

Abstract
Adenosine (ADO) is a well-known regulator of a variety of physiological functions in the heart. In stress conditions, like hypoxia or ischemia, the concentration of adenosine in the extracellular fluid rises dramatically, mainly through the breakdown of ATP. The degradation of adenosine in the ischemic myocytes induced damage in these cells, but it may simultaneously exert protective effects in the heart by activation of the adenosine receptors. The contribution of ADO to stimulation of protective effects was reported in human and animal hearts, but not in rat hearts. The aim of this study was to evaluate the role of adenosine A1 and A3 receptors (A1R and A3R), in protection of isolated cardiac myocytes of newborn rats from ischemic injury. The hypoxic conditions were simulated by exposure of cultured rat cardiomyocytes (4–5 days in vitro), to an atmosphere of a N2 (95%) and CO2 (5%) mixture, in glucose-free medium for 90 min. The cardiotoxic and cardioprotective effects of ADO ligands were measured by the release of lactate dehydrogenase (LDH) into the medium. Morphological investigation includes immunohistochemistry, image analysis of living and fixed cells and electron microscopy were executed. Pretreatment with the adenosine deaminase considerably increased the hypoxic damage in the cardiomyocytes indicating the importance of extracellular adenosine. Blocking adenosine receptors with selective A1 and A3 receptor antagonists abolished the protective effects of adenosine. A1R and A3R activation during the hypoxic insult delays onset of irreversible cell injury and collapse of mitochondrial membrane potential as assessed using DASPMI fluorochrom. Cardioprotection induced by the A1R agonist, CCPA, was abolished by an A1R antagonist, DPCPX, and was not affected by an A3R antagonist, MRS1523. Cardioprotection caused by the A3R agonist, Cl-IB-MECA, was antagonized completely by MRS1523 and only partially by DPCPX. Activation of both A1R and A3R together was more efficient in protection against hypoxia than by each one alone. Our study indicates that activation of either A1 or A3 adenosine receptors in the rat can attenuate myocyte injury during hypoxia. Highly selective A1R and A3R agonists may have potential as cardioprotective agents against ischemia or heart surgery.
Keywords: adenosine receptors, cardiomyocyte, cardioprotection, hypoxia, light and electron microscopy
Introduction
Adenosine (ADO) is a metabolite of adenine nucleotides, which has multiple physiological and pathological functions in numerous types of cells in vitro and in vivo. ADO exerts its effects via activation of four receptor subtypes that have been cloned and designated A1, A2A, A2B and A3 [1]. ADO is released during myocardial ischemia and hypoxia and is one of the plausible modulators in the protection of the ischemic myocardium [2]. The observation that a brief hypoxic period can increase the endurance against subsequent ischemic injury was termed ischemic preconditioning. Although the mechanism of preconditioning was elusive, there were various indications that activation of the ADO A1 and/or A3 receptors (A1R, A3R) was involved [3, 4]. The protection afforded by preconditioning was simulated by a brief prior exposure to nanomolar concentrations of the A3R agonist in an isolated rabbit heart [5]. This phenomenon also occurs in chick ventricular myocytes [6, 7]. Stambaugh et al. [7] have shown that activation of A1R and A3R during hypoxia can protect against injury in cardiac ventricular myocytes cultured from chick embryos. In rats, the efficiency of A3R agonists were studied in hypoxic conditions and controversy results were obtained. Lasley et al. [8], reported hemodynamic effects of A3R agonists in isolated rat hearts, that were blocked by A2AR antagonist and only partially by A3R antagonist, and these effects were not found in the isolated rabbit heart. Moreover, Cave et al. [9] did not find cardioprotective effects mediated by adenosine receptors in rat hearts. On the contrary, Nojiri et al. [10] and Headrick [3] concluded that adenosine is of primary importance in mediating the cardioprotection in rat. We have shown recently that A3R is expressed in rat’s newborn cultured cardiac myocytes and its activation by high doses (≥ 10 μM) induce apoptosis [11, 12]. The well-documented species variability in A3R activity, together with the several studies that proposed that A3R are not expressed in the ventricular myocardium in adult rats [13, 14], directed us to this investigation. The aim of the present study was to establish an experimental model of acute hypoxic condition in isolated rat cardiac myocytes and to evaluate the role of A1R and A3R in mediating the adenosine-induced protection in cardiomyocytes at the cellular and ultrastructural levels.
Materials and methods
Cell culture
Hearts of 2–3 day-old rats were removed under sterile conditions and washed 3 times in phosphate buffered saline (PBS) to remove excess blood cells. The hearts were minced and then gently agitated in a solution of proteolytic enzymes, RDB (Biological Institute, Ness-Ziona, Israel), which was prepared from a fig tree extract. The RDB was diluted 1:100 in Ca2+ and Mg2+-free PBS at 25°C for a few cycles of 10-min each, as described previously [12]. Dulbecco’s modified Eagle’s medium (DMEM) containing 10% horse serum (Biological Industries, Kibbutz Beit Haemek, Israel) was added to supernatant suspensions containing dissociated cells. The mixture was centrifuged at 300 g for 5 min. The supernatant phase was discarded, and the cells were suspended again. The suspension of the cells was diluted to 1.0 × 106 cells/ml and 1.5 ml were placed in 35-mm plastic culture dishes on collagen/gelatin-coated coverglasses. The cultures were incubated in a humidified atmosphere of 5% CO2, 95% air at 37°C. Confluent monolayer exhibiting spontaneous contractions were developed in culture within 2 days. Myocyte cultures were washed in serum-free medium BIO-MPM-1 (Kibbutz Beit Haemek, Israel) containing 5-mg/ml glucose and incubated in this medium for further 48 h. After 48 h in serum-free medium the experiments were performed.
