

Abstract
StAR‐related lipid transfer domain‐3 (STARD3) is a sterol‐binding protein that creates endoplasmic reticulum (ER)–endosome contact sites. How this protein, at the crossroad between sterol uptake and synthesis pathways, impacts the intracellular distribution of this lipid was ill‐defined. Here, by using in situ cholesterol labeling and quantification, we demonstrated that STARD3 induces cholesterol accumulation in endosomes at the expense of the plasma membrane. STARD3‐mediated cholesterol routing depends both on its lipid transfer activity and its ability to create ER–endosome contacts. Corroborating this, in vitro reconstitution assays indicated that STARD3 and its ER‐anchored partner, Vesicle‐associated membrane protein‐associated protein (VAP), assemble into a machine that allows a highly efficient transport of cholesterol within membrane contacts. Thus, STARD3 is a cholesterol transporter scaffolding ER–endosome contacts and modulating cellular cholesterol repartition by delivering cholesterol to endosomes.
Keywords: cholesterol, endoplasmic reticulum, endosome, lipid transfer protein, membrane contact site
Subject Categories: Membrane & Intracellular Transport
Introduction
Eukaryotic cells are compartmentalized into discrete organelles, which allow a spatial division of labor. A universal feature of eukaryotic cells is that the various lipid species, and in particular sterols, are unevenly distributed between organelles (Chang et al, 2006; van Meer et al, 2008). In mammalian cells, cholesterol is highly enriched in the plasma membrane (PM), and intermediate concentrations are found in early endosomes and the trans‐Golgi network (TGN), whereas the endoplasmic reticulum (ER) harbors the lowest level of cholesterol (Mesmin et al, 2013a). This precise distribution of cholesterol is ensured by vesicular and non‐vesicular transport pathways (Holthuis & Levine, 2005; Holthuis & Menon, 2014). Of note, several members of the steroidogenic acute regulatory protein (StAR)‐related lipid transfer (START) domain family are involved in non‐vesicular sterol transport (Iaea et al, 2014). These proteins possess a conserved START domain of about 210 amino acids that serves to transfer lipids between membranes (Tsujishita & Hurley, 2000; Alpy & Tomasetto, 2005). Notably, this domain defines a hydrophobic cavity able to accommodate one lipid molecule and thereby serves as a “hydrophobic bridge” across the aqueous gap between donor and acceptor organelle membranes.
STARD3 is a cholesterol‐specific START protein ubiquitously expressed and anchored at the membrane of late endosomes (Alpy et al, 2001). An early assumption was that STARD3 could act as a sterol transporter. It was legitimate in view of the sequence similarity between STARD3 and the founding member of the START protein family, StAR, alias STARD1. Indeed, STARD1 is essential for intracellular sterol transport in steroidogenic tissues as it conveys cholesterol to the mitochondria where it is metabolized into pregnenolone by the P450scc enzymatic complex (Clark et al, 1994; Clark & Stocco, 2014). For STARD3, despite numerous studies, it is not yet known whether this protein mediates a defined transport route for sterols. Because of its localization on the endosome surface, STARD3 was initially proposed to facilitate cholesterol exit from this organelle. However, this view was challenged by the fact that STARD3 overexpression does not increase sterol O‐acyltransferase‐mediated cholesterol esterification in the ER (Borthwick et al, 2010; Liapis et al, 2012) and does not rescue Niemann‐Pick type C mutant cells, which accumulate cholesterol in endosomes (Alpy et al, 2001; Holtta‐Vuori et al, 2005; Vanier, 2014). More confusingly, STARD3 expression was associated with higher cholesterol content in a number of distinct organelles such as endosomes, but also the PM and mitochondria (Alpy et al, 2001; Zhang et al, 2002; Holtta‐Vuori et al, 2005; Charman et al, 2010; Liapis et al, 2012; Vassilev et al, 2015). Up until now, how STARD3 functions in cholesterol transport is still elusive.
Interestingly, the functional characterization of STARD3 revealed that it distinguishes itself from the other START domain proteins by the presence of a conserved amino‐terminal domain named MENTAL (MLN64 NH2‐terminal) that is shared with its paralog STARD3NL (alias MENTHO) (Alpy et al, 2002). The MENTAL domain has many functions: It anchors the protein in endosome membranes; it exposes the START domain in the cytosol (Alpy et al, 2001); and it mediates homotypic as well as heterotypic interactions between STARD3 and STARD3NL (Alpy et al, 2005). Of note is that the MENTAL domain interacts with cholesterol in vivo (Alpy et al, 2005; Hulce et al, 2013) supporting the notion that this domain is a sterol reservoir. Along with this, we previously revealed that STARD3 has an FFAT (two phenylalanines in an acidic tract) motif (Loewen et al, 2003) located at the carboxy terminal end of the MENTAL domain and upstream of the START domain (Alpy et al, 2013). We then found that STARD3, anchored at late endosomes, uses this FFAT motif to interact with ER‐localized VAP‐A and VAP‐B (vesicle‐associated membrane protein‐associated protein) proteins and create zones of close apposition between the ER network and endosomes (Alpy et al, 2013). Such an ability to create contact sites is reminiscent to what is known for CERT, alias STARD11. This protein is required for sphingomyelin (SM) synthesis by acting as a ceramide transporter, which shuttles this lipid between the ER and the Golgi apparatus (Hanada et al, 2003). Although the exact mechanism is not known, it is widely accepted that CERT ensures its function in ER–Golgi contacts by connecting the Golgi apparatus and the ER via its Pleckstrin Homology (PH) domain and its FFAT motif, respectively (Hanada, 2010). The characterization of CERT which showed the association of two distinct activities within the same protein, the ability to transport lipids, and to connect membranes, brought a novel paradigm to explain how lipid fluxes are guided inside cells (Munro, 2003).
By analogy with StAR and CERT, we speculated that STARD3 connects the ER and endosomes to pilot sterol transfer between these organelles. This prompted us to clarify STARD3 function by investigating how this protein modulates intracellular cholesterol distribution. Here, using a combination of cholesterol labeling and microscopy approaches and in vitro reconstitution, we provide evidence that STARD3 transports sterols from the ER to the endosome. Furthermore, we show that its activity depends both on STARD3's ability to bind sterols and to create membrane contact sites. Finally, we show that STARD3 acts as a lipid transfer protein that redirects sterol to the endosome at the expense of the PM and favors membrane formation inside endosomes. Overall, our results describe a new pathway for sterol fluxes within eukaryotic cells and additionally provide data strengthening the concept that a reduced intermembrane distance at organelle contacts allows a highly efficient transport of lipid.
Results
STARD3 expression results in cholesterol accumulation in endosomes
Having previously established that STARD3 has an ER–endosome tethering activity and contains a sterol‐binding domain (Alpy et al, 2013; Wilhelm et al, 2016), we surmised that the creation of extended ER–endosome contacts by STARD3 might significantly alter cholesterol distribution inside cells. To explore this possibility, we devised novel assays for quantifying free cholesterol, not only at the PM, but also inside cells, using two specific fluorescent probes: the D4 fragment of perfringolysin O (θ toxin, Clostridium perfringens) fused to GFP (GFP‐D4) (Ohno‐Iwashita et al, 2004; Abe et al, 2012) and the polyene macrolide filipin from Streptomyces filipinensis (Wüstner et al, 2012). We identified experimental conditions under which these two probes can be efficiently used to label sterols in the PM or in subcellular structures such as endosomes (Fig EV1). We then used these probes to compare sterol distribution in control and STARD3‐expressing cells. Using GFP‐D4, we first observed that in parental and in control HeLa cells (thereafter named HeLa/Ctrl), which both expressed very low amounts of STARD3 (Fig 1Ab and f), endosomes labeled by Lamp1 were not enriched in cholesterol (Fig 1Aa–h). In contrast, in HeLa cells that stably expressed STARD3 (HeLa/STARD3, Fig 2B), STARD3 localized in Lamp1‐labeled endosomes, and these endosomes were also strongly labeled with the cholesterol probe (Fig 1Ai–l). In fact, most GFP‐D4‐positive vesicles were also positive for STARD3 and Lamp1 (Fig 1B), and GFP‐D4 and STARD3 signals were correlated (Fig 1E). Since STARD3 is a protein from late endosomes (Alpy et al, 2001), we characterized STARD3‐positive endosomes using a series of endosomal markers including EEA1, Rab7, CD63, and the lipid BMP. As shown in Fig EV2, STARD3 strongly co‐localizes with all late endosome markers and not with the early endosomal protein EEA1 (Fig EV2A and B). This suggested that high STARD3 expression promotes free sterol accumulation in a STARD3‐positive late endosomal compartment (Fig 1Ai–l). Moreover, as GFP‐D4 binds only to membranes containing more than 35 mol% sterol (Ohno‐Iwashita et al, 2004), this result indicated a massive accumulation in endosomes.
Figure EV1. Cholesterol staining with GFP‐D4 or filipin.
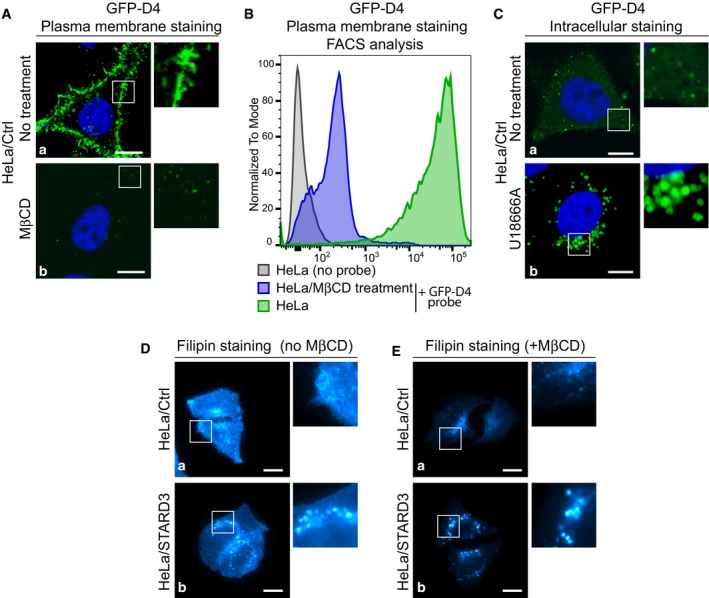
- Plasma membrane cholesterol staining with the GFP‐D4 probe. Live HeLa/Ctrl cells were left untreated (a) or treated with MβCD (b) to remove plasma membrane cholesterol (10 mM in serum‐free medium; 30 min at 37°C), and incubated with GFP‐D4 prior to fixation and nucleus staining (blue). GFP‐D4 highly stained the plasma membrane of untreated cells (a), while almost no staining was present on MβCD‐treated cells (b).
- Analysis by flow cytometry of plasma cholesterol membrane staining with the GFP‐D4 probe. HeLa cells were either left untreated and unstained (HeLa/no probe), untreated (HeLa + GFP‐D4 probe) or treated with MβCD (HeLa/MβCD treatment + GFP‐D4 probe), and next stained; cells were then analyzed by flow cytometry. These representative histograms display the number of cells analyzed (normalized to mode) as a function of GFP‐D4 fluorescence (log intensity). Note that HeLa cells are strongly labeled with the GFP‐D4 probe; MβCD treatment prior to labeling strongly decreases GFP‐D4 signal intensity.
- Intracellular cholesterol staining with GFP‐D4. HeLa/Ctrl cells were left untreated (a) or treated with U18666A (1 μg/ml; 1 h at 37°C) to promote intracellular cholesterol accumulation (b). After fixation, cells were permeabilized by freezing in liquid nitrogen and incubated with GFP‐D4. Untreated cells (a) were labeled on small discrete structures by the GFP‐D4 probe; in U18666A‐treated cells (b), cholesterol‐filled endosomes were strongly labeled by the GFP‐D4 probe.
- Whole‐cell cholesterol staining with filipin on fixed HeLa/Ctrl (a) and HeLa/STARD3 (b) cells. Filipin stains cholesterol in the plasma membrane and in intracellular compartments. Note that HeLa/STARD3 cells display intracellular puncta of filipin staining.
- Intracellular cholesterol staining with filipin. Live HeLa/Ctrl (a) and HeLa/STARD3 (b) cells were treated with MβCD (10 mM in serum‐free medium; 30 min at 37°C) prior to fixation and filipin staining. MβCD treatment removed cholesterol from the plasma membrane and allowed a better visualization of intracellular cholesterol pools.
Figure 1. STARD3 favors cholesterol accumulation in endosomes.
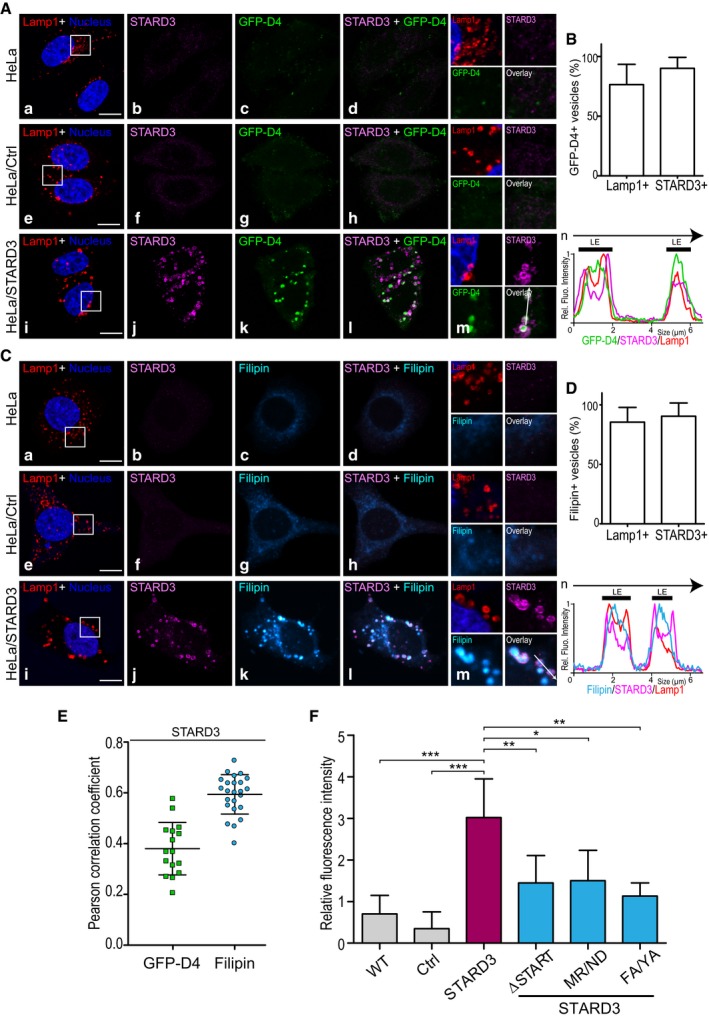
- To follow cholesterol accumulation in late endosomes, HeLa (a–d), HeLa/Ctrl (e–h), and HeLa/STARD3 (i–l) cells were labeled with anti‐Lamp1 antibodies (red), anti‐STARD3 antibodies (magenta), and with the fluorescent cholesterol probe GFP‐D4 (green). Nuclei were stained in blue. Merged image of GFP‐D4 and STARD3 signals is shown in (d, h, and l). The subpanels on the right are higher magnification (2.5×) images of the area outlined in white (a, e, i). Overlay indicates GFP‐D4 and STARD3 merged image. (n) Linescan analyses with relative fluorescence intensities of the green, magenta, and red channels along the arrow in (m) (HeLa/STARD3 cell). Black thick lines indicate the positions of late endosomes (LE).
- Colocalization between GFP‐D4‐positive vesicles and Lamp1 and STARD3 was quantified in HeLa/STARD3 cells (12 cells).
- Filipin as a second method to follow cholesterol accumulation in late endosomes. HeLa (a–d), HeLa/Ctrl (e–h) and HeLa/STARD3 (i–l) cells were labeled with anti‐Lamp1 antibodies (red), anti‐STARD3 antibodies (magenta), and with the fluorescent cholesterol probe filipin (Cyan Hot). Nuclei are stained in blue. Merged image of filipin and STARD3 signals is shown in (d, h and l). Shown on the right are higher magnification (2.5×) images of the area outlined in white (a, e, i). The filipin and STARD3 merged image is labeled Overlay. (n) Linescan analyses with relative fluorescence intensities of the cyan, magenta, and red channels along the arrow in (m) (HeLa/STARD3 cell). Black thick lines indicate the positions of late endosomes.
- Colocalization between filipin‐positive vesicles and Lamp1 and STARD3 was quantified in HeLa/STARD3 cells (10 cells).
- Pearson correlation coefficients between STARD3 and GFP‐D4 (left) or filipin (right) staining are shown. Each dot represents a single cell (GFP‐D4: 16 cells; filipin: 24 cells; from three independent experiments).
