

Summary
Tumor sequencing studies have revealed the widespread genetic diversity of melanoma. Sequencing of 108 genes previously implicated in melanomagenesis was performed on 462 patient-derived xenografts (PDX), cell lines and tumors to identify mutational and copy number aberrations. Samples came from 371 unique individuals; 263 were naïve to treatment, and 108 were previously treated with targeted therapy (34), immunotherapy (54) or both (20). Models of all previously reported major melanoma subtypes (BRAF, NRAS, NF1, KIT and WT/WT/WT) were identified. Multiple minor melanoma subtypes were also recapitulated, including melanomas with multiple activating mutations in the MAPK signaling pathway and chromatin remodeling gene mutations. These well-characterized melanoma PDX and cell lines can be used not only as reagents for large array of biological studies, but also as pre-clinical models to facilitate drug development.
Keywords: melanoma, patient-derived xenografts, massively parallel sequencing, cell lines
eTOC
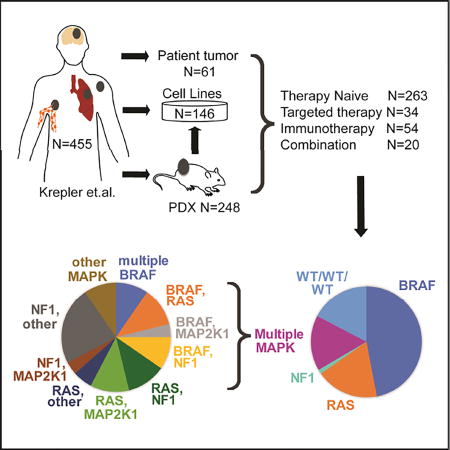
Garman et al. have characterized melanoma PDX and cell lines described in Krepler et al., identifying major and minor subtypes, some of which were previously not well-defined, targeted and immunotherapy resistance, and tumor heterogeneity, creating a set of reagents for future drug discovery and biological studies.
Introduction
Although cancer incidence overall has declined in the United States from 2002–2011, the incidence rates of melanoma continue to rise (Ryerson et al., 2016). If diagnosed early, surgical resection is curative in most melanoma patients. However, roughly 20% of patients will develop metastatic disease. Melanoma accounts for approximately 50,000 deaths per year worldwide, over 75% of skin cancer-related mortality (Corrie et al., 2014). With the cost of massively parallel sequencing technologies decreasing at a rapid rate, “precision medicine” is routinely practiced, in which the genetic profile of a patient’s melanoma is obtained and used to guide diagnosis and treatment. This practice is particularly valuable for melanoma due to the malignancy’s severity and the availability of effective targeted therapies for common mutations (Robert et al., 2015).
Melanoma is characterized by constitutive activation of the mitogen-activated protein kinase (MAPK) and/or phosphoinositide 3-kinase (PI3K) signaling pathways and disruption of the cell cycle. Approximately 45% of melanomas harbor an activating mutation affecting codon 600 of the serine/threonine-protein kinase BRAF (BRAF V600E) against which targeted inhibitors (BRAFi) were developed (Davies et al., 2002; Chapman et al., 2011; Krepler et al., 2016). BRAFi provide clinical benefit to a large percentage of advanced melanoma patients whose tumors harbor a BRAF V600E mutation. However, median progression-free survival is approximately six months (Chapman et al., 2011; Hauschild et al., 2012). Combining BRAFi with MEK inhibitor (MEKi) therapy increases responses rates and approximately doubles median progression-free survival (Robert et al., 2015). Nevertheless, drug resistance is still a major hurdle in the long-term management of melanoma with targeted therapies (Wagle et al., 2014). Simultaneously, immune checkpoint inhibitors have been increasingly used for melanoma treatment. These agents (anti-CTLA-4, anti-PD-1, and anti-CTLA-4/anti-PD1) have demonstrated increasing rates of responses in clinical trials, many of which are durable (i.e., > 2 years) (Larkin et al., 2015). Targeted and immune-therapy combinations are currently being explored.
In recent years, several large-scale massively parallel sequencing studies have provided valuable insights into the genetics of melanoma. Initial whole exome sequencing studies demonstrated that NF1, ARID2, PPP6C, RAC1, SNX31, TACC1, and STK19 are significantly mutated genes in melanoma (Hodis et al., 2012; Krauthammer et al., 2012). The Cancer Genome Atlas Skin Cutaneous Melanoma (SKCM-TCGA) exome sequencing data set identified several additional significantly mutated melanoma genes, namely MAP2K1, IDH1, RB1, and DDX3X (TCGA, 2015). The same groups classified melanomas into genetic subtypes: BRAF mutant, RAS mutant, NF1 mutant, and the triple wild-type (WT/WT/WT). Rare, low-frequency, driver mutations were identified in the WT/WT/WT subtype in KIT, CTNNB1, GNA11, and GNAQ. Additionally, an apparent increase in CNV frequency in WT/WT/WT, particularly copy number amplifications, was detected in driver genes. Further whole exome sequencing studies revealed that NF1-mutant melanomas frequently carry additional mutations in other MAPK signaling pathway genes (Krauthammer et al., 2015; Arafeh et al., 2015).
As sequencing of patient tumors continues to reveal the widespread genetic variability of melanomas, there is a critical need for genetically-annotated melanoma translational models that accurately recapitulate the biology and molecular characteristics of the patient’s original tumor for use in preclinical studies to develop personalized treatment strategies. We sequenced genes previously implicated in melanomagenesis to evaluate mutations and copy number changes in 115 human melanoma cell lines, 248 patient-derived xenografts (PDX), 31 cell lines derived from PDX (PDX CL), and 68 patient tumors (462 samples total). Two hundred and sixty-three patients with melanoma were treatment naïve, 54 previously exposed to immunotherapy with anti-CTLA-4 or anti-PD-1, 34 to targeted therapy with BRAFi and/or MEKi, and 20 to a combination of targeted and immunotherapy.
Results
Demographic and clinicopathological characteristics of sequenced cell lines, patient derived xenografts (PDX), patient tumors and PDX cell lines (CL)
Sequencing was performed on cell lines, PDX, and patient tumors. One hundred and fifteen Wistar Melanoma cell lines were generated at the Wistar Institute, partial characterization has been reported on a subset (Hoek et al., 2006; Lin et al., 2008), and 31 additional lines developed from PDX models. A further 314 tumor samples representing 253 individuals were either made into PDX or directly sequenced from patients treated at University of Pennsylvania (UPENN, 112), MD Anderson Cancer Center (MDACC, 86), Massachusetts General Hospital (23), Helen F. Graham Cancer Center (17), Jefferson (2), John Wayne Cancer Institute (8), Wills Eye Institute (4), and University of Duisburg-Essen (1). Three patients (1%) had stage II melanoma, 34 (7%) patients had stage III, 106 (42%) patients had stage IV, and for 115 (45%) the patient’s stage at biopsy was unknown. Clinicopathological characteristics are summarized in Table S1; further details can be found in the companion paper (Krepler et al., 2017). Twenty-two samples (6% of unique cohort) were non-cutaneous melanomas, with mucosal (10; 3%), acral cutaneous (7; 2%), and uveal (5; 1%) primaries included.
Variability between cell lines, PDX, PDX CL and patient tumors
Tumors were sequenced on a custom capture panel of 108 genes (MEL V1) known to be important in melanomagenesis (Table S2). The full genes (exons and introns) were sequenced for tumor suppressors, to facilitate copy number calling, with a few exceptions. Exons only were sequenced for oncogenes. We developed an in-house annotation pipeline to classify variants as deleterious, likely deleterious, and of unknown significance (VUS) (see Methods and Figure S1). Variants and copy number alterations (CNA) were identified in all 108 targeted genes. A subset (101) was sequenced on a 119 gene panel (MEL V2) (Table S3); 106 genes were shared with MEL V2. Forty-five of 101 samples (36 unique patients) were sequenced on both panels, enriched for non-BRAF V600E/K/D and non-NRAS Q61 mutant samples. The deleterious/likely deleterious variant concordance rate of MEL V1 and MEL V2 was 94% (217 out of 231 MEL V1 variants found on MEL V2), and 97% (217 out of 223 MEL V2 variants found on MEL V1) (Figure S2). Testing results for mutations in the major driver melanoma genes (BRAF, RAS, NF1, KIT) did not differ.