Hypoxic conditions
Myocyte cultures were washed in serum and glucose-free medium (DMEM) before incubating with DMEM in the presence of ligands under hypoxic conditions. The cardiomyocyte cultures were exposed to hypoxia of varied duration. In the first set of experiments myocyte damage after 1 and 2 h hypoxia were compared in the presence or absence of adenosine deaminase (ADA). The second set of experiments was performed to test the influence of hypoxia for 75 min to cultured myocytes in the presence of adenosine A1R and A3R antagonists. Finally, in the third set of experiment the effects of adenosine A1R and A3R agonists were studied after 90-min hypoxia. Hypoxic conditions were carried out in an hypoxic incubator where O2 was replaced by N2 (95%) and CO2 (5%) mixture, in glucose-free media. The hypoxic damage was characterized at the end of the hypoxic period by morphological and biochemical evaluations.
Experiments with A1R and A3R ligands
CCPA (A1R agonist), Cl-IB-MECA (A3R agonist), DPCPX (A1R antagonist), 8-SPT (A1R and A3R antagonist) and MRS1523 (A3R antagonist) at various concentrations were introduced to cell cultures 10-min prior to and during the hypoxic conditions. When both agonist and antagonist introduced to the medium, the cells were pre-treated first with the antagonist and after 10 min with the agonist. When the cells were treated with A1 and A3 agonists, both agonists were co-administered 10 min before hypoxia and as well during the hypoxia.
α-Sarcomeric actin staining
Cells on coverslips were stained for immunohistochemical demonstration of α-sarcomeric actin using mouse monoclonal anti-α-sarcomeric actin (C-5) and goat anti-mouse biotinylated immunoglobulin conjugated with extra avidin-peroxidase. The chromogen 3-amino-9-ethylcarbazole (AEC) was used as described previously [11, 12].
Transmission electron microscopy
The cells were fixed with 2.5% gluteraldehyde in 0.1 M sodium cacodylate for 1 h, postfixed in 1% osmium tetroxide in the same buffer for 1 h, en bloc [11]. The cultures were dehydrated in an ascending series of alcohols, infiltrated in Epon-Araldite epoxy resin, and heat polymerized. En face (with respect to the culture substratum) sections were cut on an LKB ultramicrotome, poststained with uranyl acetate (saturated solution) and lead citrate [11], and examined in a JEOL-1200 transmission electron microscope at an operating voltage of 80 kV.
Enzyme release – lactate dehydrogense (LDH)
Protein content and lactate dehydrogenase activity were determined according to El-Ani et al. [15]. Briefly, 25 μl of the supernatant was transferred into 96-well dish and the LDH activities were determined using LDH-L kits (Sigma, St. Louis, MO, USA) as described by the manufacturer. The product of the enzyme was measured spectrometrically at a wavelength of 340 nm as described previously [16]. The results are expressed as a fold of the control in the same experiment. Experiments were done in 4–8 replicas each and were repeated at least 3 times.
Propidium iodide assay
The assay is based on vital binding of propidium iodide to nuclei of cells whose plasma membranes have become permeable due to cell damage. The assay was performed according to Nieminen et al. [17]. Cell number was evaluated by using a Scan-Array 2 Image Analyzer (Galai, Israel). The analyzer consisted of an Axiovert 135TV microscope (Zeiss, Germany) and a black and white Sony camera interfaced to an image analysis computer [12].
Examination of mitochondria in living cells
The method was according to Shneyvays et al. [12]. The accumulation of DASPMI dye in the mitochondrial matrix space is known to be dependent on the presence of high membrane potential of the mitochondrial inner membrane. Living cells grown on round coverslips were exposed to DASPMI, dissolved in PBS (at a final concentration 10 μg/ml) for 15 min. Then, the coverslips were washed and mounted on chambers containing dye-free medium. DASPMI fluorescence was excited at 460 nm, and the emission wavelength was 540 nm.
Chemicals
The highly selective ADO A3 agonist, Cl-IB-MECA, was gift from the National Institute of Mental Health Chemical Synthesis and Drug Supply Program. The selective ADO A3 antagonist MRS1523 was synthesized as described by Li et al. [18]. DASPMI (4-Di-1-ASP, D-288) was acquired from Molecular Probes Inc., and other reagents were purchased from Sigma Chemical.
Statistical analysis
Results are expressed as mean ± S.E.M. Data were analyzed by analysis of variance (ANOVA) with application of a post-hoc Tukey-Kramer test. P < 0.05 was accepted as indicating statistical significance.
Results
To simulate ischemic conditions, the cardiomyocyte cultures were exposed to hypoxic periods of varied duration. In our system, a 2-h hypoxic exposure, but not a 1-h exposure, was sufficient to cause injury to the myocytes, as measured by morphological and biochemical criteria (LDH release). Exposure of similar cultures to the same conditions in the presence of adenosine deaminase (ADA) caused toxic effects even within 1-h of hypoxia (Fig. 1). These results may indicate the release of endogenous adenosine during hypoxic conditions, which protects the myocytes only during short-term hypoxic exposure (1 h). Preliminary results indicated that in our system, a 90-min hypoxic period is the appropriate duration for studying the effect of adenosine receptor ligands (data not shown). The effects of A1R and A3R activation on LDH release from cardiac myocytes under hypoxic conditions for 75 min in the presence of antagonist (Fig. 2a), and for 90-min in the presence of agonist (Fig. 2b) were determined. In order to characterize the cardiotoxic effects, antagonists to A1R (DPCPX), and to A3R (MRS1523), or the non-selective adenosine receptor antagonist (8-SPT) were used. The antagonists increased the LDH release, indicating that protective activity of endogenous adenosine, which released from the cells during ischemia, abolished by the antagonists (Fig. 2a). The effect of DPCPX appeared more pronounced, even at low concentrations (10 nM), than the cardiotoxicity in the presence of the MRS1523, which exhibited a lesser degree of LDH release at the same concentration (Fig. 2a). 8-SPT was only effective at higher antagonist concentrations (≥ 1 μM).
Fig. 1.
