386→Asn (D386N) mutant is a catalytically defective form of Src and is insensitive to imidazole (21). Thus, SFK-Src D386N/Y527F (Src-D/N) was used as control.
Figure 1. Domain structure of SFKs and tyrosine phosphorylation-dependent SFK activation mechanism.

(A and B) SFKs have an SH4 (Src homology 4) domain, which can be post-translationally modified with myristoylation and palmitoylation for plasma membrane targeting; an SH3 domain, which interacts with the SH1 domain; an SH1 (or kinase catalytic domain) in which tyrosine phosphorylation renders SFKs active; and an R (regulatory) domain in the C-terminal end in which phosphorylated tyrosine interacts with the SH2 domain. SH2 and SH3 participate in protein-protein interaction. Phosphorylation at the tyrosine residue in the C-terminus induces a closed conformation that is inactive (A). Dephosphorylation at the tyrosine residue in the C-terminus results in an open-conformation of SFKs, followed by autophosphorylation at the tyrosine residue in the kinase domain to become an active form of SFK (B).
Based on this background, we used the rescue approach of SFKs to investigate whether inducible temporal activation of SFKs would cause the tyrosine phosphorylation of TLR4. HEK293 cells were stably co-transfected with SFK-Src-R/A or SFK-Src-D/N in conjunction with TLR4-HA encoding construct. Our data showed that imidazole treatment clearly induced SFK-Src activation (evaluated by phospho-Src) in Src-R/A expressing cells, while Src-D/N expressing cells did not exhibit Src activation after imidazole treatment (Fig. 2A). In line with imidazole-rescued SFK activation, the immunoprecipitation results showed that imidazole treatment induced the tyrosine phosphorylation of TLR4 in Src-R/A cells, while TLR4 tyrosine phosphorylation was not observed in Src-D/N cells. Notably, the overexpression of SFK-Src-R/A or SFK-Src-D/N alone did not induce TLR4 tyrosine phosphorylation when imidazole was absent. We confirmed that the level of total Src (pan-Src) and TLR4 expression was similar between the samples. Therefore, these data indicate that temporal and specific SFK activation induces tyrosine phosphorylation in TLR4.
Figure 2. Rescued SFK activity induces TLR4 tyrosine phosphorylation.

(A and B) HEK293 cells (A) were stably transfected with HA-TLR4 WT (full length) construct together with SFK-Src R388A/Y527F (Src-R/A) or SFK-Src D386N/Y527F (Src-D/N) construct. Human colonic epithelial (NCM460) cells (B) were also stably transfected with HA-TLR5 construct and Src-D/N or Src-R/A construct. The cells were treated with fresh prepared imidazole (Imid., 10 mM) for the indicated time points, as we previously described (39). The cell lysates were subjected to immunoprecipitation (IP) assay with HA antibody, followed by immunoblot (IB) analysis with anti-phospho-tyrosine antibody (4G10) to examine the tyrosine phosphorylation of TLR4 (TLR4-Y-P). In parallel, the cell extracts were applied for IB analysis to determine imidazole-rescued SFK-Src activation represented by phospho-Src (Y416). Subsequently, total Src (pan-Src) and HA-TLR4 expression were evaluated. β-Actin was determined as a loading control. Presented are the representative from three independent experiments.
Similar to TLR4, TLR5 is a type-I transmembrane protein that recognizes bacterial flagellin and associates with MyD88 and Mal/Tirap to mediate inflammatory responses (26,29). We next tested whether rescuing SFK activity would also induce the tyrosine phosphorylation of TLR5. Since intestinal epithelial cells strongly express TLR5 and exhibit potent intracellular signaling following the binding of flagellin (24,29), human colonic epithelial cells (NCM460) were co-transfected with SFK-Src-R/A or SFK-Src-D/N with TLR5-HA constructs. In NCM460 cells transfected with SFK-Src-R/A, imidazole treatment induced Src activation, whereas SFK-Src-D/N expressing NCM460 cells did not exhibit SFK activation upon imidazole treatment (Fig. 2B, lane 1 to 6). However, the immunoprecipitation data showed that restored SFK activation did not induce the tyrosine phosphorylation of TLR5, while, as a positive control, restored SFK-Src activity in HEK293 cells still elicited TLR4 tyrosine phosphorylation (Fig. 2B, lane 7). It is worth noting that little difference was otherwise observed between imidazole-treated and vehicle-treated HEK293 or NCM460 cells. Moreover, when imidazole was absent, SFK-Src activation was not observed in HEK293 or NCM460 cells expressing Src-R/A or Src-D/N. Together, these data demonstrate that temporally restored SFK activation induces the tyrosine phosphorylation of TLR4, but not TLR5.