Following variant calling, all 115 cell lines harbored at least one deleterious mutation, compared to 236 of 248 PDX (95%), 30 of 31 PDX CL (97%) samples, and 59 of 68 patient tumors (87%). Likely deleterious mutations were found in 69 of 115 cell lines (60%), 168= of 248 PDX (67%), 18 of 31 PDX CL (58%), and 36 of 68 patient tumors (53%). Variants of unknown significance (VUS) were found in 103 of 114 cell lines (90%), 222 of 248 PDX (93%), 27 of 31 PDX CL (87%), and 55 of 68 patient tumors (81%). Table S5 lists all called variants in our cohort. Among all four sample types, the total number of calls (deleterious/likely deleterious/VUS) did not differ significantly (Figure 1B). However, the alternate allele fractions (AAF) of variants detected across all sample types, were statistically significant different (p = 2.2×10−16). Patient tumors had the lowest AAF, presumably due to admixture with non-tumor cells, with PDX CL the highest AAF (Figure 1C).
Figure 1. Variability among four different sample types (PDX CL, PDX, patient tumor, Cell Line).

(A) Average number of deleterious variants detected per sample. (B) Average number of total filtered variants detected per sample. (C) Allelic fractions across the four sample types (D) Major subtypes (BRAF hotspot, RAS hotspot, NF1 hotspot, WT/WT/WT) and their differential distribution among entire cohort.
We compared the prevalence of common mutations across the four sample types. In the samples from the 371 unique individuals, 203 (55%) had mutations in BRAF, 72 (19%) in RAS (NRAS, KRAS) and 22 (6%) in NF1 (Figure 1D). As discussed in detail below, some samples had mutations in more than one of these genes, and for this purpose they were included in the most prevalent mutation group (e.g. those with BRAF and RAS mutations in the BRAF mutant group, and those with RAS and NF1 mutations in the RAS group). Seventy-four samples (20%) did not have mutations in the above genes (WT/WT/WT). Fourteen WT/WT/WT were non-cutaneous melanomas - either acral (1; 1%), mucosal (9; 12%), or uveal (4; 6%). For nine WT/WT/WT tumor biopsies, the sample provided may be normal tissue, as we only identified one to three VUSs at 50% allele frequency in each; four did not grow in mice and five have not been tested for growth. Cell lines had a higher prevalence of both deleterious BRAF and RAS mutations than PDX, and patient tumors (p = 0.05; Figure 1D). CDKN2A mutations and homozygous deletions occurred in higher frequency (74; 68%) in cell lines, compared to 104 (52%) PDX, and 14 (24%) tumors (p = 3.3×10−7). TP53 mutations and homozygous deletions also occurred at higher frequency in cell lines (34%), as compared to PDX (23%), and tumors (21%), but not significantly. The distribution of mutations in PDX and tumors were reflective of what has been reported previously (TCGA, 2015). In contrast, cell lines were significantly more likely to be BRAF or RAS mutant with loss of CDKN2A, likely reflecting difficulties establishing cell lines from NF1 mutant or WT tumors.
Prevalence of gene mutations and predicted copy number changes
Among all sequenced samples (462), we identified deleterious/likely deleterious mutations in 101 of 108 genes. To summarize the prevalence of gene mutations, the percentage of unique patients (371) was calculated. Deleterious mutations were most prevalent in our cohort in the following genes: TERT promoter region (215; 62.5% of all samples), BRAF (200; 58.1%), NRAS (81; 23.51%), TP53 (63; 18.3%), CDKN2A (49; 14.2%), NF1 (35; 10.2%), ARID2 (28; 8.1%) and PTEN (20; 5.8%) (Figure S3A). Likely deleterious mutations were most frequently detected in: DCC (32; 19% of all samples), GRM3 (30; 17.9%), PTPRP (19; 11.3%), PREX2 (19; 11.3%), GRIN2A (19; 10%), and PTEN (17; 10.1%) (Figure S3B). Variants were not detected in CD274, MDM4, SDHD or SMARCB1.
Two hundred and ninety-four unique samples (79%) of the 371 unique samples had a homozygous loss or high amplification in at least one gene. The mean (and range) of the number of highly amplified (copy number > 3.3) and homozygously deleted genes per sample was 1 (0 to 5) and 2 (0 to 8), respectively. The most frequently highly amplified genes were: CDK6 (91; 30%), MET (79; 26.1%), DDX3X (69; 22.8%), BRAF (68; 22.4%), DYNC1I1 (55; 18.2%), EZH2 (51; 16.8%), MITF (49; 16.2%), MYC (45; 14.9%), PREX2 (45; 14.9%), STK19 (43; 14.2%), and NOTCH2 (30; 10%) (Figure S3C). The genes most frequently homozygously deleted were: CDKN2A (130; 65.3%), CDKN2B (102; 51.3%), PTEN (47; 23.6%), and TP53 (12; 6%) (Figure S3D). A complete list of CNAs can be found in Table S6.
MAPK signaling pathway mutations
The mutational landscape of our samples revealed two distinct patterns of mutations within the MAPK signaling pathway: 1) single hotspot BRAF or NRAS mutations; and 2) multiple non-hotspot variants across different genes encoding proteins within the MAPK signaling pathway, including NF1 mutations. Across our naïve and immunotherapy cohort (317 patients; 85% of unique cohort), 206 melanomas representing unique individuals (65% of naïve and immunotherapy cohort) followed Pattern 1; 148 had solitary driver mutations in BRAF (72%) and 58 (28%) in NRAS. Pattern 2 melanomas representing 52 unique individuals (14% of unique; 16% of naïve and immunotherapy cohort) had more than one deleterious or likely deleterious mutation in either a MAPK signaling gene or in a gene encoding an effector protein of the MAPK pathway, as shown in Table 1. Three Pattern 2 samples were acral (2) and mucosal (1). Eighteen samples harbored a deleterious or likely deleterious non-V600 BRAF mutation (p.H57Y, p.G464E, p.S465Y, p.G466E, p.G469E, p.L496V, p.N581S, p.N581Y, p.D594G, p.V624F, p. K601E and p.600_601del) (Figure S4). We also noted an additional six non-V600 BRAF mutations, which we designated as VUS, but may have functional significance (p.G7S, p.F294L, p.S365L, p.S365L, p.A497V, p.T740A). We also identified five BRAF non-V600 variants (p.G9A, p.L505H, p.P318S, p.P328S, p.A366P) in samples also carrying V600E mutations, which are less likely to be functional. All non-V600 BRAF mutations had concurrent deleterious/likely deleterious mutations or high-level amplifications in other MAPK signaling genes. Co-occurring RAS deleterious mutations were most frequent, found in 60% of non-hotspot BRAF mutant samples as compared to 2%, 6.3% and 21.5% of those with BRAF V600E, V600K, and other BRAF hotspot mutations, respectively (p<0.0001). Most melanomas had a single second deleterious mutation in a MAPK signaling gene; a few had three. We identified concurrent mutations or amplifications in other MAPK signaling pathway genes in all BRAF non-V600 codon mutations, except for one, which had incomplete sequencing.
Table 1.
Samples with co-occurring mutations in the MAPK signaling pathway.

|
Green - high-level amplification (>3.3 fold); red - deleterious mutations; blue - likely deleterious mutations, grey - samples and genes not sequenced on the 119-gene panel; purple – loss of wild type allele; peach – non-cutaneous melanoma.
Twenty-three RAS mutants had co-occurring mutations in the MAPK signaling pathway (Table 1, Figure S4) and therefore fell into Pattern 2, compared to 58 Pattern 1 RAS-mutant samples (53; 91% Q61, 5; 9% non-Q61). Eleven Q61 (17% of all Q61) and 13 non-Q61 (75% of all non-Q61) mutated melanoma samples harbored additional mutations in the MAPK signaling pathway (p = 2.2×10−16 for enrichment of non-Q61 mutations). Twenty-six of the 30 NF1-mutated melanomas (87%) harbored concurrent deleterious/likely deleterious MAPK signaling pathway mutations (Table 1). Two of the remaining four NF1 mutated melanomas had VUSs in MAP3K5 (WM4242 also had a deleterious mutation in ROS1 [c.780-1G>A]). Of the four possible mutations observed in RASA2 (one truncating, three likely deleterious missense), only two were observed in NF1 mutant samples. Four out of nine (44%) KIT mutant samples also carried concurrent MAPK pathway mutations. Four “wild-type” (WT/WT/WT) samples in this cohort harbored likely deleterious and deleterious mutations in MAP2K1/2, and MAP3K5, including one with a truncating deleterious mutation in MAP3K5, p.E477X (Figure S5).