- Relative fluorescence intensity of intracellular filipin signals in HeLa, HeLa/Ctrl, HeLa/STARD3, HeLa/STARD3 ΔSTART, HeLa/STARD3 MR/ND, and HeLa/STARD3 FA/YA cells. n: number of independent experiments. HeLa, HeLa/Ctrl, HeLa/STARD3, HeLa/STARD3 ΔSTART: n = 6; HeLa/STARD3 MR/ND and HeLa/STARD3 FA/YA: n = 3. Total number of cells analyzed: HeLa: 309; HeLa/Ctrl: 234; HeLa/STARD3: 295; HeLa/STARD3 ΔSTART: 238; HeLa/STARD3 MR/ND: 116 and HeLa/STARD3 FA/YA: 137. Number of cells analyzed per sample per experiment ≥ 32.
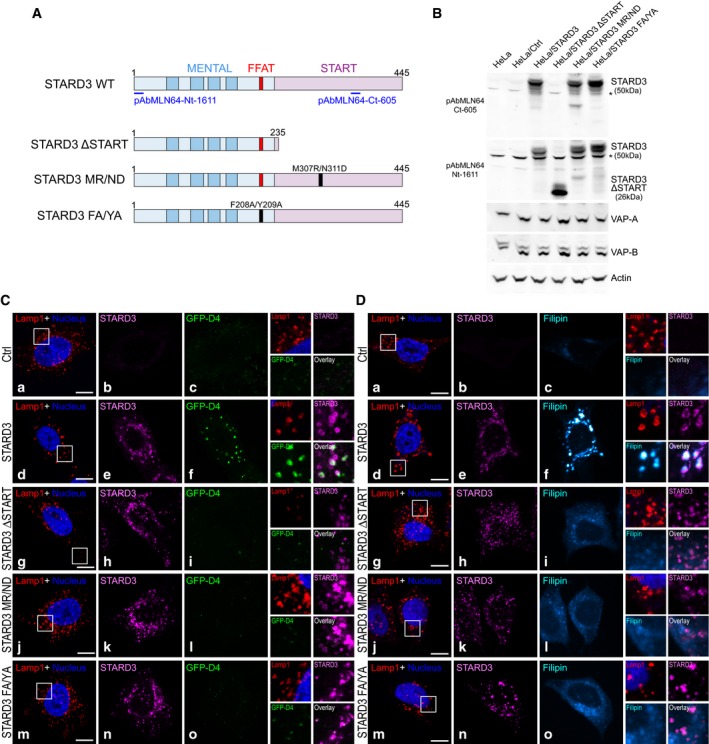
Figure 2. STARD3 needs a functional START domain and ER–endosome contacts to induce cholesterol accumulation in endosomes.
-
ASchematic representation of the different STARD3 mutants used in the study. The MENTAL domain in light blue contains 4 transmembrane helices (dark blue) and a FFAT motif (red); the START domain in pink contains two essential residues involved in cholesterol binding (M307 and N311). Positions of the epitopes recognized by the rabbit polyclonal pAbMLN64‐Nt‐1611 and pAbMLN64‐Ct‐605 antibodies are shown. Point mutation positions are labeled in black.
-
BWestern blot analysis of STARD3 expression in the different cell lines. The expression of VAP proteins is unchanged; actin was used as a loading control. *: unspecific band.
-
C, DTo mark cholesterol accumulation in endosomes, HeLa/Ctrl (a–c), HeLa/STARD3 (d–f), HeLa/STARD3 ΔSTART (g–i), HeLa/STARD3 MR/ND (j–l), and HeLa/STARD3 FA/YA (m–o) were labeled with anti‐Lamp1 antibodies (red), anti‐STARD3 antibodies (magenta), and with the fluorescent cholesterol probe GFP‐D4 (C: green) or filipin (D: Cyan Hot). Nuclei are stained in blue. Higher magnification (2.5×) images of the area outlined in white (a, d, g, j, m) are shown on the right. The GFP‐D4 and STARD3 merged image (C) and the filipin and STARD3 merged image (D) are labeled Overlay.
Figure EV2. Characterization of cholesterol‐enriched endosomes in HeLa/STARD3 cells.
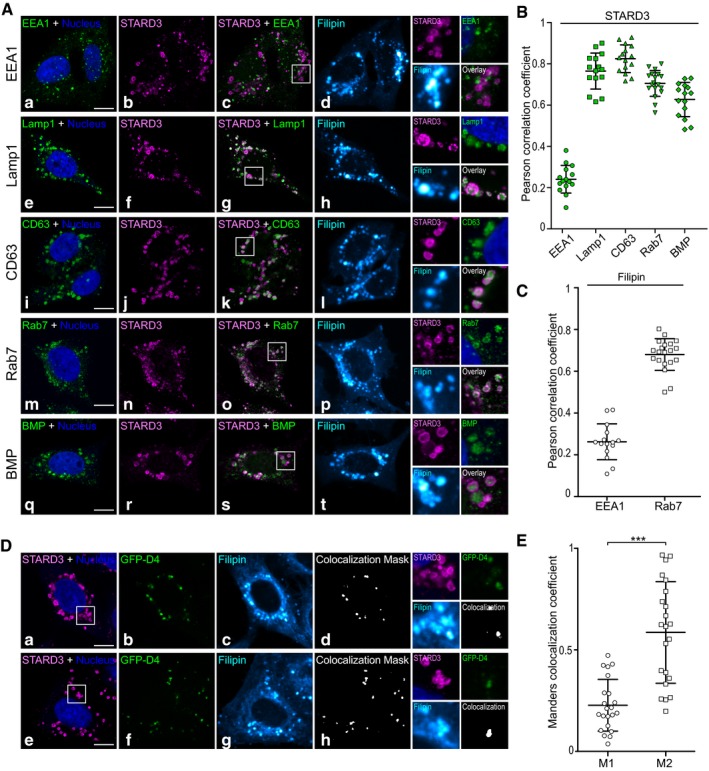
- HeLa/STARD3 cells were co‐labeled with anti‐STARD3 (magenta), with the fluorescent cholesterol probe filipin (Cyan Hot) and with anti‐EEA1 (a–d, green), anti‐Lamp1 (e–h, green), anti‐CD63 (i–l, green), anti‐Rab7 (m–p, green), or anti‐BMP (q–t, green) antibodies. Nuclei were stained in blue. Merged image of magenta and green signals is shown in (c, g, k, o, and s). The subpanels on the right are higher magnification (2.6×) images of the area outlined in white (c, g, k, o, s). Overlays show STARD3 and the endocytic markers merged images. Scale bars: 10 μm.
- Pearson correlation coefficients between STARD3 and the endocytic markers EEA1, Lamp1, CD63, Rab7, and BMP are shown; each dot represents one single cell; cells originates from three independent experiments (15 cells). The horizontal lines show the mean ± SD.
- Pearson correlation coefficients between filipin and the early endosome marker EEA1 (left) and with the late endosome marker Rab7 (right). Each dot represents one single cell (left 15 cells, right: 20 cells); cells originate from three independent experiments. The horizontal lines show the mean ± SD.
- HeLa/STARD3 cells were co‐stained with two fluorescent cholesterol probes (GFP‐D4 in green and filipin in Cyan Hot) and with anti‐STARD3 (magenta). Two similar cells are shown (bottom and top). Nuclei were stained in blue. (d, h) Pixels where the green and the Cyan Hot channels co‐localize are shown in white. The subpanels on the right are higher magnification (2.6×) images of the area outlined in white (a, e). Scale bars: 10 μm.
- Mander's correlation coefficients between filipin and GFP‐D4 are shown; each dot represents one single cell acquired from three independent experiments (22 cells). M1 corresponds to the fraction of filipin signal overlapping with GFP‐D4 signal. M2 corresponds to the fraction of GFP‐D4 signal overlapping with filipin signal. GFP‐D4‐labeled structures are strongly labeled with filipin, while filipin‐positive structures can be GFP‐D4 positive or GFP‐D4 negative. This illustrates the different properties of the two cholesterol probes: While filipin binds to all free cholesterol, GFP‐D4 only binds to cholesterol‐rich membranes. The horizontal lines show the mean ± SD. Paired two‐tailed t‐test; ***P < 0.0001.
To substantiate this finding, we measured sterol accumulation by repeating the experiments using filipin as a cholesterol probe. Immunolabeling of Lamp1 together with filipin staining in control cells confirmed that endosomes contained low cholesterol amounts (Fig 1Ca–h). By contrast, in STARD3‐expressing cells, Lamp1‐ and STARD3‐positive endosomes accumulated free cholesterol (Fig 1Ci–l and D). In fact, most filipin‐positive vesicles were also positive for Lamp1 and STARD3 (Fig 1D), and filipin and STARD3 signals were highly correlated (Fig 1E). Accordingly, filipin staining was strongly correlated with the late endosome marker Rab7 but not with the early endosome marker EEA1 (Fig EV2C). The two cholesterol probes filipin and GFP‐D4 have different properties: While filipin binds to all free cholesterol, GFP‐D4 only binds to cholesterol‐rich membranes (Ohno‐Iwashita et al, 2004). As anticipated, co‐labeling experiments using filipin and GFP‐D4 in HeLa/STARD3 cells showed that filipin labeled more endosomal vesicles than GFP‐D4 (Fig EV2D and E), thus reflecting the heterogeneity of cholesterol accumulation levels between individual endosomes. To quantify intracellular cholesterol, we set up an image analysis protocol based on a semi‐automated image segmentation and quantitative analysis of intracellular filipin staining (Fig EV3), which allowed a systematic measure of intracellular sterol levels in many cells. As shown in Fig 1F, while control cells had little intracellular filipin staining, HeLa/STARD3 cells displayed ~4 times higher intracellular filipin staining. Jointly, these results indicated that STARD3 elicits a substantial accumulation of cholesterol in endosomes.
Figure EV3. Image analysis procedure for filipin staining intensity quantification.

- Fields containing about a dozen of cells labeled with filipin, anti‐STARD3, and propidium iodide (PI) were randomly acquired by confocal microscopy in three‐channel images.
- Image analysis was performed with Fiji (http://fiji.sc/). Image segmentation was performed on the PI staining image. An intensity threshold was applied to determine the cell contours. Nuclei positions, manually selected on the PI staining image, were used to divide the image into discrete areas with a watershed algorithm (Find maxima). Combination of the thresholded and the segmented particles images allowed to build a segmentation mask where individual cell contours could be determined.
- To quantify the filipin staining intensity, an intensity threshold was first applied on raw images. This threshold allowed to focus the analysis on filipin accumulation puncta. After applying the segmentation mask onto the filipin staining thresholded image, filipin staining intensity was measured in individual cells.
STARD3 triggers cholesterol accumulation in endosomes and promotes internal membrane formation
It has been widely assumed for lipid transfer proteins notably STARD3, that the ability to reduce intermembrane distance by tethering should facilitate lipid transfer between organelles (Loewen et al, 2003). Taking advantage of the unique phenotype observed in HeLa/STARD3 cells, we explored this question at the level of ER–endosome contact sites. We first assessed whether the buildup of cholesterol in endosomes caused by STARD3 was dependent on its sterol binding START domain, and then on its ability to form ER–endosome contacts.
First, we used a mutagenesis approach to generate STARD3 mutants, which are unable to bind sterols. Two mutants were created: A mutant lacking the entire START domain (STARD3 ΔSTART); and a mutant bearing two point mutations in the START domain (STARD3 M307R/N311D, herein referred to as MR/ND) described as being deficient in sterol binding (Tsujishita & Hurley, 2000; Holtta‐Vuori et al, 2005) (Fig 2A). The sterol transport capacity of the START domain with the MR/ND mutation was assessed in vitro using liposomes (Fig EV4). Transfer was measured with a FRET assay using DHE, a fluorescent mimetic of cholesterol, and a second fluorescent lipid, DNS‐PE (John et al, 2002; Moser von Filseck et al, 2015a). As the decrease in FRET between the two lipids is a function of DHE transport from the donor to the acceptor liposome, the amount of sterol that is transported can be quantified in real time. We found that the isolated START domain of STARD3 transfers DHE with an initial rate of ~2 molecules/min (Fig EV4), which is consistent with a previous study (Zhang et al, 2002). By contrast, the MR/ND mutant was totally devoid of sterol transfer activity (Fig EV4A and B). We then established stable cell lines expressing these mutants (HeLa/STARD3 ΔSTART and HeLa/STARD3 MR/ND) to study both ER–endosome tethering and in vivo endosomal cholesterol accumulation. Mutant and wild‐type (WT) proteins were expressed at similar levels (Fig 2B). We observed that the STARD3 ΔSTART and MR/ND mutants, as they still possess the FFAT‐like motif and the MENTAL domain (Fig 2A), kept their ability to generate ER–endosome contacts (Fig EV5). In situ staining using the GFP‐D4 probe or filipin showed that intracellular accumulation of cholesterol in cells expressing the two sterol transfer‐deficient mutants was highly decreased compared to that observed with WT STARD3 (Fig 2C and D). Moreover, quantitative image analysis of filipin staining of more than 100 cells indicated that sterol accumulation was lowered in endosomes bearing mutant STARD3 (Fig 1F).
Figure EV4. In vitro sterol transport activity of WT and MR/ND mutant START domain of STARD3.
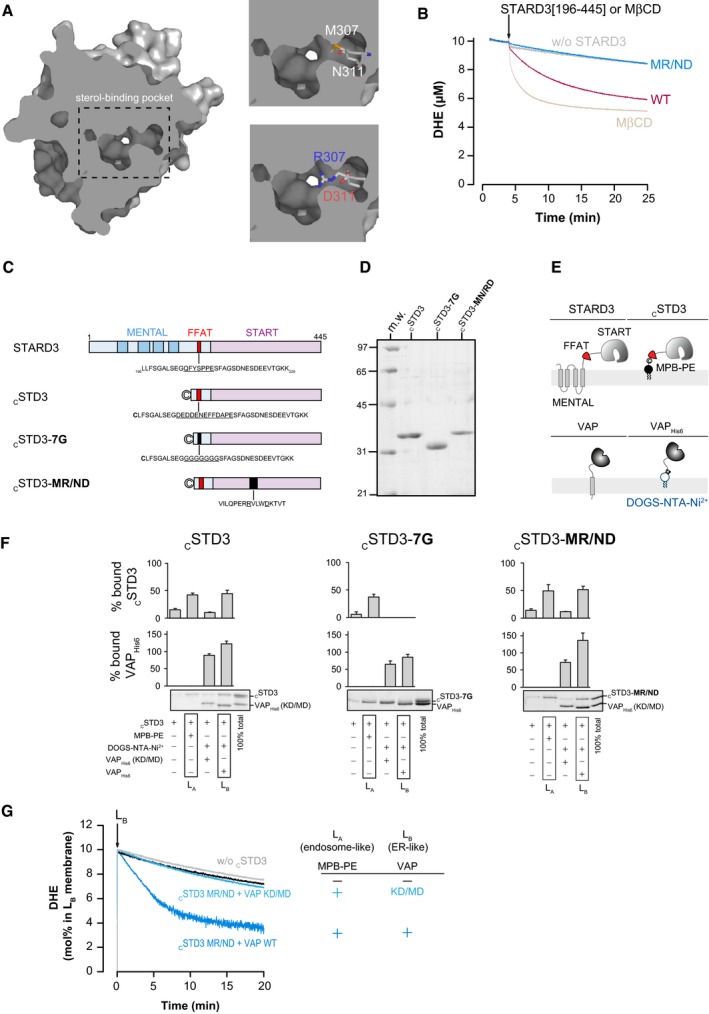
- Structure of the START domain of STARD3 (PDB entry 1EM2). Top right image: M307 and N311 residues, which are localized in the sterol‐binding pocket, are shown in stick. Bottom right image: a mutated form of the START domain is shown with the M307R/N311D double mutation. Carbon atoms are shown in white, oxygen in red, nitrogen in blue, and thiol in yellow.
- DHE transfer assay. DOPC liposomes (100 μM total lipids) containing 10 mol% DHE and 2.5 mol% DNS‐PE (donor) were mixed with a similar amount of DOPC liposome (acceptor) at 37°C. After 3 min, cSTD3 which corresponds to STARD3[196–445] (500 nM) or its mutated version (MR/ND) was injected. Negative and positive experiments are done in the absence of the protein and in the presence of 1 mM MβCD, respectively. The signal was converted into amount of DHE present in donor liposome (in μM). The slow decay observed without the protein (gray line) was due to spontaneous DHE transfer. The lipid binding mutant was totally deficient in cholesterol transport (blue line). The non‐mutated version is able to transfer sterol (magenta line). MβCD extracted sterol from donor liposomes (brown line).
- Domain organization of STARD3 and description of the different cSTD3 recombinant proteins used in the study.
- SDS–PAGE gel of purified cSTD3 constructs. The gel was stained with Sypro Orange to visualize proteins and molecular weight markers.
- Position of cSTD3 and VAPHis6 with respect to the membrane compared to the respective cytosolic region of the full‐length proteins.