3.2. Rescued SFK catalytic activity inhibits LPS-induced inflammatory responses
Next, we examined a functional outcome of SFK activation in TLR4-mediated signaling pathways. Human colonic epithelial SW480 cells are responsive to LPS with functionally active endogenous TLR4 expression (30). Therefore, we stably transfected SW480 cells with the SFK-Src-R/A encoding construct to generate SW480-Src-R/A cells. SW480-Src-R/A cells were then treated with LPS in conjunction with imidazole or vehicle. Due to the preserved endogenous TLR4-mediated signaling, LPS stimulation was able to induce ERK1/2, JNK1/2, and NFκB (represented by phosphorylated p105 and p60) activation in SW480-R/A cells without imidazole treatment (Fig. 3A). As an immediate signaling event, the activation of SFK-Src was induced by LPS stimulation in a time-dependent manner in SW480-R/A cells without imidazole treatment. However, imidazole treatment enhanced LPS-induced SFK-Src activation. In addition, LPS-induced ERK1/2 activation was further increased by imidazole treatment. Because SFK activation is able to induce ERK1/2 activation in SW480 cells (31), these signaling events indicate that SFK activity was successfully rescued. Thus, we believe that the combination of rescued SFK activity and LPS stimulation led to potentiated SFK-Src and ERK/12 activation. However, imidazole treatment inhibited LPS-stimulated JNK1/2 and NFκB activation in SW480-Src-R/A cells. We confirmed that total Src expression was similar between the samples, and that Src-R/A expression itself induced no difference in MAPKs and NFκB activation in the absence of imidazole.
Figure 3. Restored SFK activity inhibits LPS-stimulated responses in SW480 and RAW264.7 cells.

(A) Human colonic epithelial SW480 cells were stably transfected with SFK-Src-R/A construct, followed by LPS (0.5 μg/mL) and/or imidazole (10 mM) treatment for the indicated time points. Cell lysates were subjected to immunoblot analysis. (B) RAW264.7 cells were transiently co-transfected with NFκB-luciferase (Luc.) reporter and Src-R/A constructs, followed by LPS (20 ng/mL) and/or imidazole treatment for as indicated 6 hours. Cell lysates were used for measuring the luciferase activity (RLA, relative luciferase activity). (C and D) SW480-Src-R/A cells were treated with LPS (0.5 |ig/mL) and/or imidazole as indicated for 8 hours. The culture supernatants were collected to measure the cytokine production by ELISA performed in triplicate. All data presented are the representative from three independent experiments. The data in graphs were shown as mean + SEM. ***P<0.001 (Mann-Whitney U test).
Subsequently, with the transient co-transfection of NFκB-luciferase reporter and Src-R/A constructs into murine macrophages RAW264.7 cells, we found that imidazole treatment substantially reduced LPS-induced NFκB reporter activity (Fig. 3B). Furthermore, production of the LPS-stimulated cytokines IL-6 and IL-8 was markedly reduced in imidazole-treated Src-R/A cells compared to vehicle-treated Src-R/A cells (Fig. 3C and D). Imidazole treatment itself did not influence NFκB reporter activity or cytokine production.
Together, our results demonstrate that restored SFK activation inhibits LPS-induced JNK1/2 and NFκB activation, while it enhances ERK1/2 activation. Due to its inhibitory effects on JNK1/2 and NFκB activation, SFK activation diminishes cytokine production in LPS-induced inflammatory responses.
3.3. Restored SFK activation disrupts the association of MyD88 and Mal/Tirap with TLR4, butTLR4-Tram association is preserved
In order to decipher the mechanism by which SFK activation can inhibit LPS-induced responses, HEK293-Src-R/A cells were treated with LPS in the presence or absence of imidazole, followed by an immunoprecipitation assay. In the absence of imidazole, LPS stimulation induced Src activation in a time-dependent manner in Src-R/A cells; however, the presence of imidazole substantially potentiated LPS-induced SFK-Src activation, indicating that Src activation was restored by the small molecule (Fig. 4A). As expected, in the absence of imidazole, LPS stimulation induced the association of endogenous MyD88 and Mal/Tirap adaptors with TLR4. However, the interaction of MyD88 or Mal/Tirap with TLR4 was substantially reduced by imidazole treatment. Notably, LPS-induced interaction of Tram with TLR4 was preserved in vehicle- or imidazole-treated Src-R/A cells (Fig. 4B). These data indicate that the induced SFK catalytic activity disrupted the association of MyD88 and Mal/Tirap with TLR4, whereas SFK activation did not alter LPS-induced TLR4-Tram interaction.