Genetic/genomic landscape of sequenced naïve melanoma cell lines, PDX, PDX CL, and patient tumors
Deleterious mutations, homozygous losses and high-level copy number amplifications in the 225 naïve to treatment samples are shown in Figure 2; only one sample from each patient is included. Likely deleterious mutations and other genomic aberrations also are included in Figure S6. As deleterious TERT promoter mutations (Table S4) were detected in 67% of samples, and ubiquitously in all subtypes, they are not included in Figures 2 or S6A.
Figure 2. Mutational and copy number profile of naive melanoma cell lines, PDX, PDX CL and tumors.

A single sample from each of 225 unique patients is included. NMVD: no missense variants in targeted genes detected.
Within BRAF-V600E-mutant treatment-naïve samples, we observed previously well-described subtypes. Forty-four of 105 (42%) BRAF V600E-mutated samples from unique patients harbored truncating and/or deleterious missense mutations in cell cycle genes (CDKN2A, CDK4, and/or TP53). The remaining 61 (58%) BRAF V600E-mutated samples had more frequent homozygous loss of CDKN2A/B (46% vs 25%; p=0.03), but not PTEN (25% vs. 11%). Homozygous deletions in PTEN occurred almost exclusively in BRAF V600E samples (91%) (p<0.0001). Within the subset of BRAF V600E-mutated samples lacking additional CDKN2A, CDK4, and/or TP53 mutations, 10 samples (9.5% of unique patients) lacked the C>T nucleotide substitution pattern characteristic of UV sun damage (Brash, 2015), possibly due to low mutation burden. NF1-mutant samples had the highest overall mutational burden of all subtypes (p = 1×10−6), the majority of which were C>T transitions. Of 11 NF1-mutant samples from unique patients, seven samples (64%) harbored a deleterious mutation in TP53, more frequently than in other subtypes (p=0.003). Ten of 29 WT/WT/WT (34.5%) harbored previously reported rare mutations in GNAQ, GNA11, and CTNNB1, in a mutually exclusive fashion, six which were uveal PDX and cell lines. A further two WT/WT/WT samples (7%) were acral and mucosal melanomas.
Multiple samples displayed high level copy number amplifications. Nineteen of 27 (70%) high amplifications in MITF occurred in BRAF V600E-mutated samples. Of the 12 co-occurrences of high amplification in FGF3/4, and/or CCND1, nine (75%) also harbored high amplification in MITF. FGF3, FGF4 and CCND1 are co-localized at 11q13.3, explaining co-occurrence, whereas MITF is on chromosome 3, suggesting a synergistic effect. Twenty-five of 31 (80.6%) concurrent amplifications in BRAF, MET, and/or EZH2 occurred in BRAF hotspot mutant samples (p = 0.009). Three very high level BRAF amplification events (6–16 fold) were identified, two of which were in BRAF V600K mutants. Finally, 14 of 24 (58%) of high amplification events in NOTCH2 co-occurred with high amplification in NRAS, both are on 1p. Of note, WT/WT/WT patients harbored significantly lower numbers of CNAs, compared to hotspot BRAF and RAS subtypes (p < 1×10−4), but not to NF1 mutated samples.
We performed formal correlation analyses to examine co-occurrence of copy number changes that were found in more than 10% of samples (Figure S6B, C). When all samples were considered, significant correlations were identified between co-localized genes, such as deletion of CDKN2A/CDKN2B (9p21.3), and amplifications of PREX2/SNX31/MYC (8q) and BRAF/RAC1/EGFR/EZH2/GRM3/DYNC1I1/CDK6/MET (chr 7). Significant correlations between non-co-localized genes also were observed, with the most significant being between MITF amplification and CDKN2A deletion (p=0.0005), MITF and EGFR amplifications (p=0.0005), and STK19 and AKT3 amplifications (p=2×10−5) (Figure S6B). The first two correlations are due to co-enrichment in BRAF mutated samples. Within the BRAF mutant subset, the only correlation that emerged was between MYC and STK19 amplifications (p=0.0001) (Figure S6C). Little is known about STK19 in melanoma; these data suggest further functional evaluation is warranted.
Genetic landscape of melanoma cell lines, PDX, PDX CL, and patient tumors exposed to targeted therapies
Forty-nine (54 samples) unique patients had samples taken either post-progression (37) or on-treatment (12) with targeted therapy. Twenty-one (43%) were treated with a combination of BRAFi and MEKi, 27 (55%) BRAFi alone, and one MEKi alone (Figure 3A). Post-progression PDX were expanded in vivo on a continuous BRAFi or BRAFi/MEKi to maintain the resistance phenotype (Krepler et al., 2016). Twenty (41%) patients received a combination of targeted and immuno-therapy.
Figure 3. Mutational and copy number profile of PDX, PDX CL and tumors from patients that received targeted therapy with BRAFi, MEKi, or combination.

(A) A single sample from each of 49 unique patients is included. (B) Five subtypes of potential resistance mechanisms in samples that progressed post-treatment.
Potential resistance mechanisms were identified for 29 of 36 (81%) patients that progressed on treatment (Figure 3B) and classified into four categories: BRAF high-level amplifications (10; 28%), NRAS mutant (6; 17%), MAP2K1 mutant (p.C121S, p.K57E/N, p.P124S, and p.Q56P) (7; 19%), and non-MAPK pathway alterations (MITF and MET high amplification, and PTEN homozygous loss) (5; 14%). Although no deleterious mutations in MAP2K2 were identified, a VUS (p.K61E) was found in the non-MAPK pathway altered group, which may be associated with resistance. MAP2K2 p.K61E has been reported in a patient with cardio-facio-cutaneous syndrome, one of the Rasopathies, supporting a functional role (Dentici et al., 2009). Additional genetic and genomic changes were observed that also may contribute to therapeutic resistance (Figure 3B). Homozygous loss of PTEN and high-level amplification of MET were seen in 20% and 50% of patients with high-level amplification of BRAF, respectively. High-level amplification of BRAF was observed in 67% of samples also having secondary NRAS Q61/G12 mutations, along with additional CNAs in non-MAPK pathway genes. The mechanisms of resistance found in the samples did not differ between patients treated with BRAFi alone and with BRAFi/MEKi.
Genetic landscape of melanoma cell lines, PDX, PDX CL, and patient tumors from patients treated with immunotherapy
Overall, 71 unique patients (two acral) were previously exposed to immunotherapy with either anti-CTLA4 (33; 46%), anti-PD-1 (19; 27%), or anti-CTLA4/anti-PD1 (19; 27%). Twenty patients (28%) received both immunotherapy and targeted therapy. Of the 71 patients, eight (11%) patients were responders (one acral), 33 (47%) had progressive disease, seven (10%) had stable disease, and one patient (1%) had a mixed response (Figure 4). Disease outcome was unknown for 22 (31%) patients (one acral). The genetic and genomic landscape was similar to the naïve sample set, with an enrichment for non-BRAF mutated tumors. However, mutational burden (nonsynonymous variants/mb) in patients that received immunotherapy was significantly higher than the naïve cohort (p = 0.03).
Figure 4. Mutational and copy number profile of PDX, PDX CL and tumors from patients that received immune-therapy with anti-CTLA-4, anti PD-1, or a combination.

