

Abstract
CREB is a transcription factor that regulates diverse cellular responses, including proliferation, survival, and differentiation. CREB is induced by a variety of growth factors and inflammatory signals and subsequently mediates the transcription of genes containing a cAMP-responsive element. Several immune-related genes possess this cAMP-responsive element, including IL-2, IL-6, IL-10, and TNF-α. In addition, phosphorylated CREB has been proposed to directly inhibit NF-κB activation by blocking the binding of CREB binding protein to the NF-κB complex, thereby limiting proinflammatory responses. CREB also induces an antiapoptotic survival signal in monocytes and macrophages. In T and B cells, CREB activation promotes proliferation and survival and differentially regulates Th1, Th2, and Th17 responses. Finally, CREB activation is required for the generation and maintenance of regulatory T cells. This review summarizes current advances involving CREB in immune function—a role that is continually being defined.
CREB is a transcription factor that is known for its role in cell proliferation, differentiation, and survival (1-3). However, emerging evidence has revealed specific functions of CREB in immune responses, including inhibiting NF-κB activation, inducing macrophage survival, and promoting the proliferation, survival, and regulation of T and B lymphocytes. In this brief review, we summarize our understanding to date of how this key transcription factor plays a role in the regulation of immune responses.
CREB
CREB is one of the best understood phosphorylation-dependent transcription factors (1-3). Several different serine-threonine kinases have been shown to promote phosphorylation of CREB at its transcription activating site, serine 133, including 1) a cAMP-dependent protein kinase A (PKA); 2) protein kinase C (PKC; including PKCε); 3) calmodulin kinases (CaMKs; e.g., CaMK-IV) that respond to calcium fluxes from the extra-cellular environment or from intracellular calcium stores; and 4) pp90 ribosomal S6 kinase (pp90 RSK; also known as RSK2) (1-6). Once serine 133 of CREB is phosphorylated, CREB interacts with its coactivator protein, CREB-binding protein (CBP), or p300 to initiate transcription of CREB-responsive genes (1-3). CBP is a cofactor for many other transcription factors and helps to stimulate transcription by modulating chromatin through histone acetylation and recruiting factors required for RNA polymerization (1-3). CREB has been shown to be involved in a variety of cellular processes, including cell proliferation, survival, differentiation, adaptive responses, glucose homeostasis, spermatogenesis, circadian rhythms, and synaptic plasticity associated with memory (1-3). However, emerging evidence over the past decade has demonstrated that CREB plays an important role in immune responses.
The CREB family of transcription factors and their structural components
The CREB family of transcription factors consists of cAMP-responsive activators in mammalian systems including CREB, cAMP response element modulator, and activating transcription factor 1 (1-3). The CREB family is composed of specific structural components characterized by a transactivation domain that consists of a kinase inducible domain (KID) and a constitutively active glutamine-rich domain (Q2) that synergize in response to cAMP stimulation (1-3). All CREB family members have a basic region leucine zipper dimerization domain located at the carboxy-terminal end, and they bind to DNA target sequences, such as the cAMP-responsive element (CRE), by dimerization through a leucine zipper (1-3). The target sequence CRE exists as both an eight-base-pair palindrome (5′-TGACGTCA-3′) and also as a less active half-site motif (5′-CGTCA-3′) (1-3). In addition, transducers of regulated CREB activity enhance CRE-dependent transcription by interacting with the transcription factor II D, which is a key general transcription factor that contains the TATA box binding protein (7). In 2004, a genome-wide analysis of rat PC12 cells (a cell line established from a rat pheochromocytoma that is a common cell type used for studies of CREB function) identified all genes that are targets of CREB, termed the CREB regulon (8). This analysis resulted in both the confirmation of known targets and the identification of additional targets of CREB that were grouped into the following categories: transcription factors, signaling molecules, neuron-associated molecules, metabolic factors, and factors involved in cell cycle and proliferation (8). These many and vastly different targets of CREB help to explain the numerous functional cellular processes that CREB has been shown to regulate. However, because this study was performed in a rat pheochromocytoma cell line, this list of CREB targets may not reflect the behavior of CREB in all cell types. Furthermore, CREB has been shown to induce transcription of immune-related genes that possess a CRE element, including IL-2, IL-6, IL-10, TNF-α, cyclooxygenase-2, and macrophage migration-inhibitory factor (2, 9, 10).
CREB and NF-κB signaling
The innate immune system uses various types of pattern recognition receptors (PRRs) to recognize components of bacteria, fungi, and viruses called pathogen-associated molecular patterns (11, 12). These PRRs include TLRs, nucleotide-binding oligomerization domain-like receptors, retinoic acid inducible gene I-like receptors, and C-type lectin receptors (11, 12). In particular, TLRs are PRRs that mediate cellular responses to lipopeptides (TLRs 1, 2, and 6), LPS (TLR4), flagellum (TLR5), and microbial RNA and DNA nucleotide sequences (TLRs 3, 7, 8, and 9) (11, 12). PRRs, including TLRs, initiate various signaling cascades that lead to the transcription of proinflammatory mediators that promote innate immune responses, including cytokines, chemokines, adhesion molecules, and antimicrobial peptides (11, 12). One critical pathway that is triggered by PRRs is the NF-κB pathway (13, 14). The NF-κB family of transcription factors includes RelA (p65), c-Rel, RelB, NF-κB1 (p50 and p105), and NF-κB2 (p52 and p100) (13, 14). RelA, c-Rel, and RelB contain transcriptionally active domains in their C-terminal halves and mediate the majority of NF-κB–mediated gene transcription, whereas p50 and p52 do not contain transactivation domains but can modulate NF-κB activity by forming heterodimers with RelA, c-Rel, and RelB (13, 14). Prototypical activators of NF-κB are TLRs, which initiate a signaling cascade beginning with TLR adapter molecules MyD88 and TRIF, resulting in NF-κB pathway activation and the triggering of other important signaling cascades through IFN regulatory factors and MAPKs, including ERK1/2, JNKs, and p38 isoforms (11, 12). In addition to PRRs, the NF-κB pathway can also be activated by proinflammatory cytokines, such as IL-1β and TNF-α (13, 14). Under resting conditions, the NF-κB transcription factors are sequestered in the cytoplasm associated with inhibitor molecule IκB, which prevents NF-κB activation (13, 14). When NF-κB is activated, a cascade of signaling events occur that ultimately lead to IκB degradation, which allows the release of NF-κB and facilitates the translocation of NF-κB to the nucleus, where it promotes the transcription of genes involved in proinflammatory immune responses (13, 14).
