

Abstract
Brain cells normally respond adaptively to bioenergetic challenges resulting from ongoing activity in neuronal circuits, and from environmental energetic stressors such as food deprivation and physical exertion. At the cellular level, such adaptive responses include the “strengthening” of existing synapses, the formation of new synapses, and the production of new neurons from stem cells. At the molecular level, bioenergetic challenges result in the activation of transcription factors that induce the expression of proteins that bolster the resistance of neurons to the kinds of metabolic, oxidative, excitotoxic, and proteotoxic stresses involved in the pathogenesis of brain disorders including stroke, and Alzheimer's and Parkinson's diseases. Emerging findings suggest that lifestyles that include intermittent bioenergetic challenges, most notably exercise and dietary energy restriction, can increase the likelihood that the brain will function optimally and in the absence of disease throughout life. Here, we provide an overview of cellular and molecular mechanisms that regulate brain energy metabolism, how such mechanisms are altered during aging and in neurodegenerative disorders, and the potential applications to brain health and disease of interventions that engage pathways involved in neuronal adaptations to metabolic stress.
Keywords: aging, brain energetics, ketone bodies, metabolism
Subject Categories: Metabolism, Neuroscience
Introduction
The higher cognitive functions of the human brain depend upon the expansion and increased density and complexity of the neocortex during evolution (Rakic, 2009). The enhanced abilities of the human brain to plan complex behaviors, make decisions, and process emotional and social contexts came with hefty energy requirements. Although it is only 2% of the total body weight, the brain accounts for 20% of an individual's energy expenditure at rest (Kety, 1957; Sokoloff, 1960). Among brain cells, neurons expend 70–80% of the total energy, with the remaining portion being utilized by glial cells (astrocytes, oligodendrocytes, and microglia) (Harris et al, 2012; Hyder et al, 2013). Organisms allocate their available energy among the competing needs of maintenance, growth, reproduction, and, particularly in primates, higher cortical functions (communication, imagination, and creativity). A growing body of evidence suggests that metabolic adaptations within the brain and whole body played important roles in the expansion of the cerebral cortex during primate evolution. Several studies comparing the expression of genes and regulatory regions in brains of various primates have shown an up‐regulation of genes and metabolites involved in oxidative metabolism and mitochondrial functions in human brains (Grossman et al, 2001, 2004; Cáceres et al, 2003; Uddin et al, 2004; Haygood et al, 2007). Furthermore, recent evidence indicates that an increase in metabolic rate, coupled with a higher predisposition to deposit fat and changes in the allocation of energy supplies, was crucial for the evolution of brain size and complexity (Pontzer et al, 2016). Understanding the metabolic signatures of different brain cells, and their metabolic interactions, will not only advance our understanding of how the brain functions and adapts to environmental demands, but may also elucidate the propensity of the human brain to age‐related neurodegenerative disorders. In recent years, it has become evident that metabolic alterations strongly influence the instigation and progression of many neurodegenerative disorders. Decreases in glucose and oxygen metabolic rates of brain cells occur during normal aging (Hoyer, 1982a) and are further exacerbated in disorders such as Alzheimer's (AD), amyotrophic lateral sclerosis (ALS), Parkinson's (PD), and Huntington's (HD) diseases (Hoyer, 1982b).
In this review article, we summarize the current knowledge of neural cell energy metabolism in the contexts of normal brain function, adaptive neuroplasticity, and the pathogenesis of neurodegenerative disorders.
Brain barriers and metabolite transporters
Neurons in the adult brain rely mostly on glucose as an energy source (Kety, 1957; Sokoloff, 1960). However, in some circumstances neurons can use substrates other than glucose. For example, ketone bodies are utilized during brain development and in the adult during prolonged fasting periods (Owen et al, 1967; Nehlig & Pereira de Vasconcelos, 1993), while lactate utilization is increased during intense physical activity (Dalsgaard et al, 2003; van de Hall et al, 2009). Given its high metabolic demands and negligible intrinsic energy stores, the brain depends upon a continuous influx of substrates from the blood. In order to protect the brain from fluctuations in the blood composition that could impact its milieu and functions, the exchanges of molecules between blood and cerebral fluids are regulated by the blood–brain barrier (BBB), and the blood–cerebrospinal fluid barrier (BCSFB). The main function of the these barriers is to limit the free diffusion of solutes between blood and brain fluids, and to selectively transport essential nutrients, ions, and signaling molecules, while removing metabolic waste products. The BBB separates the brain interstitial fluid from the blood and is formed by capillary endothelial cells interconnected by tight and adherens junctions, their underlying basement membrane, pericytes, and the “end feet” of astrocytes (Fig 1). The BBB controls the influx of metabolites such as glucose, amino acids, and ketones from the blood into the brain, while preventing the access of blood‐borne molecules and cells (e.g., lymphocytes) that could be detrimental for neuronal functions. The BCSFB is formed by the modified epithelial cells of the choroid plexus which separates the peripheral blood from the CSF, and the arachnoid epithelium separating the cerebral blood from the CSF. In addition to filtering functions similar to the BBB, the epithelial cells of the BCSFB are also responsible for producing the CSF.
Figure 1. Nutrient transport across the blood–brain barrier.
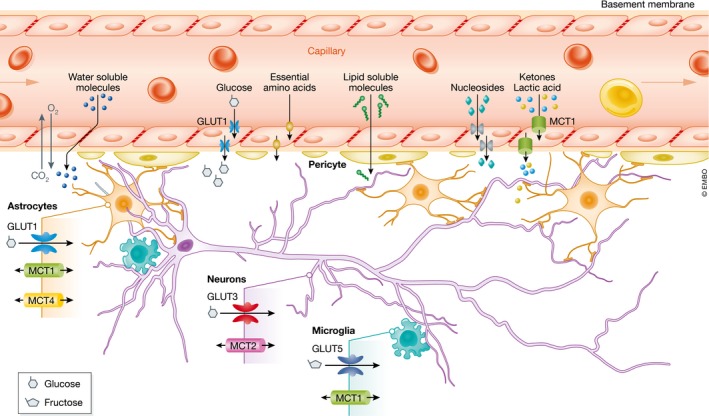
The blood–brain barrier is formed by capillary endothelial cells surrounded by basement membrane, pericytes, and the astrocyte perivascular end feet. The presence of tight junctions between the endothelial cells strongly inhibits the penetration of water‐soluble molecules. Passive diffusion is limited to gases and small nonpolar lipids. All other nutrients require passive or active mediated transporters. GLUT1‐5, glucose transporter 1‐5; MCT1‐4, monocarboxylic acid transporter 1‐4.
The modalities by which specific molecules cross through the BBB depend upon the nature of the solutes (Fig 1). Passive partition is limited to small nonpolar lipid‐soluble molecules, and to diffusible gases such as oxygen and carbon dioxide according to their concentration gradients. The presence of tight junctions restricts paracellular diffusion of polar molecules such as proteins (Zlokovic et al, 1985a,b; Zlokovic & Apuzzo, 1997), which cross the BBB by interacting with receptors or transporters expressed on both the luminal and abluminal membranes, or selectively on one side (Zloković et al, 1987; Zlokovic et al, 1990; Abbott et al, 2010). Large peptides and proteins such as hormones, growth factors, and neuroactive peptides are transferred via receptor‐mediated, adsorptive‐mediated, and carrier‐mediated transport (Zlokovic, 1995, 2008). Based on the requirement or not to hydrolyze ATP to move the solute across the membrane, two major families of transporters have been identified in the BBB: the ATP‐binding cassette (ABC) proteins and the solute carrier (SLC) proteins. The ABC transporters include multidrug resistance‐associated proteins (MRPs, ABCB1‐6), P‐glycoprotein, and breast cancer resistance protein (BRPC) (Begley, 2004). These transporters function as efflux pumps that couple ATP hydrolysis to move lipid‐soluble molecules against their concentration gradient. The solute carrier proteins comprise a large superfamily of more than 300 members; they are instrumental for ensuring a stable supply of carbohydrates, amino acids, monocarboxylic acids, nucleotides, fatty acids, and organic anions and cations (Abbott et al, 2010).
Among the SLC carriers, those that transport hexose and pentose sugars (glucose transporters; GLUTs) and monocarboxylates (monocarboxylic acid transporters; MCTs) are particularly important for brain metabolism. The intake of glucose into the brain is mediated by GLUT1, which is expressed as a 55‐kD isoform in endothelial cells of the BBB. A second 45‐kD GLUT1 isoform ensures delivery of glucose to glia, ependymal cells, and the choroid plexus. GLUT3 mediates uptake of glucose in neurons; GLUT3 is mainly concentrated in axons and dendrites. GLUT3 has a higher glucose affinity and transport capacity compared to other transporters, and so ensures that neurons receive a constant supply of glucose even when interstitial glucose concentrations are low. Other members of the glucose transporter family are expressed at much lower levels compared to GLUT1 and GLUT3 in specific cell types and/or in specialized brain regions. For example, the insulin‐sensitive GLUT4 is present in astrocytes, neurons, and endothelial cells (Kobayashi et al, 1996), and GLUT8 is located in the cytoplasm of neurons mostly in the hippocampus, amygdala, cerebellum, and hypothalamus (Reagan et al, 2001; Ibberson et al, 2002). GLUT2 is expressed in a subset of glutamatergic neurons in the hypothalamus and has recently been identified as a brain glucose sensor that triggers sugar seeking behavior under hypoglycemic conditions (Labouèbe et al, 2016). GLUT6 has been detected in neurons (Doege et al, 2000) and GLUT7 in astrocytes (Maher et al, 1994). In microglia, the most abundant transporter is GLUT5 which has a very low affinity for glucose and mostly fluxes fructose (Mantych et al, 1993).
The predominant roles of GLUT1 and GLUT3 in efficiently moving glucose from the blood across the BBB and into neurons have been clearly demonstrated in studies of gene knockout mice. GLUT1+/− mice have a reduced brain size and abnormal motor behavior (Wang et al, 2006), reminiscent of the phenotypes observed in human GLUT1 deficiency syndrome patients (De Vivo et al, 1991). GLUT3+/− mice exhibit abnormal spatial learning and working memory, in addition to perturbed social behavior (Zhao et al, 2010). GLUT8‐null homozygous mice have modest reductions of hippocampus volume (Membrez et al, 2006), and locomotion (Schmidt et al, 2008). In addition to facilitative glucose transporters, the endothelial cells of the BBB also express sodium‐dependent unidirectional transporters that are members of the solute carrier 5 family (SGLT) 1 and 2. These carriers couple the sodium electrochemical gradient to transfer glucose against its concentration gradient across the membrane. Their role under physiological conditions is not clear, but they appear to be functional during conditions of oxygen/glucose deprivation or ischemia (Yu et al, 2010). Because GLUT1 and GLUT3 transporters are constitutively located on the plasma membrane and do not respond to stimulation with insulin, brain glucose uptake is believed to be insulin‐independent.
There are 14 MCTs with particular affinities for one or more substrates. MCTs 1–4 are expressed in cells of the BBB (Fig 1) and are responsible for bidirectional passive proton‐linked transport of lactate, ketone bodies (i.e., acetoacetate and 3‐β‐hydroxybutyrate), and pyruvate. MCT1 has high affinity for pyruvate and also transports lactate and ketone bodies; it is present in endothelial cells (Gerhart et al, 1997), astrocytes (Bröer et al, 1997), oligodendrocytes (Lee et al, 2012), and microglia (Moreira et al, 2009). Only a few specific subsets of hypothalamic neurons express MCT1 (Carneiro et al, 2016). MCT2 is the major transporter in neurons (Pierre et al, 2002), and compared to MCT1 has an overall higher affinity for all the substrates (Bröer et al, 1997). MCT2 is concentrated in dendritic spines where it associates with postsynaptic density proteins, as well as the AMPA receptor subunit Glur2 (Bergensen et al, 2005). MCT3 transports lactate and is only expressed in the retinal epithelium and the choroid plexus epithelium (Philp et al, 2001). MCT4 carries lactate and is exclusively expressed in astrocytes (Pellerin et al, 2005). The specific cell distribution patterns and substrate affinities of MCTs in the brain suggest that MCTs play fundamental roles in shuttling energy substrates among different brain cell types.
Glucose metabolic pathways in neurons and astrocytes
The metabolic fate of glucose in the brain depends upon the cell type and the selective expression of metabolic enzymes. Neurons are predominantly oxidative, while astrocytes are mostly glycolytic (Hiden & Lange, 1962; Hamberger & Hyden, 1963). In addition to the production of adenosine‐5′‐triphosphate (ATP), glucose is also used to generate metabolic intermediates for the synthesis of fatty acids and other lipids required for membrane and myelin synthesis (Ramsey et al, 1971; Jones et al, 1975); amino acids for protein synthesis and neurotransmitter production (Vrba et al, 1962; Gaitonde & Richter, 1966); and 5‐carbon sugars for the synthesis of nucleotides (Gaitonde et al, 1983); and to produce glycogen in astrocytes.
In neurons, each molecule of glucose is oxidized via glycolysis, the pentose phosphate pathway (PPP), the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation, with the production of carbon dioxide, water, and 30–36 molecules of ATP depending upon the rates of proton leakage in the mitochondria (Fig 2). The glycolytic process metabolizes glucose to pyruvate, which can be actively transported into the mitochondria where it is converted to acetyl coenzyme A (acetyl‐CoA). Acetyl‐CoA is complexed with citrate which undergoes a series of regenerative enzymatic reactions producing reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) in the TCA cycle. The NADH and FADH2 produced during glycolysis and the TCA cycle are subsequently re‐oxidized in the electron transport chain (ETC). ETC. utilizes the energy produced by the transfer of electrons through its various complexes to transport protons across the inner mitochondrial membrane into the intermembrane space. The flux of protons back into the mitochondrial matrix is mediated by the enzyme ATP synthase, which utilizes the energy to generate ATP from ADP. Once inside the cell, glucose is irreversibly converted to glucose‐6‐phosphate (G6P) by hexokinase (HK). G6P can then be further metabolized via glycolysis or the pentose phosphate pathway (PPP) or can be used for glycogen synthesis.
Figure 2. Metabolic pathways of glucose utilization in neurons and astrocytes.