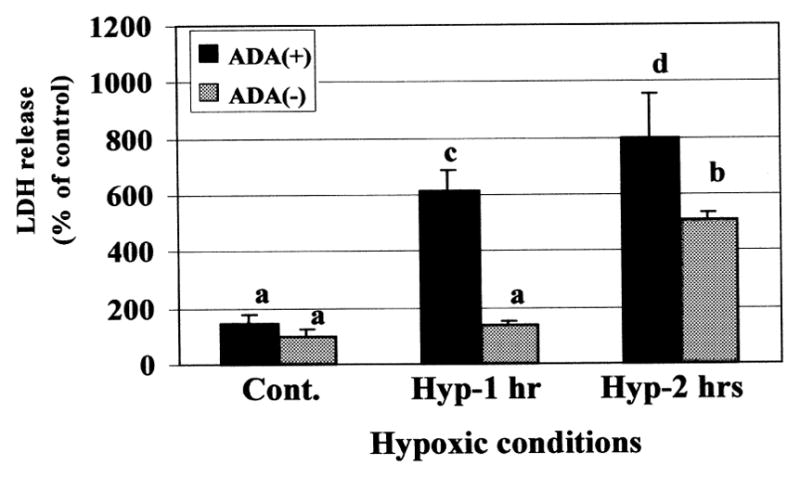
Duration of hypoxic conditions and the effect of adenosine deaminase (ADA) (5 units/ml) on endogenic adenosine release from cultured cardiomyocytes after prolonged exposure to hypoxic conditions. The LDH release was determined immediately after hypoxia. One hundred percent were considered as the release in the control cultures. Data are expressed as mean ± S.E.M. of at least 3 replicates in 3 separate experiments. Means with the same letter are not significantly different (p < 0.05) according to a post-hoc Tukey-Kramer test.
Fig. 2.
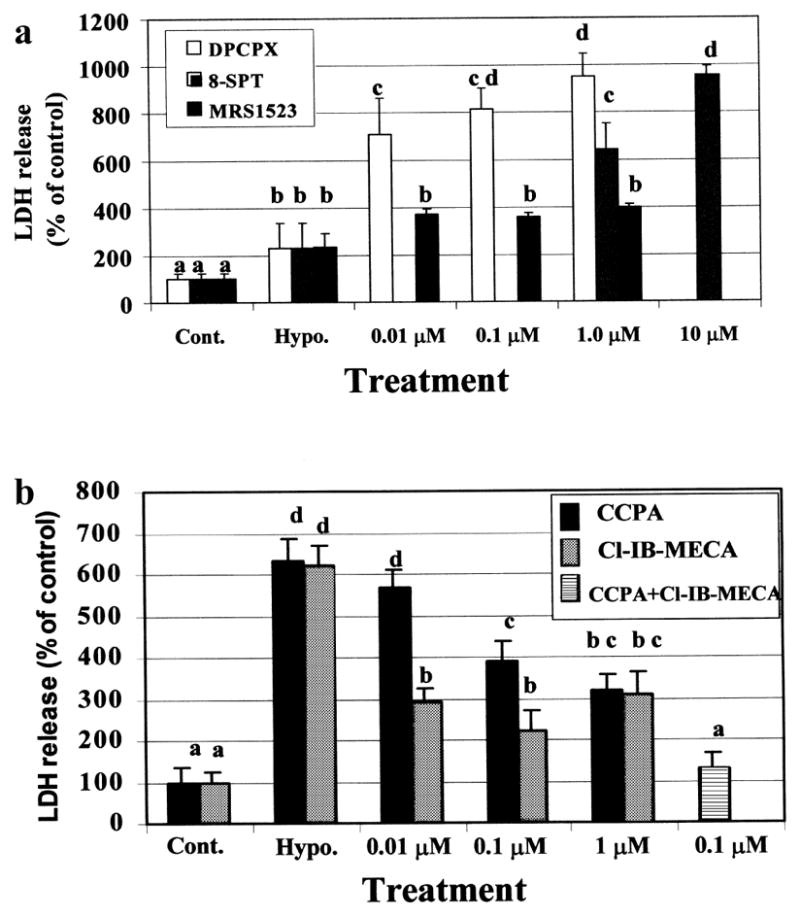
Effect of Adenosine A1R and A3R ligand in cardiac myocytes, under hypoxic conditions for 75-min in the presence of antagonist (a), and for 90-min in the presence of agonist (b). The LDH release was determined immediately after hypoxia. One hundred percent were considered as the release in the control cultures. Data are means of at least 3 replicates in 5 separate experiments. Means with the same letter are not significantly different (p < 0.05) according to a post-hoc Tukey-Kramer test.
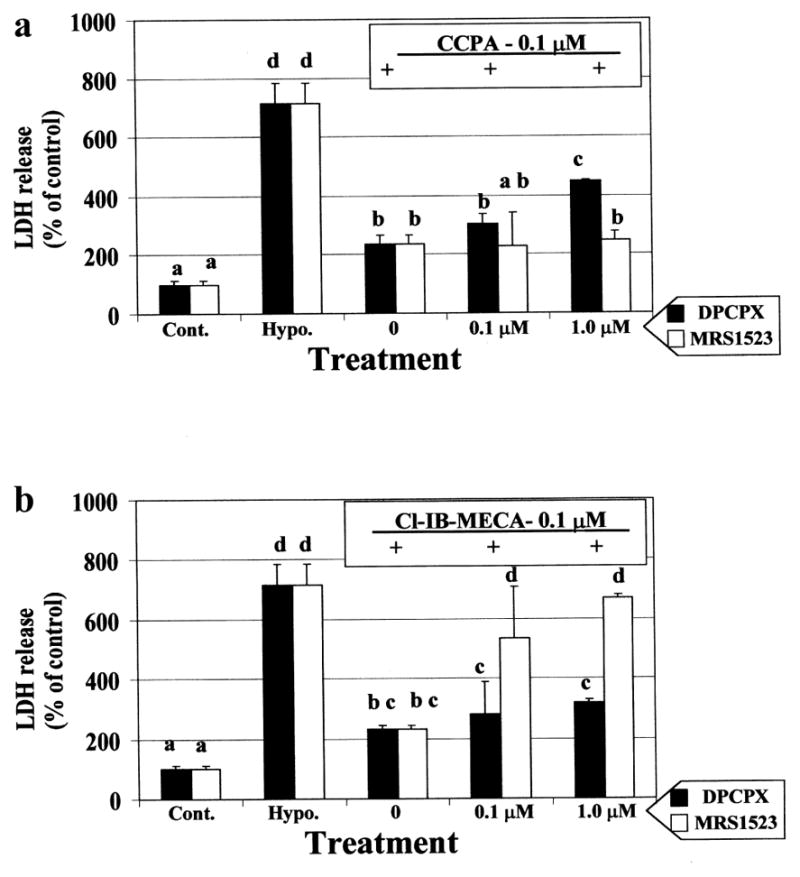
The ability to attenuate myocyte injury during prolonged hypoxia was measured by exogenously activating the A1R and A3R by specific agonists (CCPA and Cl-IB-MECA respectively, Fig. 2b). The cardioprotective ability of the A3R agonist, Cl-IB-MECA, was observed at 10 nM, while at higher concentrations (≥ 1 μM) the protective ability decreased. The A1R agonist, CCPA, exhibited protection in a concentration-dependent manner. Activation of both A1R and A3R together was more efficient in protection against hypoxia than by each other (Fig. 2b). The concentration-dependent effects and specificity of the selective antagonists DPCPX and MRS1523 on cardioprotection induced by the selective agonists, CCPA, (Fig. 3a) and Cl-IB-MECA were studied (Fig. 3b). Decrease in LDH release to the culture media suggest that both A1R and A3R agonists (CCPA and Cl-IB-MECA, respectively) prevented hypoxia-induced injury. The cardioprotection caused by A1R agonist CCPA, was abolished by A1R antagonist, DPCPX, in a dose dependent pattern, and was not affected by A3R antagonist, MRS1523. Moreover, the protection achieved by the A3R agonist Cl-IB-MECA was inhibited effectively by the A3R antagonist, MRS1523, while only slightly by high concentrations (> 1 μM) of the A1R antagonist DPCPX (Fig. 3b).