- Flotation assays. cSTD3 (750 nM) was incubated with DOPC liposomes, doped or not with 3 mol% MPB‐PE (LA liposome), or with liposomes containing 2 mol% DOGS‐NTA‐Ni2+ (LB liposome) and decorated with VAPHis6 or VAP(KD/MD)His6. After centrifugation, the liposomes were recovered by centrifugation at the top of a sucrose cushion and analyzed by SDS–PAGE. The amount of protein recovered in the top fraction (lanes 1–4) was quantified, and the fraction of liposome‐bound cSTD3 and VAPHis6 as reported in the upper panels was determined by using the content of lane 5 (total 100%) as a reference. Mean ± SEM; n = 3 independent experiments (cSTD3 MR/ND and cSTD3 7G mutants) or 4 independent experiments (cSTD3).
- DHE transport assay using mutant STARD3 MR/ND. DOPC liposomes (62.5 μM total lipids, LA) containing or not 3 mol% MPB‐PE were mixed with cSTD3‐MR/ND (475 nM). After 5 min, LB liposomes (DOPC/DOGS‐NTA‐Ni2+/DNS‐PE/DHE 85.5/2/2.5/10 mol/mol, 62.5 μM total lipids), naked or covered with 500 nM VAPHis6 (dark blue) or VAP(KD/MD)His6 (light blue), were added. It is noteworthy that connecting LA and LB liposomes is sufficient to induce some sterol transfer in the presence of mutated STARD3 (cSTD3 MR/ND) (dark blue). Possibly, a close apposition of membranes induced by the tethering allows a faster spontaneous sterol transfer.
Figure EV5. Structure/function studies of STARD3 on VAP recruitment in ER–endosome contacts, and on endosome ultrastructure.
-
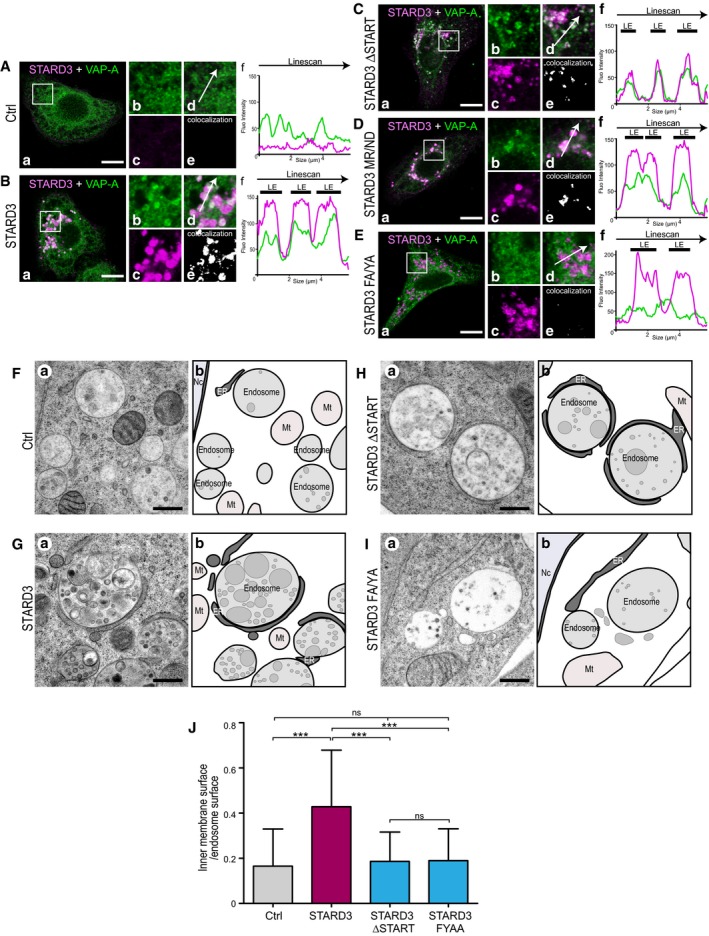
A–EHela/Ctrl (A), HeLa/STARD3 (B), HeLa/STARD3 ΔSTART (C), HeLa/STARD3 MR/ND (D), and HeLa/STARD3 FA/YA (E) cells were labeled with anti‐STARD3 (pAbMLN64‐Nt‐1611; magenta) and with anti‐VAP‐A (green) antibodies. Subpanels: (a) Merged image of green and magenta signals. Scale bars: 10 μm. (b–d) Higher magnification (3×) of the area outlined in white in (a), showing the green (b), magenta (c), and merge (d) signals. (e) Pixels where the green and the magenta channels co‐localize are shown in white. (f) Linescan analyses showing fluorescence intensities of the magenta and green channels along the arrow in (d). Black thick lines indicate the positions of LEs. WT STARD3, STARD3 ΔSTART, and STARD3 MR/ND recruit VAP‐A in ER–endosome contacts while STARD3 FA/YA does not.
-
F–ITEM images (a) of control HeLa/Ctrl (F), HeLa/STARD3 (G), HeLa/STARD3 ΔSTART (H), and HeLa/STARD3 FA/YA (I) cells. (b) Schematic representation of images shown in (a); the ER, endosomes, and inner endosomal membranes are in dark, light, and medium gray. Scale bar: 400 nm. Please note that HeLa/STARD3 and HeLa/STARD3 ΔSTART cells exhibit extensive ER–endosome contacts while HeLa/STARD3 FA/YA cells only display few ER–endosome contacts, similarly to HeLa/Ctrl cells. Mt: mitochondria; Nc: nucleus.
-
JQuantification by stereology of relative inner endosomal membrane surface on TEM sections. Thirty‐eight endosome sections were quantified for each cell line. Mean ± SD; ***P < 0.001, Kruskal–Wallis with Dunn's multiple comparison test.
Next, to directly study the contribution of membrane contact sites in the cholesterol accumulation phenotype, a STARD3 mutant lacking a functional FFAT motif was constructed. This mutant STARD3 F207A/Y208A (herein referred to as FA/YA) was obtained by alanine replacement of the two FFAT‐like motif core aromatic residues (Fig 2A). A stable HeLa/STARD3 FA/YA cell line was generated (Fig 2B). As expected, this mutant was unable to make ER–endosome contacts (Fig EV5). Intracellular cholesterol levels and distribution were measured. We observed that the FA/YA mutant did not promote cholesterol accumulation in endosomes (Figs 2C and D, and 1F). This data supported the notion that the ability of STARD3 to scaffold ER–endosome contacts is essential for an efficient sterol transfer in endosomes.
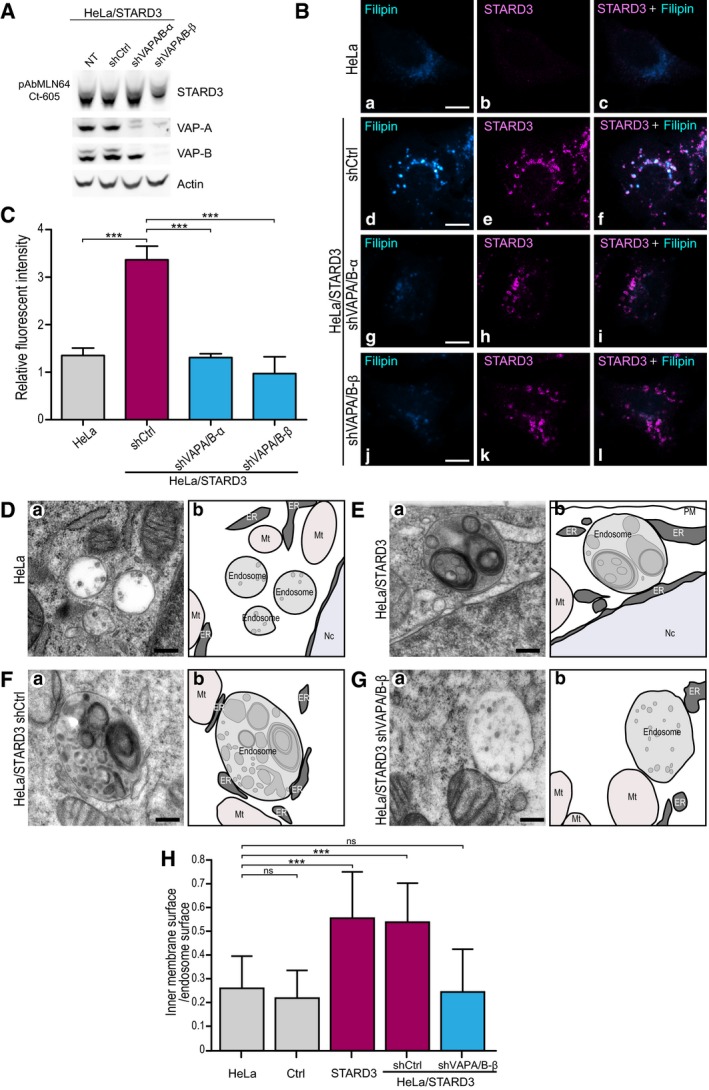
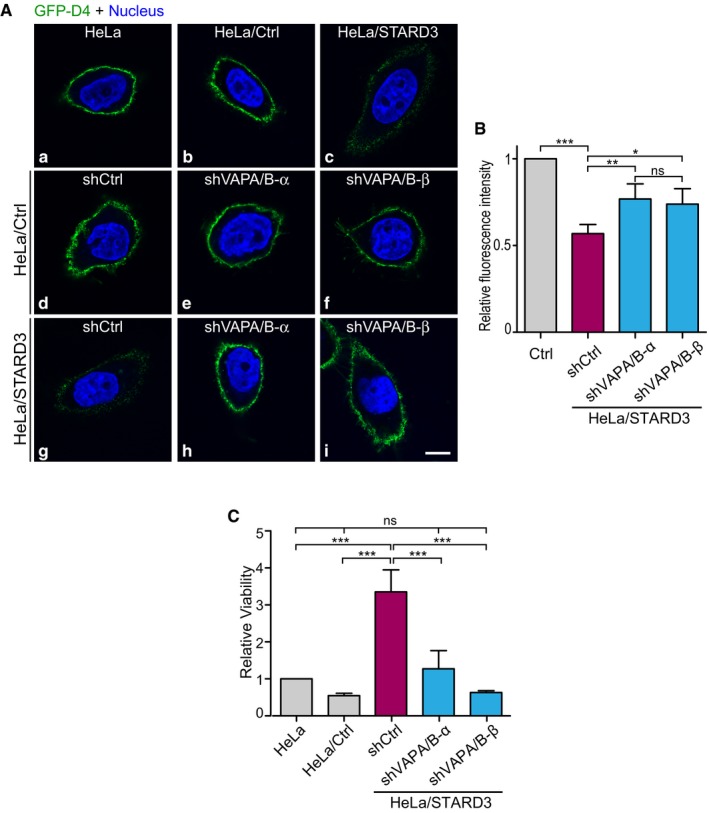
To further substantiate this idea, we knocked down the VAP proteins, the partners of STARD3 (Alpy et al, 2013), implicated in ER–endosome tethering in HeLa cells expressing STARD3 (HeLa/STARD3) (Fig 3A). The proteins VAP‐A and VAP‐B were downregulated using two sets of small hairpin RNAs (shRNAs), targeting distinct sequences (Fig 3A). Compared to non‐silenced cells (HeLa/STARD3/shCtrl), VAP‐A and VAP‐B levels were reduced by 80 and 30%, respectively, with one set of shRNAs (HeLa/STARD3/shVAPA/B‐α), and 84 and 85%, respectively, with the other set (HeLa/STARD3/shVAPA/B‐β) (Fig 3A). Compared with HeLa/STARD3 cells, HeLa/STARD3 silenced for VAP proteins did not accumulate cholesterol in endosomes as shown by in situ filipin labeling and quantification (Fig 3B and C).
Figure 3. VAP protein knockdown abolishes STARD3‐mediated cholesterol accumulation in endosomes.
-
AWestern blot analysis of VAP‐A and VAP‐B expression in untreated (NT) HeLa/STARD3 cells or HeLa/STARD3 expressing a control shRNA (shCtrl) or two pairs of shRNAs targeting VAP‐A and VAP‐B (shVAP‐A/B‐α or shVAP‐A/B‐β). Actin was used as a loading control.
-
BHeLa cells (a–c), and HeLa/STARD3 cells expressing a control shRNA (shCtrl; d–f) or two pairs of shRNAs targeting VAP‐A and VAP‐B [shVAP‐A/B‐α (g–i) or shVAP‐A/B‐β (j–l)] were labeled with anti‐STARD3 antibodies (magenta) and with the fluorescent cholesterol probe filipin (Cyan Hot). Merged images of filipin and STARD3 signals are shown in (c, f, i and l). Scale bars: 10 μm.
-
CRelative fluorescence intensity of intracellular filipin signal in HeLa, HeLa/STARD3/shCtrl, HeLa/STARD3/shVAP‐A/B‐α, and HeLa/STARD3/shVAP‐A/B‐β. n = 3 independent experiments. Total number of cells analyzed: HeLa: 84; HeLa/STARD3/shCtrl: 130; HeLa/STARD3/shVAP‐A/B‐α: 109; HeLa/STARD3/shVAP‐A/B‐β: 92. Number of cells analyzed per sample per experiment ≥ 25. Mean ± SD; ***P < 0.001, ANOVA with Tukey's multiple comparison test.
-
D–GTEM images (a) of control HeLa cells (D), HeLa/STARD3 cells (E), or HeLa/STARD3 cells expressing a control shRNA (F) or a pair of shRNAs targeting VAP‐A and VAP‐B (G). Scale bars: 200 nm. Schematic representation (b) of images shown in (a); the ER, endosomes, and intraluminal membranes are in dark, light, and median gray, respectively. Mt: mitochondria; Nc: nucleus; PM: plasma membrane.
-
HQuantification by stereology of relative intraluminal membrane surface on TEM sections. Fifty (HeLa; HeLa/Ctrl; HeLa/STARD3) and 25 (HeLa/STARD3/shCtrl; HeLa/STARD3/shVAP‐A/B‐β) endosome sections were quantified. Mean ± SD; ***P < 0.001, Kruskal–Wallis with Dunn's multiple comparison test.
To gain more insights into the biological role of STARD3 in endosomes, we characterized the ultrastructure of the endosomal compartment by transmission electron microscopy (TEM). TEM allows to get information on the inner organization of the endocytic compartment. In particular, it is known that endocytic vesicles enclose internal membranes termed intraluminal vesicles (ILVs) and multilamellar regions (Vacca et al, 2016). ILVs are small spherical structures of about 50 nm of diameter (Vacca et al, 2016). We thus addressed whether the increase in endosomal cholesterol resulting from the association between STARD3 and VAP impacts the inner membrane content of the endocytic compartment, by performing TEM analysis of control and HeLa/STARD3 cells (Fig 3D–H). We quantified internal membranes in all endocytic organelles containing at least an ILV, therefore including multivesicular bodies or endosomal carrier vesicles, and late endosomes, but excluding lysosomes. Compared to control cells, HeLa/STARD3 cells displayed endosomes with more internal membranes (Fig 3D, E and H). The overall morphology of the endocytic compartment was modified in STARD3 cells, notably MVB containing homogenous population of ILV were rarer, while pleomorphic late endosomes containing ILV and multilamellar or mixed multivesicular/multilamellar regions were frequently observed. This morphological alteration was consistent with our previous observations showing that STARD3 functions in organelle dynamics by altering the formation of endosomal tubules (Alpy et al, 2013). Strikingly, internal membrane quantification by stereology showed that compared to control cells, HeLa/STARD3 cells displayed endosomes with more inside membranes, namely ILVs or multilamellar or mixed multivesicular/multilamellar structures (Fig 3D, E and H). To understand the molecular mechanism involved in this process, we examined the ultrastructure of the endocytic compartment in VAP‐silenced cells devoid of STARD3‐mediated ER–endosome contacts (Fig 3F–H). While HeLa/STARD3/shCtrl cells also showed increased inner membrane content in endosomes, HeLa/STARD3/shVAPA/B‐β exhibited fewer inner membranes in the endosomes. To substantiate this result, we analyzed the endosome ultrastructure in cells expressing the STARD3 ΔSTART mutant (Fig EV5H), which is unable to bind sterol, and in cells expressing the STARD3 FA/YA mutant, which is unable to generate ER–endosome contacts (Fig EV5I). Compared to HeLa/STARD3 cells (Fig EV5G), which displayed endosomes with more internal membranes, HeLa/STARD3 ΔSTART and HeLa/STARD3 FA/YA cells possessed endosomes with internal membrane structures similar to control cells (Fig EV5J). These observations suggest that STARD3‐mediated cholesterol transport provides building blocks for membrane formation within endocytic vesicles.
Overall, these results demonstrate that STARD3‐mediated cholesterol buildup in endosomes directly depends on STARD3 START domain, and on its ability to scaffold ER–endosome contacts. At the ultrastructural level, STARD3 expression is associated with an alteration of the endosomal compartment with more inner membranes either in the form of ILVs or multivesicular/multilamellar structures. This increased formation of inner membranes is associated with STARD3's ability to accumulate cholesterol in endosomes.
