

SUMMARY
The generation of distinct hematopoietic cell types, including tissue-resident immune cells, distinguishes fetal from adult hematopoiesis. However, the mechanisms underlying differential cell production to generate a layered immune system during hematopoietic development are unclear. Using an irreversible lineage tracing model, we identify a definitive hematopoietic stem cell (HSC) that supports long-term multilineage reconstitution upon transplantation into adult recipients, but does not persist into adulthood in situ. These HSCs are fully multipotent, yet display both higher lymphoid cell production and greater capacity to generate innate-like B and T lymphocytes as compared to coexisting fetal HSCs and adult HSCs. Thus, these developmentally restricted HSCs define the origin and generation of early lymphoid cells that play essential roles in establishing self-recognition and tolerance, with important implications for understanding autoimmune disease, allergy, and rejection of transplanted organs.
Graphical abstract
INTRODUCTION
Developmental hematopoiesis occurs in waves, characterized by the generation of distinct cell types (Dzierzak and Speck, 2008; McGrath and Palis, 2005). The developmental origin and hierarchical relationship between the progenitors responsible for the production of specialized hematopoietic cell types is under intense investigation (Gomez Perdiguero et al., 2015; Hoeffel et al., 2015). Elegant fate-mapping and adoptive transfer experiments have confirmed that some fetal definitive HSCs, defined by their capacity for long-term multilineage reconstitution of adult recipients, persist into adulthood (Gothert et al., 2005; Medvinsky and Dzierzak, 1996; North et al., 2002; Samokhvalov et al., 2007). However, fetal and early neonatal HSCs exhibit markedly different properties as compared to adult HSCs, including differences in surface marker expression (Christensen and Weissman, 2001; Matsuoka et al., 2001), proliferative state (Bowie et al., 2007), and repopulation capability (Morrison et al., 1995). Additionally, fetal HSCs possess distinct potential for innate-like lymphocytes as compared to adult HSCs (Barber et al., 2011; Ghosn et al., 2012; Hardy and Hayakawa, 1991; Ikuta et al., 1990). It remains unclear whether these differences reflect disparities in the maturation state of HSCs in a single lineage or the existence of distinct, parallel lineages of HSCs across ontogeny (Herzenberg and Herzenberg, 1989). Recent investigation of human development supports the latter by positing the existence of HSCs that gives rise to specialized immune cells promoting tolerance in early life (Herzenberg and Herzenberg, 1989; Mold et al., 2010), but the identification of a transient HSC that specifies innate-like lymphocyte subsets has remained elusive. Here, using an irreversible lineage tracing model, we identify a definitive HSC with long-term multilineage reconstitution (LTMR) capacity that is restricted to early life and efficiently generates innate-like B and T cells.
RESULTS
GFP+ HSCs exist during fetal development
We recently established a lineage tracing model that tracks cells with a history of expression of the tyrosine kinase receptor Flk2 (Flt3). (Figure 1A). Importantly, we have shown that all adult HSCs in this “FlkSwitch” mouse model express Tomato (Tom) and that only Tom+ cells within the adult bone marrow (BM) possess LTMR potential (Boyer et al., 2012; Boyer et al., 2011; Buza-Vidas et al., 2011) (Figures S1A and S1B). Because the switch from Tom to GFP expression is due to the irreversible excision of the Tom gene, GFP+ cells cannot give rise to Tom+ cells (Figure S1C). Thus, adult HSCs cannot be derived from cells with a history of Flk2 expression. The FlkSwitch model thereby provides a unique tool with which to interrogate the developmental relationships between fetal and adult HSCs.
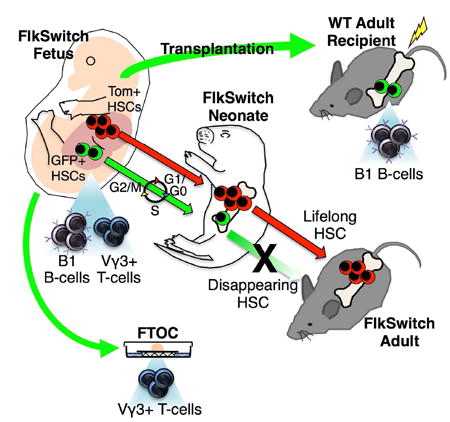
Figure 1. GFP+ cells coexist with Tom+ cells in fetal stem and progenitor compartments in FlkSwitch mice.
A, Strategy for generation of the FlkSwitch lineage tracing mouse model. Flk2-driven Cre expression leads an irreversible switch from Tomato to GFP expression in Flk2-expressing cells and their progeny.
B, Chronological depiction of developmental hematopoiesis. Primitive hematopoiesis initiates in the blood islands (BI) of the early embryonic yolk sac (YS). Definitive hematopoiesis occurs in the aorta-gonad-mesonephros region (AGM), the placenta (PL), and the fetal liver (FL) in the mid-gestation embryo. Late fetal hematopoiesis occurs primarily in the FL. The bone marrow (BM) becomes the main site of HSC residence at birth, where they persist throughout life.
C-H, Phenotypic stem and progenitor populations from E10.5-14.5 contain both Tom+ and GFP+ cells. Plots depict flow cytometric analysis of reporter activity within the hematopoietic stem cell- and progenitor-enriched compartments of the YS at E9.5 (C); the YS (D) and AGM (E) at E10.5; the FL (F) and PL (G) at E12.5; and the FL at E14.5 (H) in FlkSwitch embryos. Top row depicts gating strategy for stem and progenitor cell compartments. Bottom row depicts reporter gene fluorescence profiles of cells within gated regions shown above.
I and J, A fraction of neonatal HSCs express GFP, whereas all adult HSCs express Tom. Flow cytometric analysis of reporter activity in the P14 (I) and adult (J) BM. Plots (left) indicate the gating strategies used to define HSCs (c-kit+Lineage-Sca1+ Flk2-CD150+ CD48- BM cells) and multipotent progenitors (MPPs; KLS Flk2+CD150- CD48+ BM cells).
See also Figure S1.
As we previously demonstrated that all adult HSCs in the FlkSwitch model are Tom+ (Figure 1J, middle panel, and S1B) (Boyer et al., 2012; Boyer et al., 2011), all developmental precursors of adult HSCs must be derived via a Flk2-negative lineage. Surprisingly, investigation of reporter expression in phenotypic stem and progenitor cell compartments of FlkSwitch embryos revealed the existence of both Tom+ and GFP+ cells (Figure 1). Neither Flk2 surface protein nor GFP expression was observed prior to embryonic day (E) 10 (Figures 1C, S1D, and S1I). By E10.5, coincident with emergence of definitive hematopoiesis and Flk2 surface expression, GFP+ cells were also found., Both Tom+ and GFP+ cells were consistently observed within the phenotypic compartments enriched for primitive stem/progenitor activity in the yolk sac (YS), aorta-gonad-mesonephros region (AGM), placenta (PL), and fetal liver (FL) between E10.5 and E14.5 during fetal development (Figure 1B,and 1D-H), as well as in the neonate BM at postnatal day 14 (P14) (Figure 1I, middle panel), regardless of phenotypic markers used (Figure S1E-H). Cells appearing both Tom+ and GFP+ had undergone excision of the Tom gene and had thus recently switched from Tom to GFP expression (Figure S2A). As shown previously (Boyer et al., 2012; Boyer et al., 2011; Buza-Vidas et al., 2011), GFP+ cells were not present in the phenotypic (Figure 1J) or functional (Figure S1B) adult HSC compartment, whereas the majority of adult Flk2+ multipotent progenitors (MPPs) expressed GFP (Figure 1J). The presence of both Tom+ and GFP+ cells within the phenotypic stem cell compartments across fetal development opened the possibility of coexistence of two HSC populations during fetal and neonatal hematopoiesis.
GFP+ FL cells are capable of serial, multilineage reconstitution
To determine which fetal population contained functional HSCs, we transplanted Tom+ or GFP+ cells sorted from the cKit+Lin-Sca1+ (KLS) compartment of E14.5 FL of FlkSwitch embryos into sublethally (750 rad) irradiated adult recipients (Figure 2A). We focused on the E14.5 FL stage, as developmental HSCs are relatively abundant and well characterized at this time point. We transplanted more GFP+ cells (1000) as compared to Tom+ cells (500) based on a greater proportion of CD150+ cells in the Tom+ KLS fraction (Figure S2C) (Kim et al., 2006). Surprisingly, and in direct contrast to transplantation of the equivalent populations from the BM of adult FlkSwitch mice (Figure S1B and (Boyer et al., 2012; Boyer et al., 2011; Buza-Vidas et al., 2011), both Tom+ and GFP+ FL populations were capable of long-term multilineage reconstitution (LTMR) (Figure 2B-D and Table S1). Average reconstitution from both Tom+ and GFP+ cells was remarkably robust (6% or greater) and sustained without evidence of decline over time. GFP+ FL KLS cells similarly reconstituted lethally irradiated adult recipients in competition with co-transplanted adult cells (Figure S4A,B). Although Tom+ FL KLS cells gave rise to both Tom+ and GFP+ cells upon transplantation, as previously described (Boyer et al., 2011; Boyer et al., 2012), GFP+ FL KLS cells never gave rise to Tom+ cells (Figure S2B). The absence of Tom+ cells in mice transplanted with GFP+ cells confirmed that the switch to GFP expression within the HSC compartment is irreversible, and that Tom+ cells did not contaminate transplanted GFP+ cells. Although limiting dilution analysis indicated that GFP+ HSCs were present at lower frequency than Tom+ HSCs (Figure 2E), the efficiency of Flk2-Cre mediated recombination is incomplete during fetal development (Figure S3A), and therefore the frequency of GFP+ HSCs is therefore somewhat underestimated by the limit dilution assays of Figure 2E.