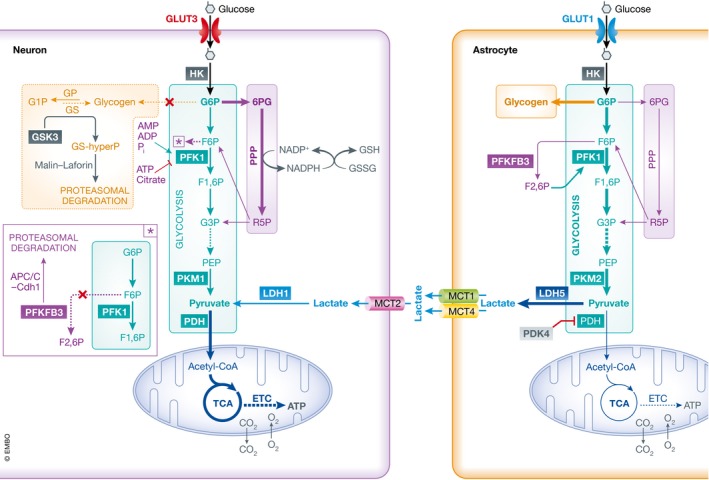
In neurons after entering the cell via glucose transporter 3 (GLUT3), glucose is phosphorylated by hexokinase (HK) to glucose‐6‐phosphate (G6P), which is subsequently routed in the glycolytic pathway and the pentose phosphate pathway (PPP). The end product of glycolysis is pyruvate that enters the mitochondria where it is metabolized through the tricarboxylic acid (TCA) cycle and oxidative phosphorylation in the electron transport chain (ETC.), generating adenosine‐5′‐triphosphate (ATP) and carbon dioxide (CO2) while consuming oxygen (O2). Pyruvate can also be generated from lactate dehydrogenase 1 (LDH1)‐dependent conversion of lactate. In the PPP, G6P is converted to 6‐phosphogluconate (6PG) that is transformed in ribulose‐5‐phosphate (R5P), with the concomitant production of reduced nicotinamide adenine dinucleotide phosphate (NADPH). NADPH is utilized to regenerate oxidized antioxidants such as glutathione (GSH) and thioredoxin. Neurons are not able to store glucose in the form of glycogen due to constitutive degradation of glycogen synthase (GS) via glycogen synthase kinase 3 (GSK3) phosphorylation, and subsequent ubiquitin‐dependent proteasomal digestion mediated by the malin–laforin complex. In astrocytes, glucose is imported trough glucose transporter 1 (GLUT1) and preferentially stored as glycogen, or metabolized via glycolysis. The pyruvate generated is converted to lactate thanks to the expression of lactate dehydrogenase 5 (LDH5), and pyruvate dehydrogenase kinase 4 (PDK4)‐dependent inhibition of pyruvate dehydrogenase (PDH). The presence of 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase 3 (Pfkfb3) allows astrocytes to generate fructose‐2,6‐bisphosphate (F2,6P) that acts as an allosteric modulator of PKF1 boosting glycolysis. Abbreviations are as follows: F6P, fructose‐6‐phosphate; PKF1, phosphofructokinase 1; F1,6P, fructose‐1,6‐diphosphate; G3P, glyceraldehyde‐3‐phosphate; Mit, mitochondrion; PEP, phosphoenolpyruvate; PKM1, pyruvate kinase M1; PKM2, pyruvate kinase M2; G1P, glucose‐1‐phosphate; GP, glycogen phosphorylase; APC/C‐Cdh1, anaphase‐promoting complex C/cytosome‐Cdh1; MCT, monocarboxylic acid transporter.
Although negligible compared to peripheral energy deposits, glycogen represents the largest energy reserve in the brain. Glycogen metabolism is regulated by two key enzymes, glycogen synthase (GS) and glycogen phosphorylase (GP). The reason why glycogen is produced and stored exclusively in astrocytes (Magistretti et al, 1993) is because in neurons GS is maintained in a constitutively inactive state by hyperphosphorylation via glycogen synthase kinase 3 (GSK3), and subsequent ubiquitin‐dependent proteasomal degradation mediated by the malin–laforin complex (Vilchez et al, 2007) (Fig 2 inset). A similar degradation process also occurs for protein targeting to glycogen (PTG), the regulatory subunit of protein phosphatase 1 that is able to activate GS by dephosphorylation, thus preventing the accumulation of glycogen in neurons (Vilchez et al, 2007). The preferred route of G6P metabolism in neurons is the PPP, an anabolic metabolic pathway that converts G6P into 5‐carbon sugars utilized for the biosynthesis of nucleotides with generation of reduced nicotinamide adenine dinucleotide phosphate (NADPH). Based on the cellular requirements, a portion of ribulose‐5‐phosphate (R5P) can be converted back into the glycolytic intermediates fructose‐6‐phosphate (F6P) and glyceraldehyde‐3‐phosphate (G3P). In neurons, this conversion is minimal, and NADPH is utilized as a cofactor for synthesis of fatty acids and myelin, for neurotransmitter turnover, and to maintain redox homeostasis. The maintenance of neuronal antioxidant potential relies on the use of NADPH as cofactor to regenerate reduced glutathione (GSH) (Fig 2) and thioredoxin by glutathione and thioredoxin reductase, respectively.
The balance between glycolysis and PPP rates in neurons is very important, and diversion of glucose utilization toward exclusive glycolysis can result in decreased availability of NADPH, increased oxidative stress and cell death (Herrero‐Mendez et al, 2009). The preferential use of G6P in the PPP in neurons, as well as their inability to up‐regulate glycolysis, is due to the selective expression of enzymes favoring such a metabolic route coupled with the absence of specific glycolysis modulators. In addition to the HK step mentioned above, the glycolytic flux is regulated by phosphofructokinase 1 (PKF1) and pyruvate kinase (PK) (Lowry & Passonneau, 1964). PKF1 catalyzes the phosphorylation of F6P to fructose‐1,6‐bisphosphate (F1,6P). Its activity is inhibited by metabolites associated with a high energy state (i.e., ATP, citrate) and enhanced by those resulting from high metabolic activity (i.e., ADP, AMP, phosphate), as well as by fructose‐2, 6‐bisphosphate (F2,6P). It was recently shown that neurons lack the enzyme responsible for the generation of F2,6P, 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase 3 (Pfkfb3) due to continuous ubiquitin‐dependent proteasomal degradation (Herrero‐Mendez et al, 2009) (Fig 2 inset). While neurons lack Pfkfb3, they express pyruvate kinase M1 (PKM1) (Zhang et al, 2014), a constitutively active enzyme with a very high affinity for phosphoenolpyruvate (PEP), thereby favoring the generation of high levels of pyruvate. This, in association with the expression in neurons of the low‐pyruvate‐affinity isoform of lactate dehydrogenase (LDH1), prevents pyruvate conversion to lactate and favors its entrance into the TCA cycle (Fig 2). Further metabolic bias toward the TCA cycle results from the lower levels of expression in neurons of pyruvate dehydrogenase kinase 4 (PDK4) which controls the activity of pyruvate dehydrogenase (PDH), and therefore the decarboxylation of pyruvate to acetyl‐CoA.
Astrocyte utilization of glucose is complementary to that of neurons. A portion of G6P is channeled into glycogen synthesis and PPP, but its predominant metabolism occurs via glycolysis with production of lactate and very low rates of mitochondrial oxidation (Itoh et al, 2003). This metabolic phenotype of astrocytes is the result of their unique expression of various enzymes and transporters. In contrast to neurons, astrocytes express very high levels of Pfkfb3 which favors glycolysis via allosteric activation of PFK by F2,6P (Herrero‐Mendez et al, 2009). Furthermore, under basal conditions the levels of PDH phosphorylation are high (Halim et al, 2010) thanks to elevated expression of PDK4 (Zhang et al, 2014), efficiently limiting the conversion of pyruvate to acetyl‐CoA (Fig 2). Astrocytes also express low levels of mitochondrial aspartate/glutamate carrier (AGC) decreasing the import of reduced equivalents (NADH) from the cytosol (Ramos et al, 2003). The expression of LDH5, which has a high affinity for pyruvate, rather than LDH1, ensures its conversion to lactate with concomitant oxidation of NADH to NAD+ thus maintaining high rates of NAD+/NADH that further favor aerobic glycolysis. The presence of PKM2 instead of PKM1 also enables astrocytes to easily up‐regulate the rate of glycolysis to increase the production of lactate, if needed.
Monocarboxylic acid metabolism
Over the past few decades, it has become clear that in addition to glucose, neurons can utilize alternate fuels, namely lactate and ketone bodies. Seminal in vitro studies of McIlwain in the 1950s demonstrated that in human cerebral cortex slices, both pyruvate and lactate could replace glucose to support respiration under basal conditions, and during electrical stimulation (McIlwain, 1953). Neurons in vitro have a preference for lactate over glucose when both substrates are provided (Itoh et al, 2003; Bouzier‐Sore et al, 2006). However, clear evidence for a role for lactate in brain metabolism in vivo has been obtained only recently. The cell type‐specific distribution of MCTs, and the intrinsic metabolic properties of astrocytes and neurons, led to the hypothesis that lactate is shuttled between the two cell types to support neuronal metabolism (Pellerin & Magistretti, 1994) (Fig 2). Such metabolic coupling of astrocytes and neurons is supported by optogenetic studies showing an in vivo lactate gradient from astrocytes to neurons (Mächler et al, 2016). Furthermore, pharmacological inhibition or genetic targeting of MCT2 irreversibly impairs long‐term memory in mice (Newman et al, 2011; Suzuki et al, 2011). Long‐term memory impairment can be reversed by intrahippocampal administration of lactate, but not glucose, in MCT4‐deficient mice (Suzuki et al, 2011). Targeted disruption of MCT1 and MCT2 impairs memory consolidation/reconsolidation in cocaine‐induced conditioned place preference and self‐administration (Zhang et al, 2016). Heterozygous MCT1 knockout mice have impaired inhibitory avoidance memory (Tadi et al, 2015). Altogether, these results strongly suggest that the neuronal uptake of lactate is important for the establishment of long‐term memories. The overall contribution of lactate to brain metabolism varies with its availability. Studies in conscious humans have shown that under resting conditions, lactate uptake by the brain provides about 8% of its energy requirements (van de Hall et al, 2009). The percentage increases up to 20% under conditions of high plasma levels of lactate such as during intense exercise (van de Hall et al, 2009). Furthermore, at various exercise intensities the metabolism of lactate in the brain is higher in trained subjects compared to controls (Kemppainen et al, 2005). This suggests the possibility of adaptive mechanisms allowing the brain to respond to changes in substrate availability. Notably, in rodents acute exercise induces brain region‐specific up‐regulation of MCTs (Takimoto & Hamada, 2014) and enhances oxidative capacity of cells in the motor cortex (McCloskey et al, 2001).
In addition to lactate, brain cells can metabolize the ketone bodies 3‐β‐hydroxybutyrate (3HB) and acetoacetate (AcAc). Ketones are recognized as an essential energy substrate for the brain during development, delivering up to 30–70% of its energy requirement (Nehlig, 2004); compared to the adult, the immature brain has high activity and levels of MCTs (Gerhart et al, 1997; Pellerin et al, 1998). Also, in rodents the brain activity of enzymes involved in ketone metabolism increases steadily through the suckling period, and then drops after weaning (Page et al, 1971; Middleton, 1973). The high level of ketone utilization during development is necessary to support energy metabolism, as well as the amino acid and lipid biosynthesis required for brain maturation (De Vivo et al, 1975; Yeh et al, 1977). In rats, incorporation of 3HB into amino acids is two‐ to threefold higher than glucose during the nursing period (De Vivo et al, 1975). Similarly, lipid synthesis, fundamental for myelination, is preferentially sustained by the use of ketones as precursors during the suckling period (Yeh et al, 1977). In addition to anabolic functions, the oxidation of ketones is also important during the early postnatal period (Fig 3). Mice with succinyl‐CoA‐3‐oxoacid CoA transferase (SCOT) deficiency have normal prenatal development, but right after birth they become ketotic, with reduced plasma levels of glucose and lactate (Cotter et al, 2011). In the adult brain, the utilization of ketones is greatly reduced in the fed state, but can increase considerably under conditions of limited glucose availability as occurs during fasting, starvation, low carbohydrate/high fat intake, and prolonged or intense exercise bouts (Fig 3). Under such conditions, the liver generates ketone bodies from fatty acid and ketogenic amino acid oxidation. Among brain cells, only astrocytes are equipped to generate ketone bodies from fatty acid β‐oxidation (Edmond, 1992), but the rates of fatty acid transport are very low compared to those in the liver. All brain cell types are, however, able to uptake ketones, mostly 3HB and AcAc, via MCTs; the ketones are then metabolized to acetyl‐CoA to support the cell energy and biosynthetic needs (Fig 3). In adults, the activity of ketone‐metabolizing enzymes is high enough that it would easily permit a complete switch from glucose to ketones to support brain energy needs (Krebs et al, 1971). Because ketones are never produced at saturating concentrations, the brain rate of utilization is strictly regulated by their blood concentration (Sokoloff, 1973). Indeed, during ketosis the brain glucose utilization has been shown to decrease by about 10% for each millimole of plasma ketones (LaManna et al, 2009). During medically supervised starvation of obese patients, ketones provide up to 60% of the energy utilized by the brain (Owen et al, 1967).
Figure 3. Schematic of ketone body oxidative and anabolic utilization in brain.
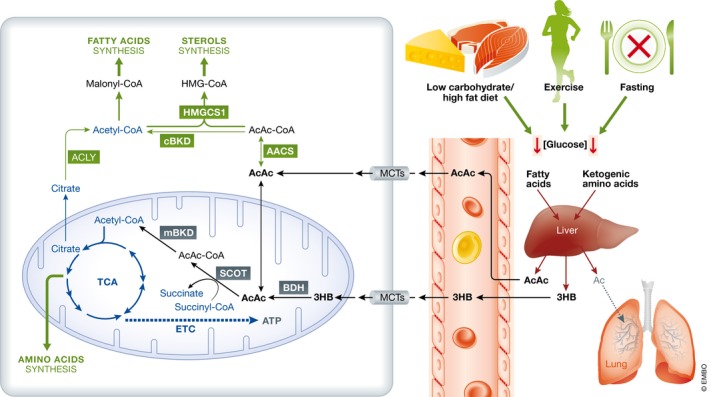
Under conditions of reduced glucose availability such as low carbohydrates/high‐fat diet, exercise, or fasting, the liver utilizes fatty acids mobilized from adipose tissue and ketogenic amino acids (i.e. leucine, lysine, phenylalanine, isoleucine, tryptophan, tyrosine, threonine) to produce acetoacetate (AcAc), 3‐β‐hydroxybutyrate (3HB), and acetone (Ac). Acetone is considered to have negligible metabolic significance and rapidly eliminated through urine and lungs. Ketone bodies cross the blood–brain barrier via monocarboxylate transporters (MCTs). Inside the cells, they may be directed toward anabolic or oxidative pathways depending on the developmental stage and cellular requirements. In the anabolic pathway taking place in the cytosol, acetoacetate is converted into acetoacetyl‐CoA (AcAc‐CoA) by acetoacetyl‐CoA synthase (AACS). AcAc‐CoA can be condensed with acetyl‐CoA to generate the precursor of sterols, 3‐hydroxy‐3‐methylglutaryl‐CoA (HMG‐CoA) by 3‐hydroxy‐3‐methylglutaryl‐CoA synthase 1 (HMGCS1). The acetyl‐CoA produced from AcAc‐CoA by cytosolic β‐ketothiolase (cBKD), or from citrate by ATP‐citrate lyase (ACLY), can be transformed in malonyl‐CoA for fatty acid synthesis. Amino acid can be synthesized utilizing intermediates of the TCA cycle. Oxidation of ketones occurs in the mitochondria (Mit) where AcAc directly taken up or generated from 3HB by 3‐β‐hydroxybutyrate dehydrogenase (BDH) is transformed into acetyl‐CoA via succinyl‐CoA‐3‐oxoacid CoA transferase (SCOT), and mitochondrial β‐ketothiolase (mBKD). The complete oxidation of AcAc yields 23 molecules of ATP, while 3HB generates 26 molecules of ATP.