A single sample from each of 49 unique patients is included. NMVD: no missense variants in targeted genes detected
Evaluation of multiple samples from the same patient
Multiple samples from 40 patients were sequenced, including cell lines, PDX, PDX cell lines, and biopsies (Figure S7, Table S7). Full mutational concordance was observed across 65 samples from 28 (70%) patients. Thirty-five samples from 12 patients were discordant. In six instances, discordance could be attributed to within patient tumor heterogeneity, three instances to within tumor heterogeneity, one instance to the development of acquired resistance mutations, two to acquired resistance mutations in cell lines adapting to targeted therapy and in two instances no potential etiology could be identified. Discordant deleterious mutations in KRAS (p.A146T, p.K117N), PTEN and TACC1 were found in two biopsies taken on the same day but different locations in patient progressing on pembrolizumab/dabrafenib (WM4420). One patient had a total of five biopsies at three different time points; discordant mutations were observed in three genes, varying over time (before and after treatment with ipilimumab) and location (WM4295). We also observed discordant mutations in five genes in biopsies taken from left and right axillary lymph node metastases (WM4413). Further, an early in transit metastasis was found to have a deleterious TP53 mutation, with two subsequent biopsies from different locations, while the patient was on BRAFi therapy for 12 months, both TP53 WT (WM4011). Interestingly, the thick primary melanoma differed remarkably from a residual lung metastasis after anti-CTLA4 therapy (WM4210). We also observed two instances in which tumor grafts from the same PDX expanded in different mice did not have the same mutational changes. These data suggest that intra-tumoral heterogeneity can lead to the outgrowth of several sub-clones during the propagation of PDX and may explain some of the heterogeneity seen in PDX efficacy studies (Krepler et al., 2016). Therapeutic pressures also can lead to new mutations conferring selective growth advantages. In the BRAF V600E mutant model WM4351, two PDX derived from therapy naïve biopsies were both NRAS WT, whereas a biopsy taken after progression on a BRAFi/MEKi had an NRAS Q61K mutation. In two cases, PDX derived from targeted therapy progressed patients did not demonstrate any acquired mutations, but were resistant to the same therapy the patient had received when dosed in vivo. When we established cell line cultures, they initially did not grow, but became resistant after several passages. Each cell line had a resistance mutation, one in NRAS (Q61K allele frequency, 0.44) and the other in MAP2K1 (C121S allele frequency 0.43) (Table S7).
Chromatin remodeling gene mutations in melanoma cell line, PDX, PDX CL, and patient tumors
Mutations in the genes that encode the SWI/SNF chromatin remodeling enzymes (ARID1A (BAF250A/SMARCF1), ARID1B (BAF250B), ARID2 (BAF200), SMARCA4 (BRG1) have been implicated in melanoma, as have those that encode other chromatin organization/histone modification proteins (EZH1, EZH2, SETD2, and TRRAP) (Hodis et al., 2012; TCGA, 2015). Overall, 65 of 371 (17.5%) samples from unique patients harbored a likely deleterious/deleterious mutation in at least one of the genes associated with chromatin remodeling or chromatin organization/histone modification (Figure 5). The most frequently mutated were ARID2 (23), followed by ARID1A (13), ARID1B (7) and SMARCA4 (4). Deleterious mutations in ARID2, ARID1A and SMARC4 were mutually exclusive (p=2.2×10−16), apart from a co-occurrence of ARID2 and SMARCA4 in one sample. However, deleterious mutations in ARID1B were found concurrently with mutations in ARID2 (1) and ARID1A (2). Restricting to naïve samples to reduce bias, BRAF V600E mutations were the least likely to be associated with chromatin remodeling gene mutations (7 of 105; 7%). Chromatin remodeling gene mutations were observed comparatively frequently with BRAF V600K (3 of 15; 20%), RAS (12 of 50; 24%) and NF1 (1 of 11; 9%) mutations (p = 0.013). One deleterious truncating mutation was detected in EZH1; deleterious/likely deleterious missense mutations were found in 12 EZH1/2-mutated samples from unique patients. Rare mutually exclusive mutations were found in SETD2 (4), TRRAP (4), IDH1 (1) and BAP1 (1).
Figure 5. Mutational and copy number profile in unique patient cell lines and PDX with a likely deleterious/deleterious mutation in chromatin remodeling genes, which reveals mutual exclusivity of mutations.

Comparison of genotypes in clinical samples and PDX
Clinical tumor sequencing data from 79 melanoma patients treated at Penn Medicine or MDACC was compared to our data (Table 2). At the Center for Personalized Diagnostics at Penn Medicine, the TruSeq Amplicon Cancer Panel (Illumina) was used for clinical sequencing (Hiemenz et al., 2016). At MDACC, CMS50 (Life Technologies) was used for clinical sequencing (Kim et al., 2017). For each patient, mutational profiles of PDX or tumor biopsy were compared to the clinical mutational profile. It is important to note that in virtually all cases a different sample was used for clinical sequencing than to establish the PDX or sent as a research tumor biopsy. Additionally, we could only compare samples for which positive results were found on either clinical or study sequencing in regions covered by both. All deleterious mutations were determined to be such by both the site and study. However, for six likely deleterious mutations and VUSs, the pathogenicity calls varied. Overall, there were 101 potentially overlapping mutations, of which 91 (91%) were found by both the site and study. We identified eight of 63 (12.6%) samples with discordant results. Of those, four (WM3407, WM4428, WM4462, WM4464) research biopsies were likely normal tissue rather than melanoma, as they had one to three VUSs at allelic frequencies of 50%. For two samples (WM4433, WM4323), although clinical sequencing was done on pre-treatment sample, we sequenced a post-treatment sample and identified presumably de novo resistance mutations. In one sample (WM4279), we identified a KIT p.L576P mutation not found in the clinical samples. Of the 16 UPENN samples with sequencing of a clinical sample and PDX, we only found one (6%) with discrepant results; interestingly, we each found different truncating mutations in PTEN. We observed that mutations tended to have higher allele frequencies in the PDX, as compared to clinical sequencing, which could be due to either admixture in the original tumor or loss of the wild-type allele during establishment of the PDX, which we have observed for ovarian cancer PDX (George et al., 2017).
Table 2.
Results from clinical and study sequencing of samples from the same patient.
| Sample ID | Sample Type | Clinical Site |
Gene | NT change | Site Variant Call | Study Variant Call |
% Tumor |
Tumor AF(%) |
PDX AF(%) |
% Increase |
Concordance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WM4428 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | - | - | Discordant | |||
| WM4433 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 22.6 | Concordant | |||
| WM4433 | Patient Biopsy | MDACC | NRAS | c.A182G:p.Q61R | - | Deleterious | 32.5 | Discordant | |||
| WM4435 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 28.4 | Concordant | |||
| WM4437 | Patient Biopsy | MDACC | BRAF | c.GT1798_1799AA:p.V600K | Deleterious | Deleterious | 33.7 | Concordant | |||
| WM4444 | Patient Biopsy | MDACC | BRAF | c.GT1798_1799AA:p.V600K | Deleterious | Deleterious | 90 | Concordant | |||
| WM4449 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 56.9 | Concordant | |||
| WM4462 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | - | - | Discordant | |||
| WM4464 | Patient Biopsy | MDACC | BRAF | c.T1790G:p.L597R | Deleterious | - | - | Discordant | |||
| WM4472 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 44.3 | Concordant | |||
| WM4478 | Patient Biopsy | MDACC | BRAF | c.A1801G:p.K601E | Deleterious | Deleterious | 42.4 | Concordant | |||
| WM4487 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 52.1 | Concordant | |||
| WM4494 | Patient Biopsy | MDACC | BRAF | c.GT1798_1799AA:p.V600K | Deleterious | Deleterious | 29.2 | Concordant | |||
