

Abstract
De novo synthesis of the sphingolipid sphingomyelin requires non-vesicular transport of ceramide from the endoplasmic reticulum to the Golgi by the multidomain protein ceramide transfer protein (CERT). CERT's N-terminal pleckstrin homology (PH) domain targets it to the Golgi by binding to phosphatidylinositol 4-phosphate (PtdIns(4)P) in the Golgi membrane, whereas its C-terminal StAR-related lipid transfer domain (START) carries out ceramide transfer. Hyperphosphorylation of a serine-rich motif immediately after the PH domain decreases both PtdIns(4)P binding and ceramide transfer by CERT. This down-regulation requires both the PH and START domains, suggesting a possible inhibitory interaction between the two domains. In this study we show that isolated PH and START domains interact with each other. The crystal structure of a PH–START complex revealed that the START domain binds to the PH domain at the same site for PtdIns(4)P-binding, suggesting that the START domain competes with PtdIns(4)P for association with the PH domain. We further report that mutations disrupting the PH–START interaction increase both PtdIns(4)P-binding affinity and ceramide transfer activity of a CERT-serine–rich phosphorylation mimic. We also found that these mutations increase the Golgi localization of CERT inside the cell, consistent with enhanced PtdIns(4)P binding of the mutant. Collectively, our structural, biochemical, and cellular investigations provide important structural insight into the regulation of CERT function and localization.
Keywords: ceramide, fluorescence resonance energy transfer (FRET), isothermal titration calorimetry (ITC), lipid transport, phosphatidylinositol, sphingolipid, X-ray crystallography, AlphaScreen, ceramide transfer protein
Introduction
Sphingolipids are fundamental components of cellular and organelle membranes. Its members are also involved in the regulation of a variety of biological processes such as membrane biogenesis, cell growth, apoptosis, senescence, migration, and inflammation (1, 2). Ceramide, the central building block for the syntheses of more complex sphingolipids, is synthesized in the endoplasmic reticulum (ER)2 and converted to glucosylceramide and sphingomyelin (SM) in the Golgi. Although the ceramide pool for glucosylceramide synthesis is delivered through vesicular trafficking, most of the ceramide used for SM synthesis is transported to the Golgi in a non-vesicular manner by the ceramide transfer protein (CERT) (3–6). The loss of CERT function leads to impaired SM synthesis in the Golgi, indicating its critical role in sphingolipid metabolism (5).
CERT contains multiple domains and motifs (Fig. 1A). The N-terminal ∼100-residue pleckstrin homology (PH) domain is responsible for targeting CERT to the Golgi apparatus by specifically recognizing phosphatidylinositol 4-monophosphate (PtdIns(4)P) (7, 8). After the PH domain is a serine- and threonine-rich sequence termed the serine-rich (SR) motif. The ceramide transfer activity of CERT is carried out by the C-terminal ∼240 residue steroidogenic acute regulatory protein (StAR)-related lipid transfer (START) domain. Between SR and the START domain is the middle region (MR). MR is not predicted to contain any globular domains but harbors a short FFAT (two phenylalanines in an acidic tract) motif that associates with an ER-resident type II membrane protein, vesicle-associated membrane protein (VAMP)-associated protein A (VAP-A) (9, 10). This association is enhanced by phosphorylation of Ser315, which is adjacent to the FFAT motif (11). On the other hand, hyperphosphorylation of SR by protein kinase D and casein kinase Iγ2 decreases the PtdIns(4)P binding and ceramide transfer activity of CERT (12–14). Under SR hyperphosphorylation, the inhibition of PtdIns(4)P binding by the PH domain requires the presence of the START domain, and the inhibition of the START domain ceramide transfer activity requires the PH domain (12). These findings suggest the possibility of an autoinhibitory interaction between the PH and START domains and prompted our investigations.
Figure 1.

Crystal structure of CERT PH and START complex at 2.4 Å resolution. A, CERT domains and motifs. B, schematic representation of the PH–START complex with the PtdIns(4)P-binding site indicated (PDB code 5JJD; PH domain in gold, START domain in blue). C, surface representation of the PH–START complex. D, electrostatic surface potential of the PH–START complex (left) and its opened-up subunits (right) are colored at ±5 kT/e (red, negative; blue, positive). The charge potential was calculated using APBS (19, 46–47). E, superposition of the PH–START complex with a HADDOCK model of diC6–PtdIns(4)P (shown as stick model) docked onto CERT PH domain (PDB code 4HHV).
Our data show that isolated PH and START domains interact with a dissociation constant (KD) of ∼9.2 μm. We also determined the crystal structure of a protein complex formed by these two domains. The structure reveals that the START domain interacts with PH domain at its PtdIns(4)P-binding site. Moreover, the β1/β2 loop of the PH domain, which interacts with the membrane interfacial region, is completely shielded by the START domain (7, 8). Competition assays indicate the START domain competes with PH domain association with PtdIns(4)P-containing liposomes. Consistent with the structural finding, disruption of the PH–START interaction in a SR phosphorylation-mimic CERT restores both PtdIns(4)P binding and ceramide transfer activity. Moreover, with these mutations CERT also exhibited increased Golgi localization. Altogether, our structural, biochemical, and cellular investigations revealed that the START domain associates with the PH domain at the same site used for PH domain membrane-binding and confers CERT functional regulation.
Results
Structure of the CERT PH and START domain complex
Phosphorylation of the Ser/Thr residues of CERT SR leads to down-regulation of PtdIns(4)P-binding by the PH domain and ceramide transfer by the START domain. This regulation is suggested to involve a direct interaction between the PH and START domains of CERT (12). To understand the structural basis of CERT functional regulation, we solved the crystal structure of a PH–START complex to 2.4 Å, limiting resolution (Table 1). Electron density allowed for building of 97 residues of the 103-residue PH domain and 235 of the 239-residue START domain. The structure contains two potential interfaces between the PH and START domains. The lower energy assembly (EBI PISA server), which also makes the most biological sense, is shown in Fig. 1B (15). The overall structure of the PH domain in the complex is nearly identical to that of the free CERT PH structure (PDB code 4HHV) (7, 8). The root mean square deviation (r.m.s.d.) of superposition across PH Cα atoms is <1 Å with the greatest differences found in the β1/β2 loop. The START domain structure in the complex is also similar to that of the free CERT START domain. The r.m.s.d. of superposition of START domain in the complex with the START domain-alone structure (PDB code 2E3M) across Cα atoms is <1 Å (16–18) (supplemental Fig. S1).
Table 1.
Crystallographic data collection and refinement statistics
| Data collectiona | |
| Space group | P212121 |
| Unit cell dimensions | |
| a, b, c (Å) | 59.761 60.914 95.804 |
| α, β, γ (°) | 90, 90, 90 |
| Wavelength (Å) | 1.000 |
| Resolution (Å) | 38.97 (2.40) |
| Completeness (%) | 97.8 (84.7) |
| Unique reflections | 13,856 |
| Redundancy (fold) | 12.3 (6.4) |
| 〈I〉/〈σI〉 | 19.9 (2.27) |
| Rmerge (%) | 12.5 (55.9) |
| Refinement | |
| Number of molecules/a.u. | 2 |
| Rwork/Rfree (%) | 20.1/24.5 |
| Number of non-hydrogen atoms | |
| Protein | 2,707 |
| Ligand | 31 |
| Ramachandran plot (%) | |
| Favored | 98 |
| Allowed | 1.5 |
| Outliers | 0.31 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.483 |
| Average B-factor (Å2) | 46.19 |
a Numbers in parentheses are for the highest-resolution shell.