Brain metabolism in aging
About 20–40% of healthy people between 60 and 78 years old experience discernable decrements in cognitive performance in several domains including working, spatial, and episodic memory, and processing speed (Mattay et al, 2006; Glisky, 2007). Semantic memory and knowledge show no decline until very late in life, while emotional, automatic, and autobiographic memory are not impacted by aging (Hedden & Gabrieli, 2004). These cognitive alterations correlate with neuroanatomical changes, including an age‐dependent decrease in gray matter volume not related to pathological conditions (Resnick et al, 2003). This thinning of the cortex is not uniform, with some regions such as the prefrontal cortex, medial temporal lobe, and hippocampus being more impacted by aging; other regions, such as the cingulate gyrus and the occipital cortex, remain relatively unaffected (Sowell et al, 2003). The loss of gray matter does not appear to be the result of neuronal loss, but instead involves a gradual decline of dendritic arborization and synapse numbers (Nakamura et al, 1985; Page et al, 2002). Aging also reduces white matter density and increases the number of white matter lesions (Guttmann et al, 1998), mostly in the prefrontal cortex and the anterior corpus callosum (O'Sullivan et al, 2001). By altering the interactions between prefrontal cortex and structures such as the hippocampus and striatum, white matter abnormalities result in poor performance in tasks requiring processing speed and immediate or delayed memory (Glisky, 2007). The brain undergoes a gradual decline in energy utilization during aging (Hoyer, 1982a). Functional neuroimaging studies have shown that glucose hypometabolism and mitochondrial dysfunction are early indicators of age‐related functional changes during normal brain aging (De Leon et al, 1983; Small et al, 2000: Mosconi et al, 2008). Positron emission tomography analyses of fluorodeoxyglucose uptake into brain cells in human subjects of different ages have revealed age‐related decrements in glucose utilization in several different brain regions (Zuendorf et al, 2003). Regional analyses revealed age‐related metabolic declines in temporal, parietal, and cerebral cortex, with a particularly rapid decline in the frontal cortex (Kuhl et al, 1984a). In rats, age‐dependent reduction in brain cell energy metabolism (glucose utilization) in the hippocampus and prefrontal cortex is associated with impaired performance in learning and memory tests (Gage et al, 1984). The current resolution of functional brain imaging is insufficient to establish a temporal sequence between hypometabolism and neuroanatomical changes. It is however tempting to speculate that the increased mitochondrial capacity and oxidative metabolism that appear to have driven expansion of the cerebral cortex during human evolution (Grossman et al, 2001, 2004; Cáceres et al, 2003; Uddin et al, 2004; Haygood et al, 2007; Pontzer et al, 2016) may have also rendered the brain susceptible to cognitive decline in aging. Synaptic spines are the site of neurotransmission, and thus fundamental for forms of synaptic plasticity such as long‐term potentiation and long‐term depression. Excitatory synapses are subcellular sites with very high rates of energy consumption as large amounts of ATP are required to support the activities of neurotransmitter transporters, and membrane Na+ and Ca2+ pumps that rapidly restore gradients of these ions after synapse activation (Attwell & Laughlin, 2001; Alle et al, 2009; Harris et al, 2012; Rangaraju et al, 2014). Accordingly, when the ability of neurons to generate sufficient ATP is compromised (e.g. aging, ischemia, and neurodegenerative disorders), synapses are vulnerable to dysfunction and degeneration (Harris et al, 2012) (Fig 4). Many factors likely contribute to the age‐dependent brain hypometabolism. Clinical studies have shown a negative correlation between cerebral blood flow and age (Schultz et al, 1999; Fabiani et al, 2014). In addition, the permeabilities of the BBB and BCSFB are greater in older compared to younger individuals (Rosenberg, 2012). Brain hypoperfusion and loss of BBB integrity can result in diminished import of nutrients, and/or removal of toxins. Furthermore, a compromised BBB allows the parenchymal accumulation of blood‐derived proteins (e.g., fibrinogen, immunoglobulins, albumin, thrombin, hemoglobin), and immune cells which can cause inflammation (Zlokovic, 2011). Studies of humans and animals have clearly shown reduced expression of glucose transporters in the brain with aging (Ding et al, 2013), as well as changes in the expression of key enzymes involved in glycolysis and oxidative phosphorylation (Meier‐Ruge et al, 1980; Ulfert et al, 1982; Bowling et al, 1993). Studies of mice have shown that levels of ATP are reduced in white matter during aging, in correlation with ultrastructural alterations in mitochondria, and a reduced association of mitochondria with endoplasmic reticulum (Stahon et al, 2016). NAD levels are critical for mitochondrial function and ATP production (Bai et al, 2011; Pittelli et al, 2011). An increase in the levels of NADH, with decreased total NAD and NAD+ levels, has been shown in human brain during normal aging (Zhu et al, 2015). Experimental evidence supporting a causative role for hypometabolism in cognitive impairment comes from recent studies showing that mice with reduced GLUT1 levels display an age‐dependent decrease in cerebral capillary density, reduced cerebral blood flow and glucose uptake, and increased BBB leakage (Winkler et al, 2015). These metabolic and vascular alterations precede dendritic spine loss in CA1 hippocampal neurons, and associated behavioral impairments (Winkler et al, 2015).
Figure 4. Age‐related cognitive decline as a result of neuroanatomical changes driven by decreased energy supply.
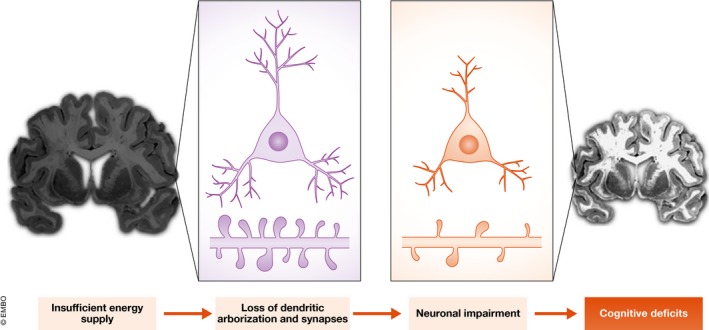
The neuronal firing patterns that play an important role in normal cognitive processing rely on the neurons' ability to exchange information across synapses. Compared to young neurons (left), aging neurons (right) are characterized by a significant reduction of the dendritic tree, as well as changes in spines size, shape, density, and turnover. Age‐dependent diminished nutrient import, as well as changes in glycolytic and oxidative phosphorylation efficiency, results in decreased ATP production. The reduced energy availability impairs the ability of aging neurons to preserve synapse homeostasis. The resulting structural changes lead to perturbations in neuronal function, and impairments in memory and learning.
Although we tend to think of age‐related metabolic decline as a “malfunction” of the brain, it is possible it represents an evolutionary adaptation. Human physiology is the result of millions of years of evolution under challenging environmental conditions and limited food availability. The drastic rapid changes in the lifestyle of modern human societies have led to an increased incidence of metabolic disorders (i.e., diabetes, obesity, metabolic syndrome, hyperlipidemia) that may be explained from an evolutionary perspective by the so‐called thrifty genotype hypothesis (Neel, 1962). The positive natural selection of genes that decreased metabolic rates while maintaining cognitive efficiency would have allowed individuals to survive times of limited food availability, but such genes may be detrimental when food is abundant (Nesse & Williams, 1998). Indeed, as described in the section on “healthy habits for a healthy brain” below, the fundamental bioenergetic challenges that were a driving force for brain evolution (i.e., fasting/starvation, and physical mental exertion) are exactly those that engage adaptive signaling pathways that promote optimal brain health, and resistance to brain injury and neurodegenerative disorders in modern humans.
Altered metabolism in neurodegenerative disorders
Neurodegenerative brain disorders are a broad spectrum of fatal conditions characterized by progressive neuronal dystrophic structural changes and loss of function. AD and PD are the most common neurodegenerative disorders, with ALS and HD being less prevalent. These diseases share several mechanistic similarities at the subcellular levels including atypical protein aggregation, failure of protein degradation pathways, impaired axonal transport, mitochondrial dysfunction, and programmed cell death (Mattson et al, 1999). Increasing evidence suggests that metabolic alterations strongly influence the initiation and progression of neurodegenerative disorders. Positron emission tomography imaging studies have documented reduced glucose utilization in brain regions affected in patients with AD, PD, ALS, and HD (Hoyer, 1982b). Epidemiological studies indicate that diabetes, obesity, high blood pressure, and atherosclerosis are all risk factors for dementia (Kivipelto et al, 2006). Because each of the latter disorders involves impaired energy metabolism, and/or adverse changes in the cerebral vasculature, reduced energy availability to neurons in the brain may contribute to increased vulnerability of the brain to cognitive impairment and dementia. Considerable evidence suggests that the BBB integrity is compromised in AD patients (Glenner, 1979, 1985; Powers et al, 1981; Zipser et al, 2007; Zlokovic, 2011). In patients with mild cognitive impairment, or early stages of AD, the age‐dependent changes of the BBB permeability are accelerated compared to neurological normal individuals (Montagne et al, 2015; van de Haar et al, 2016). This suggests that neurovascular dysfunction may be an early occurrence in the pathogenesis of AD. Additionally, changes in nutrient transporter and metabolic enzyme expression levels, and/or activities, have been reported in AD. For example, levels of GLUT1 and GLUT3 are reduced in the brains of AD patients (Simpson et al, 1994; Harr et al, 1995) and correlate with diminished brain glucose uptake and subsequent cognitive decline (Landau et al, 2010). A precipitous loss of activities of phosphofructokinase (PFK), phosphoglycerate mutase, aldolase, glucose‐6‐phosphate isomerase, and lactate dehydrogenase occurs in brain tissue samples of AD patients compared to age‐matched controls (Iwangoff et al, 1980). The activities of pyruvate dehydrogenase complex (Perry et al, 1980; Sorbi et al, 1983), cytochrome oxidase (Kish et al, 1992), and α‐ketoglutarate dehydrogenase complex (Gibson et al, 1988) are also decreased in the brains of AD patients. In mouse models of AD, reduction of GLUT1 levels worsens amyloid pathology, neurodegeneration, and cognitive function (Winkler et al, 2015), while ketone and nicotinamide supplementation reduces Aβ and p‐Tau pathologies and improves behavioral outcomes (Kashiwaya et al, 2013; Liu et al, 2013).
Glucose hypometabolism in the brains of patients with PD has been documented using magnetic resonance imaging and positron emission tomography methods (Kuhl et al, 1984b; Borghammer et al, 2010). Decreased levels of the PPP key enzymes glucose‐6‐phosphate dehydrogenase and 6‐phosphogluconate dehydrogenase occur at early stages in the putamen and cerebellum of PD patients (Dunn et al, 2014). The glycolytic enzyme glucose‐6‐phosphate isomerase that catalyzes the conversion of G6P to F6P has been recently identified as a conserved modifier of dopamine metabolism, protein aggregation, and neurodegeneration in Caenorhabditis elegans, Drosophila melanogaster, and murine neurons (Knight et al, 2014). Furthermore, it was recently shown that plasma levels of α‐synuclein regulate glucose uptake in adipocytes (Rodriguez‐Araujo et al, 2013). Importantly, mutations in multiple genes that cause early‐onset inherited forms of PD (α‐synuclein, Parkin, PINK1, LRRK2, DJ‐1) result in mitochondrial dysfunction (Pickrell & Youle, 2015). Moreover, interventions that bolster mitochondrial bioenergetics can ameliorate neuropathology and motor deficits in animal models of PD (Tieu et al, 2003; Yang et al, 2009).
ALS patients are hypercatabolic and have increased energy expenditure at rest (Desport et al, 2001; Funalot et al, 2009). Glucose intolerance (Pradat et al, 2010), insulin resistance (Reyes et al, 1984), and hyperlipidemia (Dupuis et al, 2008) have all been reported in ALS patients. At a cellular level, ALS patients exhibit altered endothelial transporter proteins (Niebroj‐Dobosz et al, 2010), astrocyte end feet degeneration (Miyazaki et al, 2011), increased permeability of the BBB/BCSFB resulting in abnormal levels of blood proteins in the CSF (Leonardi et al, 1984; Annunziata & Volpi, 1985), and IgG and complement deposits in the spinal cord and motor cortex (Donnenfeld et al, 1984). In superoxide dismutase 1 mutant mice and rats, BBB/BCSFB breakdown occurs prior to motor neuron degeneration and inflammation (Garbuzova‐Davis et al, 2007; Zhong et al, 2008; Nicaise et al, 2009; Miyazaki et al, 2011). Collectively, these findings strongly suggest that altered metabolic homeostasis plays a major role in ALS insurgence and progression.
HD is a genetic disorder caused by trinucleotide repeat (CAG) expansions in the huntingtin gene that causes early degeneration of medium spiny neurons in the striatum, resulting in continuous involuntary motor movements. Striatal metabolism is decreased well prior to atrophy, and the progression of the disease is more strongly correlated with glucose hypometabolism than the number of CAG repeats (Mazziotta et al, 1987; Grafton et al, 1992; Antonini et al, 1996). HD patients at early stages of striatum degeneration have normal total levels of glucose transporters (Gamberino & Brennan, 1994), but diminished glucose uptake in the brain (Kuhl et al, 1982; Ciarmiello et al, 2006). Immunohistochemical analysis utilizing antibody raised against an extracellular epitope of GLUT3 recently showed a diminished cell surface expression in the striatum and cortex of HD mice compared to wild‐type mice (McClory et al, 2014). The diminished ability of neurons to uptake glucose can explain the characteristic hypometabolism that precedes neuronal loss. Interestingly, higher copy numbers of SLC2A3 (Glut3) delay the age of onset in HD patients (Vittori et al, 2014). In fruit fly models of HD, overexpression of GLUT3, PFK, and G6PD protects against HD phenotypes and increases survival (Vittori et al, 2014; Besson et al, 2015). Evidence suggests that the lysine deacetylases sirtuin 1 (SIRT1) and sirtuin 3 (SIRT3) can preserve mitochondrial function and protect striatal neurons against dysfunction and degeneration (Jeong et al, 2011; Jiang et al, 2011; Fu et al, 2012). Agents that increase SIRT1 activity (e.g., SRT2104) attenuate degeneration of striatal neurons and improve functional outcome in huntingtin mutant mice (Jiang et al, 2014). It was also reported that an agent that increases SIRT3 levels (viniferin) protects neural cells against the toxicity of mutant huntingtin (Fu et al, 2012). Collectively, the emerging data suggest that interventions that bolster neuronal bioenergetics may delay disease onset or slow the progression of HD.
