

Abstract
Activated microglia release pro-inflammatory factors and calpain into the extracellular milieu, damaging surrounding neurons. Mechanistic links to progressive neurodegeneration in diseases such as Parkinson’s disease (PD) and multiple sclerosis (MS) remain however obscure. We hypothesize that persistent damaged/dying neurons may also release cytotoxic factors and calpain into the media which then activate microglia again. Thus, inflammation, neuronal damage, and microglia activation, i.e. bi-directional interaction between neurons and microglia, may be involved in the progressive neurodegeneration. We tested this hypothesis using two in vitro models: (1) the effects of soluble factors from damaged primary cortical neurons upon primary rat neuron-microglia and (2) soluble factors released from CD3/CD28 activated peripheral blood mononuclear cells (PBMCs) of MS patients on primary human neurons and microglia. The first model indicated that neurons injured with pro-inflammatory agents (IFN-γ) release soluble neurotoxic factors, including Cox-2, ROS, and calpain, thus activating microglia, which in turn released neurotoxic factors as well. This repeated microglial activation leads to persistent inflammation and neurodegeneration. The released calpain from neurons and microglia was confirmed by calpain inhibitors calpeptin or SNJ-1945 as well as μ and mcalpain knock down using siRNA technology. Our second model using activated PBMCs, a source of pro-inflammatory Th1/Th17 cytokines and calpain released from auto-reactive T cells corroborated results in human primary cell cultures and confirmed calpain to be involved in progressive MS.
These insights into reciprocal paracrine regulation of cell injury and calpain activation in the progressive phase of MS, PD, and other neurodegenerative diseases suggest potentially beneficial preventive and therapeutic strategies, including calpain inhibition
Keywords: calpain, microglia, microgliosis, multiple sclerosis, neurodegeneration, neurons
Graphical abstract
Inflammation, neuronal damage, and microglia activation, i.e. bi-directional interaction between neurons and microglia, may be involved in neurodegenerative diseases mechanisms. This study used two in vitro models to advance our understanding of the pathogenesis: (1) the effects of soluble factors from damaged primary cortical neurons upon primary rat neuron-microglia and (2) soluble factors released from CD3/CD28 activated peripheral blood mononuclear cells (PBMCs) of multiple sclerosis (MS) patients on primary human neurons and microglia. Our results indicate that neurons injured with pro-inflammatory agents (IFN-γ) release soluble neurotoxic factors, including Cox-2, reactive oxygen species (ROS), and calpain, that activate microglia, which in turn release neurotoxic factors.
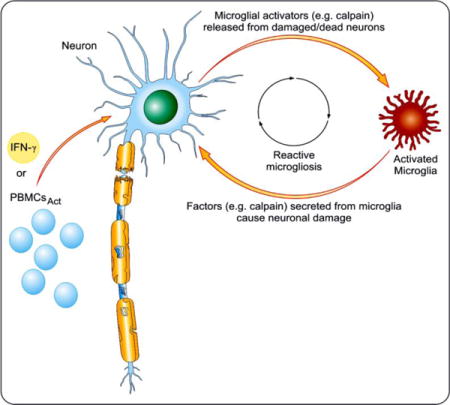
Introduction
A large body of experimental evidence suggests that inflammation is the most common factor and plays a critical role in many neurodegenerative diseases. The pathology of degenerative diseases such as Alzheimer’s (AD), multiple sclerosis (MS), and Parkinson’s disease (PD) is associated with neuronal death and axonal loss leading to progression of neurodegeneration culminating in impaired function. The dynamics of microglia-neuron communication in the neurodegenerative disease have gained attention in recent years. Our evidence indicates that the communication between neurons and microglia is bi-directional and involves several modulatory factors, including calpain. Yet many unanswered questions remain, including the neuron-microglial damage and the immediate consequence.
To this end, renewed interest in axonal and neuronal injury in multiple sclerosis (MS) has significantly shifted the focus of research into this disease toward degeneration (Bjartmar & Trapp 2001, Bitsch et al. 2000). The underlying mechanisms and nature of this degenerative process at different phases of disease progression in MS is still a mystery. Although the mechanism of persistent inflammation is accompanied by chronic neurodegeneration, the processes that contribute to neuronal damage in PD and/or cause autoimmune demyelination in progressive MS (Block & Hong 2005, McDowell et al. 2011, Samantaray et al. 2015, Smith et al. 2011) are largely unknown. No drugs have thus far been developed to significantly attenuate inflammation and progression of degeneration and resultant disability in these diseases. A major therapeutic goal, particularly in MS treatment, is to prevent the axonal loss that causes permanent neurologic disability. Because different mechanisms may contribute to axonal/neuronal damage during different stages of disease, an important goal of current and future MS research is to clarify the pathophysiology of axonal and neuronal loss in MS and other diseases.