Fig. 3.

Dose dependent effect of A1R (DPCPX) and A3R (MRS1523) antagonists on the cardioprotection induced by A1R (CCPA – 0.1 μM) (a) and A3R (Cl-IB-MECA – 0.1 μM) (b) agonists. The LDH release was determined after 90 min hypoxia. The 100% were determined as the release in the control cultures. Data are means of at least 3 replicates in 3 separate experiments. Means with the same letter are not significantly different (p < 0.05) according to a post-hoc Tukey-Kramer test.
The morphological changes in the rat myocyte cultures treated under hypoxic conditions were followed by immunohistochemical staining of α-sarcomeric actin with hematoxylin counterstaining (Fig. 4). In rat primary cardiac cultures, the flattened contractile cells exhibited strands of well organized striated myofibrils run in various directions (Fig. 4A), and the myocytes possessed one or two nuclei. Hypoxic conditions caused damage of the cardiac cells, beginning with vacuolization of cytoplasm, concentration of cytoplasm around the nucleus and the loss of striation in myofibrils (Fig. 4B). Further alterations were characterized by a complete degeneration of the myofibrils and a significant decline in the α-sarcomeric actin staining, the appearance of enlarged vacuoles, and at the end stage, disruption of the cell membranes. Some cells containing pyknotic or fragmented nuclei and punctuated α-sarcomeric actin staining patterns, edematous areas in the cytoplasm and around the nucleus. Some cells exhibited sign of complete destruction (necrotic debris), their cytoplasm and nucleus material was greatly condensed and they exhibited disintegrated α-sarcomeric actin immunoreactivity. This degenerative process was almost completely inhibited in the presence of either the A1R agonist CCPA (Fig. 4C) or A3R agonist Cl-IB-MECA (Fig. 4E). The A1R antagonist DPCPX abolished the protection by CCPA (Fig. 4D), and the A3R antagonist MRS1523, abolished the protection by A3R agonist (Fig. 4F).
Fig. 4.
Cardiotoxicity and cardioprotection demonstrated by morphological investigations. (A–F) Immunohistochemical staining of α-sarcomeric actin in rat cardiomyocyte cultures followed by hematoxylin counterstaining. (A) Control – normoxic conditions; (B) 90-min hypoxic conditions; (C) Presence of A1R agonist (CCPA – 0.1 μM) during hypoxic period; (D) CCPA was introduced together with A1R antagonist (DPCPX – 1 μM) during hypoxic period; (E) Presence of A3R agonist (Cl-IB-MECA – 0.1 μM) during hypoxic period; (F) Presence of A3R agonist (Cl-IB-MECA – 0.1 μM) together with A3R antagonist (MRS1523 – 1 μM) during the hypoxic period; (G–L) The accumulation of DASPMI dye in the mitochondrial matrix space (vital staining) in conjunction with propidium iodide binding to nuclei of cells whose plasma membranes have became permeable in rat cardiomyocyte cultures. (G) Control – normoxic conditions; (H) 90-min hypoxic conditions; (I) Presence of A1R agonist (CCPA – 0.1 μM) during hypoxic period; (J) CCPA was introduced in presence of A1R antagonist (DPCPX – 1 μM) during hypoxic period; (K) Presence of A3R agonist (Cl-IB-MECA – 0.1 μM) during the hypoxic period; (L) Presence of A3R agonist (Cl-IB-MECA – 0.1 μM) together with A3R antagonist (MRS1523 – 1 μM) during hypoxic period. The results were obtained in at least 3 replicates in 3 separate experiments.