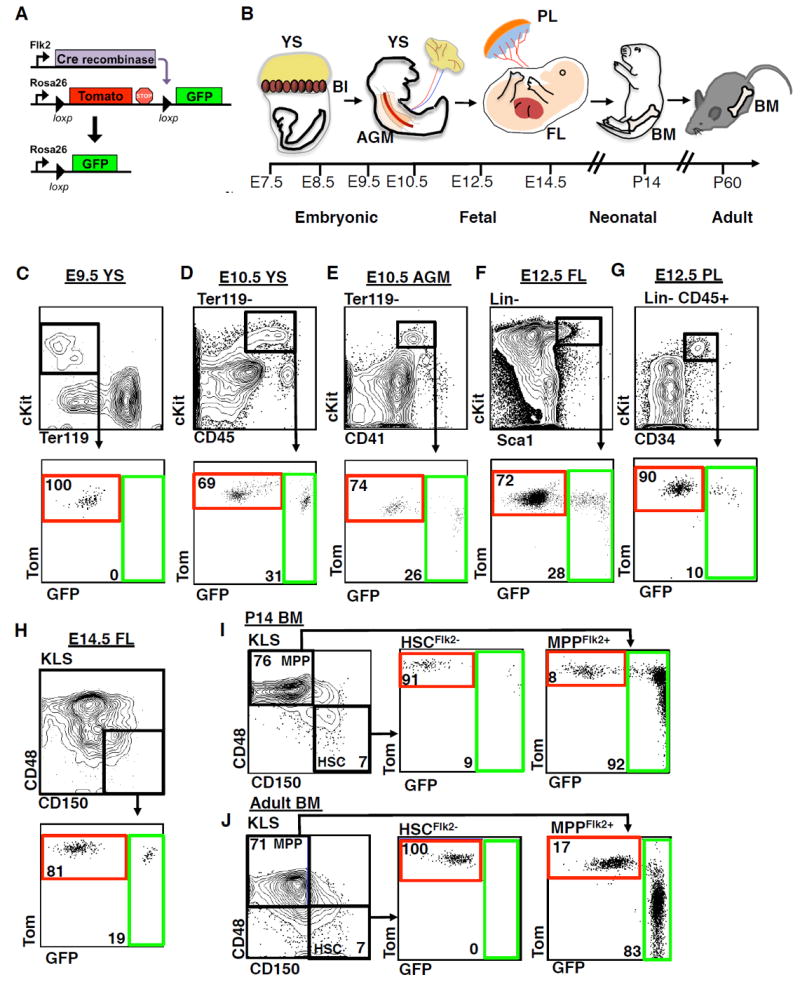
Figure 2. Both Tom+ and GFP+ fetal HSCs possess serial reconstitution potential.
A, Schematic of the experimental approach for primary and secondary transplantation and analyses. Sublethally-irradiated primary recipients were transplanted with Tom+ (500 cells) or GFP+ (1000 cells) FL KLS cells isolated from FlkSwitch fetal mice. Donor-derived chimerism was monitored in the peripheral blood (PB) over 16 weeks. After 18 weeks WBM cells from individual primary donors were transplanted into lethally irradiated secondary recipients. Chimerism was determined in the blood, peritoneal cavity (PerC), spleen, and thymus after 16 weeks in secondary recipients.
B, Proportion of primary recipients exhibiting long-term multilineage reconstitution (LTMR) following transplantation of Tom+ or GFP+ FL KLS cells. LTMR was defined as reconstitution > 0.1% in all four lineages over 16 weeks. See also Table S1.
C, Total white blood cell (WBC) contribution to the PB 16 weeks post-transplantation in primary recipients exhibiting LTMR. N=10-19 recipient mice in 4 independent experiments. Data are mean±SEM.
D, Peripheral blood (PB) contribution by Tom+ or GFP+ FL KLS cells to the granulocyte/monocyte (GM), platelet (Plt), B220+ B-cell and CD3+ T-cell lineages in the same mice from (C). Data are mean±SEM. *P<0.05.
E, Limiting dilution analysis was performed by competitive transplantation of three doses of Tom+ (500, 100, or 25) or GFP+ (1000, 200, or 50) FL KLS cells into lethally-irradiated hosts. Data are shown as the log fraction of non-engrafted (non-repopulated) mice plotted on the y axis versus the transplanted cell dose on the x axis. ELDA software (http://bioinf.wehi.edu.au/software/elda/) was used to determine HSC frequency (color-coded in bold) and assess statistical significance.
F, Donor chimerism of stem, progenitor and mature cells in the bone marrow (BM) 18 weeks post-transplantation in the mice from (D). Cell populations are defined in the online methods.
G, Proportion of primary donors that gave rise to LTMR in at least one secondary recipient after secondary transplantation. See also Table S2.
H, Total WBC donor chimerism in the PB in mice exhibiting LTMR over 16 weeks after secondary transplantation. N=21-22 recipients per cell type in 4 independent experiments. Data are mean±SEM.
I, PB contribution of donor-derived cells to GM, Plt, B-cell, and T-cell lineages over 16 weeks post-transplantation in mice from (H).
To further test the heterogeneity of the FL HSC populations, we subfractionated E14.5 FL KLS with additional cell surface markers. GFP+ and Tom+ FL HSCs expressed similar levels of CD34 (Figure S1L), CD11b (Mac1) (Figure S1M) and CD41 (Figure S1N), but the GFP+ KLS fraction expressed lower levels of CD150 as compared to the Tom+ fraction (Figure S2C). Transplantation experiments revealed that FL HSCs were enriched in the CD150+ fraction for both Tom+ and GFP+ FL KLS cells (Figure S2D-G), and that the CD150- KLS compartment contains little or no LTMR activity (Figure S2H), consistent with previous data (Kim et al., 2006). Furthermore, consistent with a previous report (Christensen and Weissman, 2001), we demonstrated both phenotypic (Figure S3E) and functional heterogeneity within the fetal KLS compartment, with LTMR capability found in both Flk2+ and Flk2- fetal KLS cells from WT (non-FlkSwitch) mice (Figure S3F-H). These data further corroborate the FlkSwitch model, as cell surface expression of Flk2, alone, is capable of distinguishing a phenotypically distinct fetal HSC. Collectively, we show that both Tom+ and GFP+ KLS cells isolated from the FlkSwitch FL were capable of LTMR upon transplantation. As all adult HSCs in the FlkSwitch model are Tom+, GFP+ HSCs appear to exist transiently during development.
To determine whether both populations with LTMR capability of the PB were also capable of repopulating the HSC compartment, we examined BM chimerism in recipients of Tom+ or GFP+ E14.5 FL KLS cells 18 weeks post-transplantation. This analysis revealed donor-derived cells of all phenotypes examined, including HSCs, in the BM of recipient mice (Figure 2F). In addition, we determined whether both populations of fetal HSCs displayed the self-renewal capability necessary to support serial reconstitution, the ‘gold standard’ for defining functional HSCs (Figure 2A). WBM cells from individual primary recipients demonstrating LTMR were transplanted into one or more secondary recipients, and donor-derived contribution to the PB was monitored over an additional 16 weeks. Remarkably, the majority of secondary recipients of either Tom+ or GFP+ FL KLS cells displayed LTMR (Figure 2G-I and Table S2). As in primary recipients, reconstitution was robust for all mature lineages (averaging 10% and above) and sustained over time, with remarkably similar reconstitution kinetics from Tom+ and GFP+ cells. Serial reconstitution was also observed when fewer cells were transferred from primary to secondary recipients, with persistence for longer than 6 months (Figure S4E-H). Consistent with the dogma that HSCs are the only cell population capable of serial reconstitution (Lemischka et al., 1986), LTMR in secondary recipients, including recipients of GFP+ KLS cells,, was accompanied by sustained BM chimerism of stem, progenitor, and mature cell compartments 18+ weeks after secondary transplantation (Figure S5), whereas recipients without sustained LTMR did not display evidence of BM chimerism (Figure S5). These data identify two coexisting populations of fetal HSCs: a Tom+ HSC that likely contributes to the adult HSC compartment, and a transient GFP+ HSC that can be distinguished from co-existing Tom+ HSCs and from adult HSCs by development via a Flk2-expressing pathway. GFP+ HSCs therefore represent a functional HSC with sustained self-renewal and multilineage engraftment potential upon transplantation, but whose existence in situ is restricted to a limited developmental window.