Figure 4. Rescued SFK activation inhibits the recruitment of MyD88 and Mal/Tirap adaptors to TLR4.

(A) HEK293 cells stably expressing SFK-Src (R/A) and HA-TLR4-WT were treated with a combination of LPS (100 ng/mL) and/or imidazole (10 mM) as indicated. Cell lysates were subjected to IP with HA antibody, followed by IB with antibody recognizing endogenous Mal/Tirap or MyD88. In parallel, the cell lysates were used for IB analysis to evaluate imidazole-induced SFK-Src activation [P-Src (Y416)] and total Src. (B) HEK293 cells expressing SFK-Src (R/A) and HA-TLR4-WT were transiently transfected with Tram-Flag encoding construct and treated with LPS and/or imidazole, followed by IP with HA and IB with Flag antibody. Cell lysates were also used for IB to evaluate SFK-Src activation and transfected gene expression. β-Actin was determined as a loading control. Presented are the representative from three independent experiments.
Together with the finding that rescued SFK activity induces the tyrosine phosphorylation of TLR4 (Fig. 2), our immunoprecipitation data suggest that TLR4 tyrosine phosphorylation by activated SFKs suppresses LPS-induced responses by dissociating MyD88 and Mal/Tirap from TLR4. Therefore, SFK activation should be regarded as a negative regulator in LPS-induced inflammatory responses.
3.4. The catalytic activity of SFKs is required to induce tyrosine phosphorylation of TLR4
We previously demonstrated that a murine TLR4 mutant with the deletion of a large part of the ectodomain (ΔTLR4) becomes a constitutively active TLR4 (22). In human TLR4 (hTLR4), the CD4-hTLR4 chimera renders hTLR4 constitutively active (32). Like the LPS-induced tyrosine phosphorylation in the full-length wild type of TLR4, the constitutively active TLR4 (both ΔTLR4 and CD4-hTLR4) is tyrosine phosphorylated when expressed in cells; however, this occurs even in the absence of LPS stimulation (5). The P712H mutation and equivalent P714A mutation in the cytoplasmic TIR domain of murine and human TLR4, respectively, not only inhibits TLR4 signaling (22,33), but also diminishes the tyrosine phosphorylation of TLR4 (5).
Among the nine SFK members, SFK-Lyn is highly expressed in macrophages/monocytes, DCs, and B cells (34). It was shown that SFK-Lyn is immediately activated by LPS stimulation and physically interacts with TLR4 in response to LPS (5). LPS induces autophosphorylation of SFK-Lyn in wild type macrophages, but not in macrophages with the TLR4 (P712H) mutation (9). In agreement with these findings, we observed that the constitutively active ΔTLR4-WT recruits SFK-Lyn in the absence of LPS stimulation, while this association is disrupted by the P712H mutation in TLR4 (Fig. 5A).
Figure 5. SFK kinase activity regulates TLR4 tyrosine phosphorylation and the interaction between TLR4 and SFK.


HEK293 cells were transiently co-transfected with the plasmid constructs in an indicated combination, followed by IP assay with HA antibody and IB analysis with Flag antibody or anti-phospho-tyrosine antibody (4G10) to evaluate the tyrosine phosphorylation of TLR4 or SFK-Lyn. (A) SFK-Lyn wild-type interacts with the constitutively active TLR4 (ΔTLR4-WT), but not with the dominant negative TLR4 [ΔTLR4 (P712H)]. (B) The kinase-dead form of SFK-Lyn [Lyn (Y397/508F)] inhibits the tyrosine phosphorylation in the constitutively active TLR4. (C and D) The kinase-active form of SFK-Lyn [Lyn (Y508F)] enhances the tyrosine phosphorylation of ΔTLR4. Cell lysates of the transfected HEK293 cells were used for two sets of the experiments (C and D) of IP with HA, followed by IB with 4G10 antibody. The blots were stripped and reprobed with Flag antibody. The expression of transfected constructs was confirmed by IB analysis. The representative set was shown from three independent experiments.