Optimal NF-κB activity at some target genes is mediated by direct interaction of the serine 276 of the RelA subunit of NF-κB with the CREB coactivators, CBP or p300 (13, 14). Furthermore, the activity of RelA is enhanced by acetylation of RelA, which can be induced by CBP/p300 (13, 14). However, the RelA component of NF-κB interacts with CBP/p300 at the same region as phosphorylated CREB, and it has been proposed that NF-κB activity is inhibited by activated CREB through competition for limiting amounts of CBP/p300 (15, 16). Thus, it could be that the balance between CREB and CBP/p300 determines whether the overall response leads to the respective inhibition (via CREB) or enhancement (via CBP/p300) of NF-κB activity and signaling (Fig. 1) (15, 16). However, the significance of this hypothetical competition in a physiologic setting is entirely unknown. Finally, a recent study demonstrated that the microRNA miR-34b inhibits CREB expression, providing a negative feedback mechanism to down-regulate CREB activity (17).
FIGURE 1.
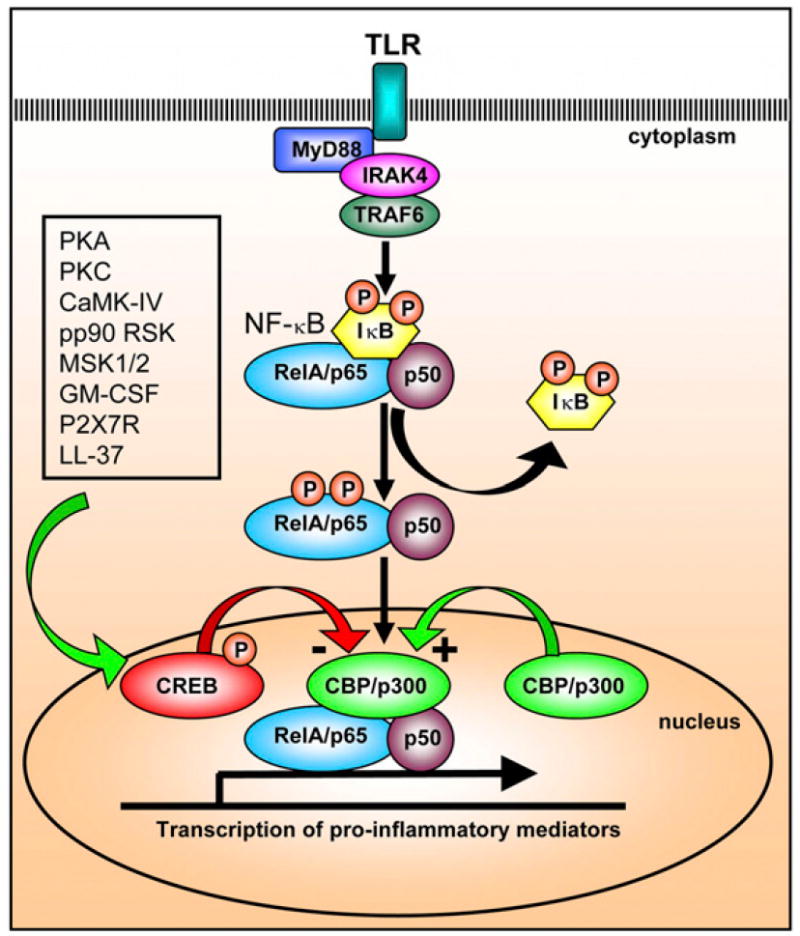
Proposed model of how CREB inhibits NF-κB activity. NF-κB activity is initiated by TLRs (and other proinflammatory signals) that trigger MyD88/IRAK-4/TRAF6 activation and subsequent phosphorylation and degradation of IκB, allowing the active RelA (p65)/p50 NF-κB complex to enter the nucleus. Optimal transcriptional activity of NF-κB for certain target genes requires interaction of the RelA subunit with CBP or p300. Phosphorylated CREB, which occurs via activation of PKA, PKC, and others as indicated, has been shown to bind to the same region as CBP/p300, and this has been proposed as a mechanism for CREB in the inhibition of NF-κB activity and signaling (15, 16).
CREB promotes a survival signal in macrophages
CREB also plays a specific role in the LPS/TLR4 pathway that mediates an NF-κB–dependent antiapoptotic response in macrophages, which promotes macrophage survival and enhancement of immune responses (18). The LPS/TLR4 anti-apoptotic response is mediated by NF-κB and p38 MAPK pathways that lead to the activation of mitogen- and stress-activated protein kinases, MSK1 and MSK2, resulting in CREB phosphorylation, which in turn induces two major antiapoptotic genes, plasminogen activator inhibitor-2 (PAI-2) and B cell lymphoma-2–related gene expressed in fetal liver-1/A1 (Bfl-1/A1) (18, 19). Thus, CREB promotes an antiapoptotic survival signal in macrophages, leading to enhanced host immune responses. This action of CREB is important because certain microbes, such as Salmonella spp., Shigellae spp., and Yersiniae spp., inhibit this survival signal and induce the apoptosis, or killing, of macrophages as a mechanism to evade host immune responses (19). Furthermore, the lethal toxin of Bacillus anthracis has been shown to directly inhibit the CREB-dependent macrophage antiapoptotic signal (18, 19). In addition to these findings, an adenylate cyclase gene in Mycobacterium tuberculosis (Rv0386) facilitated delivery of a bacteria-derived cAMP into the macrophage cytoplasm, which resulted in TNF-α production that depended on CREB phosphorylation (20). This CREB-mediated TNF-α production increased survival of M. tuberculosis within macrophages, representing an additional mechanism by which CREB promotes bacterial pathogenesis (20).