RNA-Seq reveals unique molecular regulators of GFP+ FL HSCs
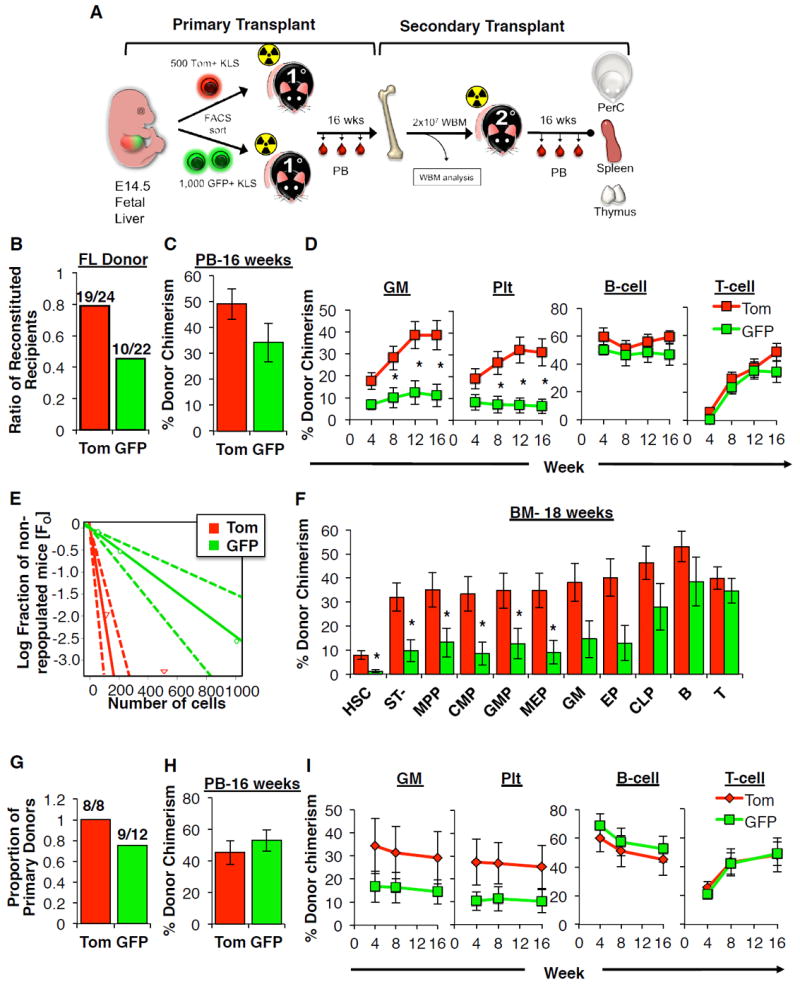
A fetal HSC capable of LTMR upon transplantation that does not persist into adulthood in situ has never before been identified. To gain further insight into the molecular regulation of this transient HSC population, we performed RNA-sequencing (RNA-seq) analysis of GFP+ and Tom+ FL HSCs (CD150+ KLS), and also compared these populations to adult HSCs. Overall, Tom and GFP+ FL HSCs had a very similar gene expression profile, as expected of two HSC populations. We identified 398 genes that were differentially expressed at least 2-fold between Tom+ and GFP+ FL HSCs, many of which were expressed at similar levels in Tom+ FL HSCs and adult HSCs (Figure 3A and (Table S3). Principal component analysis (PCA) of the same 398 differentially expressed genes identified three distinct HSC populations (Figure 3B). These analyses yielded a unique molecular profile of the distinct properties of the GFP+ HSC, despite their high degree of similarity to Tom+ FL HSCs. Furthermore, hierarchical clustering analysis revealed that Tom+ FL HSCs clustered more closely to adult HSCs (Figure 3A and data not shown), consistent with Tom+ HSCs giving rise to adult HSCs.
Figure 3. RNA-seq analysis reveals distinct molecular profile of GFP+ fetal HSCs.
A, Heat map analysis of 398 genes differentially expressed between Tom+ and GFP+ FL HSCs reveals a unique molecular signature of GFP+ HSCs. Values indicated in the color intensity scale indicate deciles of RKPM values.
B, Principal component analysis (PCA)-based comparison of Tom+ and GFP+ fetal HSCs and adult HSCs based on the expression of 398 genes described in A reveals clustering of GFP+ and Tom+ fetal HSCs and adult HSCs.
C, “Treemap” view of GO enrichment term analysis of the same genes described in (A). Each rectangle is a single cluster representative of enriched GO terms, and representatives are joined into “superclusters” of loosely related terms, visualized with different colors. Box size is proportionate to significance values.
See also Table S3.
Cell-extrinsic and cell-intrinsic mechanisms regulate the lifespan of the GFP+ HSC
RNAseq analysis revealed that genes regulating cell migration and location were differentially regulated between Tom+ and GFP+ HSCs (Figure 3C and Table S3). We therefore investigated whether GFP+ HSCs perish post-birth due to an inability to respond to CXCR4 ligands to seed the BM. However, GFP+ and Tom+ FL HSCs expressed similar levels of CXCR4 and showed equivalent capacity to migrate towards an SDF1 gradient in vitro (Figure 4A). Consistent with normal homing ability, GFP+ HSCs were capable of seeding the BM, as GFP+ FL HSCs were detected within the KLS fraction of the neonate (P14) BM by phenotypic (Figure 1I) and functional analyses (Figure 4B, C). Transplantation of 2000 GFP+ or 500 Tom+ KLS cells from P14 BM led to long-term reconstitution of all myeloid and lymphoid lineages (Figure 4B,C), in a pattern similar to that observed for FL cells (Figure 2D). GFP+ HSCs therefore arise as early as E10.5 (Figure 1D), and are capable of homing to the fetal liver and BM. However, they disappear from the BM between 2 and 8 weeks of age, coinciding with a previously described “switch” in hematopoiesis that occurs after 3 weeks of age in mice (Benz et al., 2012; Bowie et al., 2007).
Figure 4. Cell-extrinsic and cell-intrinsic mechanisms limit the developmental window of the GFP+ HSC.
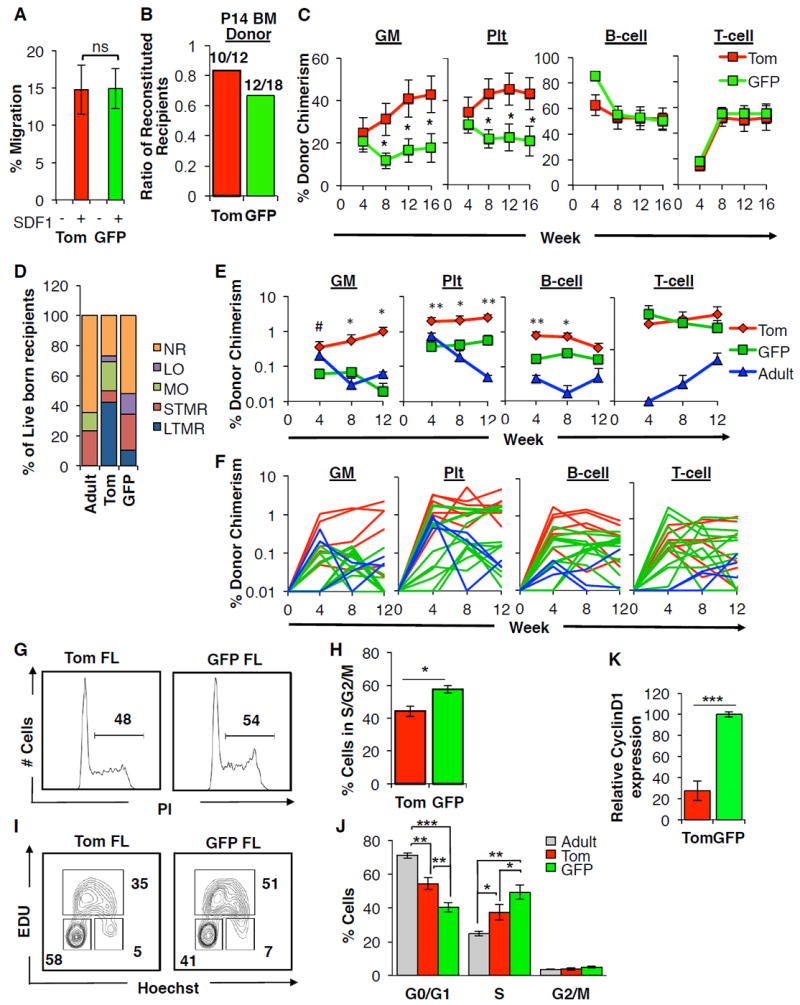
A-C, GFP+ fetal HSCs are capable of migration and seeding of the neonate BM.
A, The percentage of Tom+ or GFP+ CD150+ FL KLS cells that migrated towards an SDF1 gradient in vitro. Data are mean±SEM from 4 independent experiments performed in triplicate. ns, not significant.
B, Proportion of mice exhibiting LTMR following transplantation of either 500 Tom+ or 2000 GFP+ neonate KLS cells. Cells were isolated from the P14 BM of FlkSwitch mice and transplanted into sublethally irradiated WT recipients.
C, Peripheral blood (PB) contribution by Tom+ or GFP+ P14 BM KLS cells to the GM, Plt, B220+ B-cell and CD3+ T-cell lineages in mice exhibiting LTMR over 16 weeks post-transplantation. N=10-12 recipient mice in 3 independent experiments. Data are mean±SEM. *P<0.05.
D-F, GFP+ fetal HSCs display limited long-term engraftment following in utero transplantation.
D, The percentage of live-born recipients of Tom+ or GFP+ FL or adult KLS cells transplanted in utero into the FL of WT embryos at E14.5. Live-born recipients were classified on the basis of donor-derived chimerism within the GM, Plt, B220+ B-cell and CD3+ T-cell lineages over 12 weeks post-birth as non-reconstituted (NR), or demonstrating myeloid only (MO), lymphoid only (LO), or short-term or long-term multilineage reconstitution (STMR, LTMR). See also Table S4.
E, Peripheral blood (PB) contribution by Tom+ or GFP+ FL or adult KLS cells to GM, Plt, B-cell, and T-cell lineages over 12 weeks post-birth following in utero transplantation in recipient mice exhibiting STMR or LTMR as described in (D). Data are mean±SEM. #P< 0.1; *P<0.05; **P<0.01 for statistical differences between Tom+ and GFP+ FL populations.
F, PB chimerism in individual recipients of Tom+ or GFP+ FL or adult KLS cells to GM, Plt, B-cell, and T-cell lineages over 12 weeks following in utero transplantation in mice demonstrating STMR or LTMR as described in (D). This panel displays the reconstitution of individual mice that were shown as mean reconstitution in panel (E).
G-K, GFP+ FL HSCs are less quiescent than Tom+ FL and adult HSCs.
G-J, Analysis of cell cycle status of Tom+ and GFP+ FL and adult CD150+ KLS cells. (G-H) Representative flow cytometry plots (G) and accompanying quantification (H) of propidium iodide (PI) staining for DNA content. Brackets and values indicate the representative percentage of cells in S/G2/M. (I-J) Representative flow cytometry plots (I) and accompanying quantification (J) of cell cycle status as determined by measurement of in vivo EdU incorporation and Hoechst DNA staining. Values indicate representative frequencies of gated populations. N=8 in 3 independent experiments. Data are mean±SEM. *P< 0.05; **P< 0.01; ***P<0.001.