In the complex structure, the β6′/β7′ and β8′/β9′ loops of the START domain clamp onto the β1/β2 loop of the PH domain and buries an interface of 614 Å2 (Fig. 1, B and C). Notably, the β6′/β7′ loop of the START domain occupies the cavity of the PH domain that is used for PtdIns(4)P-binding. Moreover, the β1/β2 loop of the PH domain, which makes significant energetic contributions to CERT PH domain association with membranes, is completely sandwiched by the β6′/β7′ and β8′/β9′ loops of the START domain (7, 8). The PtdIns(4)P-binding site and the β1/β2 loop of the PH domain are collectively referred as the “basic groove” due to a significant number of basic residues in that area (7). The electrostatic surface potential maps of the PH and START domains in the complex show that the negatively charged β6′/β7′ loop of the START domain fits snuggly into the basic groove of the PH domain (Fig. 1D). Comparison of the CERT PH–START complex structure with a HADDOCK-generated structure model of the PH/diC6–PtdIns(4)P complex shows a clash between START domain and diC6–PtdIns(4)P-binding to PH domain (7, 8) (Fig. 1E). These observations suggest that PH domain-binding to START domain is incompatible with its association with PtdIns(4)P-containing membranes.
We also used nuclear magnetic resonance (NMR) chemical shift perturbation to examine the binding interface between the PH and START domains. Adding unlabeled START domain to the 15N-labeled PH domain leads to reduction or complete loss of the peak intensities of some residues, suggesting intermediate exchange in the PH domain in the presence of the START domain (supplemental Fig. S2A). The residues that vanished or showed 90% or more reduction of peak intensity are mapped onto the crystal structure of the PH domain (PDB code 4HHV) (supplemental Fig. S2B). As a comparison, shown in supplemental Fig. S2C are residues that are affected by the addition of diC6–PtdIns(4)P to 15N labeled PH domain (8). It can be observed that perturbations of the PH domain by START domain and diC6–PtdIns(4)P occur in similar regions, indicating overlapping binding interfaces. The findings from these NMR studies are consistent with the PH–START complex crystal structure (Fig. 1B).
Details of the CERT PH–START complex interface
EBI PISA and CCP4 program CONTACT were used to analyze the PH–START interface (15, 20) (Fig. 2A). The PH and START domain interface consists of 20 residues from the PH domain and 17 residues from the START domain. Specifically, Lys32, Trp33, Tyr36, Trp40, Arg43, Tyr54, Arg66, and Tyr96 of the PH domain form multiple contacts with the START domain. These PH domain interface residues are also involved in binding to PtdIns(4)P-containing membranes either through direct interactions with PtdIns(4)P or through interactions with the membrane interfacial region (7, 8), i.e. the PH domain uses the same set of residues for START domain and PtdIns(4)P-containing membrane interaction. Three residues in the β6′/β7′ loop of the START domain, Glu494, Asn495, and Glu498, form extensive hydrogen bonds with PH domain residues Lys32, Tyr36, Arg43, Tyr54, and Arg66 (Fig. 2, B and C). Phe598, the last residue of the START domain, interacts with Tyr36 through π-π stacking (Fig. 2C). Val533 and Pro535 in the β8′/β9′ loop make hydrophobic contacts with PH domain residues Trp33, Trp40, and Tyr96 (Fig. 2D). In addition, the backbone CO of Val533 forms a hydrogen bond with Trp33 indole ring NH group (Fig. 2D). The β8′/β9′ loop of the START domain, although not in direct competition with the PH domain PtdIns(4)P-binding site, shields the β1/β2 loop and would, therefore, prevent PH/membrane association. The PH–START complex structure clearly suggests that PtdIns(4)P and START domain binding by the PH domain are incompatible interactions.
Figure 2.

Detailed interactions at the CERT PH–START interface. A, residues involved in PH–START complex formation are shown as stick models (PDB code 5JJD; PH domain in gold, START domain in blue). B, key interactions between the PH domain and the β6′/β7′ loop of the START domain. Potential hydrogen bonds are shown as black dashed lines. Translucent spheres are used to indicate van der Waals contacts. C, interactions between Tyr36 of the PH domain and Phe598 and Glu498 of the START domain. D, key interactions between the PH domain and the β8′/β9′ loop of START domain.
CERT PH domain interacts with START domain with micromolar affinity
Isothermal titration calorimetry (ITC) was used to determine the affinity between the PH and START domains. ITC measurements were performed by injecting PH protein into START protein. Global fitting of three separate measurements using a single-site model yields a ΔH of −12.1 (−13.1, −11.3) kcal/mol and a KD of 9.2 (8.1, 10.5) μm (Fig. 3).
Figure 3.
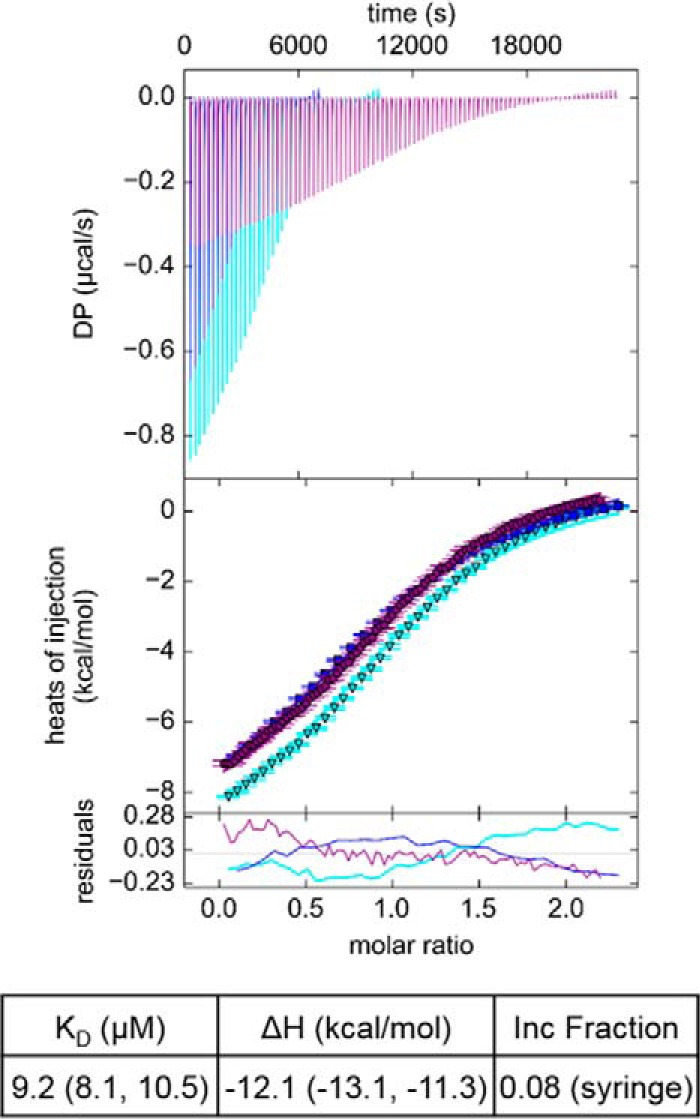
Binding affinity between CERT PH and START domains measured by ITC. Global fitting of three independent ITC experiments. The best-fit values for KD and ΔH are reported followed by the 1σ error intervals in parentheses. The incompetent (Inc) fraction reflects the difference between the stated concentration of PH domain and the concentration of binding competent material that best fits the data. DP, differential power.