STARD3 and VAP interaction couples tethering and sterol transfer between model organelles
These in vivo experiments suggested that STARD3, with the help of VAP, autonomously connects the ER network to endosomes to pilot ER‐to‐endosome cholesterol transport. To fully demonstrate that STARD3 and VAP are necessary and sufficient to efficiently transfer sterols between these two organelles, we reconstituted in vitro the tethering complex using recombinant proteins and liposomes (Fig 4A).
Figure 4. Coupling of DHE transport by STARD3 with membrane tethering.
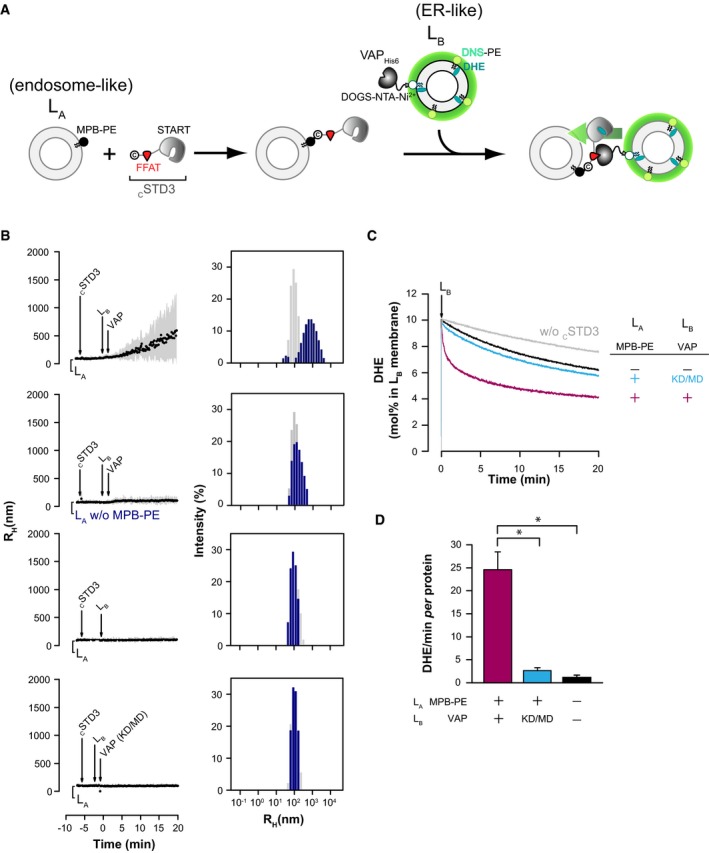
- Description of the experimental strategy. For DLS experiments, LA liposomes (endosome‐like) are decorated with cSTD3 owing to covalent links with MPB‐PE lipids, and mixed with LB liposomes (ER‐like) covered by VAPHIS6 attached to DOGS‐NTA‐Ni2+. For DHE transport experiment, LB liposomes also contain DHE and a dansylated lipid (DNS‐PE). The transport of DHE from LB to LA liposomes is followed by FRET.
- Aggregation assays in real time. Liposomes (50 μM total lipids), decorated or not with cSTD3 (380 nM), were mixed with LB liposomes (50 μM total lipids) naked or covered with VAPHis6 or VAP(KD/MD)His6 (380 nM). Aggregation was followed by DLS. Left panels: mean radius and polydispersity (shaded area) over time. Right panels: size distribution before (gray bars) and after (blue bars) the reaction. Please note that only the condition presented on the top graphs show increased polydispersity and mean radius over time.
- DHE transport assay. DOPC liposomes (62.5 μM total lipids, LA) containing or not 3 mol% MPB‐PE were mixed with cSTD3 (475 nM). After 5 min, LB liposomes (DOPC/DOGS‐NTA‐Ni2+/DNS‐PE/DHE 85.5/2/2.5/10 mol/mol, 62.5 μM total lipids), naked or covered with 500 nM VAPHis6 or VAP(KD/MD)His6, were added. FRET between DHE and DNS‐PE in the LA liposomes diminishes as DHE is transported to the LA liposomes. The signal was converted into amount of DHE present in LB liposomes (in mol%). Please note that a slow transport of DHE is observed without cSTD3 (gray line) due to spontaneous DHE transfer. The black and blue lines show the modest transfer activity of the untethered cSTD3. The magenta line show the rapid transfer activity of the tethered cSTD3.
- Initial DHE transport rate measured with cSTD3 in the presence or the absence of MPB‐PE and functional VAP (475 nM). Mean ± SEM; n = 4 independent experiments; *P < 0.05, Mann–Whitney test.
To do so, we purified a shorter STARD3 protein, termed thereafter cSTD3, devoid of the transmembrane MENTAL domain but comprising a FFAT motif and the START domain. To anchor this protein to the membrane, we introduced a cysteine residue at its N‐terminal end (Fig EV4C and D) enabling its attachment to liposomes doped with thiol‐reactive MPB‐PE lipids. Thus, using this setting, the soluble part of STARD3 is positioned on liposomes like STARD3 at endosomes surface (Fig EV4E). In parallel, we purified VAPHis6 and the VAP(KD/MD)His6 mutant in which the C‐terminal transmembrane region is substituted by a short His‐tag. The double K94D/M96D mutation of VAP‐A (herein named KD/MD) was shown to abolish FFAT binding (Kaiser et al, 2005). The His‐tag enables the attachment of the soluble part of VAP to liposomes containing NTA‐Ni2+ lipids in an orientation similar to that of VAP proteins on the ER (Fig EV4E) (Mesmin et al, 2013b).
Using flotation assays, we verified the attachment of the cSTD3 protein to a preparation of liposomes (LA) containing 3 mol% of MPB‐PE (Fig EV4F). We also assessed that VAPHis6 and its mutated form were recruited to a second type of liposomes (LB) containing 2 mol% DOGS‐NTA‐Ni2+ and, moreover, that cSTD3 was specifically recruited to these liposomes via the FFAT motif in the presence of VAPHis6 but not of VAP(KD/MD)His6 mutant (Fig EV4F).
Then, using dynamic light scattering (DLS), we monitored the tethering activity of STARD3 by recording the size of the particles formed in the presence of the two liposome populations (Fig 4A). In brief, cSTD3 was attached to LA liposomes that were then mixed with LB liposomes and VAPHis6. A rapid increase in the initial mean radius (98 nm on average) up to 600 nm (Fig 4B) was observed upon VAPHis6 addition. Size distribution analysis at the end of the kinetics was indicative of the formation of liposome aggregates (Drin et al, 2008) with high polydispersity (1,008 ± 836 nm) at the expense of free liposomes (98.2 ± 36.5 nm). This experiment was repeated with LA liposomes devoid of MPB‐PE or LB liposomes either lacking VAP or covered with the VAP(KD/MD)His6 mutant. In all case, no aggregation was seen (Fig 4B). Jointly, these data indicated that the cytosolic part of STARD3 attached by its N‐terminus to one membrane can tether a second membrane only when the latter bears VAP.
Using these established experimental conditions in which cSTD3 promoted contacts between LA and LB liposomes, we measured DHE transfer in real time by FRET (John et al, 2002; Moser von Filseck et al, 2015a). LB liposomes including both DHE (10 mol%) and DNS‐PE (2.5 mol%) and covered with VAPHis6 were added to LA liposomes decorated with cSTD3. Strikingly, we observed a very fast DHE transport within the first seconds and DHE was fully equilibrated between liposome populations after a few minutes (Fig 4C, magenta curve). The initial DHE transport rate was 24.6 ± 3.8 DHE molecules/min per cSTD3 (mean ± SEM, n = 3, Fig 4D). In contrast, with LA and LB liposomes devoid of MBP‐PE (the anchor for cSTD3) and VAP, respectively, the transport was twenty times slower (Fig 4C, black curve; Fig 4D, 1.1 ± 0.54 DHE molecules/min per cSTD3) and DHE distribution failed to reach equilibrium. Likewise, the transport was inefficient with LB liposomes covered with VAP(KD/MD) (Fig 4C, blue curve; Fig 4D). Together, these data showed that STARD3 transfers sterols far more efficiently between two membranes while connecting them at the same time.
To substantiate the link between tethering and cholesterol transport, and to provide the biochemical basis for observations done in cells, we examined in vitro the activity of two cSTD3 mutants that are either unable to make membrane contacts or to transport cholesterol. To prevent the interaction with VAP, we generated a 7G mutant in which the FFAT motif is substituted by a stretch of glycine; to block sterol transport, we introduced an MR/ND mutation in cSTD3 (Fig EV4C and D). Each mutant can be attached to LA liposomes doped with MBP‐PE but, as expected, the cSTD3 7G failed to bind to VAP proteins at the surface of LB liposomes (Fig EV4F). Then, DLS assays showed that the 7G mutant failed to cause LA and LB aggregation (Fig 5A). Naturally, the MR/ND mutant START domain remained able to interact with VAP proteins and triggered aggregation (Fig 5A). In DHE transport assays, we found that both mutants had altered kinetics of sterol transport, with initial transport rates one order of magnitude lower than that of cSTD3 (Fig 5B and C). Of interest, the transport of DHE seen with the MR/ND mutant is likely due to a greater spontaneous DHE transfer resulting from membrane apposition (Fig EV4G). Of note, the transport of the 7G mutant (Fig 5B) is very similar to the transport kinetics of the soluble STARD3 protein (Fig EV4B).
Figure 5. Tethering and sterol binding contribute to STARD3 activity in vitro .
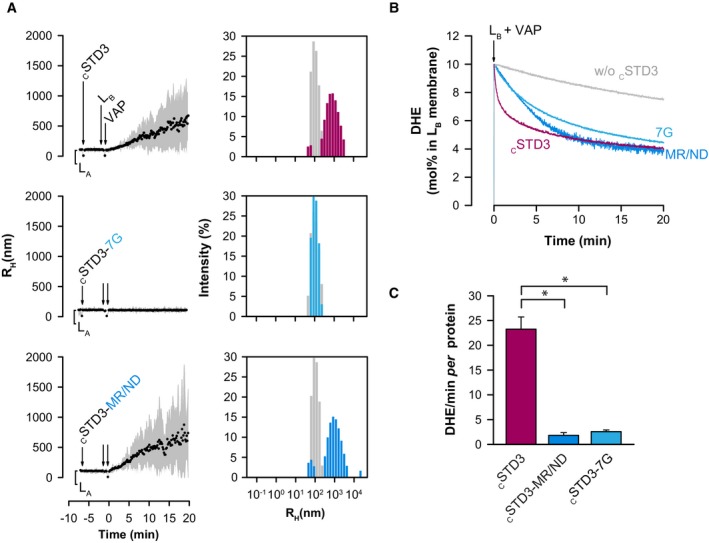
- Aggregation assays. LB liposomes (50 μM total lipids) covered with VAPHis6 (380 nM) were mixed with LA liposomes covered with cSTD3, cSTD3‐7G, or cSTD3‐MR/ND (380 nM). Left panels: mean radius and polydispersity (shaded area) over time. Right panels: size distribution before (gray bars) and after the reaction (colored bars).
- DHE transport assay. DOPC liposomes (62.5 μM total lipids, LA) containing 3 mol% MPB‐PE were mixed with cSTD3, cSTD3‐7G, or cSTD3‐MR/ND (475 nM). After 5 min, liposomes (DOPC/DOGS‐NTA‐Ni2+/DNS‐PE/DHE liposomes 85.5/2/2.5/10 mol/mol, 62.5 μM total lipids, LB), covered with 500 nM VAPHis6, were added. Please note the rapid transfer activity of the tethered cSTD3 (magenta line). A delayed and slower transport is observed both in untethered condition (light blue line) and with the lipid‐binding deficient mutant (dark blue line).
- Initial DHE transport rate measured with cSTD3, cSTD3‐7G, or cSTD3‐MR/ND. Mean ± SEM; n = 4 for cSTD3 and n = 3 for other cSTD3 constructs; *P < 0.05, Mann–Whitney test.
To conclude, using a completely defined in vitro assay enabling us to mimic organelle contacts and measure sterol transport in real time, we show that STARD3 and VAP represent an autonomous molecular machine able to rapidly transfer sterols using membrane contacts.
STARD3 expression does not activate transcriptional cholesterol sensing and favors ER cholesterol transport to endosomes
Given that STARD3 expression promoted cholesterol accumulation in endosomes, a further step was to see whether this phenotype was associated with global changes in cellular sterol levels. To address this question, the total cholesterol amount was quantified in lipid extracts originating from control cells, or cells expressing either WT STARD3 or MR/ND mutant STARD3. To ascertain the robustness of our dosage, cholesterol was also measured in lipid extracts of control cells treated with methyl‐β‐cyclodextrin (MβCD), a chemical known to remove cholesterol from the PM. As shown in Fig 6A, MβCD treatment resulted in the loss of about half of the total cholesterol in lipid extracts compared to untreated cells. In contrast, we found that the expression of STARD3 did not modify whole cholesterol levels compared to control cells or cells expressing MR/ND STARD3 (Fig 6A).
Figure 6. STARD3‐mediated cholesterol accumulation in endosomes does not alter cholesterol homeostasis.
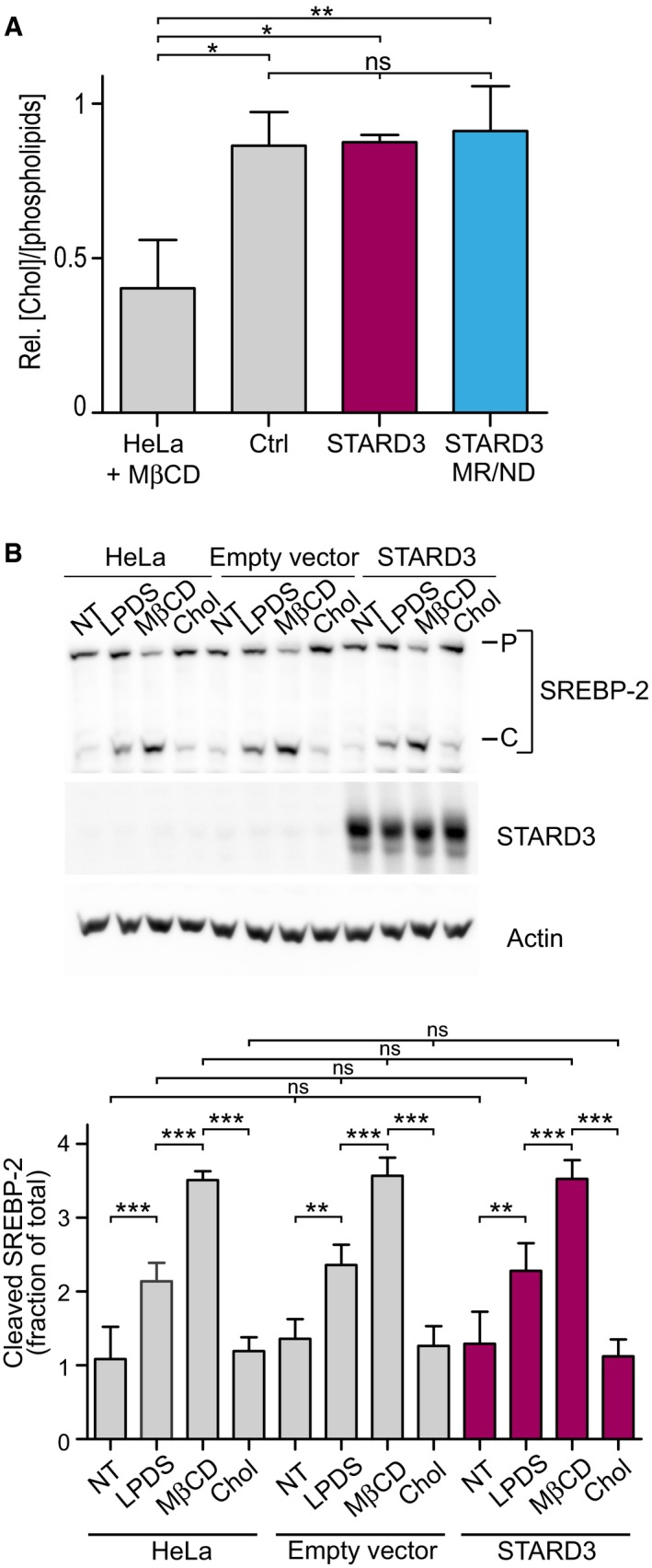
- Total free cholesterol quantification after total lipid extraction in control cells and in cells expressing STARD3 or the lipid binding mutant form of STARD3 (STARD3 MR/ND) (n = 3). Please note that control HeLa cells treated with MβCD have a significant depletion of total cholesterol (n = 2). Mean ± SD; ANOVA with Tukey's multiple comparison test.
- Western blot analysis of SREBP‐2 activation in controls (HeLa, HeLa/empty vector) or STARD3‐overexpressing HeLa cells. Cells were incubated for 2 h in DMEM culture medium supplemented with: 5% FCS (NT); 5% LPDS, 10 μM mevinolin (LPDS); 5% LPDS, 10 μM mevinolin, 1 mM MβCD (MβCD); 5% LPDS, 10 μM mevinolin, 500 μM cholesterol complexed to MβCD (MβCD‐Chol). The proteasome inhibitor MG132 (10 μM) was present in all conditions. P = precursor form of SREBP‐2; C = cleaved form of SREBP‐2. Lower panel: WB quantification in which cleaved SREBP‐2 is expressed as a fraction of total SREBP2 (P+C). Mean ± SD; n = 4 independent experiments; **P < 0.01, ***P < 0.001, ANOVA with Tukey's multiple comparison test.