K, Expression of Cyclin D1 is higher in GFP+ CD150+ KLS cells as compared to their Tom+counterparts by qRT-PCR. Data are mean±SEM. N=4 independent experiments performed in triplicate. ***P<0.001. See also Table S4.
Despite their inability to persist into adulthood in situ, GFP+ FL HSCs were able to persist when transplanted into an adult environment (Figure 2). We examined the impact of the microenvironment on the persistence of GFP+ FL HSC by determining the ability of the GFP+ FL HSCs to engraft when transplanted into a fetal environment. We performed in utero transplantation (IUT) of Tom+ or GFP+ FL or adult KLS cells into the fetal liver of unconditioned WT E14.5 embryos (Figure 4D-F). Adult BM cells performed poorly in IUT assays as compared to FL cells (Figure 4D-F), as previously described (Hayashi et al., 2003). Similarly, LTMR was only observed in a small fraction (<10%) of fetal recipients of GFP+ FL KLS cells, with the majority of fetal recipients exhibiting no or short-term reconstitution. In contrast, >40% of fetal recipients of Tom+ FL KLS cells demonstrated LTMR (Figure 4D; Table S4). The inefficiency of GFP+ FL HSCs to sustain LTMR when transplanted into an unirradiated, fetal environment is reflective of their inability to persist in this same setting in situ. These data suggest that exposure to cell-extrinsic factors within the conditioned adult environment confer long-term persistence of the GFP+ FL HSC.
To determine whether differences in quiescence may underlie the inability of the GFP+ HSC to persist in situ, we compared cell cycle parameters between Tom+ and GFP+ FL HSCs and adult HSCs. Propidium iodide staining revealed a greater proportion of GFP+ HSCs in S/G2/M phase as compared to Tom+ HSCs (Figure 4G,H). Additionally, costaining for Hoechst and EdU incorporation revealed a significantly decreased proportion of GFP+ HSCs in G0/G1 as compared to both fetal Tom+FL and adult HSCs (Figure 4I,J). Our data support previously established differences in the proliferation state between fetal HSCs and adult HSCs (Figure 4J) (Bowie et al., 2007) and further show that within the fetal HSC fraction, GFP+ HSCs are more proliferative than Tom+ HSCs. Consistent with increased proliferation, GFP+ FL HSCs also expressed higher levels of the cell cycle regulator cyclin D1 as compared to Tom+ FL HSCs (Figure 4K). The less quiescent state of GFP+ HSCs within the fetal liver suggests that they may exhaust more rapidly during development as compared to Tom+ FL HSCs, as recently suggested by studies tracing unperturbed fetal HSCs in vivo (Busch et al., 2015).
GFP+ FL HSCs are functionally distinct from Tom+ FL HSCs
We noted that despite robust and persistent generation of myeloid cells, the percent contribution to the myeloid, but not lymphoid, lineages was significantly lower in primary recipients of GFP+ FL KLS cells as compared to recipients of Tom+ FL KLS cells (Figure 2D). These differences were also reflected in significantly different distribution amongst nucleated blood cells (Figure 5A) and lower ratio of donor-derived myeloid to lymphoid cells in individual recipients of GFP+ KLS cells as compared to recipients of Tom+ KLS cells 16 weeks after transplantation (Figure 5B). Differences in lineage output could not be attributed simply to persistence of long-lasting lymphoid cells from GFP+ HSCs, as we also observed a lower ratio of myeloid to lymphoid progenitors in the BM of recipients of GFP+ HSCs as compared to recipients of Tom+ HSCs (Figure 5C). Comparison of the levels of phenotypic BM HSC chimerism in individual recipients further suggested that differences in lineage output were due to higher output of lymphoid cells, as opposed to lower myeloid output, from GFP+ FL HSCs (Figure 5D). The same conclusion was reached by using the limiting dilution data (Figure 2E) to calculate the numbers of functional Tom+ versus GFP+ HSCs delivered to each transplanted recipient (see Experimental methods). Differences in lymphoid readout between Tom+ and GFP+ cells were further exaggerated when Flk2 was included as an additional marker to account for inefficient Cre-mediated floxing (Figure S3B-D). Evidence for a lymphoid bias of GFP+ HSCs was also found when they were isolated from the P14 neonate BM (Figure 5E,F), when they were transplanted in utero (Figure 5G,H), when CD150+ GFP+ HSCs were transplanted (Figure S2F,G), and when Flk2 was used as a surface marker alone (Figure S3G,H). Remarkably, the enhanced lymphoid cell production from GFP+ FL HSCs was maintained upon serial transplantation, even when secondary transplantations were performed from primary donors with similar levels of GM chimerism (Figure 5I,J), and was also observed when transplanted in competition with adult BM cells in a lethal irradiation setting (Figures S4C-D) and in secondary transplants beyond 40 weeks post-transplantation (Figure S4G-H). Importantly, GFP+ FL HSCs exhibited transcriptional lymphoid priming, as evidenced by upregulated expression of Rag1, Rag2, IL7rα, TCR-β, and CCR9 as compared to CD150+ Tom+ FL HSCs (Figure 5K and Table S3). Increased mRNA levels of lymphoid-associated genes suggest that the lymphoid bias was intrinsic to the GFP+ HSCs themselves, and not mediated by co-transplanted progenitor cells. Thus, transient GFP+ FL HSCs are functionally distinguished from their Tom+ counterparts as they displayed transcriptional lymphoid priming and consistently exhibited greater lymphoid cell production under a variety of experimental conditions.
Figure 5. Transient GFP+ HSCs are lymphoid-biased.
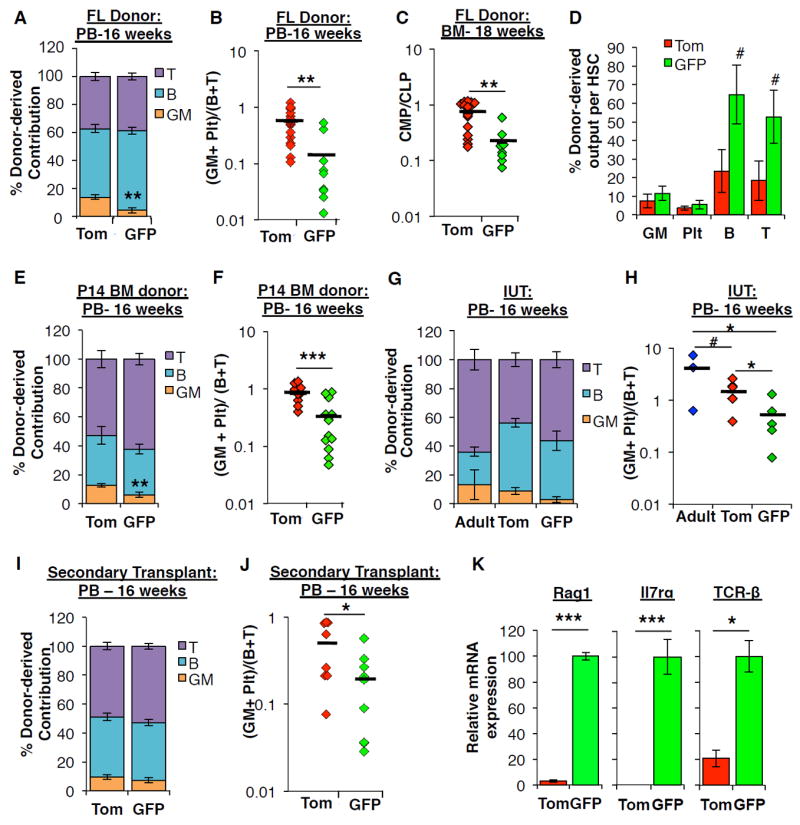
A, Distribution of donor-derived contribution to WBC lineages (GMs, B220+ B-cells, CD3+ T-cells) in the peripheral blood (PB) 16 weeks post-transplantation in primary recipients of FL KLS cells from Figure 2D. ** P < 0.01.
B, The ratio of myeloid (Plt+GM) to lymphoid (B+T) chimerism 16 weeks post-transplantation in the PB of individual primary recipients of Tom+ or GFP+ FL KLS cells from Figure 2D. ** P < 0.01.
C, The ratio of myeloid progenitors (CMP) to lymphoid progenitors (CLP) in the BM of individual primary recipients of Tom+ or GFP+ FL KLS cells from Figure 2F. ** P < 0.01.
D, Donor-derived contribution to each mature lineage calculated on a per HSC basis, based on HSC chimerism as determined in the BM of individual primary recipients of FL. # P < 0.1
E, Distribution of donor-derived contribution to WBC lineages in the PB 16 weeks post- transplantation in recipients of P14 BM KLS cells from Figure 4C. ** P < 0.01.
F, The ratio of myeloid (Plt+GM) to lymphoid (B+T) chimerism 16 weeks post-transplantation in the PB of individual recipients of Tom+ or GFP+ P14 BM KLS cells from Figure 4C. *** P < 0.001.
G, Distribution of donor-derived contribution to WBC lineages in the PB 16 weeks post- in utero transplantation (IUT) of Tom+ or GFP+ FL KLS or adult KLS cells in recipients from Figure 4E.
H, The ratio of myeloid (Plt+GM) to lymphoid (B+T) chimerism in the PB of individual recipients transplanted in utero with Tom+ or GFP+ FL KLS or adult KLS cells from Figure 4E. # P < 0.1; * P < 0.05.
I, Distribution of donor-derived contribution to WBC lineages in the PB 16 weeks post- transplantation in secondary recipients of FL KLS cells from Figure 2I.
J, The ratio of myeloid (Plt+GM) to lymphoid (B+T) chimerism in the PB 16 weeks post-transplantation in individual secondary recipients of Tom+ or GFP+ FL KLS cells from Figure 2I. * P < 0.05.