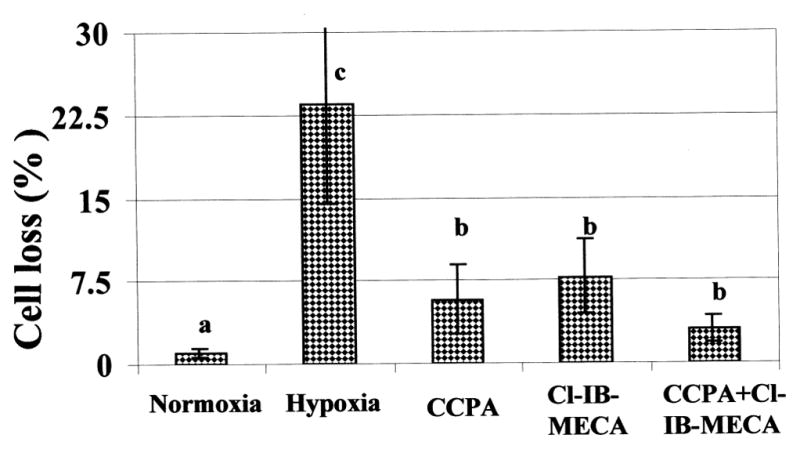
Fluorescent staining was used to differentiate between the cardiotoxicity and cardioprotective effects in living cells. Both propidium iodide (red) and DASPMI fluorescence (glossy yellow) were applied to the cells. Propidium iodide is used to identify disrupted cell membranes. DASPMI is specific for the membrane potential of mitochondria (vital staining) (Figs 4G–4L). Two types of mitochondrial patterns were observed in normoxic conditions. The first, longitudinal oriented and stretched mitochondria in subsarcolemmal areas in the cytoplasm, and the second, oval shape mitochondria in the perinuclear and intramyofibrillar regions (Fig. 4G). These cells maintained cellular integrity; propidium iodide, a marker for identification of necrotic cells, did not penetrate the cells (Fig. 4G). In hypoxic conditions the cellular membranes were damaged in many myocytes, which enabled the entrance of the propidium iodide red dyes and allows to quantify the percent of killed cells. The mitochondrial damage was characterized by the loss of the intensive glossy yellow fluorescence staining exhibited by DASPMI dye, which represent the dissipation of the mitochondrial membrane potential (Fig. 4H). Parallel changes in LDH release and percent of killed cells (Fig. 5), suggested that many cultured myocytes exhibited irreversible injury during the 90-min hypoxia. Figure 5 demonstrated that A1R agonist CCPA and A3R agonist Cl-IB-MECA, can protect cultured cardiomyocytes against hypoxia-induced injury. Activation of both A1R and A3R together was more efficient in protection against hypoxia than by each one alone. The degenerative alterations in surviving cells were almost completely inhibited in the presence of either the CCPA or Cl-IB-MECA (Figs 4I and 4K). Furthermore, the hypoxic-mediated injury was increased with the A1R antagonist, DPCPX in the presence of CCPA (Fig. 4J) or the A3R antagonist, MRS1523 in the presence of Cl-IB-MECA (Fig. 4L). The A1R antagonist, DPCPX (not shown), did not affect the protection by Cl-IB-MECA.
Fig. 5.
Effect of Adenosine A1R and A3R agonists on cardiac myocytes loss after hypoxia for 90-min. A1R agonist (CCPA – 0.1 μM), or A3R agonist (Cl-IB-MECA – 0.1 μM) were introduced 10 min before and during the 90 min hypoxia. Data are means of at least 3 replicates in 3 separate experiments. Means with the same letter are not significantly different (p < 0.05) according to a post-hoc Tukey-Kramer test.
The ultrastructural characteristics of control normoxic cells included intact nuclei with delicate heterochromatin against a pale background, dense and well-developed mitochondria (MT), and an intact sarcolemma. The myofibrils (MF) were relaxed, well developed and organized with a clear striations in longitudinal arrangements (Fig. 6A). In-contrast, the cultures exposed to hypoxic conditions (Fig. 6B) and/or exposed concomitantly to A3R agonist and antagonist (MRS1523) (Fig. 6D), exhibited severe cardiotoxic features. The extent of the damage was rather heterogeneous among the myocytes, but the presence of cellular injury was characteristic for all myocyte population. The nuclear chromatin was clumped with condensed heterochromatin and convoluted perinuclear membrane that often was split and edematous areas were evident. Many nuclei were reduced in sizes. The diameters of these shrunken nuclei decreased to half or one-third of the normal ones. The hypoxic damage was characterized by clumping of nuclear chromatin, sarcolemmal rupture, intermyofibrillar edema and increased vacuolation (Figs 6B and 6D). No signs of apoptotic degeneration of cardiomyocytes were found. The mitochondria became pale and swollen, accompanied by rupture of the internal septa and membranes. The arrangements of sarcomeres, myofibrils and myofilaments were lost and the integrity of the actin and myosin filaments was not visualized. In high magnification, holes were noted in the cell membranes (arrows). These typical oncotic and necrotic structural damage in the myocyte cultures were significantly attenuated by A3R agonist Cl-IB-MECA (100 nM) under 90-min hypoxic conditions as indicated by preservation of mitochondria, conservation of myofilaments structure, and maintenance of cellular integrity (Fig. 6C).
Fig. 6.

Electron micrographs of cardiac cells after hypoxic conditions. (A) Control cardiomyocyte in normoxic conditions; (B) Hypoxic conditions for 90-min. Gaps in the cell membrane (arrows); (C) A3R agonist (Cl-IB-MECA – 100 nM) exposure for 10-min before and during the 90-min of hypoxic conditions; (D) A3R agonist (Cl-IB-MECA – 100 nM) and A3R antagonist (MRS1523 – 1 μM) exposure for 10-min before and during the 90-min of hypoxic conditions. Nucleus (N), myofibrils (MF) and mitochondria (MT).
Discussion
Over the past decade substantial experimental data have been obtained indicating that ADO has multiple physiological and pathological functions in various tissues and organs [19]. ADO reduces reversible and irreversible ischemia/reperfusion injury by preconditioning the heart and thereby reducing the size of myocardial infarctions [20, 21]. Administration of ADO to the coronary vessels after prolonged ischemia also limited the infarct size in isolated heart [22, 23]. The ADO-mediated cardioprotection phenomena raises questions as to whether the beneficial effects are due to direct activation of ADO receptors present in cardiac myocytes or mediated through the neutrophils, vascular or neuronal elements that are implicated in the coronary vasculature exposed to ischemic conditions. ADO levels were found to be elevated in heart failure and chronic heart failure [24]. Ischemia in the intact heart [25] or hypoxia in ventricular heart cells cultured from chick embryos mediates the release of endogenous ADO, which may precondition the heart and result in cardioprotection [6].