Potential factors that may inhibit NF-κB signaling through activation of CREB
Several different factors may inhibit NF-κB activity in monocytes/macrophages through the induction of CREB. For example, GM-CSF signaling results in CREB phosphorylation (21, 22). Phosphorylaton of CREB in this setting involves activation of pp90 RSK through an MEK-dependent signaling pathway (21). Because GM-CSF can induce activation of CREB, this may be another mechanism whereby CREB inhibits NF-κB activity, thus decreasing proinflammatory responses as suggested in the proposed model (Fig. 1).
P2X7R is a ligand-gated cation channel nucleotide receptor that is present on monocytes and macrophages and is activated by extracellular ATP after tissue injury or infection (23). In particular, P2X7R triggers activation of the nucleotide-binding oligomerization domain-like receptor, nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3 inflammasome, which is a cytoplasmic protein complex required to activate caspase-1 (23). Because caspase-1 cleaves pro–IL-1β and pro–IL-18 into their active forms, P2X7R is one of the key receptors that are important for promoting IL-1β and IL-18 responses (23). Upon ligand binding, P2X7R has been shown to result in the activation of CREB and other transcription factors, including MAPKs p38 and ERK1/2, and it is also involved in the production of reactive oxygen species and IL-1 isoforms as well as the creation of nonspecific cell membrane pores (24, 25). The phosphorylation and activation of CREB by P2X7R includes a CREB/CBP complex formation and is partly mediated via an extracellular calcium influx and the MEK/ERK system (24). Although the significance of the activation of CREB by the P2X7R in immune responses is not entirely clear, one study found that P2X7R stimulation decreased LPS-stimulated inducible NO synthase and cyclooxygenase-2 expression and reduced NO release in microglia in a mechanism involving activation of CREB (26). Thus, activation of CREB by P2X7R could promote CREB inhibition of NF-κB activity as suggested in the proposed model (Fig. 1).
The antimicrobial peptide cathelidicin (also known as LL-37) has been demonstrated to have broad-spectrum microbicidal activity while also having immunomodulatory activity, which is dependent on activation of MAPKs (ERK1/2 and p38), Elk-1, and NF-κB (27, 28). When PBMCs were stimulated with LL-37 and IL-1β, there was an increase in CREB phosphorylation, suggesting that LL-37 also regulates CREB activity, potentially inducing the proposed CREB-mediated inhibition of NF-κB activity (Fig. 1) (28).
CREB induces IL-10 production
IL-10 is a potent anti-inflammatory cytokine that plays a key role in mediating a feedback inhibition loop that limits inflammation and prevents unwanted tissue damage (29). In macrophages, IL-10 is produced in response to activation of TLRs 2, 3, 4, 7, and 9 (30, 31). TLR signaling via MyD88 or TRIF results in activation of NF-κB and MAPK pathways (ERK1/2 and p38), which subsequently induces production of proinflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-10 (29). MAPKs activate MSK1 and MSK2 that phosphorylate the transcription factors CREB and AP-1 (e.g., c-Fos, c-Jun, JunB), which subsequently bind to the IL-10 promoter to induce transcription (Fig. 2) (32). This pathway is also required for the transcription of the dual-specificity protein phosphatase 1, which provides a negative feedback signal by inhibiting p38 (32). Thus, CREB plays an essential role in the production of IL-10, which in turn inhibits TLR-induced inflammation and prevents tissue damage (29). It should be noted that IL-10 can also regulate the pathway for TLR-induced NF-κB activation through the production of glycogen synthase kinase-3β (GSK-3β), which increases the binding of CREB and decreases the binding of CBP/p300 to the RelA subunit of NF-κB (Fig. 2) (33). This mechanism utilizing GSK-3β is unique from other cytokines and is regulated by the constitutively active transcription factors Sp1 and Sp3 (34-36). Finally, recognition of the yeast particle zymosan by dectin-1 induces production of IL-10 by initiating CaMK-II- and Pyk2-transduced calcium signals that activate the ERK1/2 pathway and CREB in a pathway involving reactive oxygen species (37). Thus, in addition to TLRs, dectin-1 provides another mechanism that utilizes CREB to produce IL-10 (37).
FIGURE 2.
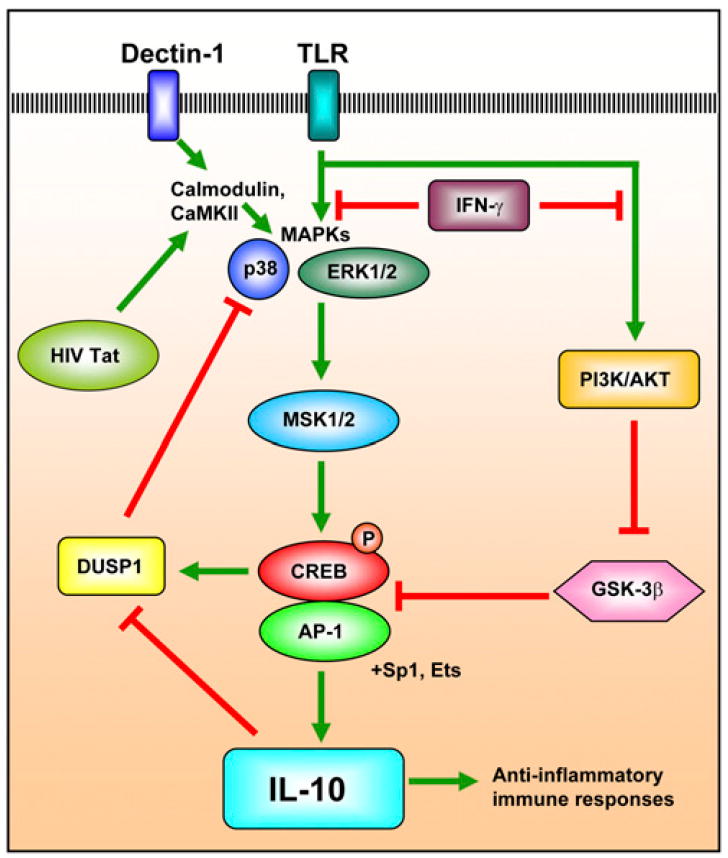
CREB-induced IL-10 production is regulated by IFN-γ. TLR signaling results in the activation of NF-κB and MAPKs (ERK1/2 and p38), which induces production of proinflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) and the anti-inflammatory cytokine IL-10 (29). MAPKs activate MSK1 and MSK2 to directly phosphorylate CREB and AP-1, which bind to the IL-10 promoter and initiate transcription (32). This same pathway also induces dual-specificity protein phosphatase 1, which feeds back to inhibit p38 (32). Dectin-1, which recognizes zymosan, and the HIV-1 Tat protein also induce IL-10 production by activating CREB, Sp1, and Ets-1 through calmodulin, CaMK-II, and MAPKs (37-40). Finally, IFN-γ inhibits IL-10 production by 1) interfering with the PI3K-AKT pathway, thereby releasing GSK-3β, which subsequently downregulates the activation and transcriptional activity of CREB and AP-1 proteins that induce IL-10 production; and 2) directly inhibiting MAPKs (45, 55).