| WM4500 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 53.8 | Concordant | |||
| WM4508 | Patient Biopsy | MDACC | BRAF | c.1799_1801del:p.600_601del | Deleterious | Deleterious | 36.4 | Concordant | |||
| WM4515 | Patient Biopsy | MDACC | NRAS | c.A182G:p.Q61R | Deleterious | Deleterious | 50.8 | Concordant | |||
| WM4528 | Patient Biopsy | MDACC | NRAS | c.C181A:p.Q61K | Deleterious | Deleterious | 41.8 | Concordant | |||
| WM4530 | Patient Biopsy | MDACC | NRAS | c.A182G:p.Q61R | Deleterious | Deleterious | 48 | Concordant | |||
| WM4532 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 47.5 | Concordant | |||
| WM4542 | Patient Biopsy | MDACC | NRAS | c.A182G:p.Q61R | Deleterious | Deleterious | 46.2 | Concordant | |||
| WM4545 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 44.6 | Concordant | |||
| WM4553 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 49.4 | Concordant | |||
| WM4558 | Patient Biopsy | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 47.7 | Concordant | |||
|
| |||||||||||
| WM3407 | PDX | MDACC | BRAF | c.G1397A:G466E | Deleterious | ND* | - | Discordant | |||
| WM3407 | PDX | MDACC | ATM | c.T728C:p.L243S | VUS‡ | VUS | 48.1 | Concordant | |||
| WM4218 | PDX | MDACC | KIT | c.T1669C:p.W557R | Deleterious | Deleterious | 47.1 | Concordant | |||
| WM4249 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 61.7 | Concordant | |||
| WM4257 | PDX | MDACC | NRAS | c.C181A:p.Q61K | Deleterious | Deleterious | 75 | Concordant | |||
| WM4257 | PDX | MDACC | TP53 | c.G629A:p.R210K | Deleterious | Deleterious | 100 | Concordant | |||
| WM4258 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 48.1 | Concordant | |||
| WM4260 | PDX | MDACC | KIT | c.A1924G:p.K642E | Deleterious | Deleterious | 78.3 | Concordant | |||
| WM4260 | PDX | MDACC | CTNNB1 | c.C134T:p.S45F | Deleterious | Deleterious | 49.9 | Concordant | |||
| WM4262 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 99.8 | Concordant | |||
| WM4264 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 64.7 | Concordant | |||
| WM4265-1 | PDX | MDACC | NRAS | c.C181A:p.Q61K | Deleterious | Deleterious | 96.8 | Concordant | |||
| WM4265-1 | PDX | MDACC | TP53 | c.C380T:p.S127F | Deleterious | Deleterious | 98 | Concordant | |||
| WM4267 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | ND | - | Discordant | |||
| WM4267 | PDX | MDACC | CDKN2A | c.G159C:p.M53I | VUS | VUS | 55.1 | Concordant | |||
| WM4276 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 0.61 | Concordant | |||
| WM4279 | PDX | MDACC | KIT | c.T1727C:p.L576P | ND | Deleterious | 86.6 | Discordant | |||
| WM4280 | PDX | MDACC | BRAF | c.GT1798_1799AA:p.V600K | Deleterious | Deleterious | 47.6 | Concordant | |||
| WM4285 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 42.2 | Concordant | |||
| WM4286-1 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 0.59 | Concordant | |||
| WM4292 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 48.6 | Concordant | |||
| WM4295 | PDX | MDACC | NRAS | c.A182T:p.Q61L | Deleterious | Deleterious | 96.3 | Concordant | |||
| WM4299-1 | PDX | MDACC | NRAS | c.A182T:p.Q61L | Deleterious | Deleterious | 0.93 | Concordant | |||
| WM4306 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 25.2 | Concordant | |||
| WM4323 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | - | - | Discordant | |||
| WM4323 | PDX | MDACC | MAP2K1 | c.1029dupA:p.I343fs | NC† | Deleterious | 46.2 | Discordant | |||
| WM4335 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 28.7 | Concordant | |||
| WM4345 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 61 | Concordant | |||
| WM4345 | PDX | MDACC | CDKN2A | c.C238T:p.R80X | Deleterious | Deleterious | 84 | Concordant | |||
| WM4351 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 75.5 | Concordant | |||
| WM4353 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 57 | Concordant | |||
| WM4367 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 51.2 | Concordant | |||
| WM4369 | PDX | MDACC | NRAS | c.G38A:p.G13D | Deleterious | Deleterious | 65.6 | Concordant | |||
| WM4370 | PDX | MDACC | BRAF | c.GT1798_1799AA:p.V600K | Deleterious | Deleterious | 92.7 | Concordant | |||
| WM4380 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 30.7 | Concordant | |||
| WM4382 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 48.6 | Concordant | |||
| WM4388 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 47 | Concordant | |||
| WM4388 | PDX | MDACC | TP53 | c.C520T:p.R174X | Deleterious | Deleterious | 91.3 | Concordant | |||
| WM4389 | PDX | MDACC | NRAS | c.G37C:p.G13R | Deleterious | Deleterious | 73.1 | Concordant | |||
| WM4404 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 67.7 | Concordant | |||
| WM4408 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 74.1 | Concordant | |||
| WM4420 | PDX | MDACC | BRAF | c.GT1798_1799AA:p.V600K | Deleterious | Deleterious | 87.7 | Concordant | |||
| WM4420 | PDX | MDACC | CTNNB1 | c.C134T:p.S45F | Deleterious | Deleterious | 65 | Concordant | |||
| WM4420 | PDX | MDACC | FBXW7 | c.C1321T:p.R441W | Deleterious | Deleterious | 52.6 | Concordant | |||
| WM4426 | PDX | MDACC | NRAS | c.A182G:p.Q61R | Deleterious | Deleterious | 84.5 | Concordant | |||
| WM4430 | PDX | MDACC | NRAS | c.A182G:p.Q61R | Deleterious | Deleterious | 100 | Concordant | |||
| WM4442 | PDX | MDACC | NRAS | c.C181A:p.Q61K | Deleterious | Deleterious | 95.7 | Concordant | |||
| WM4445 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 56.7 | Concordant | |||
| WM4451 | PDX | MDACC | NRAS | c.G35A:p.G12D | Deleterious | Deleterious | 87.4 | Concordant | |||
| WM4454 | PDX | MDACC | TP53 | c.T708G:p.C236W | Deleterious | Deleterious | 100 | Concordant | |||
| WM4454 | PDX | MDACC | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 45.3 | Concordant | |||
| WM4465 | PDX | MDACC | BRAF | c.GT1798_1799AA:p.V600K | Deleterious | Deleterious | 29.2 | Concordant | |||
|
| |||||||||||
| WM3901 | PDX | UPENN | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 44 | 67.05 | 84.9 | 26.6% | Concordant |
| WM4011 | PDX | UPENN | BRAF | c.GT1798_1799AA:p.V600K | Deleterious | Deleterious | 26 –50 | 13.82 | 76.6 | 454.3% | Concordant |
| WM4042 | PDX | UPENN | NRAS | c.A182G:p.Q61R | Deleterious | Deleterious | >50 | 34.26 | 98.9 | 188.7% | Concordant |
| WM4068 | PDX | UPENN | BRAF | c.G1406A:p.G469E | Deleterious | Deleterious | >50 | 35 | 47 | 34.2% | Concordant |
| WM4068 | PDX | UPENN | KRAS | c.G35A:p.G12D | Deleterious | Deleterious | >50 | 61.1 | 73.7 | 20.6% | Concordant |
| WM4206 | PDX | UPENN | BRAF | c.GT1798_1799AA:p.V600K | Deleterious | Deleterious | >50 | 35.06 | 65.6 | 87.1% | Concordant |
| WM4208 | PDX | UPENN | NRAS | c.A182G:p.Q61R | Deleterious | Deleterious | >50 | 19.13 | 64 | 234.6% | Concordant |
| WM4224 | PDX | UPENN | NRAS | c.A182G:p.Q61R | Deleterious | Deleterious | 44 | 10.14 | 47.7 | 370.4% | Concordant |
| WM4224 | PDX | UPENN | TP53 | c.C211T:p.R71C | Deleterious | Deleterious | 44 | 12.03 | 57.6 | 378.8% | Concordant |
| WM4224 | PDX | UPENN | CTNNB1 | c.A121G:p.T41A | Deleterious | Deleterious | 44 | 28.5 | 98.8 | 246.7% | Concordant |
| WM4231 | PDX | UPENN | RET | c.C2672T:p.S891L | VUS | Likely Deleterious | >50 | 25.68 | 50.2 | 95.5% | Concordant |
| WM4231 | PDX | UPENN | NRAS | c.A182G:p.Q61R | Deleterious | Deleterious | >50 | 30.25 | 52.5 | 73.6% | Concordant |
| WM4237 | PDX | UPENN | RB1 | c.2069_2082del:p.N690fs | Deleterious | Deleterious | 26 –50 | 38.41 | 73.8 | 92.1% | Concordant |
| WM4237 | PDX | UPENN | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 26 –50 | 21.21 | 76.3 | 259.7% | Concordant |
| WM4237 | PDX | UPENN | TP53 | c.C722T:p.S241F | Deleterious | Deleterious | 26 –50 | 40.62 | 99.2 | 144.2% | Concordant |
| WM4240 | PDX | UPENN | SMAD4 | c.G1399C:p.G467R | VUS | Likely Deleterious | 100 | 43.33 | 41.7 | - | Concordant |
| WM4243 | PDX | UPENN | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | >50 | 41.06 | 50.1 | 22.0% | Concordant |
| WM4298 | PDX | UPENN | KIT | c.T1688A:p.I563K | Likely Deleterious | VUS | >50 | 61.05 | 98 | 60.5% | Concordant |
| WM4298 | PDX | UPENN | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | >50 | 50.9 | 98.8 | 94.1% | Concordant |