The first goal of our study was to evaluate a model of hypoxia in neonatal rat cardiomyocyte cultures and to assess the cardioprotection mediated by activation of ADO receptors. Exposure of myocytes to hypoxia for 2-h resulted in significant myocyte injury. This damage was not evident after 1 h. The addition of ADO deaminase (ADA) during the 1-h hypoxic period caused pronounced myocyte injury that was augmented after 2-h. These results suggest that in our hypoxic model, endogenous ADO was released and inhibited injury to the myocytes, while the ADA caused degradation of ADO and increased cytotoxicity. Therefore, during short hypoxia, no morphological and biochemical protective changes may be evident, due to the release of endogenous ADO, which is not sufficiently beneficial during longer periods (> 2 h). These data may point the importance of evaluating the exact period of hypoxic conditions (90-min, in our system) to enable the measurements of both cardiotoxic and cardioprotective effects mediated by ADO ligands. The same hypoxic duration has been used in other in vitro studies in isolated chick ventricular cultures [6, 7, 26].
A1R and A3R stimulation has been implicated in cardioprotection in several species, including rabbit [5], human [27], chicken [6, 7, 26] and others. However, in the rat there remains a controversy about whether ischemic preconditioning and cardioprotection are mediated by these receptors [9, 10, 13, 28]. Therefore, the second goal of the present study was to assess the involvement of A1R and A3R in cardiotoxicity and cardioprotection under 90-min hypoxic conditions. In the rat cardiomyocyte cultures, the protective effect of ADO was mainly mediated through activation of the A1R and A3R, since DPCPX (A1R antagonist), MRS1523 (A3R antagonist) and 8-SPT (both A1R and A3R antagonist), blocked the cardioprotective effect of ADO.
The ability of the MRS1523 to reverse the protective effect of ADO was more pronounced than that of DPCPX at the low concentrations. It can be suggested that the cardioprotective potential of the 90-min A3R activation exceeds that of A1R activation at the same concentrations, or the difference may be due to another type of second messengers. Another explanation can be related to the high selective ability of the MRS1523 to both human and rat A3R [18] (in rat it corresponds to selectivities of 140- and 18-fold vs. A1 and A2A receptors, respectively [20]). This specificity may belong to the A3R only (within limits of selectivity of the MRS1523), while DPCPX mainly inhibits the activation of the A1R with an additional potential to inhibit the A3R at high concentrations. The ability of the 8-SPT to inhibit ADO receptor activation was observed only at high concentration (≥ 10 μM) in preconditioning experiments in the intact heart [2, 19] and in myocytes cultures [4, 6]. This phenomenon was also documented in prolonged hypoxic conditions [7]. The high concentration can be related to the low affinity of the ligand. Activation of the A1R and A3R by a low level of endogenous agonist has a protective role in hypoxic conditions. Preconditioning and chronic exposure to low concentrations of agonist may cause upregulation and sensitization of the A3R and their protection ability. In this study, the activation of both A1R and A3R by their own specific agonist (CCPA and Cl-IB-MECA, respectively) induced cardioprotective effects in prolonged hypoxic conditions and attenuated rat cardiac myocyte injury in vitro. Although low concentrations of the A3R agonist, Cl-IB-MECA, protected the cardiomyocytes, the protection declined significantly at 1 μM and higher concentrations (≥ 10 μM), which may cause toxicity and apoptosis [11, 12, 29]. Using the model of neonatal rat isolated cardiomyocytes, we have demonstrated that activation of the A1R and A3R may result in cardioprotection during 90 min hypoxic conditions. The study describes an in vitro model of hypoxic conditions where the neuronal elements, circulating blood, hemodynamic parameters and their involvement in mediating the cardioprotection are essentially precluded. The results of this study provide conclusive evidence that A1R and A3R activation in cardiomyocytes delays onset of irreversible cell injury following hypoxia. The observations of the present study confirm that in myocytes pretreated with adenosine receptor agonists, morphological injury developed more slowly than in hypoxic (non-treated) cells. Most of the alterations observed in prolonged hypoxia may be attributed to the loss of critical metabolites such as ATP. Mitochondrial membrane potential provides the driving force for ATP synthesis. The potential is generated by the supply of NADH through the matrix dehydrogenases and electron flux through electron transport chain [30]. As was shown in this study, hypoxia did not produce acute decrease in mitochondrial potential according to maintaining of DASPMI fluorescence up to terminal stage, when abrupt decrease or cessation of emission was observed, which indicate the complete collapse of the potential in dying cells (see Figs 4H and 4L). Budinger et al. [30], suggests that inhibition within the electron transport chain during hypoxia would therefore result in a decrease in electron flux and a depolarization of the membrane, while a sudden inhibition of ATP utilization or an inhibition of the ATP synthase produces hyperpolarization. Maintaining of membrane potential during acute hypoxia suggests that ATP utilization and ATP synthesis remained closely matched in surviving cells. Both sarcomeres length and mitochondrial swelling depend on the actual ATP content [31, 32]. The loss of mitochondrial function inevitably leads to cell necrosis. As was shown in this study collapse of membrane potential leads to permeability of sarcolemma to propidium iodide, sign of irreversible cell injury (see Figs 4H and 4L). Thus, for understanding cardioprotective activity of adenosine compounds we should understand the mechanism of irreversible mitochondria damage.
It is known that reactive oxygen species can be generated in ischemic/reperfused hearts in the cytosol and/or one-electron reduction process of the respiratory chain in mitochondria [33]. The oxygen radicals may induce a lipid peroxidation of mitochondrial membranes and their formation may result in mitochondrial injury and cell necrosis. Characteristic for cardiomyocytes that they contain less catalase, glutathione peroxidase and superoxide dismutase than other cells [34]. Recent evidence implicates the ADO A1Rs and A3Rs in the activation of antioxidant enzymes. It has been demonstrated that stimulation of adenosine receptors activates the antioxidant enzyme system in rat cardiac myocytes [35, 36], that might explain the cardioprotection.