Healthy habits for a healthy brain
In the not too distant past, our ancestors were regularly challenged to locate and acquire food, while avoiding hazards. Assumedly, individuals whose brains and bodies functioned well/optimally when they were in a fasted state (i.e. when they had to make critical decisions on how to acquire food) had a survival advantage over those whose brains functioned less well in a state of prolonged negative energy balance. This bioenergetic challenge‐based hypothesis of brain evolution is supported by empirical evidence that dietary energy restriction/fasting and exercise enhance synaptic plasticity, neurogenesis, and cognitive performance in animals (Mattson, 2015a). For example, running wheel exercise and food restriction each increase dendritic spine density in hippocampal neurons, and the combination of food restriction and running results in even greater increases of spine density (Stranahan et al, 2009). Hippocampal neurogenesis is also increased in response to exercise and intermittent fasting (van Praag et al, 1999; Lee et al, 2002). In Drosophila melanogaster, associative learning is performed in fasted animals. One single training is sufficient for the flies to create a “pleasant” association between a certain scent and food. However, sequential multiple trainings are needed to establish an “aversive” association between an odorant and an unpleasant stimulus (electric shock). Fasting before training has been shown to increase long‐term memory formation for both “pleasant” and “aversive” experiences (Hirano et al, 2013). The duration of fasting appears to be crucial in determining the ability of the brain to prioritize the type of memory to establish/consolidate, based on the available energy and the most pressing survival need. Short‐term fasting results in increased long‐term memory (Hirano et al, 2013), while protracted fasting prevents “aversive”, but not “pleasant”, memory formation (Hirano et al, 2013; Placais & Preat, 2013). From an evolutionary point of view, it makes sense that starving flies would channel their remaining energy in finding food, ignoring aversive/safety issues. These findings support the idea that intermittent bioenergetic challenges are beneficial for brain performance.
In this section of our article, we highlight the importance of “cerebro‐bioenergetic resiliency”, the ability of the brain to respond adaptively to bioenergetic challenges, in promoting optimal brain function and resistance to stress, injury, and disease throughout life.
Cells and organisms have evolved the ability to respond adaptively to stress by activating intra‐ and intercellular signaling pathways that increase their resistance to that specific type of stress, and stress in general. This property of biological systems is fundamental to the concept of “hormesis” which is defined by a biphasic dose–response curve in which low doses induce a stimulatory/beneficial response, while high doses are damaging/toxic (Mattson, 2008, 2015b). Numerous studies have shown that when neurons and the organism in which they reside are subjected to mild metabolic challenges, brain function is improved and resistance to dysfunction and degeneration is increased compared to those that are unchallenged. For example, when cultured neurons are first subjected to a mild metabolic stress (e.g., glutamate, 2‐deoxyglucose, or mitochondrial uncoupling agents), they become resistant to subsequent exposure to a high level of stress (e.g., metabolic, excitotoxic, or oxidative stressor) that would have killed them had they not been previously exposed to the mild stress (Marini & Paul, 1992; Lee et al, 1999; Liu et al, 2015). A classic example of neuroprotection via hormesis in vivo is ischemic preconditioning in which rats or mice that are subjected to a mild cerebral ischemia prior to full‐blown ischemic stroke exhibit reduced brain cell damage and improved functional outcome compared to animals not subjected to the preconditioning ischemia (Dirnagl et al, 2009). Similar to ischemic preconditioning, treatment of mice or rats with 2‐deoxyglucose, an analog of glucose that induces cellular metabolic stress, can protect neurons in the brain and improve functional outcome in models of ischemic stroke, excitotoxic seizures, and PD (Duan & Mattson, 1999; Lee et al, 1999; Yu & Mattson, 1999).
Lifestyle factors appear to be crucial to determine how healthily our brain will age. Lack of physical activity, excessive calorie intake, and cognitive apathy negatively influence brain aging (Mattson, 2015a) and are predisposing factors for neurodegenerative disorders, such as AD and PD (Mattson, 2015a). Conversely, healthy lifestyle habits including dietary energy restriction, macro‐ and micronutrient diet composition, physical and mental exercise, and reduction of life stress boost cognitive function (Mattson, 2015a).
Regular aerobic exercise improves executive function, attention processing, speed memory, and learning (Colcombe & Kramer, 2003; Curlik & Shors, 2013; Dresler et al, 2013). Neuroimaging studies have shown that exercise targets specific brain areas, namely prefrontal and medial temporal cortices (Berchicci et al, 2013), and hippocampus (Kerr et al, 2010; Erickson et al, 2011, 2014). Elderly people that regularly exercise have increased brain volumes in these critical network areas, compared to sedentary subjects that instead undergo a significant volume decline (Colcombe et al, 2006; Erickson et al, 2009; Kerr et al, 2010). Epidemiological and interventional studies in humans have shown that exercise can increase one's resistance to anxiety and depression, and possibly AD and PD; exercise lessens symptoms in individuals suffering from these medical conditions (Tordeurs et al, 2011; Mattson, 2012; Paillard et al, 2015). The results of studies of animal models of anxiety, depression, AD, PD, stroke, and traumatic brain injury have established broad preventative and therapeutic benefits of aerobic exercise (Greenwood & Fleshner, 2008; Yuede et al, 2009; Egan et al, 2014; Mattson, 2014; Holland & Schmidt, 2015; Ryan & Kelly, 2016). The dysfunction and degeneration of neurons in these different disorders involves impaired neuronal bioenergetics, whose onset and progression varies markedly with regard to severity and duration (insidious in AD and depression, and acute and dramatic in stroke and traumatic brain injury) (Dirnagl et al, 2009; Marazziti et al, 2011).
A second lifestyle modification that promotes brain health is dietary energy restriction that can be achieved by caloric restriction, or by intermittent fasting (IF). IF can be operationally defined as an eating pattern that includes extended periods of time (e.g. 16 h daily or 24 h twice a week) during which no or very little food is consumed. Most animal studies of IF have used alternate‐day fasting (ADF, alternating days of complete fasting and ad libitum feeding). Mice or rats maintained on ADF exhibit reduced brain neuropathology and improved functional outcomes in models of stroke, AD, PD, HD, and epilepsy (Bruce‐Keller et al, 1999; Duan & Mattson, 1999; Halagappa et al, 2007).
Age‐related cognitive decline can also be counteracted by interventions stimulating brain activity. Engaging in intellectual challenges “exercises” and reinforces neuronal circuitries. Different types of cognitive training have been shown to improve specific cognitive aspects such as learning (Bailey et al, 2010), executive functions (Basak et al, 2008), and fluid intelligence (Jaeggi et al, 2008). In animal studies, environmental enrichment enhances cognitive performance by promoting neurotrophin production, synaptogenesis, dendrite formation, and arborization (van Praag et al, 2000; Fratiglioni et al, 2004). Neuroimaging studies in humans have shown that memory training increases hippocampal volume (Engvig et al, 2012), as well as the thickness of brain areas involved in decision‐making processing (i.e., lateral and fusiform orbitofrontal cortex) (Engvig et al, 2010).
The importance of exercise, diet, and intellectual and social stimulation in brain aging is emphasized by the results of a recent study showing that changes in diet, exercise, and cognitive training slow cognitive decline in elderly subjects (Ngandu et al, 2015). An additional advantage of this healthy lifestyle habit is that their combination appears to provide synergistic benefits (Schneider & Yvon, 2013). For example, adopting an exercise routine together with cognitive training promotes memory performance (Fabre et al, 2002; Oswald et al, 2006). A recent study in elderly subjects exposed to either moderate aerobic exercise or cognitive training, or to a combination of both, showed a greater improvement in working memory, long‐term memory, and reaction times in the cohort exposed to both trainings (Shatil, 2013).
Studies of cell culture and in vivo models of bioenergetic stress‐induced neuroprotection have begun to elucidate the molecular pathways that bolster neuronal resilience. They include activation of transcription factors such as cAMP response element‐binding protein (CREB), nuclear factor κB (NF‐κB), and nuclear factor erythroid‐derived 2 (NRF2) and induction of the expression of genes encoding proteins that counteract cellular stress at multiple subcellular sites, and by different mechanisms (Mattson, 2012) (Fig 5).
Figure 5. Signaling pathways mediating adaptive responses of neurons to bioenergetic challenges.
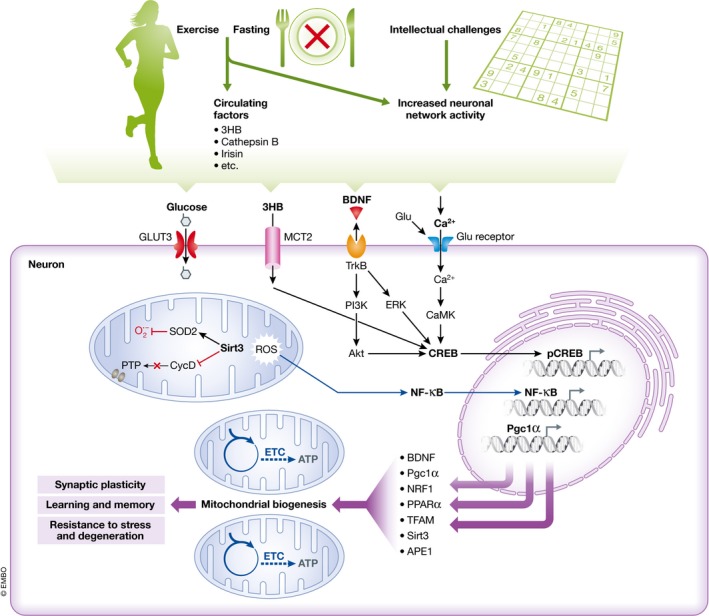
Exercise and fasting affect subcellular processes in neurons by brain‐intrinsic mechanisms mediated by increased neuronal network activity, and via signals coming from the periphery including 3‐β‐hydroxybutyrate (3HB), cathepsin B, and irisin. Intellectual challenges involve increased neuronal network activity and consequent activation of calcium‐responsive pathways. BDNF expression is up‐regulated by neuronal network activity, as well as 3HB, cathepsin B, and irisin, and BDNF is known to mediate, at least in part, the enhancement of neuronal plasticity and stress resistance by exercise, fasting, and intellectual challenges. Exercise, fasting, and intellectual challenges result in the activation of glutamate receptors at excitatory synapses, Ca2+ influx, and activation of Ca2+ calmodulin‐dependent protein kinase (CaMK) which, in turn, activates the transcription factor cyclic AMP response element‐binding protein (CREB). CREB can directly and indirectly modulate mitochondrial biogenesis via expression of several genes (i.e. BDNF, PGC‐1α, NRF1, PPARα, and TFAM). Activation of glutamate receptors also induces the expression of the mitochondrial protein sirtuin 3 (SIRT3) which can protect neurons by deacetylating superoxide dismutase 2 (SOD2) to increase its enzymatic activity, and thus reduce mitochondrial oxidative stress, and by inhibiting cyclophilin D (CycD), a protein involved in the formation of membrane permeability transition pores (PTP). 3‐β‐Hydroxybutyrate (3HB) can induce BDNF expression in neurons via the Ca2+–CREB pathway, and a pathway involving mitochondrial reactive oxygen species (ROS) and activation of the transcription factor nuclear factor κB (NF‐κB). BDNF is released from neurons and activates the receptor tropomyosin receptor kinase B (TrkB), on the same neuron and adjacent neurons, engaging downstream intracellular pathways which activate transcription factors that induce the expression of genes encoding proteins involved in synaptic plasticity, learning and memory, and neuronal stress resistance. Abbreviations are as follows: Pgc1a, peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha; NRF1, nuclear regulatory factor 1; PPARα, peroxisome proliferator‐activated receptor α; TFAM, mitochondrial transcription factor A; GLUT3, glucose transporter 3; MCT2, monocarboxylic acid transporter 2; PI3K, phosphoinositide 3 kinase; Akt, protein kinase B; ERK, extracellular signal regulated kinase; ETC., electron transport chain; ATP, adenosine‐5′‐triphosphate; APE1, apurinic/apyrimidinic endonuclease 1.
Exercise and IF can up‐regulate the expression of various proteins including antioxidant enzymes such as glutathione peroxidase, superoxide dismutase 2 (SOD2), and heme oxygenase 1; anti‐apoptotic proteins such as B‐cell lymphoma 2 family members; proteins involved in mitochondrial biogenesis and stress resistance; protein chaperones such as heat‐shock protein 70 and glucose‐regulated protein 78; neurotrophic factors such as brain‐derived neurotrophic factor (BDNF); and fibroblast growth factor 2 (Marosi et al, 2012; Mattson, 2012). Secreted neurotrophins can in turn activate cytoprotective signaling pathways in adjacent or distant neurons, thereby propagating adaptive cellular stress responses to cells that themselves had not experienced the same metabolic stress (Madinier et al, 2013). BDNF may play a significant role in several neuronal activity‐mediated effects of exercise and IF on neuronal bioenergetics and stress resistance. BDNF stimulates neuronal energy metabolism by increasing the expression of GLUT3, sodium‐dependent amino acid transport and protein synthesis (Burkahalter et al, 2003), and ketone utilization via MCT2 (Robinet & Pellerin, 2010). Furthermore, running and BDNF induce the expression of peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α) to increase mitochondrial biogenesis (Steiner et al, 1985; Cheng et al, 2012). Interestingly, exercise, moderate levels of glutamate receptor activation, and BDNF also induce the expression of the DNA repair enzyme apurinic/apyrimidinic endonuclease 1 (APE1), which plays a critical role in repairing oxidatively damaged DNA and protecting neurons against metabolic and excitotoxic stress (Yang et al, 2010, 2014).