K, Relative levels of the indicated transcripts in Tom+ and GFP+ CD150+ KLS cells isolated from the E14.5 FlkSwitch FL as quantified by qRT-PCR. N= 2-4 independent experiments performed in triplicate. * P < 0.05; *** P < 0.001.
See also Figure S2, S3, S4, and Table S3.
GFP+ FL HSCs have enhanced capacity to generate innate-like T-cells within a fetal thymic microenvironment
As the existence of GFP+ FL HSCs during a limited developmental window coincides with production of specialized hematopoietic cell types, we examined their capacity to generate developmentally regulated mature cell subsets. Expression of distinct globin genes distinguishes primitive from definitive erythropoiesis. However, we detected only definitive, adult-type globin gene expression in erythroid progenitors derived from transplanted GFP+ or Tom+ FL HSCs or adult HSCs (Figure S6), suggesting that GFP+ FL HSCs do not contribute to primitive erythropoiesis. These data are consistent with the switch from primitive to definitive erythropoiesis occurring prior to E14.5 (Sankaran et al., 2010). Based on the strong lymphoid bias of the GFP+ FL HSCs, we instead compared the ability of GFP+ and Tom+ FL HSCs to generate fetal-specified lymphoid cell subsets. Specialized B1-type B-cells, and certain γδ T-cells are efficiently generated from fetal progenitor cells, whereas adult BM cells have mostly lost this capacity (Ghosn et al., 2012; Ikuta et al., 1990). These “innate-like” lymphocyte subsets differ from conventional B- and T-cells in that they are tissue-resident and express limited immunoglobin receptors that respond primarily to conserved self-antigens.
To ensure that donor-derived cells originated from HSCs and not from co-transplanted progenitor cells, we examined the generation of innate-like lymphocytes in secondary, as opposed to primary, recipients of Tom+ or GFP+ FL KLS cells (Figure 2A) and in comparable transplants of adult HSCs. These three recipient cohorts displayed no differences in the frequency of donor-derived thymic T-cell subsets (Figures 6A-C). The vast majority of donor-derived CD3+ T-cells in the thymus expressed the adult-type T-cell receptor (TCR)-β, whereas <1% of donor-derived thymic CD3+ T-cells expressed TCR-γδ (Figures 6B and 6C), regardless of whether the transplanted HSCs were Tom+ or GFP+ FL cells or from adult donors.
Figure 6. GFP+ fetal HSCs display superior ability to give rise to developmentally-restricted T-cells in a fetal thymic microenvironment.
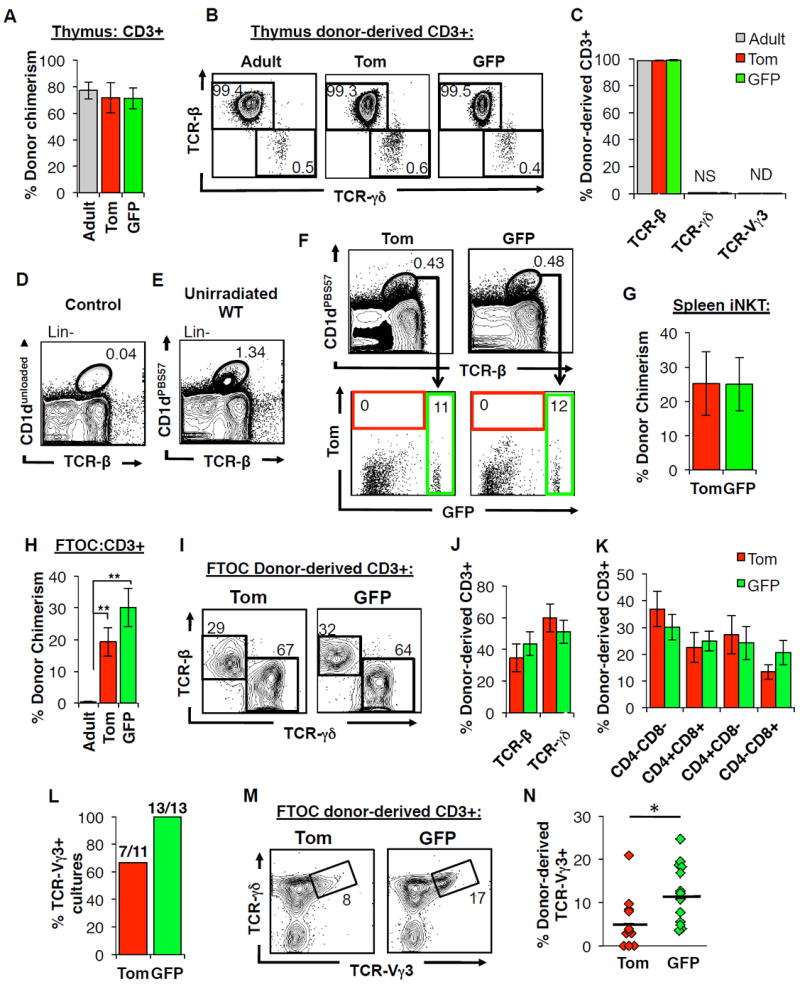
A-C, Neither fetal nor adult HSCs give rise to substantial numbers of TCRγδ-expressing T cells upon transplantation into adult recipients.
A, Donor contribution to CD3+ thymocytes within the thymus of adult, secondary recipients of Tom+ and GFP+ fetal KLS cells or comparable transplants of adult KLS cells 18-weeks post-transplantation. N=10-12 representing three independent experiments. Data are mean±SEM.
B-C, Representative FACS plots (B) and accompanying quantification (C) of donor-derived CD3+ thymocyte subsets for the mice described in (A). Numbers indicate representative frequencies of gated cell subsets. Data are mean±SEM. NS, not significant; ND, not detected.
D-G, Tom+ and GFP+ FL HSC contribute equally to minimal reconstitution of iNKTs in irradiated adult recipients
D-E, Representative FACS plots demonstrating the identification of splenic iNKT cells defined by double labeling of CD1d tetramer loaded with PBS57 (CD1dPBS57+) and antibody against TCR-β. D, Control sample labeled with an unloaded tetramer. E, Representative FACS plot indicate frequency of iNKT cells among Lin- splenocytes in an unirradiated WT mouse.
F-G, Representative flow cytometry plots (F) and accompanying quantification (G) of donor-derived contribution to splenic iNKT cells in secondary recipients of Tom+ or GFP+ FL KLS cells.
Data are shown in (G) are as mean±SEM. N=7 in 3 independent experiments.
H-N, Fetal, but not adult, HSCs efficiently produce T cells in a fetal thymic microenvironment.
H, Donor contribution to CD3+ thymocytes within fetal thymic organ cultures (FTOCs) seeded with Tom+ or GFP+ fractions of CD150+ FL KLS cells or adult CD150+ KLS cells. N=12-13 in 3 independent experiments. Data are mean±SEM. **P<0.01.
I-J, Representative flow cytometry plots (I) and quantification (J) of donor-derived contribution to TCRβ- and TCRγδ–expressing subsets in FTOCs described from (H) are shown. Numbers indicate representative frequencies of gated populations.
K, Quantification of donor-derived contribution to CD4+CD8+ (DP), CD4-CD8- (DN), CD4+, and CD8+ T-cell subsets in FTOCs described in (H).
L-N, GFP+ FL HSCs give rise to significantly more TCR-Vγ3+ T-cells in FTOCs compared to Tom+ FL HSCs. Percentage of FTOC cultures described in (H) that contained TCR-Vγ3+ cells (L). Representative flow cytometry plots (M) and quantification (N) of donor-derived contribution to TCR-Vγ3+ cells in FTOCs described in (H). Data are shown as individual data points representing individual cultures. *P<0.05.
See also Figure S6 and Table S5.
In mice, T-cells expressing the developmentally limited TCR-Vγ3 are generated only from FL, but not adult, HSCs and contribute to innate-like T-cells within the skin (Havran and Allison, 1988). Consistent with a previous report that the adult thymus does not support the generation of developmentally limited TCR-Vγ3+ cells (Ikuta et al., 1990), we observed negligible (< 0.1%) generation of TCR-Vγ3+ cells in the thymi of transplanted adult recipients (Figure 6C). Examination of donor-derived TCR-Vγ3+ cells within the epidermis in secondary recipients revealed minimal levels of chimerism (Table S5). While these results show that FL HSCs do retain the capability to generate TCR-Vγ3+ cells in the skin of adult recipients, the requirement for very high levels of overall T-cell chimerism and the absence of consistent chimerism among transplanted individuals prevented conclusive quantitative comparisons of the TCR-Vγ3+ capability of the different HSC populations within the skin of secondary recipients.
We next examined the generation of another innate-like T-cell, invariant natural killer T cells (iNKTs), which are thought to be developmentally regulated (Benlagha et al., 2002; Gapin et al., 2001). While we observed some contribution of both Tom+ and GFP+ FL KLS cells to splenic iNKT cells in secondary recipients, we did not observe significant differences in chimerism between the two donor cell types (Figure 6D-G). As the iNKT compartment did not fully recover after irradiation despite transplantation of BM cells (compare iNKT frequencies in Figures 6E and 6F), it is possible that potential differences in iNKT potential between the two HSC populations are under-appreciated in a transplantation setting.
As a previous study reported that TCR-Vγ3+ cells could be generated within a fetal thymic microenvironment (Ikuta et al., 1990), we examined differences in developmental T-cell potential using fetal thymic organ cultures (FTOCs). Adult HSCs consistently failed to contribute robustly to CD3+ T-cells in the FTOC assay, possibly because they competed poorly with resident fetal progenitors that survived the pre-conditioning by irradiation (Figure 6H). In contrast, fetal CD150+ KLS cells efficiently differentiated into T cells (Figure 6H). Tom+ or GFP+ HSCs cells yielded similar profiles of T-cell differentiation (Figures 6I-K), with the notable exception of more efficient generation of TCR-Vγ3+ T-cells by GFP+ HSCs as compared to their Tom+ counterparts (Figure 6L-N). Whereas more than 1/3 of Tom+ cultures failed to yield any TCR-Vγ3+ T-cells, all GFP+ cultures contained TCR-Vγ3+ T-cells (Figure 6L), and each GFP+ Vγ3-containing culture had significantly higher proportion of Vγ3+ T-cells compared to Tom+ FTOCs (Figures 6M and 6N). These findings show that both the progenitor cell and thymic environment must be of fetal origin to allow generation of TCR-Vγ3+ T-cells. In addition, these data suggest that GFP+ FL HSCs are an important source of TCR-Vγ3+ T-cells during development.