Subsequently, we investigated whether the catalytic activity of SFKs is responsible for inducing TLR4 tyrosine phosphorylation. We co-transfected HEK293 cells with a constitutively active TLR4 (HA-ΔTLR4-WT) together with a kinase-dead form of SFK-Lyn (Y397/508F). In contrast to the elevated tyrosine phosphorylation seen in the constitutively active ΔTLR4, expression of the kinase-dead SFK-Lyn (Y397/508F) practically eliminated the tyrosine phosphorylation of ΔTLR4, as revealed by the immunoprecipitation assay (Fig. 5B). However, tyrosine phosphorylation of ΔTLR4 was augmented by the expression of active SFK-Lyn (Y508F) (Fig. 5C, lane 3 and 4). Notably, the low levels of tyrosine phosphorylation observed in ΔTLR4 (P712H) were also increased by the expression of active SFK-Lyn (Y508F) (Fig. 5C, lane 5 and 6).
Our data clearly indicate that the kinase-dead mutant of SFK-Lyn eliminates tyrosine phosphorylation of TLR4, whereas the kinase-active SFK-Lyn enhances TLR4 tyrosine phosphorylation. Therefore, the catalytic activity of SFK is required to induce TLR4 tyrosine phosphorylation.
3.5. The open conformation of SFKs is important for TLR4 interaction, and SFK-active kinase activity promotes this interaction
The phosphorylation of either of two tyrosine residues, one found in the kinase domain and the other in the C-terminal tail region, exerts opposing regulatory effects on the activity of SFKs. A phosphorylated tyrosine at the C-terminal tail of SFK binds to its SH2 domain to lock the molecule into an inactive closed conformation, in which an essential motif for the catalytic activity is buried (Fig. 1A). Conversely, dephosphorylation at the C-terminal tyrosine residue leads to an open conformation, exposing the tyrosine residue within the kinase domain for phosphorylation in order to reconstitute fully active tyrosine kinase activity (Fig. 1B) (35,36).
These two tyrosine residues are conserved in all SFKs. In SFK-Lyn, Tyr508 at the C-terminal region is key to generating either the inactive closed or active open conformation. Thus, the Y508F mutation in SFK-Lyn prevents phosphorylation, instead forcing an open conformation. Subsequent autophosphorylation of Tyr397 in the activation loop of the kinase domain renders SFK-Lyn enzymatically active (37). Thus, the presence of both Y508F and Y397F mutations gives rise to a kinase-dead open conformation of SFK-Lyn.
Within this context, we next examined whether the catalytic activity of SFK-Lyn is required for interaction with TLR4. Compared to the interaction between ΔTLR4-WT and SFK-Lyn-WT (Fig. 5D, lane 7), ΔTLR4-WT:SFK-Lyn (Y508F) interaction was greatly enhanced (Fig. 5D, lane 4). Just like the disrupted interaction observed between ΔTLR4 (Y712H) and SFK-Lyn-WT (Fig. 5A, lane 5), the interaction between ΔTLR4 (Y712H) and SFK-Lyn (Y508F) was dramatically reduced compared to the strong ΔTLR4-WT: SFK-Lyn (Y508F) interaction (Fig. 5D, lane 4 and 5). Importantly, elevated tyrosine phosphorylation was identified in SFK-Lyn (Y508F), which exhibited a strong interaction with ΔTLR4-WT (Fig. 5D, lane 4), indicating that the key Tyr397 residue in the kinase domain of SFK-Lyn (Y508F) was phosphorylated. Together, these data suggest that the open conformation of SFK-Lyn has a greater capacity to interact with TLR4 than the wild type SFK-Lyn.
Next, we tested whether it is necessary for the SFK to be kinase-active in order to interact with TLR4. To investigate, we used an Y397F mutation in SFK-Lyn. Tyr397 in SFK-Lyn is a conserved residue within the kinase domain which must be phosphorylated for the enzyme to be active; therefore, this mutation rendered SFK-Lyn inactive. Moreover, an Y508F mutation causes SFK-Lyn to adopt an open conformation, which is normally followed by autophosphorylation at Tyr397 to confer enzymatic activity. Therefore, we used a SFK-Lyn (Y397/508F) double mutant which held an open conformation but was enzymatically inactive. We found that SFK-Lyn (Y397/508F) was still able to interact with ΔTLR4-WT (Fig. 6, lane 4). However, the degree of ΔTLR4-WT:SFK-Lyn (Y397/508F) interaction was dramatically reduced compared to the strong interaction between ΔTLR4-WT and SFK-Lyn (Y508F) (Fig. 6, lane 5). It is worth noting that, as expected, SFK-Lyn (Y397/508F) interacting with ΔTLR4-WT did not exhibit tyrosine phosphorylation, while SFK-Lyn (Y508F) strongly interacting with ΔTLR4-WT revealed strong tyrosine phosphorylation. These data indicate that the tyrosine phosphorylation detected in SFK-Lyn (Y508F) occurs at Tyr397 in the kinase domain.