Microglia, the resident immune effector of the CNS, respond to neuronal damage following their activation by releasing such cytotoxic mediators as pro-inflammatory cytokines (Balasubramaniam et al. 2009, Dheen et al. 2005), reactive oxygen species (ROS) (Colton & Gilbert 1987), nitric oxide (NO) (Liu et al. 2002, Moss & Bates 2001), and proteases (Kim et al. 2005, Levesque et al. 2010); these in turn can intensify the CNS inflammatory state (Dheen et al. 2007, Whitney et al. 2009). Numerous in vitro and in vivo studies have found that inhibition of microglia activation attenuates neurotoxic events and improves neuronal survival. In many neurodegenerative disorders, including MS (Banati et al. 2000, Peterson et al. 2001, Takeuchi et al. 2006a, Takeuchi et al. 2006b), Alzheimer’s disease (Cagnin et al. 2001, Meda et al. 1995), and Parkinsonism (Imamura et al. 2003, Long-Smith et al. 2009), over-activation of microglia is thought to be a key causative factor in the neuropathology. The regulation of microglia is multi-factorial, and their activation is suggested to be regulated by use of ‘on’ and ‘off’ signals from neurons influencing microglial activation (Biber et al. 2007). Activation of microglia as a consequence of neuronal injury augments their neurotoxicity (Polazzi & Contestabile 2002), and this microglial response to neuronal damage can be long-lived and self-propelling (Lull & Block 2010). Damaged or dying neurons themselves are potential triggers of microglial activation, and repeating cycle of neurotoxic microglial activation in response to neuron injury is a common feature of reactive microgliosis (Block et al. 2007, Streit et al. 1999).
Activation of microglia following brain insult is associated with the onset or progress of neurological disorders, either alone or in conjunction with other factors (e.g., calpain activation). Calpain, a Ca2+-dependent cysteine protease, has been implicated in the pathophysiology of many neurological disorders, including PD and MS. Ubiquitous calpain isoforms, calpain 1 (μ-calpain) and calpain 2 (m-calpain) that differ by their sensitivity to Ca2+, are abundantly expressed in the CNS (Ray & Banik 2003), and they are localized in cytosol and membranes (Banik et al. 1985). Calpain is critical for cell function and its over-activation causes cell death and degeneration. In addition, activation of calpain may profoundly affect glial function by stimulation of gliosis, infiltration of inflammatory cells, and induction of cell death. Therefore, tight regulation of calpain activity is essential for the preservation of cellular function. Dysregulation of calpain has been associated with a number of diseases, including MS (Guyton et al. 2005), optic neuritis (Shields & Banik 1998), Alzheimer’s disease (Zatz & Starling 2005), and Parkinson’s disease (Crocker et al. 2003, Levesque et al. 2010, Samantaray et al. 2015). The pathologic mechanisms underlying and consequences of calpain activation in MS are distinct from the other neurodegenerative diseases (Vosler et al. 2008).
IFN-γ, a Th1 pro-inflammatory cytokine, was recently shown to induce apoptotic cell death in VSC4.1 motoneuron and retinal ganglion cells (RGCs), which are known to be affected in MS and optic neuritis. Their exposure to IFN-γ initiated calpain-dependent apoptosis and modulation of calpain activation, which may have clinical significance by reducing neuronal death and improving clinical outcomes (Das et al. 2006b). To further support the hypothesis that IFN-γ could be one of the precipitating insults involved in the pathogenesis of MS, we have shown that exposure of VSC4.1 motoneurons to supernatant derived from IFN-γ-activated rat primary microglial cells also resulted in apoptotic cell death (McDowell et al. 2011). To further the argument that calpain is involved in the pathogenesis of MS, pro-inflammatory Th1/Th17 cytokines released from activated MS PBMCs (Smith et al. 2011, Imam et al. 2007), a source of auto-reactive T cells that contribute to the diseases initiation and perpetuation, was shown to correlate with the increased calpain activity and inhibition of calpain resulted in an abrogation of activated T cells cytokines (Das et al. 2006b, Trager et al. 2014). These studies suggest that calpain inhibition in vivo may abrogate both the direct neuronal degeneration and the indirect action of pro-inflammatory cytokine(s) (e.g., IFN-γ) mediated cell death. Thus, the potential of calpain released into the extracellular milieu to damage surrounding neurons formed the subject for further investigation.
The current study was designed to further elucidate the role of pro-inflammatory cytokine(s) (e.g., IFN-γ) initiated neuronal injury and established the mechanism contributing to progressive neuronal degeneration by focusing on neuron-microglia interaction.
Three fundamental questions were addressed: (i) why does the microglial response to neuron damage persist, (ii) why is this response toxic, and (iii) can calpain inhibition suppress neuron death and attenuate degeneration?