GFP+ FL HSCs efficiently generate innate-like B-cells in vivo
We next investigated the ability of Tom+ and GFP+ FL HSCs to give rise to innate-like B-cell subsets. As with in vivo T cell experiments, all analyses were conducted in secondary recipients. We confirmed that there were virtually no B1 progenitors (B1P) within the originally transplanted KLS fractions (Figure S7A,B), excluding these cells as a differential source of B1 cells in the primary transplant.
Supporting the reported fetal origin of peritoneal cavity (PerC) B-cells (Barber et al., 2011; Hardy and Hayakawa, 1991), we observed strikingly higher donor-derived contribution to peritoneal IgM+ B-cells in secondary recipients of fetal compared to adult HSCs (Figures 7A, 7B, and S7G). GFP+ FL HSCs gave rise to significantly higher peritoneal IgM+ B-cell chimerism than Tom+ FL HSCs, even in cohorts of mice with similar B-cell contribution in the PB (Figure 7B). To determine whether the low contribution to PerC B-cells by adult HSCs could be attributed to their overall lower lymphoid cell generation, we quantified peritoneal B cell output in recipient mice of adult HSCs or FL HSCs that displayed comparable B-cell chimerism in the PB (Figure S7I). This analysis reinforced the relative myeloid bias of adult versus fetal HSCs, and attenuated the differences in peritoneal B cell contribution between adult HSCs and Tom+ FL HSCs. Strikingly, the superior ability of GFP+ FL HSCs to generate peritoneal B-cells was still apparent (Figure S7I). Furthermore, not only was the contribution of GFP+ FL HSCs higher to all peritoneal B-cell subtypes (Figures 7B, S7G, and S7I), they also gave rise to a significantly higher proportion of B1a and B1b type B-cells compared to both adult HSCs and Tom+ FL HSCs (Figures 7C and 7D). The few donor-derived peritoneal B-cells found in recipients of adult HSCs were predominantly B2 cells (IgM+ CD5-CD11b), with very few B1a (IgM+CD5+CD11b+) or B1b (IgM+CD5-CD11b+) cells (Figures 7C and 7D). While Tom+ FL HSCs gave rise to a significantly higher proportion of B1a and B1b cells compared to adult HSCs, GFP+ FL HSCs were even more efficient in B1 cell generation (Figures 7C and 7D). Furthermore, despite lower overall reconstitution levels and limited long-term reconstitution upon in utero transplantation (IUT) (Figure 7H and Table S4), GFP+ KLS cells still retained a greater capability to seed the peritoneal IgM+ B-cell compartment and particularly B1a cells as compared to Tom+ KLS cells upon IUT (Figure 7I). Although a recent report suggested that fetal HSCs are not a major source of B1a cells (Ghosn et al., 2016), we did not observe appreciable regeneration of innate-like B-cells in the absence of LTMR and BM reconstitution (Figure S5). Secondary recipients that did not demonstrate LTMR showed limited ability to reconstitute innate-like lymphocytes in vivo (Figure S5D). Similarly, secondary transplantation of BM B-cells could not sustain PB lymphopoiesis (Figure S7C,D), nor yield appreciable innate-like B-cell reconstitution of the peritoneal cavity (Figure S7E) or spleen (Figure S7F). Collectively, our data reveal the unique capacity of the transient GFP+ HSCs to generate innate-like B cells, and in particular fetal-restricted B1 B-cells, in the peritoneum.
Figure 7. GFP+ fetal HSCs efficiently generate innate-like B-cells in vivo.
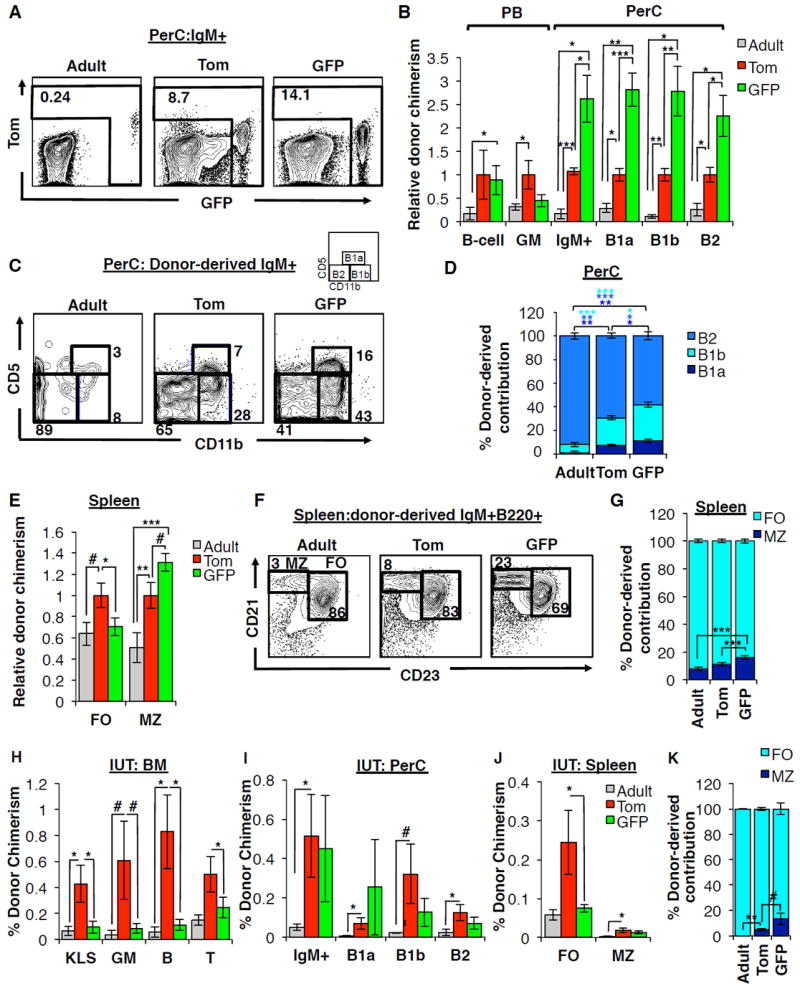
A-D, GFP+ FL HSCs generate peritoneal cavity (PerC) B cells with greater efficiency than Tom+ FL HSCs or adult HSCs.
A, Representative flow cytometry plots indicating donor-derived IgM+ cells within the peritoneal cavity in secondary recipients of adult HSCs, Tom+ FL HSCs, or GFP+ FL HSCs. Donor-derived cells from adult and Tom+ donors can be either Tom+ or GFP+; donor-derived cells from GFP+ donors are GFP+
B, Donor chimerism of PB B220+ B-cells and GM cells, and peritoneal cavity IgM+ cells, B-1a (IgM+ CD5+ Cd11b+), B-1b (IgM+ CD5- CD11b+) and B2 (IgM+ CD5- CD11b-) B-cells in secondary recipients of either Tom+ or GFP+ FL KLS cells, or adult HSCs (KLSF-). Results are displayed relative to recipients of Tom+ FL HSCs; Figure S7G displays the results as percent donor chimerism.
C-D, Representative flow cytometry plots (C) and quantification (D) of donor-derived contribution to peritoneal cavity B-cell subsets in secondary recipients of adult HSCs, Tom+ FL HSCs, or GFP+ FL HSCs.
E-G, GFP+ FL HSCs give rise to a higher proportion of marginal zone B cells in the spleen compared to Tom+ FL HSCs or adult HSCs.
E, Donor chimerism of splenic marginal zone (MZ; CD21hi CD23-) and follicular zone (FO, CD21lo CD23+) B-cells among secondary recipients of Tom+ or GFP+ FL KLS cells or comparable recipients of adult KLS cells. Results are displayed relative to recipients of Tom+ FL HSCs; Figure S7H displays the results as percent donor chimerism.
F-G, Representative flow cytometry plots (F) and quantification (G) of donor-derived contribution to splenic B-cell subsets from the same mice as in (E).
N=5-17 per group in at least three independent experiments. #P<0.1; *P<0.05; **P<0.01; ***P<0.001.
H-K, Contribution of Tom+ and GFP+ FL or adult HSCs to BM, PerC, and spleen cells upon in utero transplantation.
H, BM donor chimerism in progenitor and mature cells among fetal recipients transplanted in utero with Tom+ or GFP+ FL KLS cells or adult KLS cells. Chimerism was analyzed 12-16 weeks post-transplantation in mice exhibiting LTMR and STMR. N=3-10 group. Data are mean±SEM. Additional details in Table S4.
I, Donor chimerism of total PerC IgM+ cells, and PerC B-1a, B-1b and B2 B-cells among fetal recipients of Tom+ or GFP+ FL KLS cells or adult KLS cells 12-16 weeks post-in utero transplantation in the same mice as in (H). #P<0.10; *P<0.05.
J, Donor chimerism of splenic MZ and FO B-cells among fetal recipients of Tom+ or GFP+ FL KLS cells or adult KLS cells 12-16 weeks post-in utero transplantation in the same mice as in (h). *P<0.05.
K, Quantification of donor-derived contribution to splenic B-cell subsets among fetal recipients of Tom+ or GFP+ FL KLS cells 12-16 weeks post-in utero transplantation in the same mice as in (H). #P<0.10; *P<0.05.