Figure 6. SFK kinase activity promotes the interaction between TLR4 and SFK.

HEK293 cells were transfected with a combination of the plasmid construct as indicated. Cell extracts were subjected to IP with HA antibody, followed by IB with 4G10 antibody. The blot was stripped and reprobed with Flag antibody. The expression of transfected constructs was confirmed by IB analysis. The representative set was presented from three independent experiments.
Taken together, our data clearly indicate that the open conformation of SFK promotes the TLR4-SFK interaction, and that the kinase activity of SFK greatly potentiates the interaction between TLR4 and SFK-Lyn.
4. Discussion
LPS, a potent inflammatory endotoxin produced by Gram-negative bacteria, induces the pleiotropic production of inflammatory cytokines in various cells by binding to its specific membrane receptor, TLR4. LPS-TLR4 interaction induces diverse intracellular signaling pathways using a combination of TIR domain-containing adaptor molecules. Simultaneously, LPS stimulation immediately activates SFKs in a variety of LPS-responsive cells. Thus, SFK activation is among the major signaling events occurring immediately after LPS-TLR4 interaction. Nevertheless, its functional role in TLR4-induced responses has long been an enigma due to the preponderance of contradictory data from various gene knockout/silencing approaches in which the effect of a certain SFK deficiency might be obscured by a redundant or compensatory effect provided by other SFK members during LPS signaling.
Recently, an elegant study established the highly efficient chemical rescue approach to temporally rescue SFK catalytic activity in living cells (21). In contrast to the traditional approaches including gene knockout/silencing, overexpression of mutant constructs, and use of inhibitory molecules, the rescuing approach permits highly specific and precise temporal manipulation of SFK activation, using the relatively non-toxic and cell permeable molecule imidazole as a rescuing agent (21). Therefore, the specific and temporal rescue of SFK activity appears to be the best method to avoid potential cellular adaptation to SFK gene deficiency caused by gene knockout/silencing approaches.
Using the temporal restoration of SFK, we showed in this study that SFK activation induces the tyrosine phosphorylation of TLR4, but not TLR5. SFK activation causes the dissociation of the adaptors MyD88 and Mal/Tirap from TLR4 during LPS signaling, leading to the inhibition of LPS-induced JNK1/2 and NFκB activation and the consequent reduction of LPS-induced cytokine production. These data provide concrete evidence that SFK activation plays an inhibitory role in TLR4-induced signaling pathways. In agreement with this conclusion, an elegant study by Dr. Lowell's group recently suggested that SFK-Lyn deficient DCs could be hyper-responsive to LPS and IL-1β stimulation, in which MyD88 is a shared adaptor mediating TLR4 and IL-1R signaling (17). Similarly, another recent study suggested that RNAi transfection targeting SFK-Lyn upregulated LPS-induced TNFα production in RAW264.7 cells (15), implying a negative regulatory effect of SFK-Lyn upon LPS-induced responses in macrophages. In line with these studies, our data provide clear evidence that the catalytic activity of SFKs suppresses LPS-induced inflammatory responses.
As for the exact way in which SFKs affect TLR4-mediated responses, the relevant mechanism has not yet been clearly suggested. With an approach based upon temporal SFK activity restoration, we also demonstrate here that restoring SFK activation results in the tyrosine phosphorylation of TLR4 (Fig. 2). SFK activation disrupts the LPS-induced association of MyD88 and Mal/Tirap with TLR4, thus inhibiting LPS-induced inflammatory responses (Fig. 3 and 4). Moreover, we found that kinase-dead SFK-Lyn blocks the tyrosine phosphorylation of TLR4, while constitutively active SFK-Lyn potentiates tyrosine phosphorylation of TLR4 (Fig. 5B and C). In short, the intact kinase activity of SFK-Lyn promotes the interaction between TLR4 and SFK-Lyn (Fig. 6). Based on these results, it can be concluded that, upon LPS stimulation, SFK activation causes the tyrosine phosphorylation of TLR4. Subsequently, tyrosine phosphorylation induces the dissociation of MyD88 and Mal/Tirap from LPS-stimulated TLR4. Since LPS stimulation immediately induces SFK activation together with the activation of MyD88-dependent or -independent signaling pathways, we believe that LPS-induced SFK activation plays a critical role in a negative feedback loop of LPS-induced inflammatory responses, which would contribute to LPS tolerance. In line with our data, Dr. Lowell's group showed that DC-specific SFK-Lyn-KO mice develop autoimmune and inflammatory diseases with enhanced responsiveness in SFK-Lyn-KO DCs to LPS stimulation (17). This notion is substantiated by a study by Aki et al. which demonstrated that silencing Csk gene expression downregulates LPS-induced TNFα and IL-6 production in RAW264.7 cells (38). Csk activation phosphorylates the tyrosine residue at the C-terminus of SFKs, leading to their inactivation. Conversely, Csk deficiency results in dephosphorylation at the C-terminus of SFKs, causing their activation. Accordingly, the study by Aki et al. implied that SFK activation could inhibit LPS-induced responses. In line with these observations, our study provides compelling evidence that SFK activation negatively regulates TLR4-induced inflammatory signaling pathways.