HIV-1 Tat induces IL-10 via CREB
Interestingly, the HIV-1 transactivator protein Tat, which is required for viral replication and progression of HIV-1 disease, has been shown to induce IL-10 transcription in monocytes and macrophages by activating CREB, Sp1, and Ets-1 through calmodulin, CaMK-II, and MAPKs (ERK1/2 and p38) (38-40). Thus, during HIV-1 disease, Tat utilizes CREB to promote IL-10 production. Although the significance of this regarding HIV pathogenesis is not entirely clear, IL-10 can inhibit HIV-1 replication in monocytes and macrophages (41), suggesting that Tat/CREB-induced IL-10 production provides a negative feedback signal to prevent excess HIV-1 replication. In addition, because IL-10 can also prevent apoptosis of monocytes and macrophages (42), Tat/CREB-induced IL-10 may allow infected monocytes and macrophages to serve as cellular viral reservoirs.
IFN-γ regulates IL-10 production through multiple mechanisms involving CREB
In contrast to the anti-inflammatory effects of IL-10, IFN-γ is a potent macrophage stimulating factor that promotes various immune functions, including Ag presentation, cytokine production, and antimicrobial activity (43). In addition, IFN-γ inhibits IL-10 production and consequently blocks the immune inhibitory actions of IL-10, resulting in an enhancement of macrophage activation and immune responses (43). In particular, IFN-γ can downregulate the TLR-induced IL-10 inhibitory response (43). In addition, IFN-γ can suppress IL-10 production by two distinct mechanisms: 1) IFN-γ can interfere with the phosphoinositide 3-kinase–AKT pathway, releasing GSK-3β, which inhibits the activation and transcriptional activity of CREB and AP-1 proteins (including c-Fos, c Jun, and JunB) that promote IL-10 production; and 2) IFN-γ directly inhibits MAPKs (ERK1/2, JNK, and p38), which results in diminished CREB phosphorylation and AP-1 transcriptional activity (Fig. 2) (44, 45). Therefore, IFN-γ can inhibit IL-10 production through a number of different mechanisms that involve CREB (29).
CREB and T cells, including Th1, Th2, and Th17 cells
CRE elements have been identified in the promoters and enhancers of many T cell-specific genes, including TCRα, TCR Vβ, CD3δ, CD8α, IL-2, CD25/IL-2Rα, and IL-2Rγ, suggesting that CREB plays a role in T cell function (10, 46-51). To analyze the specific function of CREB in T cells, Barton et al. (52) engineered a transgenic mouse strain expressing a dominant negative form of CREB under the control of the T cell-specific CD2 promoter. This dominant negative CREB mutation (serine 133 to alanine 133) retained DNA-binding activity but was rendered transcriptionally inactive (52). These CREB mutant mice had normal T cell development in the thymus (52). However, activated T cells from this mouse strain had a marked defect in proliferation and IL-2 production, resulting in G1 cell-cycle arrest and apoptotic cell death (52). Another group generated a transgenic mouse strain that had the same CREB dominant-negative mutation, but under the control of the Lck promoter (53). In this case, T cells from this mouse strain did not have any defects in proliferation or IL-2 production, but instead had impaired Th cell function (53). Specifically, the CD4+ T cells were defective in their ability to produce Th-1 (IFN-γ) and Th-2 (IL-4) effector cytokines, and the mice failed to produce an Ag-specific IgM and IgG humoral immune response (53). The differences in the findings between these two studies is likely explained by the differential expression of the mutant CREB by the CD2 versus Lck promoters, but taken together these studies demonstrate that CREB plays a role in T cell proliferation and function. CREB activation also plays an important role in governing the cytokines produced by Th1 (IL-2 and IFN-γ) and Th2 cells (IL-4 and IL-13) through the regulation of IFN-γ production. One study found that the IFN-γ promoter is hypomethylated in Th1 cells, allowing CREB to induce IFN-γ production and promote Th1 responses, whereas the IFN-γ promoter is hypermethylated in Th2 cells, resulting in chromatin condensation and prevention of CREB-mediated production of IFN-γ (54). Finally, IL-17, which is produced by Th-17 cells, downregulates CREB phosphorylation and ensuing IFN-γ production in T cells obtained from patients with tuberculosis, suggesting that IL-17 can inhibit CREB-mediated IFN-γ production (55).