To evaluate the contributions of interface residues in the interaction, site-directed mutants were generated in both the PH and START domains. The abilities of these mutants to compete for binding against the wild-type PH–START interaction were assessed in an AlphaScreen competition assay (21) (Fig. 4A). IC50 values of 2.0 and 2.2 μm were obtained for the wild-type (WT) PH and START domains, respectively (Fig. 4, B–D). A PH domain R43A mutant showed a >8-fold increase in IC50 (16.8 μm) as compared with PH WT (Fig. 4, B and D). However, the IC50 for the Y54A mutant (1.5 μm) is similar to that of the PH WT, and the IC50 values for the R43A/Y54A and the R43A mutants are also close to each other (Fig. 4, B and D). One-way analysis of variance statistics indicate that the IC50 differences measured for the R43A and R43A/Y54A mutants versus PH WT domain were statistically significant, whereas Y54A was not (Fig. 4E). These results indicate that Arg43 significantly contributes to the PH–START interaction, whereas the contribution from Tyr54 is negligible. It is likely that despite the short distance, Tyr54 is not positioned at an optimal orientation for hydrogen-bond formation with Glu494. It is also conceivable that other nearby basic residues, such as Lys56, may engage in interactions with Glu494 in the Y54A mutant, thus making this residue less critical for START domain binding. We also attempted to measure the IC50 of a W33A/R43A/Y54A PH domain mutant, but the experiments were unsuccessful. Using NMR 1H,15N 2D heteronuclear single-quantum correlation (HSQC) experiments, we found that the W33A/R43A/Y54A mutant maintains the overall tertiary fold of the PH domain (supplemental Fig. S3). We speculate the difficulties with these experiments were likely caused by protein aggregation at high concentrations in the presence of the AlphaScreen beads. As an alternative to the AlphaScreen competition assay, we used 2D HSQC experiments to evaluate the effect of the W33A/R43A/Y54A mutations on START domain binding. The presence of a 1× unlabeled PH WT domain led to extensive broadening of the 15N-labeled START domain peaks, likely from intermediate binding exchange (supplemental Fig. S4A). In contrast, the addition of 1× PH W33A/R43A/Y54A mutant lead to little change in the START domain spectrum, suggesting a much-reduced affinity between the PH W33A/R43A/Y54A mutant and the START domain (supplemental Fig. S4B).
Figure 4.

CERT START domain β6′/β7′ and β8′/β9′ loops interact with PH through its PtdIns(4)P-binding site. The PH–START interface was evaluated by an AlphaScreen competition assay. A, design of the AlphaScreen competition assay. Biotinylated START and c-Myc PH domain are immobilized on streptavidin-coated donor beads and anti-c-Myc-coated acceptor beads, respectively. Illumination of donor beads produces singlet oxygen. Interaction between the tagged PH and START domains brings donor and acceptor beads into close proximity, allowing energy from singlet oxygen to be transferred to the acceptor beads and produce a light signal. The addition of untagged competitor proteins reduces the signal. B, competition for binding to biotinylated START domain by untagged PH WT and PH domain mutants. C, competition for binding to c-Myc PH domain by untagged START WT and START domain mutants. Each competitor was tested in triplicate with a representative data set shown. Data analysis and curve-fitting parameters are found under “Experimental Procedures.” Legends for panels B and C are inset. D, mean and S.D. for the IC50 values measured in B and C. E, one-way analysis of the IC50 values measured in B and C. Shown are the Sidak 99% confidence intervals for the Sidak's multiple comparisons tests.
To validate the role of the β6′/β7′ and β8′/β9′ loops of the START domain in the PH–START interaction, START domain mutants E494R/N495K and E494R/N495K/P535R/E537K and a mutant in which a (GGS)2 linker replaces the β6′/β7′ loop (Δ493–498 (GGS)2) were tested in the AlphaScreen competition assay (Fig. 4A). The E494R/N495K and the Δ493–498 (GGS)2 mutants both displayed significant decreases in affinity compared with START WT domain with an IC50 of 34.7 and 59.5 μm, respectively, demonstrating the importance of the β6′/β7′ loop in mediating the PH–START interaction (Fig. 4, C–E). The E494R/N495K/P535R/E537K mutant showed the most loss in affinity with an IC50 of 86.1 μm, confirming that both loops of the START domain contribute to PH domain binding (Fig. 4, C–E). Altogether, these competition assays establish that the START domain uses the β6′/β7′ and β8′/β9′ loops to engage with the PtdIns(4)P-binding site of the PH domain.
CERT PH–START interaction is incompatible with PH domain binding to PtdIns(4)P
The crystal structure of the PH–START complex predicts that the START domain competes with PtdIns(4)P-containing membranes for binding to the PH domain (7, 8). To test this hypothesis, an AlphaScreen competition assay in which the ability of START domain to compete against PtdIns(4)P-embedded liposomes for PH domain association was evaluated (Fig. 5A). An IC50 value of 1.5 μm was obtained for the PH WT protein, which is comparable with the reported affinity of the PH domain for PtdIns(4)P-embedded liposomes (7, 8). The START domain yielded an IC50 value of 4.4 μm, indicating that it is able to compete with the PH/liposome interaction (Fig. 5B). The START domain has been shown to interact with membranes weakly (16, 18). To evaluate how START domain association with membrane affects the competition, the assays were repeated in the presence of a START domain inhibitor (1R,3S)-N-(3-hydroxy-1-hydroxymethyl-3-phenylpropyl) dodecanamide (HPA-12), which has been shown to abolish START domain membrane-binding (22–24). The presence of HPA-12 slightly increased the IC50 of the START domain, but the difference was not significant (supplemental Fig. S5A and B), indicating START/PH interaction is likely responsible for the competition.
Figure 5.