Peripheral signals elicited in response to vigorous exercise and energy restriction/fasting may mediate some of the effects of these bioenergetic challenges on neuroplasticity and stress resistance. In addition to being used by neurons as an energy substrate, the ketone body 3HB also boosts the function, plasticity, and stress resistance of neurons in the brain by inducing the expression of BDNF in vivo (Sleiman et al, 2016) and in vitro (Marosi et al, 2016). 3HB mechanisms of action involve the generation of mitochondrial ROS and activation of the transcription factor nuclear factor κB (NF‐κB) (Marosi et al, 2016) (Fig 5), as well as the inhibition of histone deacetylases (Sleiman et al, 2016). Metabolic challenges also trigger peripheral cells to release into the circulation proteins that enter the brain where they elicit adaptive responses in neurons. Levels of cathepsin B, a predominantly lysosomal protein, are increased in skeletal muscle and plasma in response to running in mice (Moon et al, 2016). Cathepsin B induces the expression of BDNF in hippocampal neural progenitor cells, and the abilities of running to induce hippocampal neurogenesis and improve learning and memory performance are attenuated in cathepsin B‐deficient mice (Moon et al, 2016) (Fig 5). Another muscle‐derived factor that has been suggested to mediate beneficial effects of exercise on neuroplasticity is irisin, which was reported to increase BDNF levels in the brain (Wrann et al, 2013). It is therefore becoming clear that bioenergetic challenges educe a complex array of brain‐intrinsic and peripheral signaling mechanisms that coordinate adaptive responses of neurons and neural progenitor cells so as to optimize brain function and protect the brain against injury and disease.
It seems unlikely that drugs can be developed that trigger the complex, evolutionarily conserved mechanisms by which bioenergetic challenges promote brain health. However, preclinical findings and the results of some clinical trials suggest the potential for pharmacological interventions able to activate some of signaling pathways induced by exercise, fasting, and intellectual challenges. Ketogenic diets, ketone precursors (medium chain triglycerides), and 3HB have been reported in clinical studies of subjects with cognitive impairment, and AD (Reger et al, 2004; Henderson et al, 2009; Rebello et al, 2015), or PD patients (Vanitallie et al, 2005). It is not known whether improvements in cognitive function in the latter studies result from the utilization of 3HB as an energy substrate and/or the activation of adaptive stress response signaling in neurons. Caffeine, by stimulating Ca2+ release from the endoplasmic reticulum and increasing cyclic AMP levels, activates CREB (Connolly & Kingsbury, 2010) and has been shown to enhance memory consolidation in humans (Borota et al, 2014). Bitter chemicals that function as natural pesticides/antifeedants activate NRF2 and have demonstrated efficacy in animal models of stroke, AD, and PD; examples include sulforaphane, curcumin, and plumbagin (Son et al, 2008, 2010; Mattson, 2015b). Randomized controlled trials of such chemicals in human subjects with neurological disorders remain to be performed. Transcranial direct current or magnetic stimulation modulates BDNF levels (Müller et al, 2000) and can improve cognitive performance in healthy subjects and relieve symptoms in patients with depression and AD (Hsu et al, 2015; Brunoni et al, 2016). Noninvasive brain stimulation is a very exciting area because of its safety and potential for selective activation or inhibition of neuronal circuits in a brain region‐specific manner.
Although promising, such approaches should not be considered as substitutes for exercise, energy restriction, and intellectually challenging lifestyles. The adaptive cellular and molecular responses to these physiological challenges are finely tuned and are centrally and peripherally coordinated. They involve metabolic stress that occurs predominantly in excitable cells—skeletal muscle, cardiac myocytes, and neurons—and results in the activation or inhibition of numerous signaling pathways in cells throughout the brain. There is much that remains to be learned about these pathways: how they are activated, their molecular components, and how they interact to promote neuroplasticity and stress resistance. We also have little information concerning the intensities and durations of exercise and energy restriction that promote optimal brain health, nor how such regimens might vary depending upon one's age, metabolic status, or neurological disorders.
Conclusions and future directions
Emerging findings suggest that optimal brain health is promoted by intermittent bioenergetic challenges that increase activity in neuronal circuits, including intellectual challenges, restriction of energy intake, and physical exercise. Studies of animal and cell culture models have shown that such intermittent bioenergetic challenges activate signaling pathways in neurons that bolster mitochondrial health by, for example, stimulating mitochondrial biogenesis and mitophagy. The neuronal activity‐dependent and cellular stress‐responsive neurotrophic factor BDNF appears to play key roles in the neuroplasticity‐enhancing and neuroprotective actions of bioenergetic challenges. Signals from peripheral organs to brain cells may also contribute to the beneficial effects of exercise and fasting on cognitive function and neuronal resilience. During normal aging, there are decrements in the functionality of several energy metabolism‐related pathways in brain cells including glucose transport, mitochondrial electron transport, DNA repair, and neurotrophic factor signaling. Epidemiological, clinical, and experimental evidence points to important roles for impaired neuronal bioenergetics and reduced adaptation to stress in normal aging, and preclinical stages of neurodegenerative disorders such as AD and PD.
There is considerable complexity in the signaling pathways that integrate cellular energy metabolism with adaptive structural and functional responses of neuronal circuits to neuronal network activity. Future studies should be aimed at elucidating such intercellular and subcellular pathways. As new mechanisms emerge, it will be important to determine whether and how environmental and genetic factors that positively or negatively impact brain health influence brain cell energy metabolism. Translational research on cellular energy metabolism and brain health has been meager compared to efforts that focus on individual disease‐specific molecular targets. The drug development approach has thus far failed for AD, PD, and stroke. Indeed, the number of individuals living until they are in the age range for neurodegenerative disorders is rapidly increasing. The kinds of evidence from preclinical studies and human subjects described above provide a rationale for moving forward with randomized controlled trials of intermittent bioenergetic challenges achieved physiologically (e.g. intermittent fasting and exercise) or pharmacologically (e.g. mitochondrial uncoupling agents) in humans at risk of or in the early symptomatic stages of a neurodegenerative disorder, or during recovery from a stroke. As elaborated elsewhere (Mattson, 2012), it would also seem prudent to incorporate intermittent exercise and fasting protocols into physician training and healthcare practice, for disease risk reduction and early intervention in acute and chronic neurodegenerative conditions.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute on Aging.
The EMBO Journal (2017) 36: 1474–1492
Contributor Information
Simonetta Camandola, Email: camandolasi@mail.nih.gov.
Mark P Mattson, Email: mark.mattson@nih.gov.
References
- Abbott NJ, Patabendige AAK, Dolma DEM, Yusof SR, Begley DJ (2010) Structure and function of the blood‐brain barrier. Neurobiol Dis 37: 13–25 [DOI] [PubMed] [Google Scholar]
- Alle H, Roth A, Geiger JR (2009) Energy‐efficient action potentials in hippocampal mossy fibers. Science 325: 1405–1408 [DOI] [PubMed] [Google Scholar]
- Annunziata P, Volpi N (1985) High levels of C3c in the cerebrospinal fluid from amyotrophic lateral sclerosis patients. Acta Neurol Scand 72: 61–64 [DOI] [PubMed] [Google Scholar]
- Antonini A, Leenders KL, Spiegel R, Meier D, Vontobel P, Weigell‐Weber M, Sanchez‐Pernaute R, de Yébenez JG, Boesiger P, Weindl A, Maguire RP (1996) Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington's disease. Brain 119: 2085–2095 [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21: 1133–1145 [DOI] [PubMed] [Google Scholar]
- Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier‐de Murcia J, Auwerx J (2011) PARP‐1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey H, Dunlosky J, Hertzog C (2010) Metacognitive training at home: does it improve older adults' learning? Gerontology 56: 414–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak C, Boot WR, Voss MW, Kramer AF (2008) Can training in a real‐time strategy video game attenuate cognitive decline in older adults? Psychol Aging 23: 765–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley DJ (2004) ABC transporters and the blood‐brain barrier. Curr Pharm Des 10: 1295–1312 [DOI] [PubMed] [Google Scholar]
- Berchicci M, Lucci G, Di Russo F (2013) Benefits of physical exercise on the aging brain: the role of the prefrontal cortex. J Gerontol A Biol Sci Med Sci 68: 1337–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergensen LH, Magistretti PJ, Pellerin L (2005) Selective postsynaptic co‐localization of MCT2 with AMPA receptor GluR2/3 subunits at excitatory synapses exhibiting AMPA receptor trafficking. Cereb Cortex 15: 361–370 [DOI] [PubMed] [Google Scholar]
- Besson MT, Alegría K, Garrido‐Gerter P, Barros LF, Liévens JC (2015) Enhanced neuronal glucose transporter expression reveals metabolic choice in a HD Drosophila model. PLoS ONE 10: e0118765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghammer P, Chakravarty M, Jonsdottir KY, Sato N, Matsuda H, Ito K, Arahata Y, Kato T, Gjedde A (2010) Cortical hypometabolism and hypoperfusion in Parkinson's disease is extensive: probably even at early disease stages. Brain Struct Funct 214: 303–317 [DOI] [PubMed] [Google Scholar]
- Borota D, Murray E, Keceli G, Chang A, Watabe JM, Ly M, Toscano JP, Yassa MA (2014) Post‐study caffeine administration enhances memory consolidation in humans. Nat Neurosci 17: 201–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzier‐Sore AK, Voisin P, Bouchaud V, Bezancon E, Franconi JM, Pellerin L (2006) Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur J Neurosci 24: 1687–1694 [DOI] [PubMed] [Google Scholar]
- Bowling AC, Mutisya EM, Walker LC, Price DL, Cork LC, Beal MF (1993) Age‐dependent impairment of mitochondrial function in primate brain. J Neurochem 60: 1964–1967 [DOI] [PubMed] [Google Scholar]
- Bröer S, Rahman B, Pellegri G, Pellerin L, Martin JL, Verleysdonk S, Hamprecht B, Magistretti PJ (1997) Comparison of lactate transport in astroglial cells and monocarboxylate transporter 1 (MCT 1) expressing Xenopus laevis oocytes. Expression of two different monocarboxylate transporters in astroglial cells and neurons. J Biol Chem 272: 30096–30102 [DOI] [PubMed] [Google Scholar]
- Bruce‐Keller AJ, Umberger G, McFall R, Mattson MP (1999) Food restriction reduces brain damage and improve behavioral outcome following excitotoxic and metabolic insults. Ann Neurol 45: 8–15 [PubMed] [Google Scholar]
- Brunoni AR, Moffa AH, Fregni F, Palm U, Padberg F, Blumberger DM, Daskalakis ZJ, Bennabi D, Haffen E, Alonzo A, Loo CK (2016) Transcranial direct stimulation for acute major depressive episodes: meta‐analysis of individual patient data. Br J Psychiatry 208: 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkahalter J, Fiumelli H, Allaman I, Chatton JY, Martin JL (2003) Brain‐derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J Neurosci 23: 8212–8220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres M, Lachuer J, Zapala MA, Redmond JC, Kudo L, Geschwind DH, Lockhart DJ, Preuss TM, Barlow C (2003) Elevated gene expression levels distinguish human from non‐human primate brains. Proc Natl Acad Sci USA 100: 13030–13035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro L, Geller S, Fioramonti X, Hébert A, Repond C, Leloup C, Pellerin L (2016) Evidence for hypothalamic ketone body sensing: impact on food intake and peripheral metabolic responses in mice. Am J Physiol Endocrinol Metab 310: E103–E115 [DOI] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, Mattson MP (2012) Involvement of PGC‐1a in the formation and maintenance of neuronal dendritic spines. Nat Commun 3: 1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, Squitieri F (2006) Brain white‐matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington's disease. J Nucl Med 47: 215–222 [PubMed] [Google Scholar]
- Colcombe S, Kramer AF (2003) Fitness effects on the cognitive function of older adults: a meta‐analytic study. Psychol Sci 14: 125–130 [DOI] [PubMed] [Google Scholar]
- Colcombe SJ, Erickson KI, Scalf PE, Kim JS, Prakash R, McAuley E, Elavsky S, Marquez DX, Hu L, Kramer AF (2006) Aerobic exercise training increases brain volume in aging humans. J Gerontol A Biol Sci Med Sci 61: 1166–1170 [DOI] [PubMed] [Google Scholar]
- Connolly S, Kingsbury TJ (2010) Caffeine modulates CREB‐dependent gene expression in developing cortical neurons. Biochem Biophys Res Commun 397: 152–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter DG, d'Avignon DA, Wentz AE, Weber ML, Crawford PA (2011) Obligate role for ketone body oxidation in neonatal metabolic homeostasis. J Biol Chem 286: 6902–6910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curlik DM II, Shors TJ (2013) Training your brain: do mental and physical (MAP) training enhance cognition through the process of neurogenesis in the hippocampus? Neuropharmacology 64: 506–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard MK, Nybo L, Cai Y, Secher NH (2003) Cerebral metabolism is influenced by muscle ischaemia during exercise in humans. Exp Physiol 88: 297–302 [DOI] [PubMed] [Google Scholar]
- De Leon MJ, Ferris SH, George AE, Reisberg B, Christman DR, Kricheff II, Wolf AP (1983) Computed tomography and positron emission transaxial tomography evaluations of normal aging and Alzheimer's disease. J Cereb Blood Flow Metab 3: 391–394 [DOI] [PubMed] [Google Scholar]
- De Vivo DC, Rosario R, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI (1991) Defective glucose transport across the blood‐brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 325: 703–709 [DOI] [PubMed] [Google Scholar]
- Desport JC, Preux PM, Magy L, Boirie Y, Vallat JM, Beaufrère B, Couratier P (2001) Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr 74: 328–334 [DOI] [PubMed] [Google Scholar]