See also Figures S5 and S7.
Like peritoneal B1 B-cells, marginal zone (MZ) B-cells within the spleen exhibit innate-like immune responses and have a suggested fetal origin (Carey et al., 2008). We therefore examined the contribution of Tom+ and GFP+ FL HSCs and adult HSCs to MZ B-cells in the spleen by strategies described above. In mice with comparable PB B-cell chimerism, we observed a significantly lower contribution by adult HSCs to MZ B-cells (Figures 7E-G, and S7H),supporting their proposed fetal origin. GFP+ FL HSCs exhibited higher chimerism within MZ (CD21hiCD23lo), but not follicular (FO, CD21-CD23hi), splenic B-cells subsets B-cells as compared to Tom+ FL HSCs (Figure 7E and S7H). Furthermore, within the donor-derived fraction of splenic B-cells, GFP+ FL HSCs gave rise to a significantly higher ratio of MZ B-cells as compared to both Tom+ FL HSCs and adult HSCs (Figures 7F and 7G). Similarly, GFP+ KLS cells to preferentially seed the MZ of the spleen when transplanted in utero, despite yielding significantly lower overall chimerism within the spleen as compared to Tom+ KLS cells (Figures 7J and 7K). Together, these data reveal that the transient GFP+ HSCs have a unique capability to generate fetal-restricted innate-like B-cells. Retention of this ability upon transplantation into both fetal and adult recipients demonstrates that this capability is mainly regulated cell-intrinsically and does not require a fetal environment.
DISCUSSION
In summary, our Flk2-based fate mapping identified a definitive HSC that is capable of LTMR upon serial transplantation into adult recipients, but does not persist into adulthood and is not a precursor to adult HSCs. Our discovery of a ‘disappearing’ stem cell with unique lineage potential provides a new framework for understanding the specification and hierarchy of early HSCs and the mechanisms underlying developmental waves of hematopoietic cell output. In conjunction with previous fate mapping studies that have detected the persistence into adulthood of cells labeled during early development (Gothert et al., 2005; North et al., 2002; Samokhvalov et al., 2007), our discovery of a transient HSC population supports the existence of at least two distinct fetal HSC lineages: HSCs that contribute to the adult HSC compartment (Tom+), and HSCs that do not (GFP+). Though the demonstration of transient HSCs challenges the fundamental concept that stem cells contribute to tissue regeneration across the lifespan, our findings fit with a recent lineage tracing report suggesting that fetal hematopoiesis is supported by rapidly extinguishing HSCs (Busch et al., 2015). Here, we describe the prospective isolation of an HSC population that embodies this property, and demonstrate that this HSC efficiently generates fetal-specific immune cells before vanishing.
Spatiotemporal analysis and genetic fate-mapping of developmental HSCs have contributed to a model in which definitive HSCs generated within the AGM ultimately contribute to adult BM hematopoiesis (Gothert et al., 2005; North et al., 2002). The LTMR capability of prenatal cells has played a critical role in their designation as “bona fide” HSCs that give rise to the adult HSC compartment (de Bruijn et al., 2000; Medvinsky and Dzierzak, 1996). Our data suggest that not all developmental HSCs with LTMR capacity contribute to the adult HSC compartment, and therefore that adult repopulating capability cannot be used as the sole criterium to define adult HSC precursors. These findings highlight the necessity of lineage tracing models to define cell populations that ultimately contribute to the adult HSC compartment. Moreover, we show that cells that do not normally persist into adulthood can display long-term engraftment upon transplantation into a conditioned, irradiated adult microenvironment. The same cells showed limited persistence when transplanted into an unconditioned fetal microenvironment (Figure 4D-F), yet maintain enhanced capability to generate innate-like lymphocytes in the same setting (Figure 7I-K). This uncoupling of self-renewal capacity in situ with that observed upon transplantation suggests that cues associated with transplantation can induce persistence of otherwise transient cells.
Consistent with their inability to persist into adulthood under normal conditions, GFP+ HSCs express lower levels of CD150 (Figure S2C) and are less quiescent (Figure 4G-J), both properties that have been linked to lymphoid-bias (Challen et al., 2010; Morita et al., 2010). GFP+ HSCs also expressed higher levels of IL7r, Rag1, and Flk2 (Figure 5K and Table S3). Quantitative comparison of the lineage output of Tom+ and GFP+ FL HSCs with adult HSCs additionally reinforced previously described functional differences between adult and fetal HSCs (Figure 6H-N and Figure 7) (Barber et al., 2011; Benz et al., 2012; Hardy and Hayakawa, 1991; Ikuta et al., 1990). Due to the incomplete floxing efficiency in fetal FlkSwitch mice, not all Flk2+ HSCs become GFP+ (Figure S3A), leading to “contamination” of the Tom+ FL HSC fraction by cells that should be GFP+ and underestimation of the functional differences between the Tom+ and GFP+ HSCs. Therefore, the distinct properties observed between fetal and adult HSCs may be attributed mainly to the developmentally restricted HSCs, as opposed to the entire fetal HSC compartment. Because the GFP+ HSC has greater lymphoid capacity than Tom+ HSCs on a per cell basis (Figure 5D) and it is impossible for GFP+ cells to give rise to Tom+ cells (Figure S1C), the two HSC populations are unlikely to exist in a linear progenitor-progeny relationship, but rather represent two separate lineages. Collectively, our data support a model of parallel definitive hematopoietic development in which partially overlapping hematopoietic waves arise from distinct progenitor subsets.
By prospectively isolating and functionally testing a transient HSC, we provide direct evidence that discrete HSC populations are responsible for layered immune development, as proposed several decades ago by the Herzenberg Lab (Herzenberg and Herzenberg, 1989). Evidence for this mechanism in humans was recently supported by studies of human T-cell development (Mold et al., 2010). Our discovery of a fetal HSC characterized by lower CD150 expression with distinct innate-like lymphocyte potential is also consistent with a recent report demonstrating that B1 B-cell regenerative capacity mainly reside outside the conventional fetal HSC compartment (Ghosn et al., 2016). The coincidence in existence of the GFP+ HSCs with the perinatal burst in lymphoid development suggests that these HSCs play important roles in specifying innate-like B- and T-cells, while also contributing to robust establishment of adaptive immunity. Intriguingly, we found that development of distinct B-cell subsets appears to be regulated primarily cell intrinsically, as fetal HSCs generate more fetal-like B-cells than adult HSCs even within an adult environment. In contrast, T-cell development appears to be regulated both cell intrinsically and extrinsically, as both the cell of origin and the thymic microenvironment affected the generation of fetal-restricted T-cells. Together, these data indicate that both fetal-specific HSCs and the fetal niche regulate the establishment of innate-like immune cells. These findings shed new light on immune system development and have important implications for understanding the mechanisms regulating maternal-fetal tolerance during development, the development of autoimmune disorders, and determining optimal vaccination efficiency (Burt, 2013; Montecino-Rodriguez and Dorshkind, 2012). Furthermore, the demonstration of unique mature cell output from a developmentally limited HSC suggests that the potential to generate tolerogenic immune cells should be considered when choosing the optimal source of cells for transplantation, particularly for treatment of autoimmune disease.
EXPERIMENTAL PROCEDURES
Mice, cell isolation, and analysis
All mice were maintained in the UCSC vivarium according to IACUC-approved protocols. FlkSwitch mice (Boyer et al., 2012; Boyer et al., 2011; Epelman et al., 2014; Hashimoto et al., 2013) were used as donors for cell isolation and 8-12 week old WT C57Bl/6 mice were used as recipients. Adult and fetal cells were isolated and processed as previously described (Perez-Cunningham et al., 2016; Smith-Berdan et al., 2015) and analyzed using a four-laser FACSAria or LSRII flow cytometer (BD Biosciences, San Jose, CA).
Transplantation assays
Transplantations were performed as previously described (Forsberg et al., 2006; Smith-Berdan et al., 2011). Recipient mice were either sublethally irradiated (750 rad, single dose) or lethally irradiated (1000 rad, split dose). Fetal intrahepatic injections were performed as previously described (Nijagal et al., 2011). Briefly, under isofluorane-induced general anesthesia, a midline laparotomy was performed on pregnant dams, the uterus was exposed, and 5 μL of cell suspension was injected into the fetal liver of each pup using pulled glass micropipettes. The abdominal incision was closed in multiple layers. PB chimerism was analyzed by flow cytometry.
In vitro analyses
RNA-sequencing, qRT-PCR, cell cycle analysis, EdU incorporation, and fetal thymic organ culture assays were all performed as described previously (Beaudin et al., 2014); (Ugarte et al., 2015) and in the Supplemental Experimental Procedures.
Statistical analysis
Transplantation assays were randomized across age, sex, and cage for any given experiment. Investigator was blinded to treatment during preparation and analysis of samples. Unless otherwise noted, all experiments were performed in at least three independent replicates. Statistically significant differences between groups with similar variance as determined by the standard error of the mean were assessed by unpaired, two-tailed Student’s t-test, unless otherwise noted.
Supplementary Material
Acknowledgments
We thank Dr. T. Boehm for the Flt3Cre strain; Bari Nazario for flow cytometry support; Sol Katzman for bioinformatic assistance; and Cecilia Im, Chukwuemeka Ajaelo, and Elizabeth Lopez for technical assistance. This work was supported by an NIH/NIDDK award (R01DK100917), an Alex’s Lemonade Stand Foundation Innovation Award, and an American Asthma Foundation Research Scholar award to ECF; by NIH Training Grant 2T32GM008646 and an HHMI Gilliam Fellow Award to JP-C; CIRM Training grant TG2-01157 to AEB and SWB; and by CIRM Facilities awards CL1-00506 and FA1-00617-1 to UCSC. ECF is the recipient of a CIRM New Faculty Award (RN1-00540) and an American Cancer Society Research Scholar Award (RSG-13-193-01-DDC). TM is the recipient of a CIRM New Faculty Physician Scientist Translational Research Award (RN3-06532).