| WM4314 | PDX | UPENN | FGFR2 | c.811_812delinsAA:p.G271K | Likely Deleterious | Likely Deleterious | >50 | 34.96 | 46.2 | - | Discordant |
| WM4349 | PDX | UPENN | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 26 –50 | 24.63 | 51.8 | 110.3% | Concordant |
| WM4364 | PDX | UPENN | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | >50 | 42.91 | 55.6 | 29.6% | Concordant |
| WM4364 | PDX | UPENN | PTEN | c.208_209+1del | Deleterious | ND | >50 | 72.59 | ND | - | Discordant |
| WM4364 | PDX | UPENN | PTEN | c.727_728del:p.L243fs | ND | Deleterious | >50 | ND | 78.5 | - | Discordant |
| WM4543 | PDX | UPENN | KDR | c.G4066T:p.V1356F | VUS | VUS | 26 –50 | 6.84 | 44 | 543.3% | Concordant |
| WM4543 | PDX | UPENN | BRAF | c.T1799A:p.V600E | Deleterious | Deleterious | 26 –50 | 6.4 | 40.4 | 531.3% | Concordant |
ND – not detected; NC – not captured; VUS – variant of uncertain significance
Discussion
In this era of precision medicine, pre-clinical drug development of targeted oncology therapeutics relies heavily on models of cancer that have been shown to be representative of the genetic profile of the patient’s tumor. Herein, we demonstrate that targeted massively parallel sequencing of 108/119 genes previously implicated in melanomagenesis, followed by our custom analysis pipelines for mutational and copy number variation calling, is a reliable method for characterizing the genetic and genomic landscape of melanoma cell lines, tumors, PDX, and PDX-CL. To account for the lack of matched normal samples, control samples were sequenced in each lane for normalization for copy number calling and to identify common variants, which were subtracted out. We also removed sequences that more closely aligned to the mouse than human genome to decrease cross-contamination and increase accuracy of mutational and copy number calling. We used an in-house developed pipeline to classify mutations or variants as deleterious, likely deleterious and VUS, incorporating information from the literature and COSMIC, mutational type, location and effect, after filtering for a maximum population frequency greater than or equal to 0.1% to account for the lack of matched normal. We found the total number of mutation and variant calls did not differ significantly between cell lines, PDX, PDX cell lines and patient tumors, although not surprisingly there was a trend towards higher mutational rates in PDX and cell lines. We observed significantly higher rates of BRAF and RAS mutations, and CDKN2A mutations/loss in cell lines than PDX and tumor biopsies, consistent with the growth advantage conferred by those mutations. However, we did find cell lines representing all mutational groups. We also found an extremely high concordance rate between clinical sequencing results and our targeted sequencing, with only two samples of 80 (2.5%) demonstrating truly discrepant results. Taken together, the mutational profiles observed in the melanoma cell lines, PDX, and PDX CL sequenced in this study are an accurate genetic and genomic representation of the patient’s original tumor. We also identified all major previously reported melanoma subtypes, as well as the full spectrum of mutations and copy number aberrations, at roughly the same frequencies identified in large-scale original patient tumor/normal sequencing studies (Berger et al., 2012; Hodis et al., 2012; Krauthammer et al., 2015; TCGA, 2015). Thus, we have a unique genetically and genomically annotated biobank of PDX, PDX cell lines and cell lines, representative of the full spectrum of melanoma, which can be used both for functional studies and pre-clinical drug development studies in melanoma.
Our large sample set also enabled us to describe rare subtypes in greater detail. We found two mutually exclusive patterns of mutations in the MAPK signaling pathway – 1) single hotspot mutations at BRAF V600 or NRAS Q61; and 2) multiple non-hotspot variants across different genes encoding proteins within the MAPK signaling pathway, of which NF1 mutations are a subset. All deleterious/likely deleterious non-600 mutations in BRAF, 87% of the NF1, 75% of the non-Q61 RAS and 44% of KIT mutant samples harbored either a secondary mutation or high-level amplification in at least one gene encoding a MAPK signaling protein or an effector protein of the MAPK pathway. These data are consistent with functional studies that have demonstrated that kinase dead BRAF (D594 mutants) needs oncogenic RAS to drive tumor progression (Heidorn et al., 2010). Our results also suggest that BRAF mutations (e.g. at G464, G469) leading to constitutive dimerization (Yao et al., 2015) also need at least one additional MAPK signaling mutation to drive tumor progression, either a single NRAS G12/13 or multiple other mutations. These data suggest that to accurately characterize therapeutic response pre-clinically for non-V600 BRAF or non-Q61 NRAS mutations, a second MAPK signaling mutation will be needed to be included in the model. Further, we can make predictions about the functionality of uncharacterized BRAF non-V600 mutations, in that if the alteration is found without a secondary MAPK signaling pathway gene mutation or in the presence of a BRAF V600 gene mutation, it is very unlikely to have any functional significance. Similar to BRAF non-V600 mutations, RAS G12/13 mutations usually are observed with co-occurring mutations, most commonly in BRAF and NF1, suggesting they are not sufficient to drive tumorigenesis in melanoma, in contrast to other tumor types (Hobbs et al., 2016).
We identified additional MAPK pathway or co-activating gene mutations in 87% of NF1 mutant tumors. We did not observe any difference between those with one or two truncating NF1 mutations, or with accompany loss of the wild-type allele. Prior literature has suggested an enrichment for co-occurring mutations in Rasopathy genes, particularly PTPN11 and RASA2 (Arafeh et al., 2015; Cirenajwis et al., 2017; Krauthammer et al., 2015) However, we observed co-occurrence of deleterious mutations across numerous genes without specific enrichment, including those that have not been previously implicated as co-mutated with NF1, although known to be mutated in melanoma, MAP3K5 and MAP3K9. Functional studies of BRAFwt/RASwt melanoma cell lines lacking NF1 expression, or expressing NF1 at extremely low levels, have shown that not all have RAS activation and that only some were sensitive to MEKi (Krauthammer et al., 2015). This result may be explained by the co-occurrence of other MAPK signaling gene mutations. Pre-clinical modeling of response to therapies for NF1 mutated melanoma also will need to account for co-occurring mutations. The NF1 mutant cohort harboring co-mutations in MAPK3K5/9 is of particular interest as they are upstream activators of the Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase pathways (Rana et al., 2013). The current study provides the reagents to further functionally characterize this interesting rare subtype of melanoma.
Although previous sequencing studies have identified ARID1A/B, ARID2, IDH1, SMARCA4A, TRRAP, and EZH2 as chromatin remodeling genes frequently mutated in melanoma (Berger et al., 2012; Hodis et al., 2012; Zhang et al., 2016), their mutual exclusivity has not been well-described. Given this finding, it is likely that the previously described ARID1B-dependence in ARID1A-mutated ovarian cancer cells (Helming et al., 2014) is recapitulated in melanoma, as well as the EZH2-dependency in tumors with mutations in ARID1A or SMARCA4 that do not harbor co-mutations in RAS or BRAF (Kim et al., 2015). However, a few ARID1A and SMARCA4-mutated samples in our data set do have co-mutations in BRAF or RAS, which has been postulated to abolish EZH2 dependency (Kim et al., 2015), so, further investigation is needed. The biological relevance of the likely deleterious missense mutations we identified in ARID1A/B and ARID2, which occur alone and concurrently with other ARID1A/B and EZH2 mutations, also needs to be evaluated functionally. Pre-clinical studies using these PDX models may reveal other specific vulnerabilities in melanomas with a mutation in SWI/SNF components ARID1A, ARID1B, ARID2, or SMARCA4A that aid development of novel therapeutics.