Source data are available online for this figure.
Thereafter, we examined the activation of the sterol regulatory element‐binding proteins (SREBPs) in Hela cells expressing or not STARD3. SREBPs are a family of transcription factors that maintain cellular lipid homeostasis by activating genes involved in cholesterol and fatty acids synthesis and uptake (Horton et al, 2002). SREBP inactive precursors are localized in ER membranes and under low sterol conditions, are activated by cleavage (Brown & Goldstein, 1999). We analyzed the activation of SREBP‐2, which specifically controls genes involved in cholesterol metabolism (Horton et al, 2002). Under normal sterol conditions, no modification of SREBP‐2 cleavage was observed in STARD3‐expressing cells compared to control cells (Fig 6B). To study whether sterol sensing functions in STARD3 cells, we compared SREBP‐2 processing between control and STARD3‐expressing cells whose free cholesterol level was either lowered or increased. To activate the SREBP pathway by reducing cellular free cholesterol level, cells were grown in lipoprotein‐depleted medium (LPDS) or treated with MβCD. We observed a similar increase in SREBP‐2 cleavage in control and STARD3‐expressing cells (Fig 6B). Next, we increased cellular cholesterol by adding exogenous cholesterol; again this treatment had a similar effect in all cell lines, SREBP‐2 cleavage being maintained at a basal level both in STARD3‐expressing and in control cells (Fig 6B). These experiments show that endosomal cholesterol accumulation driven by STARD3 did not activate the cholesterol sensor SREBP‐2 and furthermore that cholesterol sensing was not altered in STARD3‐expressing cells (Fig 6B). Moreover, SREBP‐2 target genes (HMGCoA reductase, LDLR, and SREBF‐2) expression analysis by RT–qPCR showed that STARD3‐expressing cells responded to LDL addition in the culture medium similarly to control cells by lowering their level of expression (data not shown).
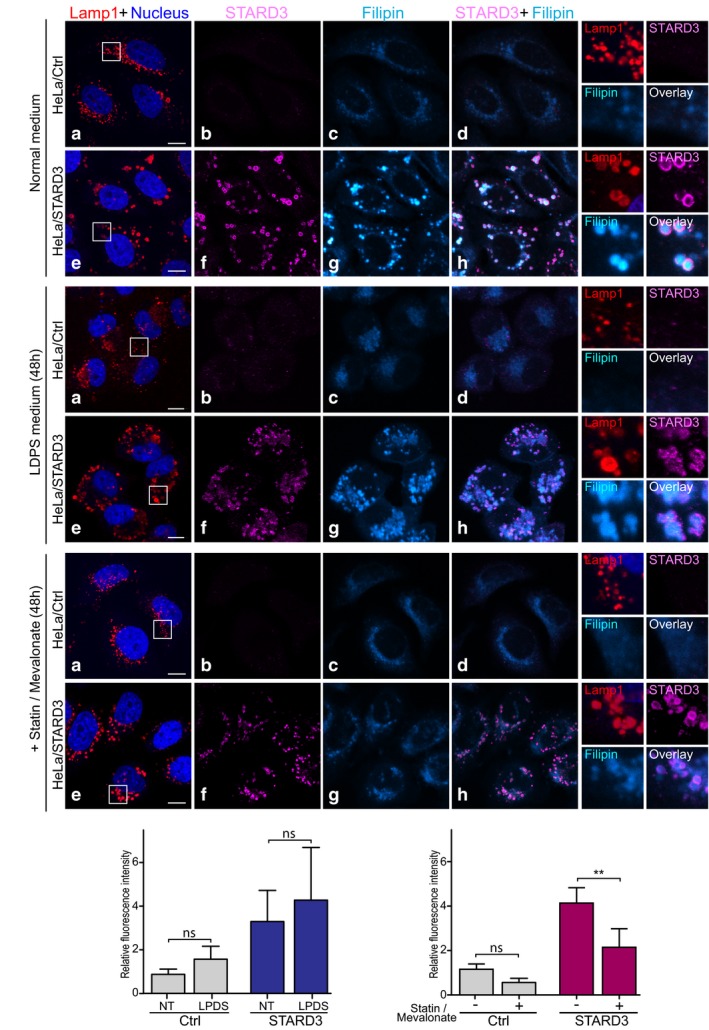
Last, we examined whether the pool of cholesterol accumulated in endosomes by STARD3 was originating from the LDL uptake or the neosynthesis pathway in the ER, both pathways providing cholesterol in HeLa cells (Ishitsuka et al, 2011). To test the contribution of the LDL pathway, cells were grown in lipoprotein‐deficient serum (LPDS) containing medium for 48 h and cholesterol localization was analyzed with filipin (Fig 7A and B). In LPDS‐treated cells, the cholesterol accumulation phenotype persisted (Fig 7D), indicating that the uptake pathway was not the main source of the cholesterol accumulated in endosomes, which therefore suggests that the ER is the major source. Next, to test the contribution of the cholesterol biosynthesis pathway, cells were treated with the cholesterol synthesis inhibitor mevinolin for 2 days and cholesterol localization was analyzed with filipin (Fig 7A–C). In control cells, no cholesterol accumulation was observed with and without inhibition. In HeLa/STARD3 cells, cholesterol accumulation was evident in normal culture conditions; contrastingly, in mevinolin treated cells, the cholesterol accumulation phenotype was markedly reduced (Fig 7E). Although intracellular cholesterol levels did not reach those of control cells, this experiment support the notion that the ER is the main source of the cholesterol accumulated in STARD3 endosomes.
Figure 7. The ER is the main source of cholesterol accumulated by STARD3 in endosomes.
-
A–CHeLa/Ctrl and HeLa/STARD3 cells were incubated in normal medium (A), LPDS‐containing medium (B) or normal medium with 50 μM mevinolin and 100 μM mevalonate (C), for 48 h. Cholesterol accumulation in endosomes was detected by filipin staining (Cyan Hot) in endosomes identified by the presence of Lamp1 (red) and STARD3 (magenta). Nuclei were stained in blue. Merged image of filipin and STARD3 signals is shown in (d and h). The subpanels on the right are higher magnification (3.5×) images of the area outlined in white (a, e). The filipin and STARD3 merged image is labeled Overlay. Scale bars: 10 μm.
-
D, ERelative fluorescence intensity of intracellular filipin in HeLa/Ctrl and HeLa/STARD3 cells incubated or not in LPDS‐containing medium (D) or treated or not with mevinolin and mevalonate (E). Mean ± SD; n = 5 (D) and n = 4 (E) independent experiments; **P < 0.01, ANOVA with Tukey's multiple comparison test.
Taken together, these data indicate that the cholesterol accumulation phenotype seen in STARD3‐expressing cells is not caused by an increase in cholesterol synthesis and/or uptake by the endocytic pathway. Rather they strongly suggest that STARD3 has the capacity to alter the intracellular cholesterol repartition by moving ER cholesterol toward endosomes.
STARD3‐VAP complex induces cholesterol accumulation in endosomes at the expense of the plasma membrane
Among the different organelles, the PM contains most of cellular cholesterol (van Meer et al, 2008). Since our data pointed to a role of STARD3 in routing cholesterol toward endosomes, we reasoned that STARD3 might indirectly act on the PM cholesterol. We thus examined whether STARD3 alters cholesterol levels in the PM. To evaluate this possibility, we measured the sensitivity of cells expressing STARD3 to the amphotericin B toxin. This molecule is a polyene antibiotic, which binds PM cholesterol, creating non‐selective ion pores that cause cell death (Kinsky, 1970). We observed that, compared to control cells, HeLa/STARD3 cells were less sensitive to amphotericin B treatment (Fig 8A). Conversely, cells expressing STARD3 mutants devoid of a functional START domain or unable to mediate ER–endosome contacts were as sensitive as control cells to the drug (Fig 8A). This experiment suggests that STARD3‐mediated sterol transfer at ER–endosome contact sites is associated with a reduction in sterol abundance in the PM.
Figure 8. STARD3‐mediated cholesterol accumulation in endosomes occurs at the expense of plasma membrane.
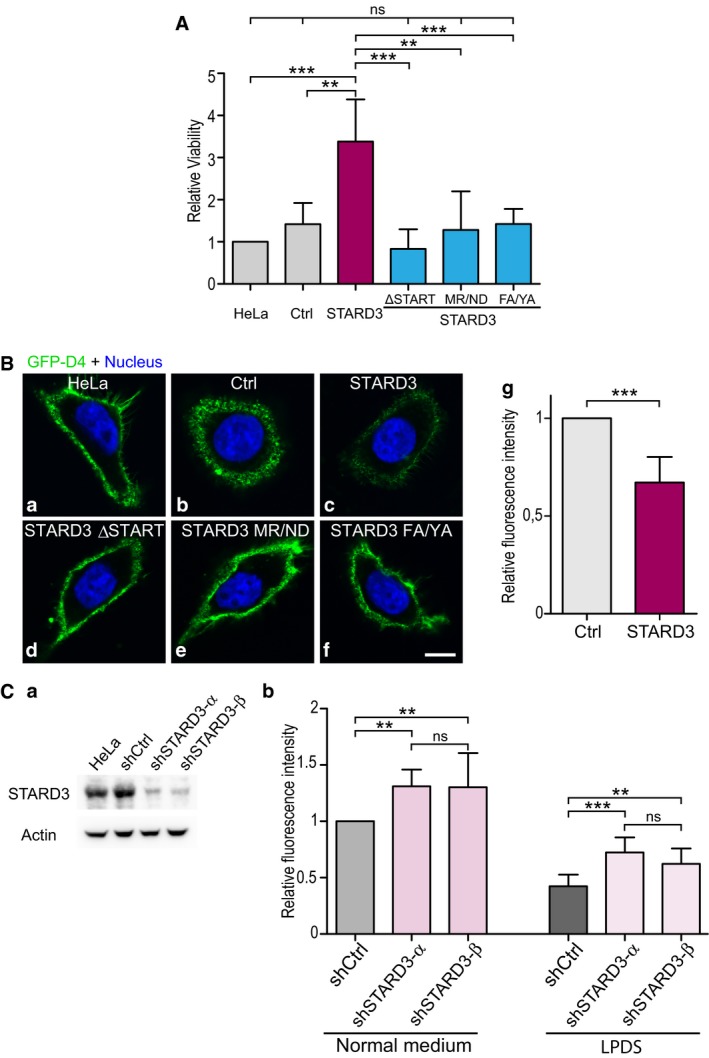
- Indirect analysis of plasma membrane cholesterol content. Cells were treated with amphotericin B (20 μg/ml; 4 h at 37°C), and cell viability was measured using a luminescent cell viability assay. Relative cell viability is represented on the graph. Mean ± SD; n: number of independent experiments; HeLa, HeLa/Ctrl, HeLa/STARD3: n = 5; HeLa/STARD3 ΔSTART; HeLa/STARD3 MR/ND; HeLa/STARD3 FA/YA: n = 4. **P < 0.01, ***P < 0.001, ANOVA with Tukey's multiple comparison test.
- Plasma membrane cholesterol labeling using the GFP‐D4 probe. Live cells were incubated with GFP‐D4 probe (green) prior to fixation and nuclei staining (blue). Both signals are merged (a–f). Scale bar: 10 μm. (g) Quantification of mCherry‐D4 membrane labeling by flow cytometry. Fluorescence mean intensities are compared with that of control cells set to one. Mean ± SD; n = 7 independent experiments; paired two‐tailed t‐test; ***P = 0.0006.
- Western blot analysis (a) of STARD3 in HeLa cells, HeLa cells expressing a control shRNA (shCtrl) and in HeLa cells expressing two shRNAs targeting STARD3 (shSTARD3‐α and shSTARD3‐β). Actin was used as a loading control. Plasma membrane cholesterol was labeled using the GFP‐D4 probe (b) under normal cell culture conditions and under LPDS culture conditions (24‐h treatment); flow cytometry quantification of the GFP‐D4 staining is expressed as mean intensity with that of control cell in normal medium set to one. Mean ± SD; n = 9 independent experiments; **P < 0.01, ***P < 0.001, ANOVA with Tukey's multiple comparison test.
To determine more directly whether cholesterol accumulation in endosomes induced by STARD3 occurs concomitantly with a decrease of PM cholesterol, we used live cell membrane cholesterol staining with the GFP‐D4 probe to image and quantify membrane cholesterol (Fig EV1). Robust membrane cholesterol labeling using the D4 probe can be measured using flow cytometry (Iwamoto et al, 1997); compared with light microscopy, it offers the opportunity to quantify membrane cholesterol labeling in a cell‐by‐cell basis on large populations of cells. Compared to control cells, HeLa/STARD3 cells showed a reduction in GFP‐D4 staining at the PM (Fig 8Ba–c and g). We addressed the mechanism of this reduction in PM cholesterol by using mutants of the START domain or the FFAT motif. Cells expressing STARD3 mutants devoid of the START domain (STARD3 ΔSTART) or with a deficient START (MR/ND mutant) exhibited membrane GFP‐D4 staining similar to that of control cells, indicating that membrane sterol levels were unaltered (Fig 8Bd–f). Likewise, the PM staining of cells expressing the STARD3 FA/YA mutant was similar to that of control cells (Fig 8B). To further support the notion that STARD3‐VAP ER–endosome contacts are modulating PM cholesterol levels, we prevented ER–endosome contact formation by removing VAP proteins (Alpy et al, 2013). First, PM was labeled with GFP‐D4 (Fig EV6A and B) in HeLa/STARD3 cells lacking VAP proteins. While HeLa/STARD3/shCtrl cells showed a reduction in GFP‐D4 staining, VAP‐silenced cells (HeLa/STARD3/shVAPA/B‐α and HeLa/STARD3/shVAPA/B‐β) had a staining similar to control cells, as shown by confocal imaging (Fig EV6A) and quantification by flow cytometry (Fig EV6B). Second, given that VAP protein reduction restores normal membrane cholesterol levels in STARD3 cells, we tested the sensitivity of HeLa/STARD3 cells lacking VAP proteins to amphotericin B treatment. While HeLa/STARD3/shCtrl cells were resistant to amphotericin B treatment, the silencing of VAP proteins in HeLa/STARD3 cells with two different shRNA (shVAPA/B‐α and shVAPA/B‐β) restored a sensitivity similar to control cells (Fig EV6C). Together, these experiments indicated that STARD3‐mediated sterol transport toward endosomes is associated with a lowering of PM cholesterol content.
Figure EV6. VAP silencing restores a normal plasma membrane cholesterol level in STARD3‐expressing cells.
- Plasma membrane cholesterol labeling using the GFP‐D4 probe as in Fig 8B. Live cells were incubated with GFP‐D4 probe (green) prior to fixation and nuclei staining (blue). Both signals are merged (a–i). Scale bar: 10 μm.
- Plasma membrane cholesterol was labeled using the GFP‐D4 probe. GFP‐D4 signal was quantified by flow cytometry. Mean ± SD; n = 4 independent experiments; *P < 0.05, **P < 0.01, ***P < 0.001, ANOVA with Tukey's multiple comparison test.
- Indirect analysis of plasma membrane cholesterol content. Cells were treated with amphotericin B (20 μg/ml; 4 h at 37°C), and cell viability was measured using a luminescent cell viability assay. Relative cell viability is represented on the graph. Mean ± SD; n = 3; ***P < 0.001, ANOVA with Tukey multiple comparison test.
To substantiate the physiological relevance of these results, we knocked down STARD3 using two different shRNAs in control HeLa cells (Fig 8C). Compared to non‐silenced cells (HeLa and HeLa/shCtrl), STARD3 was reduced to about 20% of endogenous level with shSTARD3‐α and shSTARD3‐β shRNAs (Fig 8Ca). PM cholesterol was stained on live cells with the GFP‐D4 probe and quantified by flow cytometry (Fig 8Cb). Compared to control cells (HeLa/shCtrl), HeLa cells knocked down for STARD3 displayed an increased GFP‐D4 staining at their surface (Fig 8Cb). Furthermore, to test the contribution of the LDL pathway, cells were grown in LPDS‐containing medium during 24 h and PM cholesterol was labeled with GFP‐D4. Under this condition, HeLa cells knocked down for STARD3 still displayed a higher membrane GFP‐D4 staining than control cells, further supporting the hypothesis that the ER is the main source of cholesterol transported by STARD3. As the GFP‐D4 binds cholesterol‐enriched regions, these data suggest that STARD3 knockdown either increases cholesterol level in the PM and/or modulates the local concentration of cholesterol in the PM.
Collectively, these results indicate that the expression of STARD3 is associated with a reduction in PM cholesterol. The presence of a functional cholesterol transfer START domain and ER–endosome contact sites are both required to decrease cholesterol from the PM. These results support the notion that STARD3 pilots ER cholesterol to endosomes transfer while reducing ER to PM traffic.