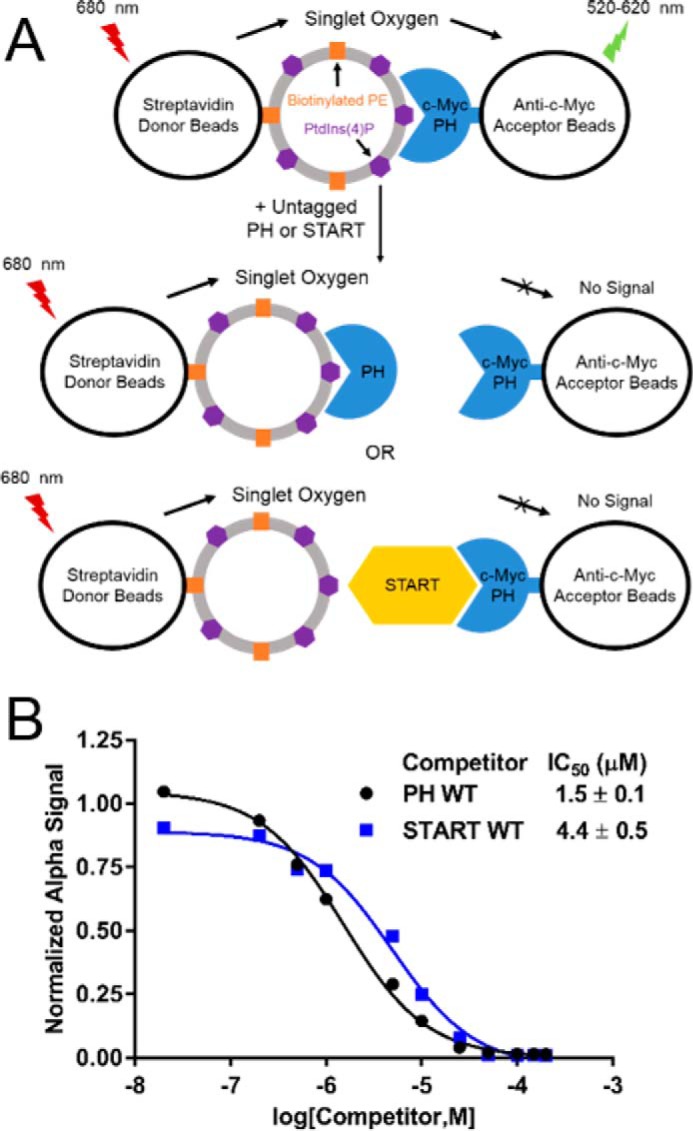
CERT START domain competes with PtdIns(4)P for binding to the PH domain. START domain competition with PtdIns(4)P for PH domain binding was evaluated by an AlphaScreen competition assay. A, design of the AlphaScreen competition assay. Design was the same as in Fig. 4 except with liposomes containing biotinylated PE and PtdIns(4)P immobilized on streptavidin-coated donor beads in place of the START domain. B, competition for binding to c-Myc PH domain by untagged PH or START domains. Each competitor was tested in triplicate with a representative data set shown. Data analysis and curve-fitting parameters are found under “Experimental Procedures.” The legend for panel B is inset.
Disrupting the PH–START interaction restores CERT SR phosphorylation mimic PtdIns(4)P-binding and ceramide transfer activity
Our investigations on isolated PH and START domains indicate that PH–START-binding competes with PH domain-binding to PtdIns(4)P-containing membranes. To further investigate the role of this interaction in the regulation of full-length CERT, we made use of a SR phosphorylation mimic of CERT, termed CERT10E, where the Ser/Thr residues of SR are mutated to Glu residues (12). We generated a CERT10E E494R/N495K/P535R/E537K mutant and investigated the effects of disrupting the PH–START interaction on CERT10E PtdIns(4)P binding and ceramide transfer activity.
Fluorescence resonance energy transfer (FRET) between protein Trp residues and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylamino-1-naphthalenesulfonyl (dansyl-PE) lipids embedded in liposomes was used to assess the binding of proteins to the liposomes (25). In the absence of PtdIns(4)P, neither CERT10E nor the CERT10E E494R/N495K/P535R/E537K mutant exhibited any detectable FRET, indicating the absence of association with the liposomes (Fig. 6A). In the presence of 4 mol% of PtdIns(4)P in the liposomes, low FRET was detected between CERT10E and liposomes, suggesting weak association. This is consistent with previous studies demonstrating that phosphorylated CERT has low affinity for PtdIns(4)P-embedded membranes (12–14). The CERT10E E494R/N495K/P535R/E537K mutant showed a clear increase in FRET signal as compared with CERT10E, indicating increased affinity for PtdIns(4)P-containing liposomes in the absence of the PH–START interaction. The data shown in Fig. 6A were performed with 40 μm total liposome concentration. The binding experiments were also carried out with 60 and 80 μm liposomes, and similar increases in PtdIns(4)P-binding of CERT10E E494R/N495K/P535R/E537K compared with CERT10E were observed (supplemental Fig. S6, A and B). This result was also consistent with prior work showing that the affinity of CERT10E for PtdIns(4)P increased after cleavage of either the PH or START domain (12).
Figure 6.

Disruption of the PH–START interaction increased both PtdIns(4)P-binding and ceramide transfer activity of a phosphorylation mimic CERT. A, binding of CERT10E and CERT10E E494R/N495K/P535R/E537K to liposomes detected by FRET between protein tryptophan residues and dansyl-PE lipids embedded in the liposomes. Shown is a representative plot of normalized FRET intensities at increasing protein concentrations with 40 μm liposomes containing 4 mol% or no PtdIns(4)P. B, ceramide transfer activity of CERT proteins as monitored by the fluorescence emission of AV-Cer. Shown are representative runs of CERT10E and CERT10E E494R/N495K/P535R/E537K activity. C, representative runs measuring the ceramide transfer activity of START WT domain and START E494R/N495K/P535R/E537K. All activity assays were repeated in triplicate for each protein. Replicates are shown in supplemental Fig. S7. Legends are inset.
To investigate if the PH–START complex interface revealed in this study is involved in the phosphorylation inhibition of CERT ceramide transfer activity, the activities of CERT10E and the CERT10E E494R/N495K/P535R/E537K mutant were compared. The transfer activity assay utilized two types of liposomes: donor liposomes that contained anthrylvinyl-ceramide (AV-Cer), a fluorescent ceramide analogue, and its quencher, 3,3′-dihexadecyloxacarbocyanine (DiO-C16), and acceptor liposomes that contain 4 mol% of PtdIns(4)P. Transfer of AV-Cer into acceptor liposomes that do not contain the quencher allowed for detection of AV-Cer emission (26, 27). CERT10E, consistent with previous results, exhibits very low ceramide transfer activity (12) (Fig. 6B and supplemental Fig. S6C). In comparison, the CERT10E E494R/N495K/P535R/E537K showed much higher ceramide transfer activity than CERT10 despite the fact that the E494R/N495K/P535R/E537K mutations decreased the intrinsic ceramide transfer activity of the isolated START domain (Fig. 6C and supplemental Fig. S6D). These results indicate that the PH–START interface identified in our structural investigation is involved in SR phosphorylation regulation of CERT ceramide transfer activity. These results are also consistent with prior studies showing that removal of the PH domain leads to an increase in the ceramide transfer activity of CERT10E (12). Ideally, mutations on the PH domain that disrupt PH–START-binding but not the intrinsic ceramide transfer activity of the START domain should have been used here. However, such mutations on the PH domain would also impact PtdIns(4)P-binding by the PH domain so as a compromise the E494R/N495K/P535R/E537K mutant that was used in these experiments.