- De Vivo DC, Leckie MP, Agrawal HC (1975) D‐beta‐Hydroxybutyrate: a major precursor of amino acids in developing rat brain. J Neurochem 25: 161–170 [DOI] [PubMed] [Google Scholar]
- Ding F, Yao J, Rettberg JR, Chen S, Brinton RD (2013) Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer's mouse brain: implication for bioenergetic intervention. PLoS ONE 8: e79977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A (2009) Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 8: 398–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege H, Bocianski A, Joost HG, Schürmann A (2000) Activity and genomic organization of human glucose transporter 9 (GLUT9), a novel member of the family of sugar‐transport facilitators predominantly expressed in brain and leucocytes. Biochem J 350: 771–776 [PMC free article] [PubMed] [Google Scholar]
- Donnenfeld H, Kascsak RJ, Bartfeld H (1984) Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J Neuroimmunol 6: 51–57 [DOI] [PubMed] [Google Scholar]
- Dresler M, Sandberg A, Ohla K, Bublitz C, Trenado C, Mroczko‐Wąsowicz A, Kühn S, Repantis D (2013) Non‐pharmacological cognitive enhancement. Neuropharmacology 64: 529–543 [DOI] [PubMed] [Google Scholar]
- Duan W, Mattson MP (1999) Dietary restriction and 2‐deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson's disease. J Neurosci Res 57: 195–206 [DOI] [PubMed] [Google Scholar]
- Dunn L, Allen GF, Mamais A, Ling H, Li A, Duberley KE, Hargreaves IP, Pope S, Holton JL, Lees A, Heales SJ, Bandopadhyay R (2014) Dysregulation of glucose metabolism is an early event in sporadic Parkinson's disease. Neurobiol Aging 35: 1111–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, Corcia P, Fergani A, Gonzalez De Aguilar JL, Bonnefont‐Rousselot D, Bittar R, Seilhean D, Hauw JJ, Lacomblez L, Loeffler JP, Meininger V (2008) Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70: 1004–1009 [DOI] [PubMed] [Google Scholar]
- Edmond J (1992) Energy metabolism in developing brain cells. Can J Physiol Pharmacol 70(Suppl): S118–S129 [DOI] [PubMed] [Google Scholar]
- Egan KJ, Janssen H, Sena ES, Longley L, Speare S, Howells DW, Spratt NJ, Macleod MR, Mead GE, Bernhardt J (2014) Exercise reduces infarct volume and facilitates neurobehavioral recovery: results from a systematic review and meta‐analysis of exercise in experimental models of focal ischemia. Neurorehabil Neural Repair 28: 800–812 [DOI] [PubMed] [Google Scholar]
- Engvig A, Fjell AM, Westlye LT, Moberget T, Sundseth Ø, Larsen VA, Walhovd KB (2010) Effects of memory training on cortical thickness in the elderly. NeuroImage 52: 1667–1676 [DOI] [PubMed] [Google Scholar]
- Engvig A, Fjell AM, Westlye LT, Skaane NV, Sundseth Ø, Walhovd KB (2012) Hippocampal subfield volumes correlate with memory training benefit in subjective memory impairment. NeuroImage 61: 188–194 [DOI] [PubMed] [Google Scholar]
- Erickson KI, Prakash RS, Voss MW, Chaddock L, Hu L, Morris KS, White SM, Wójcicki TR, McAuley E, Kramer AF (2009) Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus 19: 1030–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, Kim JS, Heo S, Alves H, White SM, Wojcicki TR, Mailey E, Vieira VJ, Martin SA, Pence BD, Woods JA, McAuley E, Kramer AF (2011) Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci USA 108: 3017–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson KI, Leckie RL, Weinstein AM (2014) Physical activity, fitness, and gray matter volume. Neurobiol Aging 35(Suppl 2): S20–S28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiani M, Low KA, Tan CH, Zimmerman B, Fletcher MA, Schneider‐Garces N, Maclin EL, Chiarelli AM, Sutton BP, Gratton G (2014) Taking the pulse of aging: mapping pulse pressure and elasticity in cerebral arteries with optical methods. Psychophysiology 51: 1072–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre C, Chamari K, Mucci P, Massé‐Biron J, Préfaut C (2002) Improvement of cognitive function by mental and/or individualized aerobic training in healthy elderly subjects. Int J Sports Med 23: 415–421 [DOI] [PubMed] [Google Scholar]
- Fratiglioni L, Paillard‐Borg S, Winblad B (2004) An active and socially integrated lifestyle in late life might protect against dementia. Lancet Neurol 3: 343–353 [DOI] [PubMed] [Google Scholar]
- Fu J, Jin J, Cichewicz RH, Hageman SA, Ellis TK, Xiang L, Peng Q, Jiang M, Arbez N, Hotaling K, Ross CA, Duan W (2012) trans‐(‐)‐ε‐Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP‐activated protein kinase (AMPK), and protects cells in models of Huntington Disease. J Biol Chem 287: 24460–24472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funalot B, Desport JC, Sturtz F, Camu W, Couratier P (2009) High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler 10: 113–117 [DOI] [PubMed] [Google Scholar]
- Gage FH, Kelly PA, Bjorklund A (1984) Regional changes in brain glucose metabolism reflect cognitive impairments in aged rats. J Neurosci 4: 2856–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaitonde MK, Richter D (1966) Changes with age in the utilization of glucose carbon in liver and brain. J Neurochem 13: 1309–1316 [DOI] [PubMed] [Google Scholar]
- Gaitonde MK, Evison E, Evans GM (1983) The rate of utilization of glucose via hexose monophosphate shunt in brain. J Neurochem 41: 1253–1260 [DOI] [PubMed] [Google Scholar]
- Gamberino WC, Brennan WA Jr (1994) Glucose transporter isoform expression in Huntington's disease brain. J Neurochem 63: 1392–1397 [DOI] [PubMed] [Google Scholar]
- Garbuzova‐Davis S, Haller E, Saporta S, Kolomey I, Nicosia SV, Sanberg PR (2007) Ultrastructure of blood‐brain barrier and blood‐spinal cord barrier in SOD1 mice modeling ALS. Brain Res 1157: 126–137 [DOI] [PubMed] [Google Scholar]
- Gerhart DZ, Enerson BE, Zhdankina OY, Leino RL, Drewes LR (1997) Expression of monocarboxylate transporter MCT1 by brain endothelium and glia in adult and suckling rats. Am J Physiol 273: E207–E213 [DOI] [PubMed] [Google Scholar]
- Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P (1988) Reduced activities of thiamine‐dependent enzymes in the brains and peripheral tissues of patients with Alzheimer's disease. Arch Neurol 45: 836–840 [DOI] [PubMed] [Google Scholar]
- Glenner GG (1979) Congophilic microangiopathy in the pathogenesis of Alzheimer's syndrome (presenile dementia). Med Hypotheses 5: 1231–1236 [DOI] [PubMed] [Google Scholar]
- Glenner GG (1985) On causative theories in Alzheimer's disease. Hum Pathol 16: 433–435 [DOI] [PubMed] [Google Scholar]
- Glisky EL (2007) Changes in cognitive function in human aging In Brain aging: models, methods, and mechanisms, Riddle DR. (ed.), Chapter 1. Boca Raton, FL: CRC Press/Taylor & Francis; [PubMed] [Google Scholar]
- Grafton ST, Mazziotta JC, Pahl JJ, St George‐Hyslop P, Haines JL, Gusella J, Hoffman JM, Baxter LR, Phelps ME (1992) Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington's disease. Arch Neurol 49: 1161–1167 [DOI] [PubMed] [Google Scholar]
- Greenwood BN, Fleshner M (2008) Exercise, learned helplessness, and the strain‐resistant brain. Neuromolecular Med 10: 81–98 [DOI] [PubMed] [Google Scholar]
- Grossman LI, Schmidt TR, Wildman DE, Goodman M (2001) Molecular evolution of aerobic energy metabolism in primates. Mol Phylogenet Evol 18: 26–36 [DOI] [PubMed] [Google Scholar]
- Grossman LI, Wildman DE, Schmidt TR, Goodman M (2004) Accelerated evolution of the electron transport chain in anthropoid primates. Trends Genet 20: 578–585 [DOI] [PubMed] [Google Scholar]
- Guttmann CR, Jolesz FA, Kikinis R, Killiany RJ, Moss MB, Sandor T, Albert MS (1998) White matter changes with normal aging. Neurology 50: 972–978 [DOI] [PubMed] [Google Scholar]
- van de Haar HJ, Burgmans S, Jansen JF, van Osch MJ, van Buchem MA, Muller M, Hofman PA, Verhey FR, Backes WH (2016) Blood‐brain barrier leakage in patients with early Alzheimer disease. Radiology 281: 527–535 [DOI] [PubMed] [Google Scholar]
- Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, Mattson MP (2007) Intermittent fasting and caloric restriction ameliorate age‐related behavioral deficits in the triple‐transgenic mouse model of Alzheimer's disease. Neurobiol Dis 26: 212–220 [DOI] [PubMed] [Google Scholar]
- Halim ND, Mcfate T, Mohyeldin A, Okagaki P, Korotchkina LG, Patel MS, Jeoung NH, Harris RA, Schell MJ, Verma A (2010) Phosphorylation status of pyruvate dehydrogenase distinguishes metabolic phenotypes of cultured rat brain astrocytes and neurons. Glia 58: 1168–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Hall G, Stromstad M, Rasmussen P, Jans O, Zaar M, Gam C, Quistorff B, Secher NH, Nielsen HB (2009) Blood lactate is an important energy source for the human brain. J Cereb Flow Metab 29: 1121–1129 [DOI] [PubMed] [Google Scholar]
- Hamberger A, Hyden H (1963) Inverse enzymatic changes in neurons and glia during increased function and hypoxia. J Cell Biol 16: 521–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr SD, Simonian NA, Hyman BT (1995) Functional alterations in Alzheimer's disease: decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J Neuropathol Exp Neurol 54: 38–41 [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75: 762–777 [DOI] [PubMed] [Google Scholar]
- Haygood R, Fedrigo O, Hanson B, Yokoyama KD, Wray GA (2007) Promoter regions of many neural‐ and nutrition‐related genes have experienced positive selection during human evolution. Nat Genet 39: 1140–1144 [DOI] [PubMed] [Google Scholar]
- Hedden T, Gabrieli JD (2004) Insights into the ageing mind: a view from cognitive neuroscience. Nat Rev Neurosci 5: 87–96 [DOI] [PubMed] [Google Scholar]
- Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC (2009) Study of the ketogenic agent AC‐1202 in mild to moderate Alzheimer's disease: a randomized, double‐blind, placebo‐controlled, multicenter trial. Nutr Metab 6: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero‐Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C‐Cdh1. Nat Cell Biol 11: 747–752 [DOI] [PubMed] [Google Scholar]
- Hiden H, Lange PW (1962) A kinetic study of the neuroglia relationship. J Cell Biol 13: 233–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Masuda T, Naganos S, Matsuno M, Ueno K, Miyashita T, Horiuchi J, Saitoe M (2013) Fasting launches CRTC to facilitate log‐term memory formation in Drosophila . Science 339: 443–446 [DOI] [PubMed] [Google Scholar]
- Holland JN, Schmidt AT (2015) Static and dynamic factors promoting resilience following traumatic brain injury: a brief review. Neural Plast 2015: 902802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer S (1982a) The young adult and normally aged brain. Its flow and oxidative metabolism. A review‐part I. Arch Gerontol Geriatr 1: 101–116 [DOI] [PubMed] [Google Scholar]
- Hoyer S (1982b) The abnormally aged brain. Its blood flow and oxidative metabolism. A review‐ part II. Arch Gerontol Geriatr 1: 195–207 [DOI] [PubMed] [Google Scholar]
- Hsu WY, Ku Y, Zanto TP, Gazzaley A (2015) Effects on non invasive brain stimulation on cognitive function in healthy aging and Alzheimer's disease: a systematic review. Neurobiol Aging 36: 2348–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyder F, Rothman DL, Bennett MR (2013) Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc Natl Acad Sci USA 110: 3549–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibberson M, Riederer BM, Uldry M, Guhl B, Roth J, Thorens B (2002) Immunolocalization of GLUTX1 in the testis and to specific brain areas and vasopressin‐containing neurons. Endocrinology 143: 276–284 [DOI] [PubMed] [Google Scholar]
- Itoh Y, Esaki T, Shimoji K, Cook M, Law MJ, Kaufman E, Sokoloff L (2003) Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo . Proc Natl Acad Sci USA 100: 4879–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwangoff P, Armbruster R, Enz A, Meier‐Ruge W (1980) Glycolytic enzymes from human autoptic brain cortex: normal aged and demented cases. Mech Ageing Dev 14: 203–209 [DOI] [PubMed] [Google Scholar]
- Jaeggi SM, Buschkuehl M, Jonides J, Perrig WJ (2008) Improving fluid intelligence with training on working memory. Proc Natl Acad Sci USA 105: 6829–6833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Cohen DE, Cui L, Supinski A, Savas JN, Mazzulli JR, Yates JR III, Bordone L, Guarente L, Krainc D (2011) Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med 18: 159–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wang J, Fu J, Du L, Jeong H, West T, Xiang L, Peng Q, Hou Z, Cai H, Seredenina T, Arbez N, Zhu S, Sommers K, Qian J, Zhang J, Mori S, Yang XW, Tamashiro KL, Aja S et al (2011) Neuroprotective role of Sirt1 in mammalian models of Huntington's disease through activation of multiple Sirt1 targets. Nat Med 18: 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Zheng J, Peng Q, Hou Z, Zhang J, Mori S, Ellis JL, Vlasuk GP, Fries H, Suri V, Duan W (2014) Sirtuin 1 activator SRT2104 protects Huntington's disease mice. Ann Clin Transl Neurol 1: 1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JP, Nicholas HJ, Ramsey RB (1975) Rate of sterol formation by rat brain glia and neurons in vitro and in vivo . J Neurochem 24: 12312–12316 [DOI] [PubMed] [Google Scholar]
- Kashiwaya Y, Bergman C, Lee JH, Wan R, King MT, Mughal MR, Okun E, Clarke K, Mattson MP, Veech RL (2013) A ketone ester diet exhibits anxiolytic and cognition‐sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer's disease. Neurobiol Aging 34: 1530–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemppainen J, Aalto S, Fujimoto T, Kalliokoski KK, Långsjö J, Oikonen V, Rinne J, Nuutila P, Knuuti J (2005) High intensity exercise decreases global brain glucose uptake in humans. J Physiol 568: 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr AL, Steuer EL, Pochtarev V, Swain RA (2010) Angiogenesis but not neurogenesis is critical for normal learning and memory acquisition. Neuroscience 171: 214–226 [DOI] [PubMed] [Google Scholar]
- Kety SS (1957) The general metabolism of the brain in vivo In Metabolism of the nervous system, Richter D. (ed.), pp 221–237. London: Pergamon; [Google Scholar]
- Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang LJ, Wilson JM, DiStefano LM, Nobrega JN (1992) Brain cytochrome oxidase in Alzheimer's disease. J Neurochem 59: 776–779 [DOI] [PubMed] [Google Scholar]
- Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soininen H, Tuomilehto J (2006) Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population‐based study. Lancet Neurol 5: 735–741 [DOI] [PubMed] [Google Scholar]
- Knight AL, Yan X, Hamamichi S, Ajjuri RR, Mazzulli JR, Zhang MW, Daigle JG, Zhang S, Borom AR, Roberts LR, Lee SK, DeLeon SM, Viollet‐Djelassi C, Krainc D, O'Donnell JM, Caldwell KA, Caldwell GA (2014) The glycolytic enzyme, GPI, is a functionally conserved modifier of dopaminergic neurodegeneration in Parkinson's models. Cell Metab 20: 145–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Nikami H, Morimatsu M, Saito M (1996) Expression and localization of insulin‐regulatable glucose transporter (GLUT4) in rat brain. Neurosci Lett 213: 103–106 [DOI] [PubMed] [Google Scholar]