Discussion
Previous reports were suggestive of the ability of STARD3 to promote cholesterol accumulation in endosomes (Alpy et al, 2001; Zhang et al, 2002; Holtta‐Vuori et al, 2005; Liapis et al, 2012). In contrast, other studies argued that STARD3 functions in cholesterol transport to the mitochondria or to the PM (Charman et al, 2010; Vassilev et al, 2015). By using novel methods allowing the visualization and the quantification of intracellular cholesterol both in vitro and in vivo, our study provides new evidence that STARD3 acts on intracellular cholesterol repartition by mediating cholesterol transport from the ER to endosomes at the expense of the PM. Moreover, this study shows that STARD3 has a built‐in capacity to efficiently transfer sterols between apposed membranes. Indeed, in vivo and in vitro data indicate that the mechanism of STARD3 action relies both on its ability to create contact sites between the ER and endosomes via the direct interaction with VAP proteins, and on the sterol exchange capacity of the START domain (Fig 9).
Figure 9. STARD3, a molecular tether transporting cholesterol within ER–endosome contacts.
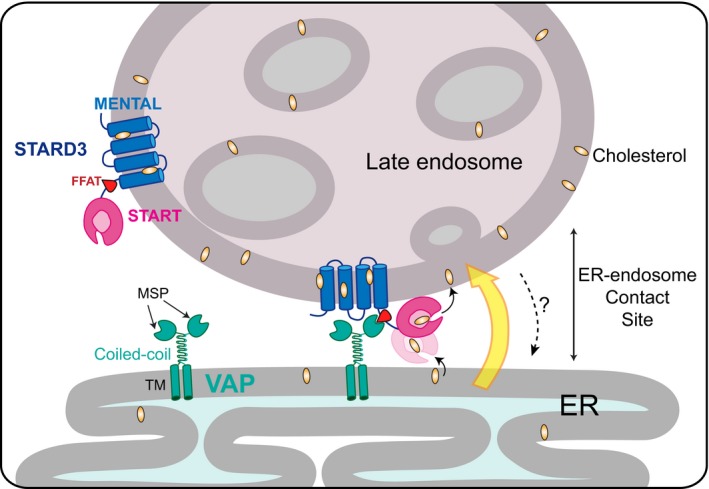
Schematic representation of STARD3 mode of action in cholesterol routing. STARD3, which is composed of a lipid‐transfer START domain and a transmembrane MENTAL domain, interacts with VAP proteins to form ER–endosome membrane appositions. Within contacts, the START domain transfers cholesterol from the ER to the endosome. TM: transmembrane domain. MSP: major sperm protein domain.
Sterol homeostasis is achieved by a number of molecular mechanisms governing the synthesis, uptake, storage, and efflux of this sterol. The PM, the ER, and endosomes are key organelles in this homeostasis (Maxfield & van Meer, 2010). Indeed, cholesterol is synthesized in the ER and trafficked to the PM or esterified by cholesterol acyl transferase in the ER to be stored as lipid droplets (Khor et al, 2013; Rogers et al, 2015). Beside this, the endocytic compartment controls cholesterol uptake via the LDL/LDL‐R pathway (low density lipoprotein–receptor) that is followed by its cellular redistribution from endosomes (Brown & Goldstein, 1979). Consequently, the interface between the ER and endosomes is thought to represent a central crossroad between these two major pathways. While cholesterol exit from endosomes has been studied for many years (Ikonen, 2008; Mesmin et al, 2013a), the ER‐to‐endosome sterol transport route was poorly documented. In this work, we propose that STARD3 localizes at this major interface to efficiently route ER cholesterol to endosome membranes. Interestingly, it has been recently reported that the endosomal oxysterol‐binding protein ORP1L might exert a similar role but in a different context, namely in the case of membrane receptor traffic (Eden et al, 2016). After epidermal growth factor (EGF) treatment, its receptor (EGFR) is activated and then endocytosed. ER–endosome contacts are then formed and provide sites of interaction for the ER‐localized phosphatase PTP1B with EGFR (Eden et al, 2010, 2016); PTP1B stops EGFR signaling by both dephosphorylating the receptor and promoting EGF‐stimulated ILV formation which sequester EGFR. In this context, ILVs formation involves the transport of cholesterol from the ER, which is mediated by the interaction between VAP and ORP1L.
The fact that the STARD3‐VAP complex might be also involved in ER to endosome cholesterol transport but independently of ORP1L (Alpy et al, 2013) reinforces the notion that multiple distinct contact sites likely exist between endosomes and the ER network (Friedman et al, 2013; Eden et al, 2016). Distinct tethering complexes, as for example STARD3‐VAP and ORP1L‐VAP, which are formed under different cellular conditions, can share a similar function and transport sterols to endosomes. Besides Niemann‐Pick C proteins (NPC2 and NPC1) which efflux cholesterol from endosomes, other proteins acting in contact sites might possibly be involved in sterol traffic in this direction (Vanier, 2014; Chu et al, 2015). The formation of MCSs provides a physical basis for inter‐organelle communication and sterol exchange which will vary depending on the cell type and the cellular physiological state. This might explain the conflicting results found in different cell lines (Charman et al, 2010; Vassilev et al, 2015). In addition, one cannot exclude that upon regulatory mechanisms the same machinery could transport cholesterol in a bidirectional manner.
At the cellular level, cholesterol level needs to be constantly tuned (Steck & Lange, 2010). The regulation of cellular cholesterol occurs at the DNA level through the transcription factors from the SREBP family (Horton et al, 2002). SREBP‐2 notably regulates the expression of genes involved in cholesterol synthesis and uptake in the ER and endosomes, respectively. One surprising finding of this study was the lack of SREBP‐2 control over the cholesterol accumulation phenotype mediated by STARD3. This suggests that intracellular cholesterol levels can be modulated in discrete organelles without activation of the transcription machinery. Such a local regulation mode offers advantages in term of reactivity and precision, compared to the global regulation mode through transcription. This mode of regulation could also offer the advantage of sparing cellular energy by avoiding cholesterol synthesis and capture. The distribution of cholesterol mediated by STARD3 at MCS might represent an economic and fast way to modulate sterol content in specific subcellular territories such as in late endosomes internal membranes.
It is increasingly clear that contact sites between organelles are zones of active non‐vesicular lipid traffic. Notably, recent studies showed that members of the oxysterol‐binding protein‐related protein (ORP)/oxysterol‐binding homology (Osh) family counter‐exchange two lipid species within contact sites; OSBP in ER–Golgi contacts and ORP5/ORP8 in ER–PM contacts exchange PtdIns(4)P with sterols or PS, respectively (Mesmin et al, 2013b; Chung et al, 2015; Moser von Filseck et al, 2015b). These proteins contain an OSBP‐related domain (ORD) lipid‐binding domain which, like the START domain, bears one lipid molecule and shields it from the hydrophilic medium through which it is transported (Raychaudhuri & Prinz, 2010; Alpy & Tomasetto, 2014; Olkkonen, 2015). Nevertheless, ORD domains can bind two ligands alternately, PtdIns(4)P and another lipid such as a sterol or PS (de Saint‐Jean et al, 2011; Maeda et al, 2013; Moser von Filseck et al, 2015b). This dual binding specificity coupled to PtdIns(4)P gradient maintained by spatially distant enzymes at ER/Golgi or ER/PM interface drives lipid transport (Mesmin et al, 2013b; Drin et al, 2016). A membrane tethering ability appears to be essential for the lipid exchange activity of some ORP proteins such as ORP5 and ORP8 which connect the ER and the PM (Chung et al, 2015). Besides, cell experiments suggest that OSBP's ability to transport sterols in exchange for PI4P relies on its lipid‐binding domain but also on its tethering capacity (Mesmin et al, 2013b; Wang et al, 2014). Mechanistic data are yet lacking for the newly discovered sterol‐binding proteins in yeast that have a dual ability to tether ER and PM and possess a START‐like domain (Gatta et al, 2015; Murley et al, 2015). Our mechanistic approach, based notably on transport or tethering‐deficient STARD3 mutants, shows that tethering distinct membranes can guide sterol transport.
In addition, we prove using a controlled in vitro system that STARD3 speeds up sterol transport only under tethering conditions. In this assay, the soluble parts of STARD3 and VAP were attached through chemical anchors to endosome‐like or ER‐like liposomes, respectively. This mediated an efficient connection between the two distinct liposome populations. Remarkably, we observed that tethering accelerated the sterol transfer rate by one order of magnitude. The kinetics calculated for the soluble and tethered protein was 2 and over 25 molecules of sterol/min per STARD3, respectively. Interestingly, the transport kinetics measured for the different soluble and isolated START domains fall into a similar range; for instance, STARD4 transports ~7 sterols/min (Iaea et al, 2015), whereas CERT/STARD11 transfers 4 ceramides/min (Hanada et al, 2003). The physiology tells a different kinetics, early work on STARD1 indicated that to promote an acute synthesis of steroids in response to trophic hormones, STARD1 must handle several lipid molecules in a very short time (Jefcoate, 2002; Clark & Stocco, 2014). Indeed, STARD1 transfers over 400 molecules of cholesterol/min (Artemenko et al, 2001). Our finding that tethering the START domain to specific contact sites promotes rapid and efficient cholesterol transfer reconciles the structural organization and lipid binding mode of the START domain (1 mole of ligand per mole of protein) with its biological activity. If acceptor and donor sites are brought together, one START protein could mediate such a rapid and efficient exchange of many ligand molecules. The pool of accessible ligand, the vicinity of high‐affinity acceptors such as metabolizing enzymes or lipids, will impose the rate and direction of transport.
A long‐standing question is whether a shorter distance between membranes is an asset for an efficient lipid transport. Previous in vitro studies suggested that the activity of OSBP is increased when the protein is positioned between ER and Golgi‐like membranes (Mesmin et al, 2013b). Yet it remains difficult to prove that this higher activity solely arises from a reduced distance between membranes. Indeed, the anchoring points for OSBP on the ER (VAP) and the Golgi membrane (PtdIns(4)P), which control its tethering activity, play also an active role in its exchange activity. Here, our study showed that a simple connection of membranes, thus a shortened distance, seems critical for the activity of a lipid transporter. Interestingly, we were able to observe in vitro that spontaneous sterol transport seems faster when membranes are closer. This stated, it appears clear that the presence of a functional sterol‐binding domain is crucial to guarantee transport velocities compatible with cellular timescales.
To summarize, we report that STARD3, by making contact sites with VAP, modulates cholesterol traffic. Our results show that STARD3 and VAP make an autonomous molecular machine able to transfer cholesterol very rapidly and without energy, using a mechanism different from ORPs. Elucidating the molecular mechanism of STARD3 function provides new insights into the mechanism of action of START domain proteins. It is tempting to speculate that the other STARD proteins are functioning likewise. Future work should provide evidence on how these proteins orchestrate their subcellular localizations with their lipid transfer activities.
Materials and Methods
Cloning and constructs
The retroviral expression vector for STARD3 (pQCXIP STARD3) was previously described (Alpy et al, 2013). STARD3 mutants were constructed in the pQCXIP vector: The STARD3 ΔSTART (Q14849‐1, residues 1–235), STARD3 M307R/N311D (referred to as MR/ND mutant), and STARD3 F207A/Y208A (referred to as FA/YA mutant) double mutants were constructed by site‐directed mutagenesis.
STARD3 cDNA was subcloned into the retroviral pMXPIE vector (kind gift from Bernardo Reina San‐Martin) to generate a bicistronic vector encoding STARD3 and the GFP.
A STARD3[196–445] fragment was PCR‐amplified from an initial plasmid using primers incorporating a BamHI and NotI restriction sites and inserted into a pGEX‐4T‐3 vector to code for a soluble form of STARD3 in fusion with a N‐terminal GST protein. A single mutation was performed to introduce a cysteine at position 196, which corresponds to the end of the MENTAL domain, in place of a leucine residue. As such, the soluble part of STARD3 should be positioned on MPB‐PE containing liposomes in a manner similar to the authentic STARD3. Hereafter, the STARD3[196–445](L196C) construct is called CSTD3. The non‐conventional FFAT motif QFYSPPE (segment 206–212) was replaced by a DEDDENEFFDAPE in CSTD3 or a GGGGGGG sequence to obtain CSTD3‐7G, respectively. A double mutation (M307R/N311D) was introduced in the sterol‐binding pocket of CSTD3 construct to obtain the CSTARD3 (MR/ND) construct.
A human VAP‐A [8–212] fragment was subcloned into pET‐21b for expression as C‐terminal His‐tag proteins in E. coli. The position of the C‐terminal tag corresponds to the beginning of the C‐terminal transmembrane region. As for STARD3, the soluble part of VAP construct should be positioned on DOGS‐NTA‐Ni2+ containing liposomes in a manner similar to the full‐length VAP protein. The mutations and deletion/insertions of sequences were done using by the Quikchange kit (Stratagene).
To silence VAP proteins, we used the previously described shRNA expression vectors targeting VAP‐A (target sequence α: 5′‐GCGAAATCCATCGGATAGAAA‐3′ or β: 5′‐CACTTAATGATACCGAAACAA‐3′) or VAP‐B (α: 5′‐GCAGAGAATGATAAACCACAT‐3′ or β: 5′‐CCAGTTCTGTTTGACTATGTA‐3′) (Alpy et al, 2013). To generate shRNA expression vectors targeting STARD3 (target sequence α: 5′‐GACCTGGTTCCTTGACTTCAA‐3′ or β: 5′‐CGGCAAGACGTTTATCCTGAA‐3′), oligonucleotides were cloned into the pLKO.1 vector (Moffat et al, 2006).
The expression vectors encoding the GFP‐D4 and mCherry‐D4 probes were previously described (Ohno‐Iwashita et al, 2004; Abe et al, 2012).
All constructs were verified by DNA sequencing.
Cell culture, transfections, and infections
HeLa cells (American Type Culture Collection CCL‐2) were maintained in DMEM with 5% fetal calf serum (FCS) and 40 μg/ml gentamycin. 293T (ATCC CRL‐3216) cells were maintained in DMEM with 10% fetal calf serum (FCS), penicillin 100 UI/ml, and streptomycin 100 μg/ml. LPDS medium is composed of DMEM supplemented with 40 μg/ml gentamycin an 5% LPDS purified as described in Goldstein et al (1983).
Cells were transfected using X‐tremeGENE 9 DNA Transfection Reagent (Roche). For retroviral infection, pQCXIP vectors were co‐transfected with pCL‐Ampho vector (Imgenex) into 293T retroviral packaging cell line. Retroviral infections were used to generate HeLa/Ctrl, HeLa/STARD3, HeLa/STARD3 ΔSTART, HeLa/STARD3 MR/ND, and HeLa/STARD3 FA/YA cell lines. The HeLa/Ctrl cell line was obtained using the empty pQCXIP or pMXPEI plasmid.
For lentiviral infection, pLKO.1 vectors were co‐transfected with three packaging plasmids pLP1, pLP2, and pLP/VSVG (Invitrogen) into the 293T cell line. Viral particles supplemented with 10 μg/ml polybrene and 20 mM Hepes were then incubated with HeLa cells.
HeLa selection was performed using 1 μg/ml puromycin or 4 μg/ml blasticidin.
Antibody production
The CSTD3 recombinant protein was used to produce a mouse monoclonal antibody as described before (Moog‐Lutz et al, 1997). The 3G11 hybridoma, which recognizes STARD3 by Western blot and immunofluorescence, was used in this study.
Immunofluorescence
Immunofluorescence was performed as previously described (Alpy et al, 2002). Primary antibodies were rabbit anti‐STARD3 pAbMLN64‐Nt‐1611 (1:1,000; Alpy et al, 2001), goat anti‐VAP‐A (1:500; K‐15—Santa Cruz Biotechnology, sc‐48698), mouse anti‐Lamp1 H4A3 (1:50; DSHB), mouse anti‐EEA1 (1:1,000; 610457 BD Biosciences), mouse anti‐CD63 H5C6 (1:1,000; DSHB), mouse anti‐BMP (1:10; 6C4 (Kobayashi et al, 1998), and rabbit anti‐Rab7L1 (1:1,000, Abcam ab199644). Slides were mounted in ProLong Gold (Invitrogen). Observations were made with a confocal microscope (Leica SP8 UV, 63×, NA 1.4).
Colocalization analysis
Linescans were drawn using ImageJ software (plot profile function; https://imagej.nih.gov/ij/). Colocalization was visualized using the colocalization highlighter plugin for ImageJ.
Colocalization coefficients (Pearson correlation coefficient or Manders M1 and M2) were determined using the JACoP plugin (Bolte & Cordelières, 2006) in Fiji software. For Pearson correlation coefficient determination, auto‐thresholding was performed using the Costes method (Costes et al, 2004). For Manders colocalization coefficient determination, thresholding was performed using Yen algorithm (Fiji).