We profiled 37 PDX, PDX-cell lines and tumor biopsies from patients that progressed on targeted therapy (either BRAFi or BRAFi/MEKi). Our evaluation for resistance mechanisms was limited by lack of matched pre-treatment or normal samples and RNA to evaluate for splice variants or potential fusions. However, we identified mutations in NRAS, MAP2K1, and BRAF amplification at rates similar to other series (Johnson et al., 2015). Amplification of BRAF was enriched in this set (40%), as compared to the naïve group (15%, p= 2×10−4). For the two PDX with high-level BRAF amplifications, it likely is the primary mechanism of resistance. For other samples with BRAF amplification, it is a potential mechanism of resistance, but cannot not be definitively proven as we lack matched pre-treatment samples. For 51% of samples, we did not identify a clear mechanism of resistance; in half of those we found amplifications and drivers outside the MAPK signaling pathway that may be associated with resistance. We also profiled PDX, PDX-cell lines and tumor biopsies from 71 patients that had received checkpoint blockade therapy. These genetic and genomic landscape of these samples was similar to naïve samples, albeit with increased mutational burden and enrichment for non-BRAF mutations. These post-treatment PDX are ideal for further studies to identify potential resistance mechanisms and pre-clinical studies of potential therapeutics for tumors resistant to either targeted or checkpoint therapy.
Our study has several limitations, when compared to prior tumor-based analyses. Although the samples are derived from human tumors, they are established in culture or as PDX models in T-cell deficient (nude) mice, so are not subject to an intact immune system, which may lead to differential selective pressures for mutations or copy number aberrations. Additionally, intra-tumor heterogeneity observed in PDX expansion (Tentler et al., 2012) can result in potentially inharmonious PDX/PDX CL and tumor mutational profiles. We observed a high consistency between clinical testing and our profiling, likely because the former mainly included major driver genes. However, when we sequenced multiple samples from the same individual, we found several instances of both within patient and within tumor heterogeneity. Further, several of the tumor biopsies (but neither PDX nor cell lines) that we sequenced that fall into the WT/WT/WT group may be normal tissue, as histopathology was not done on these research samples. Platform and analytical differences also may lead to differences among mutational and copy number rates among studies. As we did not have matched normal sequence for subtraction, we used population based data, non-matched normal and stringent calling metrics to identify deleterious and likely deleterious mutations, but these are imperfect controls. We also chose to only report out high level amplifications and homozygous deletions, to be conservative. Thus, for some genes, our data appears different than prior studies. For example, mutations in GRIN2A and TRRAP are reported in 22% and 12%, respectively, of melanomas in a meta-analysis of somatic mutations across studies (Zhang et al., 2016), but since we classified most variants in these genes as VUS, our reported rates of deleterious/likely deleterious mutations are much lower at 6% and 5.6%. Additionally, we are limited by the genes and regions included on our panel at the time of design, and so have not interrogated recently identified recurrently mutated promoter regions, genes associated with resistance to checkpoint blockage and the Rasopathy genes in all samples. Performing unbiased whole exome or whole genome sequencing on 462 samples was not possible due to cost restrictions.
This unparalleled biobank of melanoma cell lines, PDX and PDX-cell lines in this study provides a set of reagents for not only future melanoma drug discovery and development efforts, but extensive biological studies. We have characterized 146 cell lines (31 derived from PDX), 248 PDX and 68 tumor biopsies, which include both those naïve to treatment and resistant to targeted therapy and checkpoint blockade. We have been able to identify all major and minor subtypes of melanoma, thus providing reagents that in some cases were previously unavailable for functional and biological studies. Although further evaluation will need to be done in some instances to characterize the reagents (e.g. targeted therapy progression samples for which no mechanism of resistance was identified), the current genetic and genomic copy number data provides a strong basis for future studies. These reagents enable thoughtful pre-clinical trials to be designed to determine the in vivo efficacy of novel single-agent and combination therapies in genetically-defined melanoma subsets, as demonstrated in the companion paper (Krepler et al., 2017).
Experimental Procedures
Sample acquisition
Acquisition of patient samples for the purposes of establishing PDX and cell lines was approved by the corresponding institutions’ institutional review boards, and informed consent was obtained from each participant for use of his or her sample in genetic studies. Tumors were provided from the following institutions: Perelman School of Medicine at the University of Pennsylvania, MD Anderson Cancer Center, Helen F. Graham Cancer Center, Massachusetts General Hospital, the John Wayne Cancer Institute, the Center for Melanoma and Cancer Immunotherapy at Hadassah Hebrew University Medical Center’s Sharett Institute of Oncology, and the University of Duisburg-Essen.
A full description of PDX development is given in our companion paper (Krepler et al., 2016). One hundred and fourteen human melanoma cell lines, 246 PDX, and 60 PDX CL, and 68 patient tumors were sequenced (total number of samples: 462) as part of this study (Supplementary Table 1). In addition to melanoma cell lines, PDX, and PDX CL, 36 unmatched anonymous germline blood samples were sequenced simultaneously, which were used for normalization for copy number calling.
Processing of sequencing data
Short-read sequences were aligned to the GRCh37 human reference genome using the Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2009). Duplicate reads were flagged, as well as reads that map equally to more than one location. Human reads were further disambiguated from mouse by aligning to the mm10 reference genome using the following Python script (https://github.com/AstraZeneca-NGS/disambiguate), which takes the human_aligned.bam and the mouse_aligned.bam as input. Reads that aligned more confidently to the mouse genome, as well as ambiguous reads between the two species, were discarded. To achieve acceptable data quality assurance, the Broad Institute’s Genome Analysis Toolkit (GATK) “Best Practices” guidelines were followed. Single Nucleotide Variant (SNV) and small insertion and deletion (indel) variant calling was performed by GATK UnifiedGenotyper (DePristo et al., 2011; McKenna et al., 2010), VarDict (Lai et al., 2016), and Freebayes (Garrison and Marth, 2012). Variants with a read depth less than 20 and alternative allele read depth less than five, as well as all synonymous variants, and/or variants present in the germline samples, were excluded. However, variants which were called by more than one variant caller, and had sequencing depth of less than 20, were not excluded. Variants were annotated with customized version of ANNOVAR (Wang et al., 2010). Variants were removed if the minor allele frequency was greater than or equal to 0.1% in the population databases 1000 Genomes (Abecasis et al., 2012) and/or Exome Aggregation Consortium (ExAC) (Lek et al., 2016) or found in normal germline samples sequenced on our capture. The remaining annotated variants were classified as outlined in Supplementary Figure 1. Variant classification was confirmed with cBioPortal for Cancer Genomics, wherever possible (Cerami et al., 2012) and using ClinVar for the Rasopathy genes (www.ncbi.nlm.nih.gov/clinvar). Integrative Genomics Viewer was used for visual confirmation of the majority of calls (Thorvaldsdottir et al., 2013).
Copy number variation prediction
Copy number variation from sequencing data was profiled using CODEX (Jiang et al., 2015). CODEX normalizes depth of coverage using a Poisson latent factor model that removes biases due to GC content, exon capture and amplification efficiency, and latent systemic artifacts. Six Poisson latent factors were included in the normalization model for this dataset, which corresponds to sample- and target-wise biases and artifacts that cannot be directly measured or quantified. Segmentation was restricted to exons for all genes. Only homozygous loss (copy number < 0.7) and high amplification (copy number > 3.3) calls are reported. Visual confirmation of CNV calls was done in Nexus 7.5 (BioDiscovery, Inc.) software.
Biostatistical Analysis
RStudio version 1.0.136 (RStudio, Inc.) was used to analyze the data. One-way ANOVA was used to compare the means of variant calls in cell line, PDX, and PDX CL. Paired t-test (along with 95% confidence interval for the difference in means) was used to compare allelic fractions of all variants, the number of all filtered variants among all sample types, the mutational burden between patients that received immunotherapy and naïve ones, and mutational burden comparison of naïve NF1 mutants, with other naïve subtypes. Chi-squared test, Fisher’s exact test or an unpaired t-test were used to make other statistical comparisons, as appropriate. For cluster analysis based on correlations, a gene with CNV<1 or >1 was selected for data analysis if it is shown from more than 10% of study samples. Spearman correlation coefficients were calculated between each pair of selected genes, and hierarchical clustering by Euclidian distance and complete linkage using the heatmap.2 function available from the R Foundation for Statistical Computing (http://www.R-project.org) was further performed to group the genes based on their correlations. For all analyses, p<0.05 was considered statistically significant.