CREB phosphorylation in T cells has been shown to involve several signaling molecules and pathways, including PKA, PKC (including PCKα and PKCθ), Ras, ERK1/2 MAPKs, and pp90 RSK (10, 56, 57). In addition, the costimulatory molecule CD28 was shown to optimally activate CREB through p38 and CaMK-IV (58, 59). Although these different studies suggest that several signaling pathways lead to CREB phosphorylation in T cells, a large microarray analysis of con A/anti-CD28 stimulated T cells revealed a greater than 100-fold increase in phosphorylated CREB, which was reduced 50% when the cells were treated with a PKC inhibitor and completely blocked with a PKA inhibitor, suggesting that PKC and PKA are the major pathways that promote CREB phosphorylation (10). More recently, Kaiser et al. (60) demonstrated that MSK1/2 directly phosphorylated CREB in T cells and led to T cell proliferation and production of IL-2 in a pathway that was downstream of both ERK1/2 and p38. Collectively, CREB phosphorylation in T cells is mediated by triggering PKA and PKC, resulting in activation of MAPKs ERK1/2 and p38 and subsequent MSK1/2-mediated CREB phosphorylation that promotes T cell activation and proliferation.
CREB and FoxP3 regulatory T cells
Recently, there has been an intense interest on the role that CD4+CD25+ regulatory T cells (Tregs) play in promoting peripheral tolerance and downregulating pathogenic T cell responses, including autoimmune responses and allograft rejection (61). The function of Tregs is dependent on TGF-β–dependent expression of the transcription factor FoxP3, which is required for the development and function of Tregs (61). Emerging evidence has suggested a role for CREB, which is activated by TCR activation, in TGF-β/FoxP3–dependent Treg generation and maintenance (Fig. 3). Several studies have defined a Treg-specific demethylated region (TSDR) in the FoxP3 locus, which contains a CREB-activating transcription factor site overlapping a CpG island (61-64). The CpG island is found demethylated in Tregs and methylated in conventional T cells (61-64). TGF-β, as well as treatment with azacytidine, can induce demethylation of this locus, allowing CREB to stabilize FoxP3 expression, thus promoting and maintaining the Treg phenotype (61-64). In addition, a recent study demonstrated that transcription of FoxP3 and development of Tregs depends on the formation of the “c-Rel enhanceosome” at the FoxP3 promoter, which, in addition to CREB, contains c-Rel, p65, NFAT, and Smad3 (65). Together, the CREB-induced generation and maintenance of Tregs represents an additional example of how CREB inhibits immune responses.
FIGURE 3.
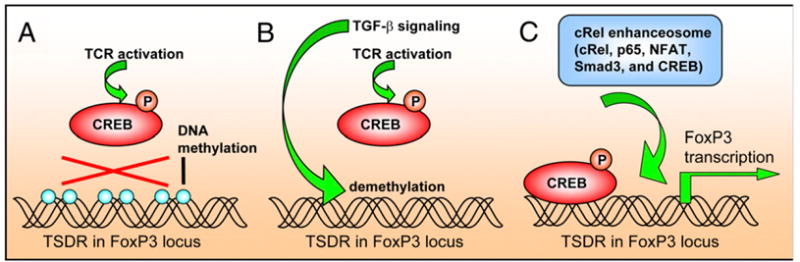
CREB promotes TGFβ-mediated generation and maintenance of FoxP3 Tregs. The TSDR in the FoxP3 locus is found methylated in conventional T cells. A, The methylation prevents phosphorylated CREB, which is induced by TCR activation, from binding to this region. B, TGF-β induces demethylation of the TSDR. C, Demethylation of the TSDR allows phosphylated CREB to bind to the FoxP3 locus to promote FoxP3 expression and the development and stabilization of Tregs (62-64). Transcription of FoxP3 also involves the formation of a “c-Rel enhanceosome”, which contains c-Rel, p65, NFAT, and Smad3 in addition to CREB (65).
CREB and B cells
Previous studies in mature and immature B cells have demonstrated that BCR stimulation, which promotes B cell activation and proliferation, involved ERK1/2- and Elk-1–induced CREB phosphorylation (66). Furthermore, BCR induction of CREB phosphorylation was dependent on PKCδ and pp90 RSK signaling pathways (67). Thus, B cell activation and proliferation through the BCR involve signaling pathways that include PKCδ and pp90 RSK, resulting in ERK1/2- and Elk-1–dependent activation of CREB (66, 67).
Conclusions
CREB plays many different roles in immune function (Table I). CREB often promotes anti-inflammatory immune responses, such as through the inhibition of NF-κB activity, the induction of IL-10, and the generation of Tregs. These anti-inflammatory responses could be protective by inhibiting unwanted inflammation, tissue damage, and autoimmune responses, or they could be pathogenic in the context of infection and tumor immunosurveillance. However, CREB also promotes activation and proliferation of T and B cells and differentially regulates Th1, Th2, and Th17 responses. Future investigation into the role of CREB in immune function will lead to an increased understanding of how this transcription factor regulates specific immune responses. One challenge is to apply innovative technology to uncover the roles of CREB in immune responses, such as chromatin immunoprecipitation coupled to massively parallel sequencing to monitor histone modifications and nucleosome dynamics to characterize the events required for CREB binding and activation of immune-related genes (68, 69). Although CREB appears to be involved in an amazingly diverse range of processes in immune cells and other cell types, further research could reveal signaling pathways or collaborating proteins that are selectively involved in its proinflammatory or its anti-inflammatory functions. Therefore, a better understanding of the activation mechanisms and mechanisms of action of CREB in different cell types and settings could suggest therapeutic strategies for selective manipulation of immune responses.
Table I.