To quantify the co‐labeling of filipin or GFP‐D4‐positive vesicles with Lamp1 or STARD3, an automatic threshold (Yen, Fiji) was applied to the different channels. The percentage of co‐labeled vesicles (filipin+/Lamp1+ or filipin+/STARD3+ or GFP‐D4+/Lamp1+ or GFP‐D4+/STARD3+) was determined manually using thresholded images.
Filipin staining and quantification
Cells were grown on glass coverslips. In order to allow a better visualization of intracellular cholesterol pools, cells were treated with 10 mM methyl‐β‐cyclodextrin (MβCD, Sigma) for 30 min at 37°C to remove cholesterol from the plasma membrane (Fig EV1). Cells were then fixed for 10 min at RT with 4% paraformaldehyde in phosphate‐buffered saline (PBS). Cells were incubated with a solution of filipin (0.1 mg/ml, F‐9765 Sigma) for 30 min. After washing and blocking in 1% bovine serum albumin (BSA) in PBS, cells were incubated with the primary antibodies. After three washes in PBS, cells were incubated for 1 h with Cy3‐ and Alexa488‐conjugated secondary antibodies (Jackson ImmunoResearch and Invitrogen‐Molecular Probes, respectively). After three washes, cells were re‐incubated in the filipin solution for 30 min. Finally, after two washes, cells were counterstained with propidium iodide (PI, 0.35 μg/ml; Sigma) or with TO‐PRO‐3 iodide (1:3,000; ThermoFisher scientific) for 5 min. Slides were mounted in ProLong Gold (Invitrogen). Image acquisition was performed with a Leica SP8 UV (63×, NA 1.4) confocal microscope using the photon counting mode; a 355 nm optically pumped semiconductor laser (Coherent) was used to excite filipin. The Fiji software was used to quantify filipin fluorescence intensity (http://fiji.sc/). Cells were segmented based on the PI staining (Fig EV3). Filipin staining intensity in individual cells was measured after an intensity thresholding. The threshold value was determined in control cells as the 99th percentile of pixel intensity values.
GFP‐D4 and mCherry‐D4 production and staining
Production
Proteins were overexpressed overnight in E. coli BL21(DE3) at 16°C in the presence of 0.4 mM isopropyl‐β‐D‐thiogalactopyranoside (IPTG, Sigma). The cell pellet was lysed and sonicated in 50 ml of lysis buffer (50 mM NaH2PO4/Na2HPO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) complemented with EDTA‐free cOmplete protease inhibitor (Roche). The lysate was centrifuged at 10,000 g for 20 min at 4°C. The supernatant was incubated with 1.5 ml of His‐Select Nickel Affinity Gel (Sigma). The resin was washed with lysis buffer. Bound proteins were eluted with elution buffer (20 mM NaH2PO4/Na2HPO4, 250 mM imidazole, pH 7.4). Eluted fraction was dialyzed into phosphate buffer (20 mM NaH2PO4/Na2HPO4 pH 7.4, EDTA‐free cOmplete).
Plasma membrane staining
For confocal acquisitions, cells were grown on glass coverslips, washed with PBS, and incubated in GFP‐D4 or mCherry‐D4 solution (1:200 in 1× PBS, 1% BSA) for 30 min. After a rapid wash, cells were fixed with 4% paraformaldehyde in PBS and nuclei were counterstained with Hoescht‐33258 dye. Slides were mounted in ProLong Gold (Invitrogen).
For flow cytometry analysis, 6.105 cells were plated in a T175 flask 24 h before the experiment. Cells were trypsinized and incubated in a solution of GFP‐D4 or mCherry‐D4 probe (1:200 or 1:1,000 in 1× PBS, 1% BSA) for 30 min at 37°C. Cells were then fixed for 10 min at RT with 2% paraformaldehyde in PBS, centrifuged, and finally resuspended in PBS before FACS analysis (FACSCalibur and LSRFortessa X‐20, BD biosciences). At least 2.104 cells were analyzed in duplicate. For cells expressing the pMXPIE construct encoding STARD3 or the empty pMXPIE vector, GFP‐positive cells were gated (FlowJo) and the mean mCherry‐D4 fluorescence was measured in this cell population.
Intracellular staining
Cells were grown on glass coverslips, treated or not with 1 μg/ml of U18666A ((3β)‐3‐{2‐(Diethylamino)ethoxy}androst‐5‐en‐17‐one, 662015 Calbiochem) during 1 h, fixed 10 min at RT with 4% paraformaldehyde (PFA) in phosphate‐buffered saline (PBS), and permeabilized in a nitrogen bath for 30 s. After blocking in 1% BSA in PBS, cells were incubated at RT with GFP‐D4 solution in PBS for 1 h. Then, cells were rapidly washed and re‐fixed with PFA before a second blocking step. Primary antibodies, rabbit anti‐STARD3 (1:1,000; Alpy et al, 2001) and mouse anti‐Lamp1 H4A3 (1:50; DSHB), were incubated overnight at 4°C, and after three washes in PBS, cells were incubated for 1 h with Cy3‐ and Cy5‐conjugated secondary antibodies (Jackson ImmunoResearch). After two washes, nuclei were counterstained with Hoescht‐33258 dye and slides were mounted in ProLong Gold (Invitrogen). Observations were made with confocal microscope (Leica SP8 UV, 63×, NA 1.4).
GFP‐D4 and filipin co‐staining
Cells were firstly stained with the GFP‐D4 probe as described above (intracellular staining protocol). After incubation with primary (anti‐STARD3) and secondary antibodies, cells were re‐incubated with filipin solution during 1 h at RT. After 1 wash, nuclei were counterstained with TO‐PRO‐3 iodide and slides were mounted.
Statin treatment
Cells were treated with 50 μM mevinolin (M‐2147, Sigma) and 100 μM DL‐mevalonolactone (SC‐211365, Santa Cruz) diluted in normal medium during 48 h at 37°C. Cells were washed, and filipin staining was performed as described above.
SDS–PAGE and Western blot analysis
SDS–PAGE and Western blot analysis were performed as previously described (Alpy et al, 2005) using the following antibodies: STARD3: pAbMLN64‐Ct‐605 (Moog‐Lutz et al, 1997); pAbMLN64‐Nt‐1611 (Alpy et al, 2001); VAP‐A: K‐15 sc‐48698 (Santa Cruz biotechnology); VAP‐B: rabbit anti‐VAP‐B (Kabashi et al, 2013); SREBP‐2 1C6 (sc‐13552; Santa Cruz biotechnology); actin: ACT‐2D7 (Euromedex).
Lipids extraction and quantification
One million cells were plated in 150‐mm petri dishes 24 h before the experiment. Cells were treated with 10 mM methyl‐β‐cyclodextrin (MβCD, C4555 Sigma) for 30 min for the positive control. Cells were washed three times and scraped in 500 μl of PBS. Cells in PBS (500 μl) were mixed with 2 ml of EtOH/chloroform (2/1) solution in glass tubes. After 5 min of centrifugation at 1,400 g, the supernatant was transferred into a new tube. Then, 0.5 ml of 50 mM citric acid, 1 ml of water, and 0.5 ml of chloroform were added. After 20 min of centrifugation at 1,400 g, the lower phase‐containing lipids were transferred in new glass tubes and dried in a stream of nitrogen. For the lipids quantification, dried lipids were solubilized in EtOH 95% solution. Cholesterol quantification was performed with the Amplex Red cholesterol assay (A12216 Invitrogen), and phospholipids quantification was performed with LabAssay phospholipid (296‐63801 Wako Chemicals) according to the manufacturer instructions.
SREBP‐2 processing
HeLa cells transduced with the empty pMXPIE vector (containing an IRES‐GFP) or the pMXPIE construct encoding STARD3 were sorted by flow cytometry (FACSAria II BD biosciences). Three hundred thousand GFP‐positive cells were seeded in a 6‐well plate and maintained 24 h in culture. Cells were then incubated for 2 h at 37°C in DMEM medium with 5% of FCS (NT condition) or 5% LPDS, 10 μM mevinolin, 10 μM MG132 (Sigma), which was supplemented with 1 mM MβCD or 500 μM MβCD/cholesterol (C4951 Sigma) to deplete or load the cells with cholesterol, respectively. Cells were subsequently harvested at 0°C and lysed in 150 mM NaCl, 50 mM Tris–HCl pH 7.5, 1% Triton X‐100 complemented with EDTA‐free cOmplete protease inhibitor (Roche).
Amphotericin B sensitivity assay
Five thousand cells were plated in a 96‐well plate (5,000 cells/well). Twenty‐four hours later, cells were treated or not with 20 μg/ml amphotericin B (A4888; Sigma) during 4 h at 37°C. Cell viability was evaluated by the CellTiterGlo Luminescent Cell viability Assay (Promega), according to the manufacturer instructions.
Protein expression and purification
GST‐CSTD3 and VAP[8‐212]His6 were expressed in E. coli at 37°C for 3 h upon induction with 1 mM IPTG (at an optical density (OD) equal to 0.6 at λ = 600 nm). For GST‐tagged proteins, all purification steps were conducted in 50 mM Tris, pH 7.4, 120 mM NaCl (TN buffer) with 2 mM DTT. For VAPHis6, a buffer containing 50 mM Tris, pH 7.5, 300 mM NaCl, 5 mM imidazole was used. All buffers were supplemented during the first purification steps with PMSF (1 mM), bestatin (10 μM), pepstatin (10 μM), phosphoramidon (10 μM), and protease inhibitor tablets (Roche). Cells were lysed by a French press, and the lysate was centrifuged at 200,000 g for 1 h.
For GST‐CSTD3, the supernatant was applied to Glutathione Sepharose 4B beads. After three washing steps with TN buffer containing 2 mM DTT, the beads were incubated with thrombin at 4°C overnight to cleave the GST fusion and allow the release of CSTD3. The protein was recovered by centrifugation, and the beads were washed three times with TN buffer. The fractions were pooled and concentrated. All constructs contain an N‐terminal GS sequence deriving from the thrombin cleavage site. Stock concentration was estimated with a BCA assay (Pierce).
For VAPHis6 constructs, the supernatant was mixed with NTA‐Ni2+ Agarose beads (QIAGEN). After three washing steps, the proteins were eluted from the beads with buffer containing first 250 mM, then 500 mM imidazole.
The day of experiment, in order to attach CSTD3 covalently to MPB‐PE‐containing liposomes, 100 μl from stock protein (adjusted at ~100 μM) was applied onto an illustra NAP‐10 column (GE Healthcare) and eluted with freshly degassed TN buffer according to manufacturer's indications to remove DTT. The concentration of the eluted protein was determined with a BCA assay.
Lipids
DOPC (1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine), POPC (1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine), DNS‐PE (1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(5‐dimethylamino‐1‐naphthalenesulfonyl)), NBD‐PE (1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(7‐nitro‐2‐1,3‐benzoxadiazol‐4‐yl)), DOGS‐NTA‐Ni2+ (1,2‐dioleoyl‐sn‐glycero‐3‐[(N‐(5‐amino‐1‐carboxypentyl)iminodiacetic acid)succinyl]), and 18:1/18:1 MPB‐PE (1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[4‐(p‐maleimidophenyl)butyramide]) were purchased from Avanti Polar Lipids. Dehydroergosterol (DHE) was from Sigma‐Aldrich. The concentration of DHE in stock solution in methanol was determined by UV‐spectroscopy using an extinction coefficient of 13,000 M−1.cm−1.
Liposome preparation
Lipids, stored in stock solutions in CHCl3 or methanol, were mixed at the desired molar ratio. The solvent was removed in a rotary evaporator under vacuum. For lipid films containing DOGS‐NTA‐Ni2+ or MPB‐PE, the mix was pre‐warmed to 33°C for 5 min prior to drying. The films were hydrated in TN buffer to obtain a suspension of multilamellar liposomes. The suspensions were extruded through polycarbonate filters of 0.2 μm pore size using a mini‐extruder (Avanti Polar Lipids). Liposomes were stored at 4°C and in the dark when containing light‐sensitive lipids (DHE, DNS‐PE, NBD‐PE) and used within 2 days.
Dynamic light scattering measurements of liposome aggregation
All experiments were performed at 25°C in a Dynapro apparatus (Protein Solutions). The sample initially contained LA liposomes (50 μM total lipids) in TN buffer freshly degassed in a small quartz cell (volume 20 μl). A first set of about 12 autocorrelation curves was acquired to measure the size distribution of initial liposome suspension. Then, CSTD3 construct (380 nM final concentration) was added manually and mixed thoroughly. After an incubation of 5 min, LB liposomes were added (50 μM followed by the injection of VAPHIS6 (380 nM). The kinetics of aggregation was followed by acquiring one autocorrelation curve every 10 s. At the end of the experiment, a set of 12 autocorrelation functions was acquired. The data were analyzed using two different algorithms provided by the Dynamics v6.1 software (Protein Solutions). During the aggregation process, the autocorrelation functions were fitted assuming that the size distribution is a simple Gaussian function. This mode, referred as the monomodal or cumulant algorithm, gives a mean radius, R, and the width (or polydispersity). The polydispersity is represented in the kinetics measurements by the shaded area and can reach very large values because of the simultaneous presence of free liposomes and of liposome aggregates of various sizes. Before and after the aggregation process, the autocorrelation functions were fitted using a more refined algorithm, referred as a regularization algorithm. This algorithm is able to resolve several populations of different size, such as free liposomes and liposome aggregates.
DHE transport assay
Experiments were carried out in a Shimadzu RF 5301‐PC fluorimeter equipped with a cylindrical quartz cuvette. A suspension (570 μl) of LA liposomes (62.5 μM total lipids final concentration) made of DOPC and containing 3 mol% MPB‐PE was incubated with 475 nM CSTD3 at 37°C under constant stirring in TN buffer. After 5 min, 30 μl of LB liposomes, containing 10 mol% DHE, 2.5 mol% DNS‐PE, and 2 mol% DOGS‐NTA‐Ni2+ (62.5 μM total lipids final concentration), pre‐incubated or not with VAPHis6 (500 nM final concentration) was added. Lipid transport was measured by recording the DNS‐PE signal at 525 nm (bandwidth 10 nm) upon DHE excitation at 310 nm (bandwidth 1.5 nm). The quantity of DHE transported from LB to LA membrane is expressed in term of mol% DHE in LB liposomes. It is equal to 10 × ((F–F 0)/(F max‐F 0)) where F max is the signal measured in the absence of cSTD3 upon the addition of LB liposome and F 0 is the signal measured upon total DHE extraction by 10 mM methyl‐β‐cyclodextrin (Sigma). Liposomes and proteins were injected from stock solutions with Hamilton syringes through a guide in the cover of the fluorimeter.
Flotation experiments
Proteins (750 nM) were incubated with NBD‐PE containing liposomes (750 μM total lipids) in 150 μl TN buffer at room temperature for 5 min. The suspension was adjusted to 30% sucrose by mixing 100 μl of a 75% (w/v) sucrose solution in HK buffer and overlaid with 200 μl HK containing 25% (w/v) sucrose and 50 μl sucrose‐free HK. The sample was centrifuged at 240,000 g in a swing rotor (TLS 55 Beckmann) for 1 h. The bottom (250 μl), middle (150 μl), and top (100 μl) fractions were collected. The top fractions were analyzed by SDS–PAGE using Sypro Orange staining and a FUJI LAS‐3000 fluorescence imaging system.
Electron microscopy and stereology
Electron microscopy was performed as previously described (Alpy et al, 2013). Briefly, cells grown on type I collagen‐coated Aclar film were cryoprotected with a solution of 20% BSA in culture medium and immediately frozen at high pressure (EMPACT‐2 Leica). Alternatively, cells grown on carbon‐coated sapphire disks were cryoprotected with DMEM containing 10% FCS and frozen at high pressure (HPM 10 Abra Fluid AG). Samples were then freeze‐substituted and embedded in Lowicryl HM20. Thin sections were collected on formvar–carbon‐coated nickel slot grids and stained with uranyl acetate and lead citrate. Imaging was performed with a transmission electron microscope (Philips CM12) coupled to an Orius 1000 CCD camera (Gatan). Inner endosomal membranes quantification was performed by stereology (Mayhew, 1991). A virtual array of points separated by 31.6 nm was randomly placed on TEM images. The number of points over inner endosomal membranes from a given endosome section was divided by the total number of points over the endosome section to get an estimate of the volume to volume percentage of internal membranes per endosome.
Statistical analyses
Unless otherwise specified, statistical analyses were performed using the parametric test ANOVA. All conditions were compared by the Tukey's multiple comparison test (Prism, Graphpad and SigmaPlot, Systat software).
P‐values < 0.05, < 0.01, and < 0.001 are identified with 1, 2, and 3 asterisks, respectively. ns: P > 0.05.