In order to clarify the controversy whether adenosine can protect rat cardiomyocytes in hypoxia [9, 10, 13, 27, 28], we have characterized the specificity of the A1R and A3R. The presence of CCPA (A1R selective agonist) during 90-min hypoxic conditions resulted in cardioprotection. This effect was abolished by the A1R antagonist DPCPX and not affected by the A3R antagonist MRS1523. Furthermore, the protection achieved by A3R agonist Cl-IB-MECA was inhibited by pretreatment with A3R antagonist, but not by the A1R antagonist. Activation of each of ADO receptors A1R and A3R resulted in delaying of irreversible cardiomyocyte damage. Cave et al. [9], showed the lack of protection against contractile dysfunction in isolated rat hearts, but found a significant decrease in creatine kinase leakage by adenosine pretreatment, that confirms our results in culture. Protection against contractile dysfunction seems to involve a combination of several receptors and downstream signaling cooperation in heart tissue and not only ADO receptors.
In conclusion, low doses of highly selective ADO agonist may have therapeutic potential in treatment of cardiac ischemia and other cardiac disorders. Highly selective A1R and A3R ligands may represent novel potent cardioprotective agents and co-activation of both A1R and A3R was more potent than that produced by activation of either receptor individually. These agonists may have therapeutic potential during infarct-producing ischemia and may reduce the size of myocardial infarction.
Acknowledgments
This study was supported (in part) by Grant No. 4390 from the Chief Scientist’s Office of the Ministry of Health, Israel.
Abbreviations
- ADO
adenosine
- A1R, A3R
adenosine receptor subtype 1 or 3
- CCPA
2-chloro-N6-cyclopentyladenosine
- C1-IB-MECA
2-chloro-N6-(3-iodobenzyl) adenosine-5′-N-methyluronamide
- CK
creatine kinase
- DASPMI
2-(p-dimethylaminostyryl) pyridyl methyl iodide
- DPCPX
8-cyclopentyl-1-3-dipropylxanthine
- LDH
lactate dehydrogenase
- MRS1623
5-propyl-2-ethyl-4-propyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate
- 8-SPT
8-sulphophenyl-theophylline
References
- 1.Tucker AL, Robeva AS, Taylor HE, Holeton D, Bockner M, Lynch KR, Linden J. A1 adenosine receptors. Two amino acids are responsible for species differences in ligand recognition. J Biol Chem. 1994;269:27900–27906. [PubMed] [Google Scholar]
- 2.Berne RM. Cardiac nucleotides in hypoxia: Possible role in regulation of coronary blood flow. Am J Physiol. 1963;204:317–322. doi: 10.1152/ajplegacy.1963.204.2.317. [DOI] [PubMed] [Google Scholar]
- 3.Headrick JP. Ischemic preconditioning: Bioenergetic and metabolic changes and the role of endogenous adenosine. J Mol Cell Cardiol. 1996;28:1227–1240. doi: 10.1006/jmcc.1996.0113. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Drake L, Sajjadi F, Firestein GS, Mullane KM, Bullough DA. Dual activation of adenosine A1 and A3 receptors mediates preconditioning of isolated cardiac myocytes. Eur J Pharmacol. 1997;320:241–248. doi: 10.1016/s0014-2999(96)00901-6. [DOI] [PubMed] [Google Scholar]
- 5.Tracey WR, Magee W, Masamune H, Kennedy SP, Knight DR, Buchholz RA, Hill RJ. Selective adenosine A3 receptor stimulation reduces ischemic myocardial injury in the rabbit heart. Cardiovasc Res. 1997;33:410–415. doi: 10.1016/s0008-6363(96)00240-4. [DOI] [PubMed] [Google Scholar]
- 6.Strickler J, Jacobson KA, Liang BT. Direct preconditioning of cultured chick ventricular myocytes. Novel functions of cardiac adenosine A2a and A3 receptors. J Clin Invest. 1996;98:1773–1779. doi: 10.1172/JCI118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stambaugh K, Jacobson KA, Jiang JL, Liang BT. A novel cardioprotective function of adenosine A1 and A3 receptors during prolonged simulated ischemia. Am J Physiol. 1997;273:H501–H505. doi: 10.1152/ajpheart.1997.273.1.H501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lasley RD, Narayan P, Jahania MS, Partin EL, Kraft KR, Mentzer RMJ. Species-dependent hemodynamic effects of adenosine A3-receptor agonists IB-MECA and C1-IB-MECA. Am J Physiol. 1999;276:H2076–H2084. doi: 10.1152/ajpheart.1999.276.6.H2076. [DOI] [PubMed] [Google Scholar]
- 9.Cave AC, Collis CS, Downey JM, Hearse DJ. Improved functional recovery by ischaemic preconditioning is not mediated by adenosine in the globally ischaemic isolated rat heart. Cardiovasc Res. 1993;27:663–668. doi: 10.1093/cvr/27.4.663. [DOI] [PubMed] [Google Scholar]
- 10.Nojiri M, Tanonaka K, Yabe K, Kawana K, Iwai T, Yamane M, Yoshida H, Hayashi J, Takeo S. Involvement of adenosine receptor, potassium channel and protein kinase C in hypoxic preconditioning of isolated cardiomyocytes of adult rat. Jpn J Pharmacol. 1999;80:15–23. doi: 10.1254/jjp.80.15. [DOI] [PubMed] [Google Scholar]
- 11.Shneyvays V, Nawrath H, Jacobson KA, Shainberg A. Induction of apoptosis in cardiac myocytes by an A3 adenosine receptor agonist. Exp Cell Res. 1998;243:383–397. doi: 10.1006/excr.1998.4134. [DOI] [PubMed] [Google Scholar]
- 12.Shneyvays V, Jacobson KA, Li AH, Nawrath H, Zinman T, Isaac A, Shainberg A. Induction of apoptosis in rat cardiocytes by A3 adenosine receptor activation and its suppression by isoproterenol. Exp Cell Res. 2000;257:111–126. doi: 10.1006/excr.2000.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norton GR, Woodiwiss AJ, McGinn RJ, Lorbar M, Chung ES, Honeyman TW, Fenton RA, Dobson JG, Jr, Meyer TE. Adenosine A1 receptor-mediated antiadrenergic effects are modulated by A2a receptor activation in rat heart. Am J Physiol. 1999;276:H341–H349. doi: 10.1152/ajpheart.1999.276.2.H341. [DOI] [PubMed] [Google Scholar]