Summary of the many different roles of CREB in immune function
| Role of CREB | Cell Type | Associated Factors | Overall Role in Immune Function | References |
|---|---|---|---|---|
| Inhibition of NF-κB signaling | Monocytes and endothelial cells | CREB interacts with the RelA (p65) component of NF-κB at the same region as CBP/p300 | Anti-inflammatory (may be protective or pathogenic depending on the context) | 15, 16 |
| Antiapoptotic survival signal | Macrophages | TLR4 triggers NF-κB/MAPKs and MSK1/2, which activate PAI-2 and Bfl-1/A1 to phosphorylate CREB | Pathogenic (mechanism for pathogens to evade host immune responses) | 18, 19 |
| TNF-α–mediated survival signal | Macrophages | M. tuberculosis-derived adenylate cylcase (Rv0386) phosphorylates CREB | Pathogenic (promotes survival of M. tuberculosis in macrophages) | 20 |
| Induction of IL-10 | Macrophages and myeloid dendritic cells | MAPKs (ERK1/2 and p38) trigger MSK1/2 to activate CREB | Anti-inflammatory (may be protective or pathogenic depending on the context) | 29-32 |
| Induction of IL-10 | Macrophages | TLR2 activation by Candida albicans triggers IL-10 production | Pathogenic (promotes survival of Tregs, leading to disseminated candidiasis) | 31 |
| Induction of IL-10 by HIV-1 Tat | Macrophages | Sp1, Ets-1, calmodulin, CaMK-II, and MAPKs | Unknown, may be pathogenic (macrophages could act as cellular viral reservoirs) | 38-40 |
| Promotes T cell activation and proliferation and Th cell activity | T cells | PKA, PKC, Ras, ERK1/2, pp90 RSK and CD28 (via p38 and CaMK-IV) | May be protective or pathogenic (depending on the Ag) | 52, 53, 56-59 |
| Promotes IFN-γ production | T cells | IFN-γ promoter is hypomethylated in Th1 cells, allowing CREB binding | Protective (promotes Th1 responses that are protective in tuberculosis) | 54, 55 |
| Induction of FoxP3, promoting Treg generation and maintenance | T cells | TSDR in the FoxP3 locus, which allows CREB to promote FoxP3 expression | Anti-inflammatory (may be pathogenic relating to infections and tumor immunosurveillance and protective relating to autoimmunity) | 61-65 |
| Promotes B cell activation and proliferation | B cells | ERK1/2, E1K-1, PKCδ, pp90 RSK | May be protective or pathogenic (depending on the Ag) | 66, 67 |
Acknowledgments
This work was supported by National Institutes of Health Grants T32 HD07512 (to A.Y.W.); R01 HL83077, HL78526, and HL097561 (to K.M.S.); and R01 AI078910 and R03 AR054534 (to L.S.M.). K.M.S. is also a Scholar of the Leukemia and Lymphoma Society of America. K.M.S. has received research support from Abbott Laboratories, Inc.
Abbreviations used in this paper
- Bfl-1/A1
B cell lymphoma-2–related gene expressed in fetal liver-1/A1
- CaMK
calmodulin kinase
- CBP
CREB-binding protein
- CRE
cAMP-responsive element
- GSK-3β
glycogen synthase kinase-3β
- PAI-2
plasminogen activator inhibitor-2
- PKA
protein kinase A
- PKC
protein kinase C
- pp90 RSK
pp90 ribosomal S6 kinase
- PRR
pattern recognition receptor
- Treg
regulatory T cell
- TSDR
Treg-specific demethylated region
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 2.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 3.Sakamoto KM, Frank DA. CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin Cancer Res. 2009;15:2583–2587. doi: 10.1158/1078-0432.CCR-08-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brindle P, Linke S, Montminy M. Protein-kinase-A-dependent activator in transcription factor CREB reveals new role for CREM repressors. Nature. 1993;364:821–824. doi: 10.1038/364821a0. [DOI] [PubMed] [Google Scholar]
- 5.Enslen H, Tokumitsu H, Soderling TR. Phosphorylation of CREB by CaM-kinase IV activated by CaM-kinase IV kinase. Biochem Biophys Res Commun. 1995;207:1038–1043. doi: 10.1006/bbrc.1995.1289. [DOI] [PubMed] [Google Scholar]
- 6.Gubina E, Luo X, Kwon E, Sakamoto K, Shi YF, Mufson RA. betac cytokine receptor-induced stimulation of cAMP response element binding protein phosphorylation requires protein kinase C in myeloid cells: a novel cytokine signal transduction cascade. J Immunol. 2001;167:4303–4310. doi: 10.4049/jimmunol.167.8.4303. [DOI] [PubMed] [Google Scholar]
- 7.Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M. TORCs: transducers of regulated CREB activity. Mol Cell. 2003;12:413–423. doi: 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 8.Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 9.Brenner S, Prösch S, Schenke-Layland K, Riese U, Gausmann U, Platzer C. cAMP-induced Interleukin-10 promoter activation depends on CCAAT/enhancer-binding protein expression and monocytic differentiation. J Biol Chem. 2003;278:5597–5604. doi: 10.1074/jbc.M207448200. [DOI] [PubMed] [Google Scholar]
- 10.Hughes-Fulford M, Sugano E, Schopper T, Li CF, Boonyaratanakornkit JB, Cogoli A. Early immune response and regulation of IL-2 receptor subunits. Cell Signal. 2005;17:1111–1124. doi: 10.1016/j.cellsig.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 12.Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771–776. doi: 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- 14.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 15.Ollivier V, Parry GC, Cobb RR, de Prost D, Mackman N. Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J Biol Chem. 1996;271:20828–20835. doi: 10.1074/jbc.271.34.20828. [DOI] [PubMed] [Google Scholar]
- 16.Parry GC, Mackman N. Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol. 1997;159:5450–5456. [PubMed] [Google Scholar]
- 17.Pigazzi M, Manara E, Baron E, Basso G. miR-34b targets cyclic AMP-responsive element binding protein in acute myeloid leukemia. Cancer Res. 2009;69:2471–2478. doi: 10.1158/0008-5472.CAN-08-3404. [DOI] [PubMed] [Google Scholar]