- Krebs HA, Williamson DH, Bates MW, Page MA, Hawkins RA (1971) The role of ketone bodies in caloric homeostasis. Adv Enzyme Regul 9: 387–409 [Google Scholar]
- Kuhl DE, Phelps ME, Markham CH, Metter EJ, Riege WH, Winter J (1982) Cerebral metabolism and atrophy in Huntington's disease determined by 18FDG and computed tomographic scan. Ann Neurol 12: 425–434 [DOI] [PubMed] [Google Scholar]
- Kuhl DE, Metter EJ, Riege WH, Hawkins RA (1984a) The effect of normal aging on patterns of local cerebral glucose utilization. Ann Neurol 15(Suppl): S133–S137 [DOI] [PubMed] [Google Scholar]
- Kuhl DE, Metter EJ, Riege WH, Markham CH (1984b) Patterns of cerebral glucose utilization in Parkinson's disease and Huntington's disease. Ann Neurol 15(Suppl): S119–S125 [DOI] [PubMed] [Google Scholar]
- Labouèbe G, Boutrel B, Tarussio D, Thorens B (2016) Glucose‐responsive neurons of the paraventricular thalamus control sucrose‐seeking behavior. Nat Neurosci 19: 999–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaManna JC, Salem N, Puchowicz M, Erokwu B, Koppaka S, Flask C, Lee Z (2009) Ketones suppress brain glucose consumption. Adv Exp Med Biol 645: 301–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, Petersen RC, Shaw LM, Trojanowski JQ, Jack CR Jr, Weiner MW, Jagust WJ (2010) Alzheimer's disease neuroimaging initiative. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 75: 230–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Bruce‐Keller AJ, Kruman Y, Chan SL, Mattson MP (1999) 2‐Deoxy‐D‐glucose protects hippocampal neurons against excitotoxic and oxidative injury: evidence for the involvement of stress proteins. J Neurosci Res 57: 48–61 [DOI] [PubMed] [Google Scholar]
- Lee J, Duan W, Mattson MP (2002) Evidence that brain‐derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem 82: 1367–1375 [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487: 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi A, Abbruzzese G, Arata L, Cocito L, Vische M (1984) Cerebrospinal fluid (CSF) findings in amyotrophic lateral sclerosis. J Neurol 231: 75–78 [DOI] [PubMed] [Google Scholar]
- Liu D, Pitta M, Jiang H, Lee JH, Zhang G, Chen X, Kawamoto EM, Mattson MP (2013) Nicotonamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol Aging 34: 1564–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Zhang Y, Gharavi R, Park HR, Lee J, Siddiqui S, Telljohann R, Nassar MR, Cutler RG, Becker KG, Mattson MP (2015) The mitochondrial uncoupler DNP triggers brain cell mTOR signaling network reprogramming and CREB pathway‐up‐regulation. J Neurochem 134: 677–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Passonneau JV (1964) The relationships between substrates and enzymes of glycolysis in brain. J Biol Chem 239: 31–42 [PubMed] [Google Scholar]
- Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber‐Castell A, Kaelin V, Zuend M, San Martín A, Romero‐Gómez I, Baeza‐Lehnert F, Lengacher S, Schneider BL, Aebischer P, Magistretti PJ, Barros LF, Weber B (2016) In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab 23: 94–102 [DOI] [PubMed] [Google Scholar]
- Madinier A, Bertrand N, Rodier M, Quirié A, Mossiat C, Prigent‐Tessier A, Marie C, Garnier P (2013) Ipsilateral versus contralateral spontaneous post‐stroke neuroplastic changes: involvement of BDNF? Neuroscience 231: 169–181 [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Sorg O, Yu N, Martin JL, Pellerin L (1993) Neurotransmitters regulate energy metabolism in astrocytes: implications for the metabolic trafficking between neural cells. Dev Neurosci 15: 306–312 [DOI] [PubMed] [Google Scholar]
- Maher F, Vannucci SJ, Simpson IA (1994) Glucose transporter proteins in brain. FASEB J 8: 1003–1011 [DOI] [PubMed] [Google Scholar]
- Mantych GJ, James DE, Devaskar SU (1993) Jejunal/kidney glucose transporter isoform (Glut‐5) is expressed in the human blood brain barrier. Endocrinology 132: 35–40 [DOI] [PubMed] [Google Scholar]
- Marazziti D, Baroni S, Picchetti M, Landi P, Silvestri S, Vatteroni E, Catena Dell'Osso M (2011) Mitochondrial alterations and neuropsychiatric disorders. Curr Med 18: 4715–4721 [DOI] [PubMed] [Google Scholar]
- Marini AM, Paul SM (1992) N‐methyl‐D‐aspartate receptor‐mediated neuroprotection in cerebellar granule cells requires new RNA and protein synthesis. Proc Natl Acad Sci USA 89: 6555–6559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marosi K, Bori Z, Hart N, Sárga L, Koltai E, Radák Z, Nyakas C (2012) Long‐term exercise treatment reduces oxidative stress in the hippocampus of aging rats. Neuroscience 226: 21–28 [DOI] [PubMed] [Google Scholar]
- Marosi K, Kim SW, Moehl K, Scheibye‐Knudsen M, Cheng A, Cutler R, Camandola S, Mattson MP (2016) 3‐hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J Neurochem 139: 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattay VS, Fera F, Tessitore A, Hariri AR, Berman KF, Das S, Meyer‐Lindenberg A, Goldberg TE, Callicott JH, Weinberger DR (2006) Neurophysiological correlates of age‐related changes in working memory capacity. Neurosci Lett 392: 32–37 [DOI] [PubMed] [Google Scholar]
- Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S (1999) Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann N Y Acad Sci 893: 154–175 [DOI] [PubMed] [Google Scholar]
- Mattson MP (2008) Awareness of hormesis will enhance future research in basic and applied neuroscience. Crit Rev Toxicol 38: 633–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP (2012) Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab 16: 706–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP (2014) Interventions that improve body and brain bioenergetics for Parkinson's disease risk reduction and therapy. J Parkinsons Dis 4: 1–13 [DOI] [PubMed] [Google Scholar]
- Mattson MP (2015a) Lifelong brain health is a lifelong challenge: from evolutionary principles to empirical evidence. Ageing Res Rev 20: 37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP (2015b) What doesn't kill you. Sci Am 313: 40–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazziotta JC, Phelps ME, Pahl JJ, Huang SC, Baxter LR, Riege WH, Hoffman JM, Kuhl DE, Lanto AB, Wapenski JA, Markham CH (1987) Reduced cerebral glucose metabolism in asymptomatic subjects at risk for Huntington's disease. N Engl J Med 316: 357–362 [DOI] [PubMed] [Google Scholar]
- McClory H, Williams D, Sapp E, Gatune LW, Wang P, DiFiglia M, Li X (2014) Glucose transporter 3 is a rab11‐dependent trafficking cargo and its transport to the cell surface is reduced in neurons of CAG140 Huntington's disease mice. Acta Neuropathol Commun 2: 179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloskey DP, Adamo DS, Anderson BJ (2001) Exercise increases metabolic capacity in the motor cortex and striatum, but not in the hippocampus. Brain Res 891: 168–175 [DOI] [PubMed] [Google Scholar]
- McIlwain H (1953) Substances which support respiration and metabolic response to electrical impulses in human cerebral tissues. J Neurol Neurosurg Psychiatry 16: 257–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier‐Ruge W, Iwangoff P, Reichlmeier K, Sandoz P (1980) Neurochemical findings in the aging brain. Adv Biochem Psychopharmacol 23: 323–338 [PubMed] [Google Scholar]
- Membrez M, Hummler E, Beermann F, Haefliger JA, Savioz R, Pedrazzini T, Thorens B (2006) GLUT8 is dispensable for embryonic development but influences hippocampal neurogenesis and heart function. Mol Cell Biol 26: 4268–4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton B (1973) The acetoacetyl‐coenzyme A thiolases of rat brain and their relative activities during postnatal development. Biochem J 132: 731–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Ohta Y, Nagai M, Morimoto N, Kurata T, Takehisa Y, Ikeda Y, Matsuura T, Abe K (2011) Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J Neurosci Res 89: 718–728 [DOI] [PubMed] [Google Scholar]
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV (2015) Blood‐brain barrier breakdown in the aging human hippocampus. Neuron 85: 296–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon HY, Becke A, Berron D, Becker B, Sah N, Benoni G, Janke E, Lubejko ST, Greig NH, Mattison JA, Duzel E, van Praag H (2016) Running‐induced systemic cathepsin B secretion is associated with memory function. Cell Metab 24: 332–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira TJ, Pierre K, Maekawa F, Repond C, Cebere A, Liljequist S, Pellerin L (2009) Enhanced cerebral expression of MCT1 and MCT2 in a rat ischemia model occurs in activated microglial cells. J Cereb Blood Flow Metab 29: 1273–1283 [DOI] [PubMed] [Google Scholar]
- Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, Laska E, Rusinek H, de Leon MJ (2008) Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol Aging 29: 676–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller MB, Toschi N, Kresse AE, Post A, Keck ME (2000) Long‐term repetitive transcranial magnetic stimulation increases the expression of brain‐derived neurotrophic factor and cholecystokinin mRNA, but not neuropeptide tyrosine mRNA in specific areas of rat brain. Neuropsychopharmacology 23: 205–215 [DOI] [PubMed] [Google Scholar]
- Nakamura S, Akiguchi I, Kameyama M, Mizuno N (1985) Age‐related changes of pyramidal cell basal dendrites in layers III and V of human motor cortex: a quantitative Golgi study. Acta Neuropathol 65: 281–284 [DOI] [PubMed] [Google Scholar]
- Neel JV (1962) Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 14: 353–362 [PMC free article] [PubMed] [Google Scholar]
- Nehlig A, Pereira de Vasconcelos A (1993) Glucose and ketone body utilization by the brain of neonatal rats. Prog Neurobiol 40: 163–221 [DOI] [PubMed] [Google Scholar]
- Nehlig A (2004) Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot Essent Fatty Acids 70: 265–275 [DOI] [PubMed] [Google Scholar]
- Nesse RM, Williams GC (1998) Evolution and the origins of disease. Sci Am 279: 86–93 [DOI] [PubMed] [Google Scholar]
- Newman LA, Korol DL, Gold PE (2011) Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE 6: e28427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, Bäckman L, Hänninen T, Jula A, Laatikainen T, Lindström J, Mangialasche F, Paajanen T, Pajala S, Peltonen M, Rauramaa R, Stigsdotter‐Neely A, Strandberg T, Tuomilehto J, Soininen H et al (2015) A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at‐risk elderly people (FINGER): a randomised controlled trial. Lancet 385: 2255–2263 [DOI] [PubMed] [Google Scholar]
- Nicaise C, Mitrecic D, Demetter P, De Decker R, Authelet M, Boom A, Pochet R (2009) Impaired blood‐brain and blood‐spinal cord barriers in mutant SOD1‐linked ALS rat. Brain Res 1301: 152–162 [DOI] [PubMed] [Google Scholar]
- Niebroj‐Dobosz I, Janik P, Sokołowska B, Kwiecinski H (2010) Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur J Neurol 17: 226–231 [DOI] [PubMed] [Google Scholar]
- O'Sullivan M, Jones DK, Summers PE, Morris RG, Williams SC, Markus HS (2001) Evidence for cortical “disconnection” as a mechanism of age‐related cognitive decline. Neurology 57: 632–638 [DOI] [PubMed] [Google Scholar]
- Oswald W, Gunzelmann T, Rupprecht R, Hagen B (2006) Differential effects of single versus combined cognitive and physical training with older adults: the SimA study in a 5‐year perspective. Eur J Ageing 3: 179‐192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF Jr (1967) Brain metabolism during fasting. J Clin Invest 46: 1589–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page MA, Krebs HA, Williamson DH (1971) Activities of enzymes of ketone‐body utilization in brain and other tissues of suckling rats. Biochem J 121: 49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page TL, Einstein M, Duan H, He Y, Flores T, Rolshud D, Erwin JM, Wearne SL, Morrison JH, Hof PR (2002) Morphological alterations in neurons forming corticocortical projections in the neocortex of aged Patas monkeys. Neurosci Lett 317: 37–41 [DOI] [PubMed] [Google Scholar]
- Paillard T, Rolland Y, de Souto Barreto P (2015) Protective effects of physical exercise in Alzheimer's disease and Parkinson's disease: a narrative review. J Clin Neurol 11: 212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 91: 10625–10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Martin JL, Magistretti PJ (1998) Expression of monocarboxylate transporter mRNAs in mouse brain: support for a distinct role of lactate as an energy substrate for the neonatal vs. adult brain. Proc Natl Acad Sci USA 95: 3990–3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Bergersen LH, Halestrap AP, Pierre K (2005) Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J Neurosci Res 79: 55–64 [DOI] [PubMed] [Google Scholar]
- Perry EK, Perry RH, Tomlinson BE, Blessed G, Gibson PH (1980) Coenzyme A‐acetylating enzymes in Alzheimer's disease: possible cholinergic “compartment” of pyruvate dehydrogenase. Neurosci Lett 18: 105–110 [DOI] [PubMed] [Google Scholar]
- Philp NJ, Yoon H, Lombardi L (2001) Mouse MCT3 gene is expressed preferentially in retinal pigment and choroid plexus epithelia. Am J Physiol Cell Physiol 280: C1319–C1326 [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85: 257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre K, Magistretti PJ, Pellerin L (2002) MCT2 is a major neuronal monocarboxylate transporter in the adult mouse brain. J Cereb Blood Flow Metab 22: 586–595 [DOI] [PubMed] [Google Scholar]
- Pittelli M, Felici R, Pitozzi V, Giovannelli L, Bigagli E, Cialdai F, Romano G, Moroni F, Chiarugi A (2011) Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol Pharmacol 80: 1136–1146 [DOI] [PubMed] [Google Scholar]
- Placais PY, Preat T (2013) To favor survival under food shortage, the brain disables costly memories. Science 339: 440–442 [DOI] [PubMed] [Google Scholar]
- Pontzer H, Brown MH, Raichlen DA, Dunsworth H, Hare B, Walker K, Luke A, Dugas LR, Durazo‐Arvizu R, Schoeller D, Plange‐Rhule J, Bovet P, Forrester TE, Lambert EV, Thompson ME, Shumaker RW, Ross SR (2016) Metabolic acceleration and the evolution of human brain size and life history. Nature 533: 390–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers JM, Schlaepfer WW, Willingham MC, Hall BJ (1981) An immunoperoxidase study of senile cerebral amyloidosis with pathogenetic considerations. J Neuropathol Exp Neurol 40: 592–612 [DOI] [PubMed] [Google Scholar]
- van Praag H, Christie BR, Sejnowski TJ, Gage FH (1999) Running enhances neurogenesis, learning, and long term potentiation in mice. Proc Natl Acad Sci USA 96: 13427–13431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH (2000) Neural consequences of environmental enrichment. Nat Rev Neurosci 1: 191–198 [DOI] [PubMed] [Google Scholar]