In summary, in addition to the immediate recruitment of the TIR-domain containing adaptor proteins to the cytoplasmic region of TLR4 to mediate LPS-induced immune and inflammatory responses, LPS stimulation also immediately induces the association of SFK with TLR4 to induce the tyrosine phosphorylation of TLR4. The tyrosine phosphorylation results in the dissociation of the adaptor molecules MyD88 and Mal/Tirap, from TLR4, leading to suppressed LPS-induced inflammatory responses (Fig. 7). Using chemical rescue of a mutant SFK, our study, therefore, provides clear mechanistic evidence for how SFK activation plays an inhibitory role in TLR4-mediated signaling pathways; and provides significant insight into the molecular mechanism of LPS tolerance.
Figure 7. The activation of SFK by LPS results in the dissociation of MyD88 and Mal/Tirap from TLR4, leading to suppressed LPS-induced inflammatory responses.

(A) LPS recognition by TLR4 immediately induces the recruitment of TIR-domain containing adaptor molecules, including MyD88 and Mal/Tirap, to the cytoplasmic region of TLR4, leading to immune and inflammatory responses. Simultaneously, LPS-stimulation elicits SFK binding to TLR4, which induces the tyrosine phosphorylation of TLR4. (B) The tyrosine phosphorylation of TLR4 by SFK dissociates MyD88 and Mal/Tirap from TLR4, leading to suppressed LPS-induced inflammatory responses.
Acknowledgments
This research was supported by a grant from Oakland University and the National Institutes of Health (DK079015, S.H.R) and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1D1A1B03932222 to E.I.).
Abbreviations
- BMMCs
bone marrow-derived mast cells
- Csk
C-terminal Src kinase
- IRF
IFN-regulated factor
- Mal
myeloid differentiation factor 88 (MyD88)-adapter-like
- SFK
Src-family kinase
- TIR domain
Toll/IL-1R (TIR) domain
- Tirap
toll-interleukin 1 receptor (TIR) domain containing adaptor protein
- TLRs
Toll-like receptors
- Tram
TRIF-related adaptor molecule
- TRIF
Toll/IL-1R homologous region (TIR)-domain-containingadapter-inducing IFN-β
Footnotes
Conflict of Interest: The authors declare that they have no conflicts of interest with the contents of this article.