- 18.Park JM, Greten FR, Wong A, Westrick RJ, Arthur JS, Otsu K, Hoffmann A, Montminy M, Karin M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis—CREB and NF-kappaB as key regulators. Immunity. 2005;23:319–329. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Hsu LC, Park JM, Zhang K, Luo JL, Maeda S, Kaufman RJ, Eckmann L, Guiney DG, Karin M. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 2004;428:341–345. doi: 10.1038/nature02405. [DOI] [PubMed] [Google Scholar]
- 20.Agarwal N, Lamichhane G, Gupta R, Nolan S, Bishai WR. Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase. Nature. 2009;460:98–102. doi: 10.1038/nature08123. [DOI] [PubMed] [Google Scholar]
- 21.Cheng JC, Kinjo K, Judelson DR, Chang J, Wu WS, Schmid I, Shankar DB, Kasahara N, Stripecke R, Bhatia R, et al. CREB is a critical regulator of normal hematopoiesis and leukemogenesis. Blood. 2008;111:1182–1192. doi: 10.1182/blood-2007-04-083600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwon EM, Raines MA, Blenis J, Sakamoto KM. Granulocyte-macrophage colony-stimulating factor stimulation results in phosphorylation of cAMP response element-binding protein through activation of pp90RSK. Blood. 2000;95:2552–2558. [PubMed] [Google Scholar]
- 23.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 24.Gavala ML, Pfeiffer ZA, Bertics PJ. The nucleotide receptor P2RX7 mediates ATP-induced CREB activation in human and murine monocytic cells. J Leukoc Biol. 2008;84:1159–1171. doi: 10.1189/jlb.0907612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potucek YD, Crain JM, Watters JJ. Purinergic receptors modulate MAP kinases and transcription factors that control microglial inflammatory gene expression. Neurochem Int. 2006;49:204–214. doi: 10.1016/j.neuint.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 26.Brautigam VM, Frasier C, Nikodemova M, Watters JJ. Purinergic receptor modulation of BV-2 microglial cell activity: potential involvement of p38 MAP kinase and CREB. J Neuroimmunol. 2005;166:113–125. doi: 10.1016/j.jneuroim.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 27.Bowdish DM, Davidson DJ, Speert DP, Hancock RE. The human cationic peptide LL-37 induces activation of the extracellular signal-regulated kinase and p38 kinase pathways in primary human monocytes. J Immunol. 2004;172:3758–3765. doi: 10.4049/jimmunol.172.6.3758. [DOI] [PubMed] [Google Scholar]
- 28.Yu J, Mookherjee N, Wee K, Bowdish DM, Pistolic J, Li Y, Rehaume L, Hancock RE. Host defense peptide LL-37, in synergy with inflammatory mediator IL-1beta, augments immune responses by multiple pathways. J Immunol. 2007;179:7684–7691. doi: 10.4049/jimmunol.179.11.7684. [DOI] [PubMed] [Google Scholar]
- 29.Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 30.Boonstra A, Rajsbaum R, Holman M, Marques R, Asselin-Paturel C, Pereira JP, Bates EE, Akira S, Vieira P, Liu YJ, et al. Macrophages and myeloid dendritic cells, but not plasmacytoid dendritic cells, produce IL-10 in response to MyD88- and TRIF-dependent TLR signals, and TLR-independent signals. J Immunol. 2006;177:7551–7558. doi: 10.4049/jimmunol.177.11.7551. [DOI] [PubMed] [Google Scholar]
- 31.Netea MG, Sutmuller R, Hermann C, Van der Graaf CA, Van der Meer JW, van Krieken JH, Hartung T, Adema G, Kullberg BJ. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol. 2004;172:3712–3718. doi: 10.4049/jimmunol.172.6.3712. [DOI] [PubMed] [Google Scholar]
- 32.Ananieva O, Darragh J, Johansen C, Carr JM, McIlrath J, Park JM, Wingate A, Monk CE, Toth R, Santos SG, et al. The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat Immunol. 2008;9:1028–1036. doi: 10.1038/ni.1644. [DOI] [PubMed] [Google Scholar]
- 33.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brightbill HD, Plevy SE, Modlin RL, Smale ST. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J Immunol. 2000;164:1940–1951. doi: 10.4049/jimmunol.164.4.1940. [DOI] [PubMed] [Google Scholar]
- 35.Chan MM, Cheung BK, Li JC, Chan LL, Lau AS. A role for glycogen synthase kinase-3 in antagonizing mycobacterial immune evasion by negatively regulating IL-10 induction. J Leukoc Biol. 2009;86:283–291. doi: 10.1189/jlb.0708442. [DOI] [PubMed] [Google Scholar]
- 36.Tone M, Powell MJ, Tone Y, Thompson SA, Waldmann H. IL-10 gene expression is controlled by the transcription factors Sp1 and Sp3. J Immunol. 2000;165:286–291. doi: 10.4049/jimmunol.165.1.286. [DOI] [PubMed] [Google Scholar]
- 37.Kelly EK, Wang L, Ivashkiv LB. Calcium-activated pathways and oxidative burst mediate zymosan-induced signaling and IL-10 production in human macrophages. J Immunol. 2010;184:5545–5552. doi: 10.4049/jimmunol.0901293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gee K, Angel JB, Mishra S, Blahoianu MA, Kumar A. IL-10 regulation by HIV-Tat in primary human monocytic cells: involvement of calmodulin/calmodulin-dependent protein kinase-activated p38 MAPK and Sp-1 and CREB-1 transcription factors. J Immunol. 2007;178:798–807. doi: 10.4049/jimmunol.178.2.798. [DOI] [PubMed] [Google Scholar]
- 39.Gee K, Angel JB, Ma W, Mishra S, Gajanayaka N, Parato K, Kumar A. Intracellular HIV-Tat expression induces IL-10 synthesis by the CREB-1 transcription factor through Ser133 phosphorylation and its regulation by the ERK1/2 MAPK in human monocytic cells. J Biol Chem. 2006;281:31647–31658. doi: 10.1074/jbc.M512109200. [DOI] [PubMed] [Google Scholar]
- 40.Li JC, Lau AS. A role for mitogen-activated protein kinase and Ets-1 in the induction of interleukin-10 transcription by human immunodeficiency virus-1 Tat. Immunology. 2007;121:337–348. doi: 10.1111/j.1365-2567.2007.02580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akridge RE, Oyafuso LK, Reed SG. IL-10 is induced during HIV-1 infection and is capable of decreasing viral replication in human macrophages. J Immunol. 1994;153:5782–5789. [PubMed] [Google Scholar]