Although the PH–START complex crystal structure provides a clear explanation for the inhibition of PtdIns(4)P-binding by the START domain, it is not immediately clear how this interaction affects the ceramide transfer activity of the START domain. To address this question, the ceramide transfer activity of the isolated START domain in the presence of PH WT or PH W33A/R43A/Y54A mutant was compared. Due to the moderate affinity between the isolated PH and START domains and the relatively low concentration of the START domain used in the transfer assay, an excess amount of PH domain was used so that ∼90% of the START domain would exist in the PH domain-bound form. The activity of the START domain in the presence of PH WT was decreased, although not significantly, compared with the activity in the presence of PH W33A/R43A/Y54A (supplemental Fig. S7, A, D, and E). However, the excess of both PH WT and mutant proteins caused unexpected changes to the activity of the START domain (supplemental Fig. S7B). Because non-vesicular lipid transfer mediated by lipid transfer proteins is a multistep process that requires the transfer protein to adopt different conformations during lipid extraction, lipid transport, and lipid release, the high concentrations of protein in solution may have led to modulations of the conformational equilibrium of the START domain and its affinity for membranes, which could result in changes in the transfer activity. To determine if these changes to activity were the result of nonspecific effects from the high concentrations of PH proteins used in this assay, the ceramide transfer activity of START domain was measured in the presence of excess B1 immunoglobulin-binding domain of streptococcal protein G (GB1), which does not interact with START domain and is not related to the PH domain (28–30) (supplemental Fig. S7, B and C). Indeed, the presence of GB1 altered the rate for transfer in a manner nearly identical to PH W33A/R43A/Y54A protein, which also does not bind the START domain, indicating a nonspecific effect on START activity (supplemental Fig. S7, D and E). Altogether, these results show that the START domain is not inhibited by binding to the isolated PH domain alone. Therefore, although the PH–START interaction does play a role in the phosphorylation inhibition of CERT ceramide transfer activity, the interaction is not directly responsible for inhibiting the START domain, and additional factors are involved in the SR phosphorylation inhibition of CERT function.
Disrupting the PH–START interaction changes the cellular distribution of CERT
The FRET-based liposome-binding experiments show that disrupting the PH–START interaction increased PtdIns(4)P binding of the CERT SR phosphorylation mimic, CERT10E (Fig. 6A). Based on this result, one would predict that disrupting the PH–START interaction would increase the Golgi localization of CERT. To test this hypothesis, immunofluorescence microscopy was used to assess the role of the PH–START interaction in the cellular localization of CERT. HEK293 cells were transiently transfected with HA-tagged CERT or CERT E494R/N495K/P535R/E537K mutant. Using GM130 as a Golgi marker, the cells were visualized by indirect immunofluorescence with anti-HA and anti-GM130 antibodies (Fig. 7). HA-CERT exhibited even distribution in the perinuclear region, likely due to association with vesicle-associated membrane protein (VAMP)-associated protein A (VAP-A) in the ER membrane (5) (Fig. 7). In contrast, HA-CERT E494R/N495K/P535R/E537K showed clear preferential co-localization with GM130, indicating release of inhibition of CERT binding to the Golgi membrane. Interestingly, the cellular distribution of HA-CERT E494R/N495K/P535R/E537K is very similar to that of the CERT S132A mutant, where SR cannot be phosphorylated, suggesting HA-CERT E494R/N495K/P535R/E537K resembles the non-phosphorylated CERT (31). These similarities further illustrate that the PH–START interaction is responsible for SR phosphorylation inhibition of CERT association with the Golgi membrane. Our study provides a structural explanation for the previous observations that the inhibition of SR phosphorylated CERT-binding to the Golgi membrane requires the START domain (12).
Figure 7.

Disruption of the PH–START interaction changes CERT cellular distribution. HEK293 cells were transfected with plasmids encoding either HA-CERT (top row) or HA-CERT-E494R/N495K/P535R/E537K (bottom row). The transfectants were incubated with primary antibodies, rat anti-HA, and mouse anti-GM130 (Golgi marker), then stained with secondary antibodies, anti-rat IgG labeled with Alexa Fluor® 488 (green), and anti-mouse IgG labeled with Alexa Fluor® 594 (red). Scale bars: 25 μm.
Discussion
In this study we showed that the CERT PH domain interacts with the START domain through the PtdIns(4)P-binding site of PH domain (7, 8) (Figs. 1–4, supplemental Fig. S2). As a consequence, isolated START domain competes with PtdIns(4)P-embedded liposomes for PH domain association (Fig. 5). These findings on isolated PH and START domains were further investigated in a full-length CERT SR phosphorylation mimic, CERT10E, which has much reduced PtdIns(4)P-binding affinity as well as low ceramide transfer activity (12). Introducing the E494R/N495K/P535R/E537K mutations, which abolish the PH–START interaction, into CERT10E led to recovery of both PtdIns(4)P-binding and ceramide transfer activity of CERT10E (Fig. 6). We also used immunofluorescence to compare the cellular localization of wild-type CERT and a CERT E494R/N495K/P535R/E537K mutant. Although the wild-type CERT exhibits even distribution in the perinuclear region, the mutant CERT protein preferentially concentrates on the Golgi (Fig. 7).
The PH–START-binding interface did not overlap with the ceramide-binding cavity of the START domain (supplemental Fig. S1). It is also distant from the two residues (Trp473 and Trp562) shown to be critical for ceramide extraction and release (16, 18) (supplemental Fig. S1). Not surprisingly, we found that in the context of isolated domains, PH domain-binding has a negligible effect on START domain activity (supplemental Fig. S7). This finding implies that the inhibition of CERT transfer activity by SR phosphorylation is not a direct result of PH domain-binding to the START domain. We speculate that there are additional factors involved in the phosphorylation inhibition of CERT function. Indeed, previous studies showed that the ceramide transfer activity of a CERT SR phosphorylation mimic increased with removal of the PH domain, but further removal of the MR led to an even greater increase in ceramide transfer activity (5, 7). Consistent with these previous results from other laboratories, we also observed that in our FRET-based ceramide transfer assays, the START domain alone is much more active than the full-length CERT proteins (Fig. 6, B and C, and supplemental Fig. S7). These observations suggest the possibility that MR is also involved in the suppression of START domain activity. Further investigations are needed to fully understand the structural basis of CERT functional regulation.
In summary, our combined structural, biochemical and cellular investigations establish that the CERT PH and START domain interaction competes with PH domain binding to PtdIns(4)P-containing membranes and plays an important role in the regulation of CERT cellular localization and ceramide transfer. Our findings provide structural insights into a previous study that shows PH and START domains are required for SR phosphorylation inhibition of CERT-binding to PtdIns(4)P and ceramide transfer (12). Our investigation also raises additional questions regarding the structural mechanism of START domain inhibition in CERT under SR hyperphosphorylation. We are currently investigating these questions.
Lastly, we would like to comment on a practical aspect of research on CERT based on our findings. In various investigations it is often necessary to add either N- or C-terminal tags to the protein of interest for purification or detection purposes. Our studies here show that the very last residue of CERT, Phe598, which belongs to the START domain, makes intimate contact with Tyr36 of the PH domain and contributes to PH–START interaction (Fig. 2, A and C). As a result, a C-terminal tag in CERT could potentially interfere with PH–START-binding and lead to experimental artifacts. To avoid this, the sequence of C-terminal tag on CERT needs to be carefully chosen.
Experimental procedures
Materials
Lipids were obtained from Avanti Polar Lipids unless otherwise specified. AlphaScreen technology materials and equipment were purchased from PerkinElmer Life Sciences. The primary rat monoclonal anti-HA antibody was purchased from Sigma. The primary mouse polyclonal antibody to GM130 and the secondary antibodies, anti-rat IgG labeled with Alexa Fluor® 488 and anti-mouse IgG labeled with Alexa Fluor® 594, were purchased from Abcam.