Author Contributions: SHR and EI conceived and designed all experiments. SHR and EI wrote the paper. SHR, EI, JM, and SJK performed and analyzed all experiments. JM, SJK, AS, BV, and BS revised the paper for intellectual content and context. All authors reviewed the results and approved the final version of the manuscript. SHR has overall responsibility for the published work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124:2013–2024. doi: 10.1182/blood-2014-01-453134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badolia R, Inamdar V, Manne BK, Dangelmaier C, Kunapuli SP. Gq pathway regulates proximal C-type lectin-like receptor 2 (CLEC-2) signaling in platelets. J Biol Chem. 2017 doi: 10.1074/jbc.M117.791012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu Y, Span LM, Nygren P, Zhu H, Moore DT, Cheng H, Roder H, DeGrado WF, Bennett JS. The Tyrosine Kinase c-Src Specifically Binds to the Active Integrin alphaIIbbeta3 to Initiate Outside-in Signaling in Platelets. J Biol Chem. 2015;290:15825–15834. doi: 10.1074/jbc.M115.648428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamerman JA, Lanier LL. Inhibition of immune responses by ITAM-bearing receptors. Sci STKE. 2006;2006:re1. doi: 10.1126/stke.3202006re1. [DOI] [PubMed] [Google Scholar]
- 5.Medvedev AE, Piao W, Shoenfelt J, Rhee SH, Chen H, Basu S, Wahl LM, Fenton MJ, Vogel SN. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol Chem. 2007;282:16042–16053. doi: 10.1074/jbc.M606781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stefanova I, Corcoran ML, Horak EM, Wahl LM, Bolen JB, Horak ID. Lipopolysaccharide induces activation of CD14-associated protein tyrosine kinase p53/56lyn. J Biol Chem. 1993;268:20725–20728. [PubMed] [Google Scholar]
- 7.Beaty CD, Franklin TL, Uehara Y, Wilson CB. Lipopolysaccharide-induced cytokine production in human monocytes: role of tyrosine phosphorylation in transmembrane signal transduction. Eur J Immunol. 1994;24:1278–1284. doi: 10.1002/eji.1830240606. [DOI] [PubMed] [Google Scholar]
- 8.Gong P, Angelini DJ, Yang S, Xia G, Cross AS, Mann D, Bannerman DD, Vogel SN, Goldblum SE. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J Biol Chem. 2008;283:13437–13449. doi: 10.1074/jbc.M707986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henricson BE, Carboni JM, Burkhardt AL, Vogel SN. LPS and Taxol activate Lyn kinase autophosphorylation in Lps(n), but not in Lpsd), macrophages. Mol Med. 1995;1:428–435. [PMC free article] [PubMed] [Google Scholar]
- 10.Orlicek SL, Hanke JH, English BK. The src family-selective tyrosine kinase inhibitor PP1 blocks LPS and IFN-gamma-mediated TNF and iNOS production in murine macrophages. Shock. 1999;12:350–354. doi: 10.1097/00024382-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Lee JY, Lowell CA, Lemay DG, Youn HS, Rhee SH, Sohn KH, Jang B, Ye J, Chung JH, Hwang DH. The regulation of the expression of inducible nitric oxide synthase by Src-family tyrosine kinases mediated through MyD88-independent signaling pathways of Toll-like receptor 4. Biochem Pharmacol. 2005;70:1231–1240. doi: 10.1016/j.bcp.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 12.Smolinska MJ, Horwood NJ, Page TH, Smallie T, Foxwell BM. Chemical inhibition of Src family kinases affects major LPS-activated pathways in primary human macrophages. Molecular immunology. 2008;45:990–1000. doi: 10.1016/j.molimm.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 13.Avila M, Martinez-Juarez A, Ibarra-Sanchez A, Gonzalez-Espinosa C. Lyn kinase controls TLR4-dependent IKK and MAPK activation modulating the activity of TRAF-6/TAK-1 protein complex in mast cells. Innate Immun. 2012;18:648–660. doi: 10.1177/1753425911435265. [DOI] [PubMed] [Google Scholar]
- 14.English BK, Ihle JN, Myracle A, Yi T. Hck tyrosine kinase activity modulates tumor necrosis factor production by murine macrophages. J Exp Med. 1993;178:1017–1022. doi: 10.1084/jem.178.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borzecka-Solarz K, Dembinska J, Hromada-Judycka A, Traczyk G, Ciesielska A, Ziemlinska E, Swiatkowska A, Kwiatkowska K. Association of Lyn kinase with membrane rafts determines its negative influence on LPS-induced signaling. Mol Biol Cell. 2017;28:1147–1159. doi: 10.1091/mbc.E16-09-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keck S, Freudenberg M, Huber M. Activation of murine macrophages via TLR2 and TLR4 is negatively regulated by a Lyn/PI3K module and promoted by SHIP1. J Immunol. 2010;184:5809–5818. doi: 10.4049/jimmunol.0901423. [DOI] [PubMed] [Google Scholar]