Supplementary Material
Table S4. TERT promoter mutations and VUS detected in sequenced melanoma cell lines, PDX, PDX CL and patient tumors. Related to Figures 2, 3, 4
Table S5. Complete list of variant calls for all sequenced melanoma cell lines, PDX, PDX CL and patient tumors. Related to all Figures and Tables
Table S6. Complete list of high amplification and homozygous loss calls for all sequenced melanoma cell lines, PDX, PDX CL and patient tumors. Related to all Figures and Tables
Table S7. Duplicate samples of 10 patients with melanoma tumors, PDX, and PDX cell lines displaying discordant mutations. Related to Figures 2, 3, 4
Highlights.
Melanoma cell lines and PDX more likely to be BRAF/NRAS mutant than patient tumors
Mutations in melanoma PDXs are concordant with tumors from which they are derived
Contrasting MAPK pathway mutation patterns: one high-activity, several low-activity
Recurrent disease displays intra- and inter-tumor mutational heterogeneity
Acknowledgments
We express our sincere gratitude to the patients who have provided samples for this research. We would like to thank Drs. Takami Sato and Alexander Roesch for sample contribution and Dr. Michal Lotem for providing cell lines M230, M331, and M450. This work was supported by NIH grants P01 CA114046, P01 CA025874, R01 CA047159 to M.H.; the P50 CA174523 SPORE on Skin Cancer to the Wistar Institute and the University of Pennsylvania; the Melanoma Research Alliance (K.L.N.); philanthropic contributions to the Melanoma Moon Shots Program at the University of Texas MD Anderson Cancer Center; and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (D.S.H., M.H., M.A.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Number
The accession number for the targeted sequencing data in this study is Sequence Read Archive (SRA): SRP110573.
Author Contributions
C. K., K. S., M. B., M. X., B. S., A. W., A. V. participated in PDX establishment, expansion, banking, and in vivo experiments. B. G., I. N. A., B. W., M. A. W., K. L. N. developed, performed, and analyzed targeted sequencing. X. Y., Q. L., N. M., Y. J., N. Z. performed statistical analysis. W. X., G. K., M. F., X. X., R. A., T. C. G., D. E. E., D. A. T., L. S., L. H., J. A. W., M. D., Y. L., G. M., D. T. F., M. B., K. T. F., D. S. H., M. G., J. B., N. J. P., C. L. S., T. S., A. A., A. R., performed tissue and clinical data collection. C.K., L. S., M. H., K. L. N. participated in conception and design of the project. I. N. A., B. G. and K. L. N. wrote the manuscript with input from all authors. K. L. N., C. K. and M. H. supervised the work.
References
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arafeh R, Qutob N, Emmanuel R, Keren-Paz A, Madore J, Elkahloun A, Wilmott JS, Gartner JJ, Di Pizio A, Winograd-Katz S, et al. Recurrent inactivating RASA2 mutations in melanoma. Nat Genet. 2015;47:1408–1410. doi: 10.1038/ng.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash DE. UV signature mutations. Photochem Photobiol. 2015;91:15–26. doi: 10.1111/php.12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirenajwis H, Lauss M, Ekedahl H, Torngren T, Kvist A, Saal LH, Olsson H, Staaf J, Carneiro A, Ingvar C, et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol Oncol. 2017;11:438–451. doi: 10.1002/1878-0261.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrie P, Hategan M, Fife K, Parkinson C. Management of melanoma. Br Med Bull. 2014;111:149–162. doi: 10.1093/bmb/ldu019. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dentici ML, Sarkozy A, Pantaleoni F, Carta C, Lepri F, Ferese R, Cordeddu V, Martinelli S, Briuglia S, Digilio MC, et al. Spectrum of MEK1 and MEK2 gene mutations in cardio-facio-cutaneous syndrome and genotype-phenotype correlations. Eur J Hum Genet. 2009;17:733–740. doi: 10.1038/ejhg.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. 2012 In arXiv. [Google Scholar]
- George E, Kim H, Krepler C, Wenz B, Makvandi M, Tanyi JL, Brown E, Zhang R, Brafford P, Jean S, et al. A patient-derived-xenograft platform to study BRCA-deficient ovarian cancers. JCI Insight. 2017;2:e89760. doi: 10.1172/jci.insight.89760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Jr, Kaempgen E, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. ARID1B is a specific vulnerability in ARID1Amutant cancers. Nat Med. 2014;20:251–254. doi: 10.1038/nm.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemenz MC, Kadauke S, Lieberman DB, Roth DB, Zhao J, Watt CD, Daber RD, Morrissette JJ. Building a robust tumor profiling program: Synergy between next-generation sequencing and targeted single-gene testing. PLoS One. 2016;11:e0152851. doi: 10.1371/journal.pone.0152851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129:1287–1292. doi: 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, Schlegel NC, Brafford P, Sucker A, Ugurel S, Kumar R, Weber BL, Nathanson KL, Phillips DJ, Herlyn M, et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Melanoma Res. 2006;19:290–302. doi: 10.1111/j.1600-0749.2006.00322.x. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Oldridge DA, Diskin SJ, Zhang NR. CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 2015;43:e39. doi: 10.1093/nar/gku1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51:2792–2799. doi: 10.1016/j.ejca.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Haydu LE, Joon AY, Bassett RL, Jr, Siroy AE, Tetzlaff MT, Routbort MJ, Amaria RN, Wargo JA, McQuade JL, et al. Clinicopathological features and clinical outcomes associated with TP53 and BRAF non-V600 mutations in cutaneous melanoma patients. Cancer. 2017;123:1372–1381. doi: 10.1002/cncr.30463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, et al. SWI/SNF-mutant cancers depend on catalytic and noncatalytic activity of EZH2. Nat Med. 2015;21:1491–1496. doi: 10.1038/nm.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, McCusker JP, Ma S, Cheng E, Straub R, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet. 2015;47:996–1002. doi: 10.1038/ng.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepler C, Sproesser K, Brafford P, Beqiri M, Garman B, Xiao M, Shannon B, Watters M, Perego M, Zhang G, et al. A comprehensive patient-derived xenograft collection representing the heterogeneity of melanoma. 2017 doi: 10.1016/j.celrep.2017.10.021. [Companion Manuscript] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepler C, Xiao M, Sproesser K, Brafford PA, Shannan B, Beqiri M, Liu Q, Xu W, Garman B, Nathanson KL, et al. Personalized preclinical trials in BRAF inhibitor-resistant patient-derived xenograft models identify second-line combination therapies. Clin Cancer Res. 2016;22:1592–1602. doi: 10.1158/1078-0432.CCR-15-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, Johnson J, Dougherty B, Barrett JC, Dry JR. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44:e108. doi: 10.1093/nar/gkw227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Rana B, Mishra R, Sondarva G, Rangasamy V, Das S, Viswakarma N, Kanthasamy A. Mixed lineage kinase-c-jun n-terminal kinase axis: a potential therapeutic target in cancer. Genes Cancer. 2013;4:334–341. doi: 10.1177/1947601913485415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- Ryerson AB, Eheman CR, Altekruse SF, Ward JW, Jemal A, Sherman RL, Henley SJ, Holtzman D, Lake A, Noone AM, et al. Annual Report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer. 2016;122:1312–1337. doi: 10.1002/cncr.29936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61–68. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI, Rosen N. BRAF mutants evade Erk-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28:370–383. doi: 10.1016/j.ccell.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Dutton-Regester K, Brown KM, Hayward NK. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016;29:266–283. doi: 10.1111/pcmr.12459. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4. TERT promoter mutations and VUS detected in sequenced melanoma cell lines, PDX, PDX CL and patient tumors. Related to Figures 2, 3, 4
Table S5. Complete list of variant calls for all sequenced melanoma cell lines, PDX, PDX CL and patient tumors. Related to all Figures and Tables
Table S6. Complete list of high amplification and homozygous loss calls for all sequenced melanoma cell lines, PDX, PDX CL and patient tumors. Related to all Figures and Tables
Table S7. Duplicate samples of 10 patients with melanoma tumors, PDX, and PDX cell lines displaying discordant mutations. Related to Figures 2, 3, 4