Protein expression and purification
The human CERT PH domain-containing residues 20–122 and START domain-containing residues 360–598 were cloned into the prokaryotic overexpression vector pHis6-GB1 as previously described (8). DNA molecules encoding the CERT PH domain with an N-terminal c-Myc epitope tag and the START domain with a Cys residue added to the N terminus were purchased from GenScript in the pHis6-GB1 vector. The site-specific biotinylated START domain was produced using the EZ-Link Maleimide-PEG2-Biotin reagent (Thermo Scientific) according to the manufacturer's instructions. Mass spectrometry confirmed single or double biotinylation of the START domain. The full-length, phosphorylation mimic CERT (CERT10E) was created by site-directed mutagenesis of wild-type human CERT (CERT) to mutate Ser132, Ser135, Ser138, Ser141, Ser144, Thr146, Ser147, Thr148, Ser149, and Ser150 to glutamate (12). CERT10E was cloned into the pHis6-GB1 vector with six extra amino acids (GEFKGL) added at the N terminus after cloning. All additional mutations were created by site-directed mutagenesis, and individual clones were confirmed by DNA sequencing. Proteins were expressed and purified as previously described (8). The PH and START domain mutants were uniformly labeled with 15N, and 2D 1H,15N NMR HSQC spectra show the mutants maintain the overall fold of the wild-type protein (data not shown).
Crystallization, X-ray diffraction data collection, structure solution, and refinement
Crystallization experiments were carried out by vapor diffusion of hanging drops. CERT PH–START co-crystals appeared in 5–7 days at 20 °C using 0.1 m MES bis-tris (pH 6.0), 6% (w/v) polyethylene glycol 10,000, and 10 μm calcium chloride. Individual drops consisted of 1 μl of PH–START complex sample (8 mg/ml) and 1 μl of precipitant solution. Crystals were cryoprotected by a brief incubation in precipitant solution containing 24% (w/v) polyethylene glycol 400. Crystal samples were preserved by flash-cooling in liquid nitrogen.
X-ray diffraction data were collected at −173 °C using beamline 22-BM of the Advanced Photon Source (Argonne National Laboratory). Individual reflections were indexed, integrated, and scaled using HKL2000 (32). Initial-phase information was obtained by maximum-likelihood molecular replacement using the program Phaser using available crystal structures of the PH (PDB code 4HHV) and START (PDB code 2E3M) domains (33, 34). An initial model was built in PHENIX.AUTOBUILD. The final model was obtained after manual building in COOT followed by additional rounds of refinement using the refine program of Phenix (20, 35–37). The model was analyzed and validated using MOLPROBITY before PDB deposition with the accession code 5JJD (38). Structure representations were generated using PyMOL (39). Additional structural information and refinement statistics are presented in Table 1.
Isothermal titration calorimetry
CERT PH and START domains were exchanged into buffer containing 25 mm Hepes (pH 7.5), 100 mm sodium chloride, and 2 mm β-mercaptoethanol. All ITC experiments were carried out on a MicroCal VP-ITC instrument (Northampton, MA). The first set of ITC data were collected by injecting 290 μl of 300 μm PH domain into 1.4 ml of 30 μm START domain with 10-μl injections. Later experiments were performed with 3-μl and 7-μl injections of 400 μm PH domain into 40 μm START domain. Protein solutions were held at 30 °C with a 290-rpm stirring speed and a 120-s delay between injections.
Each data set was first analyzed with the NITPIC program which determined the injection peak baselines, integrated the areas of injection heats, prepared an isotherm, and estimated the initial parameter values (40, 41). Global fitting of all data sets was carried out using SEDPHAT 10.58d (42, 43). ITC data plots were then prepared with GUSSI 1.0.8d (44).
AlphaScreen competition assay
Binding between CERT PH and START domains or PtdIns(4)P-embedded liposomes was evaluated using a luminescent microbead AlphaScreen technology (21). In this assay c-Myc PH domain was mixed with equimolar biotinylated START domain or biotinylated-PE- and PtdIns(4)P-embedded liposomes and increasing concentrations of untagged competitor proteins. Acceptor beads coated with anti-c-Myc antibody and streptavidin-coated donor beads were added to adsorb the c-Myc PH domain and biotinylated START domain or biotinylated-PE and PtdIns(4)P-embedded liposomes, respectively. Upon illumination at 680 nm, a photosensitizer in the donor beads converts ambient oxygen to singlet oxygen. If the acceptor beads are within close proximity to the donor beads, energy is transferred from the singlet oxygen to thioxene derivatives on the acceptor bead, resulting in emission at 520–620 nm.
Using the AlphaScreen c-Myc detection kit from PerkinElmer Life Sciences, an equilibrium competition-binding assay was carried out in 96-well format ½ area opaque plates with the following procedures. Competition assays with CERT PH and START domains were carried out in a final reaction volume of 25 μl. Each component was added to the following final concentrations in buffer containing 25 mm Hepes (pH 7.5), 100 mm sodium chloride, and 0.1% (w/v) BSA: 200 nm c-Myc PH domain, 200 nm biotinylated START domain, 20 μg/ml anti-c-Myc AlphaScreen acceptor beads, and 20 μg/ml AlphaScreen Streptavidin donor beads. A dilution series of the untagged competitor protein was prepared with final concentrations ranging from 20 nm to 200 μm. The reaction began by mixing c-Myc PH domain, biotinylated START domain, and varying concentrations of the unlabeled competitor. After incubation for 1 h at room temperature, acceptor beads were added and incubated for 1 h. Donor beads were then added, and after a 1-h incubation the AlphaScreen signal was read on an EnSpire multimode plate reader. Competition assays with c-Myc PH domain and biotinylated-PE and PtdIns(4)P-embedded liposomes were carried out with the same procedure but with a final concentration of 200 nm liposomes in place of biotinylated START domain. Liposomes containing 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS), porcine brain PtdIns(4)P, and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (biotinyl-cap PE) were prepared at a molar ratio of 84.6/9.4/4/2 by the extrusion method using 0.1-μm pore size membranes (Avanti Polar Lipids).
The AlphaScreen signal was normalized to wells without competitor. A dose-response curve was generated by plotting normalized signal (%) versus log[competitor (M)]. IC50 values were calculated by nonlinear curve-fitting to a one-site competition model using GraphPad Prism7 software (GraphPad; La Jolla, CA). IC50 values are reported as averages ± S.D. from two or three experiments.
Fluorescence resonance energy transfer measurement of protein-liposome interaction
FRET between dansyl-PE embedded in liposomes and Trp residues of CERT10E or CERT10E E494R/N495K/P535R/E537K protein was used to monitor the protein/liposome interaction. Liposomes that contain either DOPC/DOPS/dansyl-PE/PtdIns(4)P at a molar ratio of 77.4/8.6/10/4 or DOPC/DOPS/dansyl-PE at a molar ratio of 81/9/10 were prepared by the extrusion method using 0.1-μm pore size membranes. Liposomes were suspended in a total volume of 1.2 ml of buffer containing 25 mm Tris (pH 7.5), 100 mm NaCl, 1 mm EDTA, and 1 mm tris(2-carboxyethyl)phosphine. The buffer also contained 8.25 μm HPA-12 to abolish START domain interaction with liposome (22–24). CERT10E or CERT10E E494R/N495K/P535R/E537K protein was added to the vesicle suspension under constant stirring at 25 °C. Trp residues were excited at 290 nm and dansyl-PE emission was monitored at 400–550 nm. Emission intensity at 530 nm was chosen for FRET analyses to ensure the absence of intensity contribution from the protein. The normalized FRET intensity (ΔF) at a given protein concentration was calculated with the following equation: ΔF = (Fi − F0)/F0, where F0 is the intensity in the absence of protein and Fi is the intensity after protein addition. The titration series were performed with liposome concentrations of 40, 60, and 80 μm. For each vesicle concentration, the normalized FRET values were plotted against protein concentration to compare the binding of CERT10E and CERT10E E494R/N495K/P535R/E537K to liposomes.