- 17.Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA. Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci U S A. 2013;110:E3311–3320. doi: 10.1073/pnas.1300617110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng F, Lowell CA. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J Exp Med. 1997;185:1661–1670. doi: 10.1084/jem.185.9.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verollet C, Gallois A, Dacquin R, Lastrucci C, Pandruvada SN, Ortega N, Poincloux R, Behar A, Cougoule C, Lowell C, Al Saati T, Jurdic P, Maridonneau-Parini I. Hck contributes to bone homeostasis by controlling the recruitment of osteoclast precursors. FASEB J. 2013;27:3608–3618. doi: 10.1096/fj.13-232736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams DM, Wang D, Cole PA. Chemical rescue of a mutant protein-tyrosine kinase. J Biol Chem. 2000;275:38127–38130. doi: 10.1074/jbc.C000606200. [DOI] [PubMed] [Google Scholar]
- 21.Qiao Y, Molina H, Pandey A, Zhang J, Cole PA. Chemical rescue of a mutant enzyme in living cells. Science. 2006;311:1293–1297. doi: 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]
- 22.Rhee SH, Hwang D. Murine TOLL-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NF kappa B and expression of the inducible cyclooxygenase. J Biol Chem. 2000;275:34035–34040. doi: 10.1074/jbc.M007386200. [DOI] [PubMed] [Google Scholar]
- 23.Choi YJ, Im E, Pothoulakis C, Rhee SH. TRIF modulates TLR5-dependent responses by inducing proteolytic degradation of TLR5. J Biol Chem. 2010;285:21382–21390. doi: 10.1074/jbc.M110.115022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi YJ, Im E, Chung HK, Pothoulakis C, Rhee SH. TRIF Mediates Tolllike Receptor 5-induced Signaling in Intestinal Epithelial Cells. J Biol Chem. 2010;285:37570–37578. doi: 10.1074/jbc.M110.158394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhee SH, Keates AC, Moyer MP, Pothoulakis C. MEK is a key modulator for TLR5-induced interleukin-8 and MIP3alpha gene expression in non-transformed human colonic epithelial cells. J Biol Chem. 2004;279:25179–25188. doi: 10.1074/jbc.M400967200. [DOI] [PubMed] [Google Scholar]
- 26.Choi YJ, Jung J, Chung HK, Im E, Rhee SH. PTEN regulates TLR5-induced intestinal inflammation by controlling Mal/TIRAP recruitment. FASEB J. 2013;27:243–254. doi: 10.1096/fj.12-217596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H. CSK: a protein-tyrosine kinase involved in regulation of src family kinases. J Biol Chem. 1991;266:24249–24252. [PubMed] [Google Scholar]
- 28.Kaplan KB, Bibbins KB, Swedlow JR, Arnaud M, Morgan DO, Varmus HE. Association of the amino-terminal half of c-Src with focal adhesions alters their properties and is regulated by phosphorylation of tyrosine 527. EMBO J. 1994;13:4745–4756. doi: 10.1002/j.1460-2075.1994.tb06800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhee SH, Kim H, Moyer MP, Pothoulakis C. Role of MyD88 in phosphatidylinositol 3-kinase activation by flagellin/toll-like receptor 5 engagement in colonic epithelial cells. J Biol Chem. 2006;281:18560–18568. doi: 10.1074/jbc.M513861200. [DOI] [PubMed] [Google Scholar]
- 30.Tang XY, Zhu YQ, Wei B, Wang H. Expression and functional research of TLR4 in human colon carcinoma. Am J Med Sci. 2010;339:319–326. doi: 10.1097/MAJ.0b013e3181cef1b7. [DOI] [PubMed] [Google Scholar]
- 31.Su N, Peng L, Xia B, Zhao Y, Xu A, Wang J, Wang X, Jiang B. Lyn is involved in CD24-induced ERK1/2 activation in colorectal cancer. Mol Cancer. 2012;11:43. doi: 10.1186/1476-4598-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 33.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 34.Gross AJ, Lyandres JR, Panigrahi AK, Prak ET, DeFranco AL. Developmental acquisition of the Lyn-CD22-SHP-1 inhibitory pathway promotes B cell tolerance. J Immunol. 2009;182:5382–5392. doi: 10.4049/jimmunol.0803941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 36.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Molecular cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 37.Harder KW, Parsons LM, Armes J, Evans N, Kountouri N, Clark R, Quilici C, Grail D, Hodgson GS, Dunn AR, Hibbs ML. Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity. 2001;15:603–615. doi: 10.1016/s1074-7613(01)00208-4. [DOI] [PubMed] [Google Scholar]
- 38.Aki D, Mashima R, Saeki K, Minoda Y, Yamauchi M, Yoshimura A. Modulation of TLR signalling by the C-terminal Src kinase (Csk) in macrophages. Genes Cells. 2005;10:357–368. doi: 10.1111/j.1365-2443.2005.00839.x. [DOI] [PubMed] [Google Scholar]
- 39.Im E, Kazlauskas A. Src family kinases promote vessel stability by antagonizing the Rho/ROCK pathway. J Biol Chem. 2007;282:29122–29129. doi: 10.1074/jbc.M702637200. [DOI] [PubMed] [Google Scholar]