- 42.Zhou JH, Broussard SR, Strle K, Freund GG, Johnson RW, Dantzer R, Kelley KW. IL-10 inhibits apoptosis of promyeloid cells by activating insulin receptor substrate-2 and phosphatidylinositol 3′-kinase. J Immunol. 2001;167:4436–4442. doi: 10.4049/jimmunol.167.8.4436. [DOI] [PubMed] [Google Scholar]
- 43.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 44.Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 45.Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem. 2001;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solomou EE, Juang YT, Tsokos GC. Protein kinase C-theta participates in the activation of cyclic AMP-responsive element-binding protein and its subsequent binding to the -180 site of the IL-2 promoter in normal human T lymphocytes. J Immunol. 2001;166:5665–5674. doi: 10.4049/jimmunol.166.9.5665. [DOI] [PubMed] [Google Scholar]
- 47.Yeh JH, Lecine P, Nunes JA, Spicuglia S, Ferrier P, Olive D, Imbert J. Novel CD28-responsive enhancer activated by CREB/ATF and AP-1 families in the human interleukin-2 receptor alpha-chain locus. Mol Cell Biol. 2001;21:4515–4527. doi: 10.1128/MCB.21.14.4515-4527.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mayall TP, Sheridan PL, Montminy MR, Jones KA. Distinct roles for P-CREB and LEF-1 in TCR alpha enhancer assembly and activation on chromatin templates in vitro. Genes Dev. 1997;11:887–899. doi: 10.1101/gad.11.7.887. [DOI] [PubMed] [Google Scholar]
- 49.Anderson SJ, Miyake S, Loh DY. Transcription from a murine T-cell receptor V beta promoter depends on a conserved decamer motif similar to the cyclic AMP response element. Mol Cell Biol. 1989;9:4835–4845. doi: 10.1128/mcb.9.11.4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao MH, Kavathas PB. Functional importance of the cyclic AMP response element-like decamer motif in the CD8 alpha promoter. J Immunol. 1993;150:4376–4385. [PubMed] [Google Scholar]
- 51.Gupta A, Terhorst C. CD3 delta enhancer. CREB interferes with the function of a murine CD3-delta A binding factor (M delta AF) J Immunol. 1994;152:3895–3903. [PubMed] [Google Scholar]
- 52.Barton K, Muthusamy N, Chanyangam M, Fischer C, Clendenin C, Leiden JM. Defective thymocyte proliferation and IL-2 production in transgenic mice expressing a dominant-negative form of CREB. Nature. 1996;379:81–85. doi: 10.1038/379081a0. [DOI] [PubMed] [Google Scholar]
- 53.Zhang F, Rincon M, Flavell RA, Aune TM. Defective Th function induced by a dominant-negative cAMP response element binding protein mutation is reversed by Bcl-2. J Immunol. 2000;165:1762–1770. doi: 10.4049/jimmunol.165.4.1762. [DOI] [PubMed] [Google Scholar]
- 54.Yano S, Ghosh P, Kusaba H, Buchholz M, Longo DL. Effect of promoter methylation on the regulation of IFN-gamma gene during in vitro differentiation of human peripheral blood T cells into a Th2 population. J Immunol. 2003;171:2510–2516. doi: 10.4049/jimmunol.171.5.2510. [DOI] [PubMed] [Google Scholar]
- 55.Pasquinelli V, Townsend JC, Jurado JO, Alvarez IB, Quiroga MF, Barnes PF, Samten B, García VE. IFN-gamma production during active tuberculosis is regulated by mechanisms that involve IL-17, SLAM, and CREB. J Infect Dis. 2009;199:661–665. doi: 10.1086/596742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grady GC, Mason SM, Stephen J, Zúñiga-Pflücker JC, Michie AM. Cyclic adenosine 5′-monophosphate response element binding protein plays a central role in mediating proliferation and differentiation downstream of the pre-TCR complex in developing thymocytes. J Immunol. 2004;173:1802–1810. doi: 10.4049/jimmunol.173.3.1802. [DOI] [PubMed] [Google Scholar]
- 57.Muthusamy N, Leiden JM. A protein kinase C-, Ras-, and RSK2-dependent signal transduction pathway activates the cAMP-responsive element-binding protein transcription factor following T cell receptor engagement. J Biol Chem. 1998;273:22841–22847. doi: 10.1074/jbc.273.35.22841. [DOI] [PubMed] [Google Scholar]
- 58.Yu CT, Shih HM, Lai MZ. Multiple signals required for cyclic AMP-responsive element binding protein (CREB) binding protein interaction induced by CD3/CD28 costimulation. J Immunol. 2001;166:284–292. doi: 10.4049/jimmunol.166.1.284. [DOI] [PubMed] [Google Scholar]
- 59.Hsueh YP, Liang HE, Ng SY, Lai MZ. CD28-costimulation activates cyclic AMP-responsive element-binding protein in T lymphocytes. J Immunol. 1997;158:85–93. [PubMed] [Google Scholar]
- 60.Kaiser M, Wiggin GR, Lightfoot K, Arthur JS, Macdonald A. MSK regulate TCR-induced CREB phosphorylation but not immediate early gene transcription. Eur J Immunol. 2007;37:2583–2595. doi: 10.1002/eji.200636606. [DOI] [PubMed] [Google Scholar]
- 61.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 62.Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–1663. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 63.Polansky JK, Schreiber L, Thelemann C, Ludwig L, Kruüger M, Baumgrass R, Cording S, Floess S, Hamann A, Huehn J. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J Mol Med. 2010;88:1029–1040. doi: 10.1007/s00109-010-0642-1. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. 2007;204:1543–1551. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yasuda T, Sanjo H, Pagès G, Kawano Y, Karasuyama H, Pouysségur J, Ogata M, Kurosaki T. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity. 2008;28:499–508. doi: 10.1016/j.immuni.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 67.Blois JT, Mataraza JM, Mecklenbraüker I, Tarakhovsky A, Chiles TC. B cell receptor-induced cAMP-response element-binding protein activation in B lymphocytes requires novel protein kinase Cdelta. J Biol Chem. 2004;279:30123–30132. doi: 10.1074/jbc.M402793200. [DOI] [PubMed] [Google Scholar]
- 68.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 69.Smale ST. Seq-ing LPS-induced enhancers. Immunity. 2010;32:296–298. doi: 10.1016/j.immuni.2010.03.011. [DOI] [PubMed] [Google Scholar]