- Pradat PF, Bruneteau G, Gordon PH, Dupuis L, Bonnefont‐Rousselot D, Simon D, Salachas F, Corcia P, Frochot V, Lacorte JM, Jardel C, Coussieu C, Le Forestier N, Lacomblez L, Loeffler JP, Meininger V (2010) Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler 11: 166–171 [DOI] [PubMed] [Google Scholar]
- Rakic P (2009) Evolution of the neocortex: perspective from developmental biology. Nat Rev Neurosci 10: 724–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos M, del Arco A, Pardo B, Martínez‐Serrano A, Martínez‐Morales JR, Kobayashi K, Yasuda T, Bogónez E, Bovolenta P, Saheki T, Satrústegui J (2003) Developmental changes in the Ca2+‐regulated mitochondrial aspartate‐glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Brain Res Dev Brain Res 143: 33–46 [DOI] [PubMed] [Google Scholar]
- Ramsey RB, Jones JP, Naqvi SH, Nicholas HJ (1971) The biosynthesis of cholesterol and other sterols by brain tissue. I. Subcellular biosynthesis in vitro . Lipids 6: 154–161 [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA (2014) Activity‐driven local ATP synthesis is required for synaptic function. Cell 156: 825–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan LP, Gorovits N, Hoskin EK, Alves SE, Katz EB, Grillo CA, Piroli GG, McEwen BS, Charron MJ (2001) Localization and regulation of GLUTx1 glucose transporter in the hippocampus of streptozotocin diabetic rats. Proc Natl Acad Sci USA 98: 2820–2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebello CJ, Keller JN, Liu AG, Johnson WD, Greenway FL (2015) Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: a randomized controlled trial. BBA Clin 3: 123–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger MA, Henderson ST, Hale C, Cholerton B, Baker LD, Watson GS, Hyde K, Chapman D, Craft S (2004) Effects of beta‐hydroxybutyrate on cognition in memory‐impaired adults. Neurobiol Aging 25: 311–314 [DOI] [PubMed] [Google Scholar]
- Resnick SM, Pham DL, Kraut MA, Zonderman AB, Davatzikos C (2003) Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J Neurosci 23: 3295–3301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes ET, Perurena OH, Festoff BW, Jorgensen R, Moore WV (1984) Insulin resistance in amyotrophic lateral sclerosis. J Neurol Sci 63: 317–324 [DOI] [PubMed] [Google Scholar]
- Robinet C, Pellerin L (2010) Brain‐derived neurotrophic factor enhances the expression of the monocarboxylate transporter 2 through translational activation in mouse cultured cortical neurons. J Cereb Blood Flow Metab 30: 286–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Araujo G, Nakagami H, Hayashi H, Mori M, Shiuchi T, Minokoshi Y, Nakaoka Y, Takami Y, Komuro I, Morishita R, Kaneda Y (2013) Alpha‐synuclein elicits glucose uptake and utilization in adipocytes through the Gab1/PI3K/Akt transduction pathway. Cell Mol Life Sci 70: 1123–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA (2012) Neurological diseases in relation to the blood‐brain barrier. J Cereb Blood Flow Metab 32: 1139–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SM, Kelly AM (2016) Exercise as a pro‐cognitive, pro‐neurogenic and anti‐inflammatory intervention in transgenic mouse models of Alzheimer's disease. Ageing Res Rev 27: 77–92 [DOI] [PubMed] [Google Scholar]
- Schmidt S, Gawlik V, Hölter SM, Augustin R, Scheepers A, Behrens M, Wurst W, Gailus‐Durner V, Fuchs H, Hrabé de Angelis M, Kluge R, Joost HG, Schürmann A (2008) Deletion of glucose transporter GLUT8 in mice increases locomotor activity. Behav Genet 38: 396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider N, Yvon C (2013) A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging 17: 252–257 [DOI] [PubMed] [Google Scholar]
- Schultz SK, O'Leary DS, Boles Ponto LL, Watkins GL, Hichwa RD, Andreasen NC (1999) Age‐related changes in regional cerebral blood flow among young to mid‐life adults. NeuroReport 10: 2493–2496 [DOI] [PubMed] [Google Scholar]
- Shatil E (2013) Does combined cognitive training and physical activity training enhance cognitive abilities more than either alone? A four‐condition randomized controlled trial among healthy older adults. Front Aging Neurosci 5: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Vannucci SJ, Maher F (1994) Glucose transporters in mammalian brain. Biochem Soc Trans 22: 671–675 [DOI] [PubMed] [Google Scholar]
- Sleiman SF, Henry J, Al‐Haddad R, El Hayek L, Abou Haidar E, Stringer T, Ulja D, Karuppagounder SS, Holson EB, Ratan RR, Ninan I, Chao MV (2016) Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β‐hydroxybutyrate. Elife 5: pii:e15092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, Lavretsky H, Miller K, Siddarth P, Rasgon NL, Mazziotta JC, Saxena S, Wu HM, Mega MS, Cummings JL, Saunders AM, Pericak‐Vance MA, Roses AD, Barrio JR, Phelps ME (2000) Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer's disease. Proc Natl Acad Sci USA 97: 6037–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff L (1960) The metabolism of the central nervous system in vivo In Handbook of physiology, section I, neurophysiology, Field J, Magoun HW, Hall VE. (eds), Vol. 3, pp 1843–1864. Washington, DC: American Physiological Society; [Google Scholar]
- Sokoloff L (1973) Changes in enzyme activities in neural tissues with maturation and development of the nervous system In: The neurosciences, third study program, Schmidt FO. (ed.), pp 885–898. Cambridge, USA: MIT Press; [Google Scholar]
- Son TG, Camandola S, Mattson MP (2008) Hormetic dietary phytochemicals. Neuromolecular Med 10: 236–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son TG, Camandola S, Arumugam TV, Cutler RG, Telljohann RS, Mughal MR, Moore TA, Luo W, Yu QS, Johnson DA, Johnson JA, Greig NH, Mattson MP (2010) Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J Neurochem 12: 1316–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbi S, Bird ED, Blass JP (1983) Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol 13: 72–78 [DOI] [PubMed] [Google Scholar]
- Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW (2003) Mapping cortical change across the human life span. Nat Neurosci 6: 309–315 [DOI] [PubMed] [Google Scholar]
- Stahon KE, Bastian C, Griffith S, Kidd GJ, Brunet S, Baltan S (2016) Age‐related changes in axonal and mitochondrial ultrastructure and function in white matter. J Neurosci 36: 9990–10001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner JL, Murphy EA, McClellan JL, Carmichael MD, Davis JM (1985) Exercise training increases mitochondrial biogenesis in the brain. J Appl Physiol 111: 1066–1071 [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP (2009) Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus 19: 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM (2011) Astrocyte‐neuron lactate transport is required for long‐term memory formation. Cell 144: 810–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadi M, Allaman I, Lengacher S, Grenningloh G, Magistretti PJ (2015) Learning‐induced gene expression in the hippocampus reveals a role of neuron ‐astrocyte metabolic coupling in long term memory. PLoS ONE 10: e0141568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto M, Hamada T (2014) Acute exercise increases brain region‐specific expression of MCT1, MCT2, MCT4, GLUT1, and COX IV proteins. J Appl Physiol 116: 1238–1250 [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson‐Lewis V, Ramasamy R, Przedborski S (2003) D‐beta‐hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest 112: 892–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordeurs D, Janne P, Appart A, Zdanowicz N, Reynaert C (2011) Effectiveness of physical exercise in psychiatry: a therapeutic approach? Encephale 37: 345–352 [DOI] [PubMed] [Google Scholar]
- Uddin M, Wildman DE, Liu G, Grossman LI, Goodman M (2004) Sister grouping of chimpanzees and humans as revealed by genome‐wide phylogenetic analysis of brain gene expression profiles. Proc Natl Acad Sci USA 101: 2957–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulfert G, Schmidt U, Hoyer S (1982) Glucose and energy metabolism of rat cerebral cortex during aging. Exp Brain Res Suppl 5: 102–111 [DOI] [PubMed] [Google Scholar]
- Vanitallie TB, Nonas C, Di Rocco A, Boyar K, Hyams K, Heymsfield SB (2005) Treatment of Parkinson disease with diet‐induced hyperketonemia: a feasibility study. Neurology 64: 728–730 [DOI] [PubMed] [Google Scholar]
- Vilchez D, Ros S, Cifuentes D, Pujadas L, Vallès J, García‐Fojeda B, Criado‐García O, Fernández‐Sánchez E, Medraño‐Fernández I, Domínguez J, García‐Rocha M, Soriano E, Rodríguez de Córdoba S, Guinovart JJ (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci 10: 1407–1413 [DOI] [PubMed] [Google Scholar]
- Vittori A, Breda C, Repici M, Orth M, Roos RA, Outeiro TF, Giorgini F, Hollox EJ, REGISTRY investigators of the European Huntington's Disease Network (2014) Copy‐number variation of the neuronal glucose transporter gene SLC2A3 and age of onset in Huntington's disease. Hum Mol Genet 23: 3129–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrba R, Gaitonde MK, Richter D (1962) The conversion of glucose carbon into protein in the brain and other organs of the rat. J Neurochem 9: 465–475 [DOI] [PubMed] [Google Scholar]
- Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, Yoo J, Noebels JL, De Vivo DC (2006) A mouse model for Glut‐1 haploinsufficiency. Hum Mol Genet 15: 1169–1179 [DOI] [PubMed] [Google Scholar]
- Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S, Kong P, Nelson AR, Sullivan JS, Zhao Z, Meiselman HJ, Wenby RB, Soto J, Abel ED, Makshanoff J, Zuniga E, De Vivo DC, Zlokovic BV (2015) GLUT1 reductions exacerbate Alzheimer's disease vasculo‐neuronal dysfunction and degeneration. Nat Neurosci 18: 521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrann CD, White JP, Salogiannnis J, Laznik‐Bogoslavski D, Wu J, Ma D, Lin JD, Greenberg ME, Spiegelman BM (2013) Exercise induces hippocampal BDNF through a PGC‐1a/FNDC5 pathway. Cell Metab 18: 649–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Calingasan NY, Wille EJ, Cormier K, Smith K, Ferrante RJ, Beal MF (2009) Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson's and Huntington's diseases. J Neurochem 109: 1427–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JL, Tadokoro T, Keijzers G, Mattson MP, Bohr VA (2010) Neurons efficiently repair glutamate‐induced oxidative damage by a process involving CREB‐mediated production of apurinic/apyrimidinic endonuclease 1. J Biol Chem 285: 28191–28199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JL, Lin YT, Chuang PC, Bohr VA, Mattson MP (2014) BDNF and exercise enhance neuronal DNA repair by stimulating CREB‐mediated production of apurinic/apyrimidinic endonuclease 1. Neuromolecular Med 16: 161–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh YY, Streuli VL, Zee P (1977) Ketone bodies serve as important precursors of brain lipids in the developing rat. Lipids 12: 957–964 [DOI] [PubMed] [Google Scholar]
- Yu ZF, Mattson MP (1999) Dietary restriction and 2‐deoxyglucose administration reduce focal ischemic brain damage and improve behavioral outcome: evidence for a preconditioning mechanism. J Neurosci Res 57: 830–839 [PubMed] [Google Scholar]
- Yu AS, Hirayama BA, Timbol G, Liu J, Basarah E, Kepe V, Satyamurthy N, Huang SC, wright EM, Barrio JR (2010) Functional expression of SGLTs in rat brain. Am J Physiol Cell Physiol 299: C1277–C1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuede CM, Zimmerman SD, Dong H, Kling MJ, Bero AW, Holtzman DM, Timson BF, Csernansky JG (2009) Effects of voluntary and forced exercise on plaque deposition, hippocampal volume, and behavior in the Tg2576 mouse model of Alzheimer's disease. Neurobiol Dis 35: 426–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xue Y, Meng S, Luo Y, Liang J, Li J, Ai S, Sun C, Shen H, Zhu W, Wu P, Lu L, Shi J (2016) Inhibition of lactate transport erases drug memory and prevents drug relapse. Biol Psychiatry 79: 928–939 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Fung C, Shin D, Shin BC, Thamotharan S, Sankar R, Ehninger D, Silva A, Devaskar SU (2010) Neuronal glucose transporter isoform 3 deficient mice demonstrate features of autism spectrum disorders. Mol Psychiatry 15: 286–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O'Banion MK, Stojanovic K, Sagare A, Boillee S, Cleveland DW, Zlokovic BV (2008) ALS‐causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci 11: 420–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W (2015) In vivo NAD assay reveals the intracellular NAD content and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci USA 112: 287628–287681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM, Vitek MP, Hovanesian V, Stopa EG (2007) Microvascular injury and blood‐brain barrier leakage in Alzheimer's disease. Neurobiol Aging 28: 977–986 [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Segal MB, Begley DJ, Davson H, Racik L (1985a) Permeability of the blood‐cerebrospinal fluid and blood‐brain barriers to thyrotropin‐releasing hormone. Brain Res 358: 191–199 [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Begley DJ, Chain‐Eliash DG (1985b) Blood‐brain barrier permeability to leucine‐enkephalin, D‐alanine2‐D‐leucine5‐enkephalin and their N‐terminal amino acid (tyrosine). Brain Res 336: 125–132 [DOI] [PubMed] [Google Scholar]
- Zloković BV, Lipovac MN, Begley DJ, Davson H, Rakić L (1987) Transport of leucine‐enkephalin across the blood‐brain barrier in the perfused guinea pig brain. J Neurochem 49: 310–315 [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Hyman S, McComb JG, Lipovac MN, Tang G, Davson H (1990) Kinetics of arginine‐vasopressin uptake at the blood‐brain barrier. Biochim Biophys Acta 1025: 191–198 [DOI] [PubMed] [Google Scholar]
- Zlokovic BV (1995) Cerebrovascular permeability to peptides: manipulations of transport systems at the blood‐brain barrier. Pharm Res 12: 1395–1406 [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Apuzzo ML (1997) Cellular and molecular neurosurgery: pathways from concept to reality–part I: target disorders and concept approaches to gene therapy of the central nervous system. Neurosurgery 40: 789–803 [DOI] [PubMed] [Google Scholar]
- Zlokovic BV (2008) The blood‐brain barrier in health and chronic neurodegenerative disorders. Neuron 57: 178–201 [DOI] [PubMed] [Google Scholar]
- Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 12: 723–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuendorf G, Kerrouche N, Herholz K, Baron JC (2003) Efficient principal component analysis for multivariate 3D voxel‐based mapping of brain functional imaging data sets as applied to FDG‐PET and normal aging. Hum Brain Mapp 18: 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]