Footnotes
Conflicts of interest: The authors have no conflicts to declare.
AUTHOR CONTRIBUTIONS
AEB and ECF conceived the study, designed experiments, and co-wrote the paper. AEB and SWB performed experiments and analyzed the data. TM designed experiments. SCD, GEH, CJ, EA, and JP-C performed experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barber CL, Montecino-Rodriguez E, Dorshkind K. Reduced production of B-1-specified common lymphoid progenitors results in diminished potential of adult marrow to generate B-1 cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(1):3700–13704. doi: 10.1073/pnas.1107172108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin AE, Boyer SW, Forsberg EC. Flk2/Flt3 promotes both myeloid and lymphoid development by expanding non-self-renewing multipotent hematopoietic progenitor cells. Experimental hematology. 2014;42:218–229. e214. doi: 10.1016/j.exphem.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benlagha K, Kyin T, Beavis A, Teyton L, Bendelac A. A thymic precursor to the NK T cell lineage. Science. 2002;296:553–555. doi: 10.1126/science.1069017. [DOI] [PubMed] [Google Scholar]
- Benz C, Copley MR, Kent DG, Wohrer S, Cortes A, Aghaeepour N, Ma E, Mader H, Rowe K, Day C, et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell stem cell. 2012;10:273–283. doi: 10.1016/j.stem.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Bowie MB, Kent DG, Dykstra B, McKnight KD, McCaffrey L, Hoodless PA, Eaves CJ. Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5878–5882. doi: 10.1073/pnas.0700460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer SW, Beaudin AE, Forsberg EC. Mapping stem cell differentiation pathways from hematopoietic stem cells using Flk2/Flt3L lineage tracing. Cell Cycle. 2012;11:3180–3188. doi: 10.4161/cc.21279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer SW, Schroeder AV, Smith-Berdan S, Forsberg EC. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell stem cell. 2011;9:64–73. doi: 10.1016/j.stem.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt TD. Fetal regulatory T cells and peripheral immune tolerance in utero: implications for development and disease. Am J Reprod Immunol. 2013;69:346–358. doi: 10.1111/aji.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Hofer T, Rodewald HR. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015 doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- Buza-Vidas N, Woll P, Hultquist A, Duarte S, Lutteropp M, Bouriez-Jones T, Ferry H, Luc S, Jacobsen SE. FLT3 expression initiates in fully multipotent mouse hematopoietic progenitor cells. Blood. 2011;118:1544–1548. doi: 10.1182/blood-2010-10-316232. [DOI] [PubMed] [Google Scholar]
- Carey JB, Moffatt-Blue CS, Watson LC, Gavin AL, Feeney AJ. Repertoire-based selection into the marginal zone compartment during B cell development. The Journal of experimental medicine. 2008;205:2043–2052. doi: 10.1084/jem.20080559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell stem cell. 2010;6:265–278. doi: 10.1016/j.stem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JL, Weissman IL. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14541–14546. doi: 10.1073/pnas.261562798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruijn MF, Speck NA, Peeters MC, Dzierzak E. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. Embo J. 2000;19:2465–2474. doi: 10.1093/emboj/19.11.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzierzak E, Speck NA. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nature immunology. 2008;9:129–136. doi: 10.1038/ni1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg EC, Serwold T, Kogan S, Weissman IL, Passegue E. New evidence supporting megakaryocyte-erythrocyte potential of flk2/flt3+ multipotent hematopoietic progenitors. Cell. 2006;126:415–426. doi: 10.1016/j.cell.2006.06.037. [DOI] [PubMed] [Google Scholar]
- Gapin L, Matsuda JL, Surh CD, Kronenberg M. NKT cells derive from double-positive thymocytes that are positively selected by CD1d. Nature immunology. 2001;2:971–978. doi: 10.1038/ni710. [DOI] [PubMed] [Google Scholar]
- Ghosn EE, Waters J, Phillips M, Yamamoto R, Long BR, Yang Y, Gerstein R, Stoddart CA, Nakauchi H, Herzenberg LA. Fetal Hematopoietic Stem Cell Transplantation Fails to Fully Regenerate the B-Lymphocyte Compartment. Stem Cell Reports. 2016;6:137–149. doi: 10.1016/j.stemcr.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosn EE, Yamamoto R, Hamanaka S, Yang Y, Herzenberg LA, Nakauchi H, Herzenberg LA. Distinct B-cell lineage commitment distinguishes adult bone marrow hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5394–5398. doi: 10.1073/pnas.1121632109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothert JR, Gustin SE, Hall MA, Green AR, Gottgens B, Izon DJ, Begley CG. In vivo fate-tracing studies using the Scl stem cell enhancer: embryonic hematopoietic stem cells significantly contribute to adult hematopoiesis. Blood. 2005;105:2724–2732. doi: 10.1182/blood-2004-08-3037. [DOI] [PubMed] [Google Scholar]
- Hardy RR, Hayakawa K. A developmental switch in B lymphopoiesis. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(1):1550–11554. doi: 10.1073/pnas.88.24.11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havran WL, Allison JP. Developmentally ordered appearance of thymocytes expressing different T-cell antigen receptors. Nature. 1988;335:443–445. doi: 10.1038/335443a0. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Abdulmalik O, Peranteau WH, Ashizuka S, Campagnoli C, Chen Q, Horiuchi K, Asakura T, Flake AW. Mixed chimerism following in utero hematopoietic stem cell transplantation in murine models of hemoglobinopathy. Experimental hematology. 2003;31:176–184. doi: 10.1016/s0301-472x(02)01024-x. [DOI] [PubMed] [Google Scholar]
- Herzenberg LA, Herzenberg LA. Toward a layered immune system. Cell. 1989;59:953–954. doi: 10.1016/0092-8674(89)90748-4. [DOI] [PubMed] [Google Scholar]
- Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC, et al. C-myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. 2015;42:665–678. doi: 10.1016/j.immuni.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, Chien YH, Weissman IL. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell. 1990;62:863–874. doi: 10.1016/0092-8674(90)90262-d. [DOI] [PubMed] [Google Scholar]
- Kim I, He S, Yilmaz OH, Kiel MJ, Morrison SJ. Enhanced purification of fetal liver hematopoietic stem cells using SLAM family receptors. Blood. 2006;108:737–744. doi: 10.1182/blood-2005-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemischka IR, Raulet DH, Mulligan RC. Developmental potential and dynamic behavior of hematopoietic stem cells. Cell. 1986;45:917–927. doi: 10.1016/0092-8674(86)90566-0. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ebihara Y, Xu M, Ishii T, Sugiyama D, Yoshino H, Ueda T, Manabe A, Tanaka R, Ikeda Y, et al. CD34 expression on long-term repopulating hematopoietic stem cells changes during developmental stages. Blood. 2001;97:419–425. doi: 10.1182/blood.v97.2.419. [DOI] [PubMed] [Google Scholar]
- McGrath KE, Palis J. Hematopoiesis in the yolk sac: more than meets the eye. Experimental hematology. 2005;33:1021–1028. doi: 10.1016/j.exphem.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Medvinsky A, Dzierzak E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell. 1996;86:897–906. doi: 10.1016/s0092-8674(00)80165-8. [DOI] [PubMed] [Google Scholar]
- Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, Weinberg K, Stoddart CA, McCune JM. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. 2010;330:1695–1699. doi: 10.1126/science.1196509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino-Rodriguez E, Dorshkind K. B-1 B cell development in the fetus and adult. Immunity. 2012;36:13–21. doi: 10.1016/j.immuni.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y, Ema H, Nakauchi H. Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. The Journal of experimental medicine. 2010;207:1173–1182. doi: 10.1084/jem.20091318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Hemmati HD, Wandycz AM, Weissman IL. The purification and characterization of fetal liver hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:10302–10306. doi: 10.1073/pnas.92.22.10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijagal A, Le T, Wegorzewska M, Mackenzie TC. A mouse model of in utero transplantation. Journal of visualized experiments : JoVE. 2011 doi: 10.3791/2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, Robin C, Binder M, Dzierzak E, Speck NA. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity. 2002;16:661–672. doi: 10.1016/s1074-7613(02)00296-0. [DOI] [PubMed] [Google Scholar]
- Perez-Cunningham J, Boyer SW, Landon M, Forsberg EC. Hematopoietic stem cell-specific GFP-expressing transgenic mice generated by genetic excision of a pan-hematopoietic reporter gene. Experimental hematology. 2016;44:755–764. e751. doi: 10.1016/j.exphem.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samokhvalov IM, Samokhvalova NI, Nishikawa S. Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature. 2007;446:1056–1061. doi: 10.1038/nature05725. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Orkin SH. Advances in the understanding of haemoglobin switching. Br J Haematol. 2010;149:181–194. doi: 10.1111/j.1365-2141.2010.08105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Berdan S, Nguyen A, Hassanein D, Zimmer M, Ugarte F, Ciriza J, Li D, Garcia-Ojeda ME, Hinck L, Forsberg EC. Robo4 cooperates with CXCR4 to specify hematopoietic stem cell localization to bone marrow niches. Cell stem cell. 2011;8:72–83. doi: 10.1016/j.stem.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Berdan S, Nguyen A, Hong MA, Forsberg EC. ROBO4-mediated vascular integrity regulates the directionality of hematopoietic stem cell trafficking. Stem Cell Reports. 2015;4:255–268. doi: 10.1016/j.stemcr.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte F, Sousae R, Cinquin B, Martin EW, Krietsch J, Sanchez G, Inman M, Tsang H, Warr M, Passegue E, et al. Progressive Chromatin Condensation and H3K9 Methylation Regulate the Differentiation of Embryonic and Hematopoietic Stem Cells. Stem Cell Reports. 2015;5:728–740. doi: 10.1016/j.stemcr.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.