- 14.Auchampach JA, Bolli R. Adenosine receptor subtypes in the heart: Therapeutic opportunities and challenges. Am J Physiol. 1999;276:H1113–H1116. doi: 10.1152/ajpheart.1999.276.3.H1113. [DOI] [PubMed] [Google Scholar]
- 15.El-Ani D, Jacobson KA, Shainberg A. Characterization of adenosine receptors in intact cultured heart cells. Biochem Pharmacol. 1994;48:727–735. doi: 10.1016/0006-2952(94)90050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waisberg M, Shainberg A. Characterization of muscarinic cholinergic receptors in intact myocardial cells in vitro. Biochem Pharmacol. 1992;43:2327–2334. doi: 10.1016/0006-2952(92)90310-f. [DOI] [PubMed] [Google Scholar]
- 17.Nieminen AL, Gores GJ, Bond JM, Imberti R, Herman B, Lemasters JJ. A novel cytotoxicity screening assay using a multiwell fluorescence scanner. Toxicol Appl Pharmacol. 1992;115:147–155. doi: 10.1016/0041-008x(92)90317-l. [DOI] [PubMed] [Google Scholar]
- 18.Li AH, Moro S, Melman N, Ji XD, Jacobson KA. Structure-activity relationships and molecular modeling of 3, 5-diacyl-2,4-dialkylpyridine derivatives as selective A3 adenosine receptor antagonists. J Med Chem. 1998;41:3186–3201. doi: 10.1021/jm980093j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Downey JM, Liu GS, Thornton JD. Adenosine and the anti-infarct effects of preconditioning. Cardiovasc Res. 1993;27:3–8. doi: 10.1093/cvr/27.1.3. [DOI] [PubMed] [Google Scholar]
- 20.Jacobson KA. Adenosine A3 receptors: Novel ligands and paradoxical effects. Trends Pharmacol Sci. 1998;19:184–191. doi: 10.1016/s0165-6147(98)01203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babbitt DG, Virmani R, Forman MB. Intracoronary adenosine administered after reperfusion limits vascular injury after prolonged ischemia in the canine model. Circulation. 1989;80:1388–1399. doi: 10.1161/01.cir.80.5.1388. [DOI] [PubMed] [Google Scholar]
- 22.Olafsson B, Forman MB, Puett DW, Pou A, Cates CU, Friesinger GC, Virmani R. Reduction of reperfusion injury in the canine preparation by intracoronary adenosine: importance of the endothelium and the no-reflow phenomenon. Circulation. 1987;76:1135–1145. doi: 10.1161/01.cir.76.5.1135. [DOI] [PubMed] [Google Scholar]
- 23.Funaya H, Kitakaze M, Node K, Minamino T, Komamura K, Hori M. Plasma adenosine levels increase in patients with chronic heart failure. Circulation. 1997;95:1363–1365. doi: 10.1161/01.cir.95.6.1363. [DOI] [PubMed] [Google Scholar]
- 24.Ely SW, Berne RM. Protective effects of adenosine in myocardial ischemia. Circulation. 1992;85:893–904. doi: 10.1161/01.cir.85.3.893. [DOI] [PubMed] [Google Scholar]
- 25.Mubagwa K, Mullane K, Flameng W. Role of adenosine in the heart circulation. Cardiovasc Res. 1996;32:797–813. [PubMed] [Google Scholar]
- 26.Liang BT, Jacobson KA. A physiological role of the adenosine A3 receptor: Sustained cardioprotection. Proc Natl Acad Sci USA. 1998;95:6995–6999. doi: 10.1073/pnas.95.12.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carr CS, Hill RJ, Masamune H, Kennedy SP, Knight DR, Tracey WR, Yellon DM. Evidence for a role for both the adenosine A1 and A3 receptors in protection of isolated human atrial muscle against simulated ischaemia. Cardiovasc Res. 1997;36:52–59. doi: 10.1016/s0008-6363(97)00160-0. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Downey JM. Ischemic preconditioning protects against infarction in rat heart. Am J Physiol. 1992;263:H1107–H1112. doi: 10.1152/ajpheart.1992.263.4.H1107. [DOI] [PubMed] [Google Scholar]
- 29.Kohno Y, Yoshitatsu S, Koshiba M, Kim HO, Jacobson KA. Induction of apoptosis in HL-60 human promyelocytic leukemia cells by adenosine A3 receptor agonists. Biochem Biophys Res Commun. 1996;219:904–910. doi: 10.1006/bbrc.1996.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Budinger GR, Duranteau J, Chandel NS, Schumacker PT. Hibernation during hypoxia in cardiomyocytes. Role of mitochondria as the O2 sensor. J Biol Chem. 1998;273:3320–3326. doi: 10.1074/jbc.273.6.3320. [DOI] [PubMed] [Google Scholar]
- 31.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 32.Piper HM, Schwartz P, Spahr R, Hutter JF, Spieckermann PG. Absence of reoxygenation damage in isolated heart cells after anoxic injury. Pflügers Arch. 1984;401:71–76. doi: 10.1007/BF00581535. [DOI] [PubMed] [Google Scholar]
- 33.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M, et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem. 1993;268:18532–18541. [PubMed] [Google Scholar]
- 34.Olson RD, Mushlin PS. Doxorubicin cardiotoxicity: Analysis of prevailing hypotheses. FASEB J. 1990;4:3076–3086. [PubMed] [Google Scholar]
- 35.Maggirwar SB, Dhanraj DN, Somani SM, Ramkumar V. Adenosine acts as an endogenous activator of the cellular antioxidant defense system. Biochem Biophys Res Commun. 1994;201:508–515. doi: 10.1006/bbrc.1994.1731. [DOI] [PubMed] [Google Scholar]
- 36.Ramkumar V, Nie Z, Rybak LP, Maggiewar SB. Adenosine, antioxidant enzymes and cytoprotection. Trends Pharmacol Sci. 1995;16:283–285. doi: 10.1016/s0165-6147(00)89051-3. [DOI] [PubMed] [Google Scholar]