FRET measurement of ceramide transfer activity
The ceramide transfer activities of CERT10E and START proteins were measured using donor liposomes containing ceramide and acceptor liposomes that do not (26, 27). The donor liposomes were prepared with DOPC, DOPS, DiO-C16 (Genolite Biotek), N-octanoyl-d-erythro-sphingosine (C8-Cer), and a fluorescent ceramide analogue called AV-Cer (Cayman Chemical) at a molar ratio of DOPC/DOPS/DiO-C16/C8-Cer/AV-Cer (79.2/8.8/3/8/1) (27). The acceptor liposomes were composed of DOPC/DOPS/PtdIns(4)P at a molar ratio of 86.4/9.6/4.0. Liposomes were prepared by the extrusion method using 0.1-μm pore-size membranes. AV-Cer emission was quenched by DiO-C16 in the donor liposomes but became detectable upon transfer to the acceptor liposomes.
The ceramide transfer assays were performed in buffer containing 25 mm Tris (pH 7.5), 100 mm sodium chloride, 1 mm EDTA, and 1 mm tris(2-carboxyethyl)phosphine with a 1.4-ml total volume at 25 °C with constant stirring. The emission of AV-Cer at 420 nm was monitored after excitation at 370 nm. 6 μm donor, and 60 μm acceptor liposomes were added first. After ∼100 s, ceramide transfer was initiated by adding 80 nm protein. No spontaneous transfer was observed in the absence of protein addition. At the end of the assay, 3.5% (v/v) Triton X-100 was added to disrupt the liposomes and release all AV-Cer molecules to give the maximum fluorescence. Fluorescence intensities were corrected for background by subtracting the intensity before the initiation of ceramide transfer. The percent of AV-Cer transferred at each time point was calculated by dividing the corresponding intensity by the maximum intensity. Ceramide transfer activity was measured in triplicate for each protein.
Immunofluorescence microscopy of CERT cellular localization
An HA-epitope tag was added to the N terminus of CERT and CERT E494R/N495K/P535R/E537K by PCR. HEK293 cells were transfected with either HA-CERT or HA-CERT-E494R/N495K/P535R/E537K in pcDNA6/V5-His expression plasmids by calcium phosphate precipitation. After 24 h, the transfected cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100 in PBS for 20 min, and then incubated in BlockAid™ Blocking Solution (Molecular Probes) for 1 h at room temperature. The cells were then incubated with primary antibodies, rat anti-HA, and mouse anti-GM130 (Golgi marker), or control IgG, diluted in PBS at 4 °C overnight. The cells were briefly rinsed with PBS then incubated with secondary antibodies, anti-rat IgG labeled with Alexa Fluor® 488, and anti-mouse IgG labeled with Alexa Fluor® 594 for 1 h at room temperature. After intensive washing with PBS, the slides were mounted by cold Vectashield Hard Set mounting medium and visualized by an Olympus Fluoview 300 confocal microscope. The images were acquired with the PlanApo 100× objective using the Olympus FluoView 5.0 software. Images for Fig. 7 were prepared with ImageJ (45).
Author contributions
X. Y. and J. P. designed the experiments and wrote the manuscript. J. P. performed or participated in all the experiments. S. B. participated in the crystal structure determination and edited the manuscript. M. F., Y. L., and X. Y. carried out the immunofluorescence microscopy experiments. D. B. carried out the HPA-12 synthesis.
Supplementary Material
Acknowledgments
We thank Dr. Dominika Borek for help with X-ray crystallography, Dr. Andrew Keightley for help with mass spectrometry, Linda Feng and Dr. John Laity for help with ITC and NMR experiments, Drs. Thomas Scheuermann and Chad Brautigam for help with the ITC data analysis. We also thank Drs. Jin-Yuan Price, Margaret Kincaid, and Christopher Nauman for help with fluorescence imaging. The X-ray diffraction data were collected at the Advanced Photon Source at the Argonne National Laboratory through the SER-CAT consortium.
This work was supported by National Institutes of Health Grant R15GM113200-0. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Experimental Procedures and Figs. S1–S7.
The atomic coordinates and structure factors (code 5JJD) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ER
- endoplasmic reticulum
- PH
- pleckstrin homology
- PtdIns(4)P
- phosphatidylinositol 4-phosphate
- SM
- sphingomyelin
- SR
- serine-rich
- CERT
- ceramide transfer protein
- StAR
- steroidogenic acute regulatory protein
- START
- StAR-related lipid transfer domain
- MR
- middle region
- r.m.s.d.
- root mean square deviation
- ITC
- isothermal titration calorimetry
- HSQC
- heteronuclear single-quantum correlation
- HPA-12
- 1R,3S)-N-(3-hydroxy-1-hydroxymethyl-3-phenylpropyl) dodecanamide
- AV-Cer
- anthrylvinyl-ceramide
- DiO-C16
- 3,3′-dihexadecyloxacarbocyanine
- GB1
- B1 immunoglobulin-binding domain of streptococcal protein G
- bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PE
- phosphatidylethanolamine
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPS
- 1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- dansyl-PE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylamino-1-naphthalenesulfonyl.