Author contributions
CT, FA, and GD conceived and supervised the project. LPW performed and analyzed all cell biology experiments, with the help of CW, CT, and FA. GD performed and analyzed all in vitro tethering/transport experiments. TK helped cholesterol labeling experiment design. BV performed gene expression analyses and some biochemical experiments. M‐PC assisted in designing and interpreting experiments. CT, FA, GD, and LPW wrote the manuscript. All authors commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Figure 6
Review Process File
Acknowledgements
We thank Alastair McEwen for his critical reading of the manuscript. We thank Marie‐Christine Rio, Françoise Hullin‐Matsuda, Karim Hnia, Shankar Pattabhiraman, Bernardo Reina San‐Martin, Marc Ruff, Robert Drillien, Christophe Lamaze, and the members of the Molecular and Cellular Biology of Breast Cancer team (IGBMC) for helpful advice and discussions. We wish to thank Dr Luc Dupuis for providing the anti‐VAP‐B antibody. We thank the IGBMC cell culture service (Betty Heller) and the imaging center (EM: Coralie Spiegelhalter and light microscopy: Basile Gurchenkov and Pascal Kessler), the polyclonal antibody facility (Gilles Duval), the monoclonal antibody facility (Mustapha Oulad‐Abdelghani), and the Flow Cytometry facility (Claudine Ebel and Muriel Philipps) for their excellent technical assistance. We are grateful to Danièle Spehner and to the EM Core Facility, European Molecular Biology Laboratory (EMBL), Heidelberg, Germany, especially to Yannick Schwab and Androniki Kolovou, for their help with EM. We thank Lolita Maufoux for her help in devising preliminary in vitro assays. L.P.W. received an allocation from the Ministère de l'Enseignement Supérieur et de la Recherche (France; http://www.enseignementsup-recherche.gouv.fr/) and a fellowship from the Fondation pour la Recherche Médicale. F.A. received the «Prix espoir de l'Université de Strasbourg» (Initiative d'excellence IDEX). This work was supported by grants from the Institut National Du Cancer INCa (INCA_9269), the Ligue Contre le Cancer (Conférence de Coordination Interrégionale du Grand Est), The Ara Parseghian Medical Research Fund and Vaincre les maladies lysosomales. We also acknowledge funds from the Institut National de Santé et de Recherche Médicale, the Centre National de la Recherche Scientifique, the Université de Strasbourg and the grant ANR‐10‐LABX‐0030‐INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d'Avenir ANR‐10‐IDEX‐0002‐02.
The EMBO Journal (2017) 36: 1412–1433
Contributor Information
Catherine Tomasetto, Email: catherine-laure.tomasetto@igbmc.fr.
Fabien Alpy, Email: fabien.alpy@igbmc.fr.
References
- Abe M, Makino A, Hullin‐Matsuda F, Kamijo K, Ohno‐Iwashita Y, Hanada K, Mizuno H, Miyawaki A, Kobayashi T (2012) A role for sphingomyelin‐rich lipid domains in the accumulation of phosphatidylinositol‐4,5‐bisphosphate to the cleavage furrow during cytokinesis. Mol Cell Biol 32: 1396–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpy F, Stoeckel ME, Dierich A, Escola JM, Wendling C, Chenard MP, Vanier MT, Gruenberg J, Tomasetto C, Rio MC (2001) The steroidogenic acute regulatory protein homolog MLN64, a late endosomal cholesterol‐binding protein. J Biol Chem 276: 4261–4269 [DOI] [PubMed] [Google Scholar]
- Alpy F, Wendling C, Rio M‐C, Tomasetto C (2002) MENTHO, a MLN64 homologue devoid of the START domain. J Biol Chem 277: 50780–50787 [DOI] [PubMed] [Google Scholar]
- Alpy F, Tomasetto C (2005) Give lipids a START: the StAR‐related lipid transfer (START) domain in mammals. J Cell Sci 118: 2791–2801 [DOI] [PubMed] [Google Scholar]
- Alpy F, Latchumanan VK, Kedinger V, Janoshazi A, Thiele C, Wendling C, Rio M‐C, Tomasetto C (2005) Functional characterization of the MENTAL domain. J Biol Chem 280: 17945–17952 [DOI] [PubMed] [Google Scholar]
- Alpy F, Rousseau A, Schwab Y, Legueux F, Stoll I, Wendling C, Spiegelhalter C, Kessler P, Mathelin C, Rio M‐C, Levine TP, Tomasetto C (2013) STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J Cell Sci 126: 5500–5512 [DOI] [PubMed] [Google Scholar]
- Alpy F, Tomasetto C (2014) START ships lipids across interorganelle space. Biochimie 96: 85–95 [DOI] [PubMed] [Google Scholar]
- Artemenko IP, Zhao D, Hales DB, Hales KH, Jefcoate CR (2001) Mitochondrial processing of newly synthesized steroidogenic acute regulatory protein (StAR), but not total StAR, mediates cholesterol transfer to cytochrome P450 side chain cleavage enzyme in adrenal cells. J Biol Chem 276: 46583–46596 [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232 [DOI] [PubMed] [Google Scholar]
- Borthwick F, Allen A‐M, Taylor JM, Graham A (2010) Overexpression of STARD3 in human monocyte/macrophages induces an anti‐atherogenic lipid phenotype. Clin Sci Lond Engl 1979(119): 265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (1979) Receptor‐mediated endocytosis: insights from the lipoprotein receptor system. Proc Natl Acad Sci USA 76: 3330–3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (1999) A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci USA 96: 11041–11048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T‐Y, Chang CCY, Ohgami N, Yamauchi Y (2006) Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol 22: 129–157 [DOI] [PubMed] [Google Scholar]
- Charman M, Kennedy BE, Osborne N, Karten B (2010) MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann‐Pick type C1 protein. J Lipid Res 51: 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B‐B, Liao Y‐C, Qi W, Xie C, Du X, Wang J, Yang H, Miao H‐H, Li B‐L, Song B‐L (2015) Cholesterol transport through lysosome‐peroxisome membrane contacts. Cell 161: 291–306 [DOI] [PubMed] [Google Scholar]
- Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, Camilli PD (2015) PI4P/phosphatidylserine countertransport at ORP5‐ and ORP8‐mediated ER–plasma membrane contacts. Science 349: 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BJ, Wells J, King SR, Stocco DM (1994) The purification, cloning, and expression of a novel luteinizing hormone‐induced mitochondrial protein in MA‐10 mouse Leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR). J Biol Chem 269: 28314–28322 [PubMed] [Google Scholar]
- Clark BJ, Stocco DM (2014) The Steroidogenic Acute Regulatory Protein (StAR) In Cholesterol transporters of the START domain protein family in health and disease, Clark BJ, Stocco DM. (eds), pp 15–47. New York: Springer; [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S (2004) Automatic and quantitative measurement of protein‐protein colocalization in live cells. Biophys J 86: 3993–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drin G, Morello V, Casella JF, Gounon P, Antonny B (2008) Asymmetric tethering of flat and curved lipid membranes by a golgin. Science 320: 670–673 [DOI] [PubMed] [Google Scholar]
- Drin G, von Filseck JM, Čopič A (2016) New molecular mechanisms of inter‐organelle lipid transport. Biochem Soc Trans 44: 486–492 [DOI] [PubMed] [Google Scholar]
- Eden ER, White IJ, Tsapara A, Futter CE (2010) Membrane contacts between endosomes and ER provide sites for PTP1B–epidermal growth factor receptor interaction. Nat Cell Biol 12: 267–272 [DOI] [PubMed] [Google Scholar]
- Eden ER, Sanchez‐Heras E, Tsapara A, Sobota A, Levine TP, Futter CE (2016) Annexin A1 tethers membrane contact sites that mediate ER to endosome cholesterol transport. Dev Cell 37: 473–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, DiBenedetto JR, West M, Rowland AA, Voeltz GK (2013) Endoplasmic reticulum–endosome contact increases as endosomes traffic and mature. Mol Biol Cell 24: 1030–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta AT, Wong LH, Sere YY, Calderón‐Noreña DM, Cockcroft S, Menon AK, Levine TP (2015) A new family of StART domain proteins at membrane contact sites has a role in ER‐PM sterol transport. eLife 4: e07253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Basu SK, Brown MS (1983) Receptor‐mediated endocytosis of low‐density lipoprotein in cultured cells. Methods Enzymol 98: 241–260 [DOI] [PubMed] [Google Scholar]
- Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M (2003) Molecular machinery for non‐vesicular trafficking of ceramide. Nature 426: 803–809 [DOI] [PubMed] [Google Scholar]
- Hanada K (2010) Intracellular trafficking of ceramide by ceramide transfer protein. Proc Jpn Acad Ser B 86: 426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis JCM, Levine TP (2005) Lipid traffic: floppy drives and a superhighway. Nat Rev Mol Cell Biol 6: 209–220 [DOI] [PubMed] [Google Scholar]
- Holthuis JCM, Menon AK (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510: 48–57 [DOI] [PubMed] [Google Scholar]
- Holtta‐Vuori M, Alpy F, Tanhuanpaa K, Jokitalo E, Mutka AL, Ikonen E (2005) MLN64 is involved in actin‐mediated dynamics of late endocytic organelles. Mol Biol Cell 16: 3873–3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109: 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF (2013) Proteome‐wide mapping of cholesterol‐interacting proteins in mammalian cells. Nat Methods 10: 259–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaea DB, Mao S, Maxfield FR (2014) Steroidogenic Acute Regulatory Protein‐related lipid transfer (START) proteins in non‐vesicular cholesterol transport In Cholesterol transporters of the START domain protein family in health and disease, Clark BJ, Stocco DM. (eds), pp 173–188. New York: Springer; [Google Scholar]
- Iaea DB, Dikiy I, Kiburu I, Eliezer D, Maxfield FR (2015) STARD4 membrane interactions and sterol binding. Biochemistry 54: 4623–4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E (2008) Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 9: 125–138 [DOI] [PubMed] [Google Scholar]
- Ishitsuka R, Saito T, Osada H, Ohno‐Iwashita Y, Kobayashi T (2011) Fluorescence image screening for chemical compounds modifying cholesterol metabolism and distribution. J Lipid Res 52: 2084–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M, Morita I, Fukuda M, Murota S, Ando S, Ohno‐Iwashita Y (1997) A biotinylated perfringolysin O derivative: a new probe for detection of cell surface cholesterol. Biochim Biophys Acta BBA ‐ Biomembr 1327: 222–230 [DOI] [PubMed] [Google Scholar]
- Jefcoate C (2002) High‐flux mitochondrial cholesterol trafficking, a specialized function of the adrenal cortex. J Clin Invest 110: 881–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John K, Kubelt J, Müller P, Wüstner D, Herrmann A (2002) Rapid transbilayer movement of the fluorescent sterol dehydroergosterol in lipid membranes. Biophys J 83: 1525–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Oussini HE, Bercier V, Gros‐Louis F, Valdmanis PN, McDearmid J, Mejier IA, Dion PA, Dupre N, Hollinger D, Sinniger J, Dirrig‐Grosch S, Camu W, Meininger V, Loeffler J‐P, René F, Drapeau P, Rouleau GA, Dupuis L (2013) Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum Mol Genet 22: 2350–2360 [DOI] [PubMed] [Google Scholar]
- Kaiser SE, Brickner JH, Reilein AR, Fenn TD, Walter P, Brunger AT (2005) Structural basis of FFAT motif‐mediated ER targeting. Structure 13: 1035–1045 [DOI] [PubMed] [Google Scholar]
- Khor VK, Shen W‐J, Kraemer FB (2013) Lipid droplet metabolism. Curr Opin Clin Nutr Metab Care 16: 632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsky SC (1970) Antibiotic interaction with model membranes. Annu Rev Pharmacol 10: 119–142 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J (1998) A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 392: 193–197 [DOI] [PubMed] [Google Scholar]
- Liapis A, Chen FW, Davies JP, Wang R, Ioannou YA (2012) MLN64 transport to the late endosome is regulated by binding to 14‐3‐3 via a non‐canonical binding site. PLoS ONE 7: e34424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewen CJR, Roy A, Levine TP (2003) A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J 22: 2025–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M, Gavin A‐C (2013) Interactome map uncovers phosphatidylserine transport by oxysterol‐binding proteins. Nature 501: 257–261 [DOI] [PubMed] [Google Scholar]
- Maxfield FR, van Meer G (2010) Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol 22: 422–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew TM (1991) The new stereological methods for interpreting functional morphology from slices of cells and organs. Exp Physiol 76: 639–665 [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9: 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B, Antonny B, Drin G (2013a) Insights into the mechanisms of sterol transport between organelles. Cell Mol Life Sci 70: 3405–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Moser von Filseck J, Lacas‐Gervais S, Drin G, Antonny B (2013b) A four‐step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER‐Golgi tether OSBP. Cell 155: 830–843 [DOI] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high‐content screen. Cell 124: 1283–1298 [DOI] [PubMed] [Google Scholar]
- Moog‐Lutz C, Tomasetto C, Régnier CH, Wendling C, Lutz Y, Muller D, Chenard MP, Basset P, Rio MC (1997) MLN64 exhibits homology with the steroidogenic acute regulatory protein (STAR) and is over‐expressed in human breast carcinomas. Int J Cancer 71: 183–191 [DOI] [PubMed] [Google Scholar]
- Moser von Filseck J, Vanni S, Mesmin B, Antonny B, Drin G (2015a) A phosphatidylinositol‐4‐phosphate powered exchange mechanism to create a lipid gradient between membranes. Nat Commun 6: 6671 [DOI] [PubMed] [Google Scholar]
- Moser von Filseck J, Čopič A, Delfosse V, Vanni S, Jackson CL, Bourguet W, Drin G (2015b) INTRACELLULAR TRANSPORT. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4‐phosphate. Science 349: 432–436 [DOI] [PubMed] [Google Scholar]
- Munro S (2003) Cell biology: earthworms and lipid couriers. Nature 426: 775–776 [DOI] [PubMed] [Google Scholar]
- Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA, Nunnari J (2015) Ltc1 is an ER‐localized sterol transporter and a component of ER–mitochondria and ER–vacuole contacts. J Cell Biol 209: 539–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno‐Iwashita Y, Shimada Y, Waheed AA, Hayashi M, Inomata M, Nakamura M, Maruya M, Iwashita S (2004) Perfringolysin O, a cholesterol‐binding cytolysin, as a probe for lipid rafts. Anaerobe 10: 125–134 [DOI] [PubMed] [Google Scholar]
- Olkkonen VM (2015) OSBP‐related protein family in lipid transport over membrane contact sites. Lipid Insights 8: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Prinz WA (2010) The diverse functions of oxysterol‐binding proteins. Annu Rev Cell Dev Biol 26: 157–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers MA, Liu J, Song B‐L, Li B‐L, Chang CCY, Chang T‐Y (2015) Acyl‐CoA:cholesterol acyltransferases (ACATs/SOATs): enzymes with multiple sterols as substrates and as activators. J Steroid Biochem Mol Biol 151: 102–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Saint‐Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B, Drin G (2011) Osh4p exchanges sterols for phosphatidylinositol 4‐phosphate between lipid bilayers. J Cell Biol 195: 965–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck TL, Lange Y (2010) Cell cholesterol homeostasis: mediation by active cholesterol. Trends Cell Biol 20: 680–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujishita Y, Hurley JH (2000) Structure and lipid transport mechanism of a StAR‐related domain. Nat Struct Biol 7: 408–414 [DOI] [PubMed] [Google Scholar]
- Vacca F, Scott C, Gruenberg J (2016) The late endosome In Encyclopedia of cell biology, Bradshaw RA, Stahl PD. (eds), pp 201–210. Waltham: Academic Press; [Google Scholar]
- Vanier MT (2014) Complex lipid trafficking in Niemann‐Pick disease type C. J Inherit Metab Dis 38: 187–199 [DOI] [PubMed] [Google Scholar]
- Vassilev B, Sihto H, Li S, Hölttä‐Vuori M, Ilola J, Lundin J, Isola J, Kellokumpu‐Lehtinen P‐L, Joensuu H, Ikonen E (2015) Elevated levels of StAR‐related lipid transfer protein 3 alter cholesterol balance and adhesiveness of breast cancer cells: potential mechanisms contributing to progression of HER2‐positive breast cancers. Am J Pathol 185: 987–1000 [DOI] [PubMed] [Google Scholar]
- Wang H, Perry JW, Lauring AS, Neddermann P, De Francesco R, Tai AW (2014) Oxysterol‐binding protein is a phosphatidylinositol 4‐kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 146: 1373–1385‐11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm LP, Tomasetto C, Alpy F (2016) Touché! STARD3 and STARD3NL tether the ER to endosomes. Biochem Soc Trans 44: 493–498 [DOI] [PubMed] [Google Scholar]
- Wüstner D, Lund FW, Solanko LM (2012) Quantitative fluorescence studies of intracellular sterol transport and distribution In Fluorescent methods to study biological membranes, Mély Y, Duportail G. (eds), pp 185–213. Berlin, Heidelberg: Springer Berlin Heidelberg; [Google Scholar]
- Zhang M, Liu P, Dwyer NK, Christenson LK, Fujimoto T, Martinez F, Comly M, Hanover JA, Blanchette‐Mackie EJ, Strauss JF (2002) MLN64 mediates mobilization of lysosomal cholesterol to steroidogenic mitochondria. J Biol Chem 277: 33300–33310 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Figure 6
Review Process File