References
- 1. van Meer G., and Holthuis J. C. (2000) Sphingolipid transport in eukaryotic cells. Biochim. Biophys. Acta 1486, 145–170 [DOI] [PubMed] [Google Scholar]
- 2. Hannun Y. A., and Obeid L. M. (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 3. van Meer G., and Sprong H. (2004) Membrane lipids and vesicular traffic. Curr. Opin. Cell Biol. 16, 373–378 [DOI] [PubMed] [Google Scholar]
- 4. Giussani P., Colleoni T., Brioschi L., Bassi R., Hanada K., Tettamanti G., Riboni L., and Viani P. (2008) Ceramide traffic in C6 glioma cells: evidence for CERT-dependent and independent transport from ER to the Golgi apparatus. Biochim. Biophys. Acta 1781, 40–51 [DOI] [PubMed] [Google Scholar]
- 5. Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., and Nishijima M. (2003) Molecular machinery for non-vesicular trafficking of ceramide. Nature 426, 803–809 [DOI] [PubMed] [Google Scholar]
- 6. Yamaji T., Kumagai K., Tomishige N., and Hanada K. (2008) Two sphingolipid transfer proteins, CERT and FAPP2: their roles in sphingolipid metabolism. IUBMB Life 60, 511–518 [DOI] [PubMed] [Google Scholar]
- 7. Sugiki T., Takeuchi K., Yamaji T., Takano T., Tokunaga Y., Kumagai K., Hanada K., Takahashi H., and Shimada I. (2012) Structural basis for the Golgi association by the pleckstrin homology domain of the ceramide trafficking protein (CERT). J. Biol. Chem. 287, 33706–33718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prashek J., Truong T., and Yao X. (2013) Crystal structure of the pleckstrin homology domain from the ceramide transfer protein: implications for conformational change upon ligand binding. PLoS ONE 8, e79590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawano M., Kumagai K., Nishijima M., and Hanada K. (2006) Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 281, 30279–30288 [DOI] [PubMed] [Google Scholar]
- 10. Peretti D., Dahan N., Shimoni E., Hirschberg K., and Lev S. (2008) Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol. Biol. Cell 19, 3871–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumagai K., Kawano-Kawada M., and Hanada K. (2014) Phosphoregulation of the ceramide transport protein CERT at serine 315 in the interaction with VAMP-associated protein (VAP) for inter-organelle trafficking of ceramide in mammalian cells. J. Biol. Chem. 289, 10748–10760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kumagai K., Kawano M., Shinkai-Ouchi F., Nishijima M., and Hanada K. (2007) Interorganelle trafficking of ceramide is regulated by phosphorylation-dependent cooperativity between the PH and START domains of CERT. J. Biol. Chem. 282, 17758–17766 [DOI] [PubMed] [Google Scholar]
- 13. Fugmann T., Hausser A., Schöffler P., Schmid S., Pfizenmaier K., and Olayioye M. A. (2007) Regulation of secretory transport by protein kinase D-mediated phosphorylation of the ceramide transfer protein. J. Cell Biol. 178, 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomishige N., Kumagai K., Kusuda J., Nishijima M., and Hanada K. (2009) Casein kinase Iγ2 down-regulates trafficking of ceramide in the synthesis of sphingomyelin. Mol. Biol. Cell 20, 348–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 16. Kudo N., Kumagai K., Matsubara R., Kobayashi S., Hanada K., Wakatsuki S., and Kato R. (2010) Crystal structures of the CERT START domain with inhibitors provide insights into the mechanism of ceramide transfer. J. Mol. Biol. 396, 245–251 [DOI] [PubMed] [Google Scholar]
- 17. Tsujishita Y., and Hurley J. H. (2000) Structure and lipid transport mechanism of a StAR-related domain. Nat. Struct. Biol. 7, 408–414 [DOI] [PubMed] [Google Scholar]
- 18. Kudo N., Kumagai K., Tomishige N., Yamaji T., Wakatsuki S., Nishijima M., Hanada K., and Kato R. (2008) Structural basis for specific lipid recognition by CERT responsible for nonvesicular trafficking of ceramide. Proc. Natl. Acad. Sci. U.S.A. 105, 488–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bielefeld-Sevigny M. (2009) AlphaLISA immunoassay platform- the “no-wash” high-throughput alternative to ELISA. Assay Drug. Dev. Technol. 7, 90–92 [DOI] [PubMed] [Google Scholar]
- 22. Yasuda S., Kitagawa H., Ueno M., Ishitani H., Fukasawa M., Nishijima M., Kobayashi S., and Hanada K. (2001) A novel inhibitor of ceramide trafficking from the endoplasmic reticulum to the site of sphingomyelin synthesis. J. Biol. Chem. 276, 43994–44002 [DOI] [PubMed] [Google Scholar]
- 23. Nakamura Y., Matsubara R., Kitagawa H., Kobayashi S., Kumagai K., Yasuda S., and Hanada K. (2003) Stereoselective synthesis and structure-activity relationship of novel ceramide trafficking inhibitors. (1R,3R)-N-(3-hydroxy-1-hydroxymethyl-3-phenylpropyl)dodecanamide and its analogues. J. Med. Chem. 46, 3688–3695 [DOI] [PubMed] [Google Scholar]
- 24. Ďuriš A., Wiesenganger T., Moravčíková D., Baran P., Kožíšek J., Daïch A., and Berkeš D. (2011) Expedient and practical synthesis of CERT-dependent ceramide trafficking inhibitor HPA-12 and its analogues. Org Lett. 13, 1642–1645 [DOI] [PubMed] [Google Scholar]
- 25. Corbin J. A., Dirkx R. A., and Falke J. J. (2004) GRP1 pleckstrin homology domain: activation parameters and novel search mechanism for rare target lipid. Biochemistry 43, 16161–16173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mattjus P., Molotkovsky J. G., Smaby J. M., and Brown R. E. (1999) A fluorescence resonance energy transfer approach for monitoring protein-mediated glycolipid transfer between vesicle membranes. Anal. Biochem. 268, 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tuuf J., Kjellberg M. A., Molotkovsky J. G., Hanada K., and Mattjus P. (2011) The intermembrane ceramide transport catalyzed by CERT is sensitive to the lipid environment. Biochim. Biophys. Acta 1808, 229–235 [DOI] [PubMed] [Google Scholar]
- 28. Gronenborn A. M., Filpula D. R., Essig N. Z., Achari A., Whitlow M., Wingfield P. T., and Clore G. M. (1991) A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science 253, 657–661 [DOI] [PubMed] [Google Scholar]
- 29. Achari A., Hale S. P., Howard A. J., Clore G. M., Gronenborn A. M., Hardman K. D., and Whitlow M. (1992) 1.67-A X-ray structure of the B2 immunoglobulin-binding domain of streptococcal protein G and comparison to the NMR structure of the B1 domain. Biochemistry 31, 10449–10457 [DOI] [PubMed] [Google Scholar]
- 30. Gallagher T., Alexander P., Bryan P., and Gilliland G. L. (1994) Two crystal structures of the B1 immunoglobulin-binding domain of streptococcal protein G and comparison with NMR. Biochemistry 33, 4721–4729 [PubMed] [Google Scholar]
- 31. Saito S., Matsui H., Kawano M., Kumagai K., Tomishige N., Hanada K., Echigo S., Tamura S., and Kobayashi T. (2008) Protein phosphatase 2Cϵ is an endoplasmic reticulum integral membrane protein that dephosphorylates the ceramide transport protein CERT to enhance its association with organelle membranes. J. Biol. Chem. 283, 6584–6593 [DOI] [PubMed] [Google Scholar]
- 32. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 33. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 36. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeLano W. L. (2002) The PyMOL Molecular Graphic System, Version 1.8, Schrodinger, LLC, New York [Google Scholar]
- 40. Keller S., Vargas C., Zhao H., Piszczek G., Brautigam C. A., and Schuck P. (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84, 5066–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scheuermann T. H., and Brautigam C. A. (2015) High-precision, automated integration of multiple isothermal titration calorimetric thermograms: new features of NITPIC. Methods 76, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Houtman J. C., Brown P. H., Bowden B., Yamaguchi H., Appella E., Samelson L. E., and Schuck P. (2007) Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 16, 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brautigam C. A., Deka R. K., Schuck P., Tomchick D. R., and Norgard M. V. (2012) Structural and thermodynamic characterization of the interaction between two periplasmic Treponema pallidum lipoproteins that are components of a TPR-protein-associated TRAP transporter (TPAT). J. Mol. Biol. 420, 70–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brautigam C. A. (2015) Calculations and publication-quality illustrations for analytical ultracentrifugation data. Methods Enzymol. 562, 109–133 [DOI] [PubMed] [Google Scholar]
- 45. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Holst M., and Saied F. (1993) Multigrid solution of the Poisson-Boltzmann equation. J. Comput. Chem. 14, 105–113 [Google Scholar]
- 47. Holst M. J., and Saied F. (1995) Numerical solution of the nonlinear Poisson–Boltzmann equation: Developing more robust and efficient methods. J. Comput. Chem. 16, 337–364 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.