Data from two primary culture models – rat and human – demonstrated that extracellular calpain released from both damaged neurons and activated microglia is a key signal driving reactive microgliosis. Furthermore, this calcium-dependent cysteine protease released upon neuron/microglia damage activates microglia and damages naïve neurons to produce ROS and NO followed by increased activity of calpain substrates cyclooxygenase-2 (COX-2) and caspase 3, culminating in apoptosis, and calpain inhibition prevents damage. These findings indicate that the damaged neurons themselves are culpable in propagating neurotoxicity via pro-inflammatory signals to microglia.
Materials and Methods
Ethics Statement
Human fetal tissues were obtained following written approval from adult female patients undergoing therapeutic abortion at 10–14 weeks gestational age at the University of Washington, WA, USA (IRB approval #11449). The use of human fetal tissue was approved by the University of South Carolina, SC, USA (USCeRA#:HSA4636), and is IRB-exempt [45 CFR 46.102(d)]. All primary human fetal neuron cell culture studies were done according to university guidelines in a biocontainment facility approved by the Institutional Biosafety Committee (IBC) of the University of South Carolina.
PBMCs were collected from human subjects enrolled according to protocols approved by the Medical University of South Carolina Institutional Review Board (IRB #9481). Microglial cells were isolated from human adult brain tissues collected from the Medical University of South Carolina operating room during surgical procedures on non-affected/not-damaged brain according to protocols approved by the Medical University of South Carolina, SC, USA (IRB #19380). All experimental protocols were reviewed and approved by the Institutional Review Board of the Medical University of South Carolina, and experiments were undertaken with the understanding and written consent of each subject. The study conforms to the Code of Ethics of the World Medical Association (Declaration of Helsinki), printed in the British Medical Journal (18 July 1964).
Rat Cell Cultures
Rat cortical cultures (neurons and microglia) were established from two-day-old Spraque-Dawley pups from dams (Harlan) as previously reported (Banker & Goslin 1988, Jana et al. 2007). Briefly, cortices were dissected and dissociated by trituration and trypsinization. The dissociated cells were re-suspended in B-27-supplemented neurobasal media (Invitrogen) and plated onto a poly-D-lysine-coated T-flask. Neurons (adherent cells) were treated with 5 μM of the anti-mitotic inhibitor arabinose-C and cultured for 7 days prior to the experiment. Non-adherent cells contained mixed glial cells and were mechanically shaken for 30 min at 200 rpm on a gyratory shaker to isolate microglia (Bhat et al. 1992). The purity of rat cortical neurons and microglia cultures was confirmed by immunochemistry with 90–95% positive staining for neuron-specific nuclear protein (NeuN) and CD11b, respectively. Ventral spinal cord 4.1 (VSC4.1) motoneuron cell line, a generous gift from Dr. S. Appel (Baylor College of Medicine, TX, USA), was grown under previously described conditions (Das et al. 2005).
Primary Human Brain Cell Cultures
Primary human brains at 8 to 12 weeks gestational age obtained from the University of Washington, Seattle, were cultured as described earlier (Chauhan et al. 2003, Mehla & Chauhan 2015). Briefly, meninges and blood vessels were removed from the cortex. The tissue was washed twice with opti-MEM containing 1% penicillin-streptomycin and amphotericin-B (GibcoBRL). Washed tissue was mechanically disrupted by one passage through a 20-ml syringe without a needle. Disrupted cortex tissue was centrifuged at 1,200 rpm for 20 minutes at 4°C. For differentiation to neurons, pellets were resuspended in opti-MEM medium and cultured in Opti-MEM supplemented with 5% FCS, 0.2% N2 supplement (Gibco BRL), and antibiotics. The purity of neuron and astrocyte cultures was verified by immunostaining using antibody against microtubule-associated protein-2 (Sigma) or glial fibrillary acidic protein (GFAP), respectively. The purity of the neuron cultures were 90–95%. Primary human fetal neuron cultures were maintained for at least a month before use in experiments. To reduce the chance of mycoplasma contamination in these cultures, plasmocin, an anti-mycoplasma agent (Invitrogen), was used during the initial culture passages. Human primary neurons were isolated from 12- to 14-week-old fetal brains, as previously described (Chauhan et al. 2003, Mehla & Chauhan 2015). Microglial cells were isolated from human adult brain tissues collected from the Medical University of South Carolina operating room during surgical procedures on non-affected/not-damaged brain according to the previously published method (De Groot et al. 2000).
Isolation and Stimulation of PBMCs from MS Patients
PBMCs from MS patients were isolated as described previously (Smith et al. 2011). Cells were stimulated with 10 μg/ml of anti-CD3 and 5 μg/ml of anti-CD28 (BD, Franklin Lakes, NJ) following pre-treatment with the calpain inhibitor calpeptin (CP, 5 μM) in DMSO for 72 h. After treatment, supernatants were collected and stored at −80°C until use.
Treatment of Rat Primary Cortical Neurons
Primary rat neurons (RN) from cortex were stimulated with IFN-γ (100 units/ml) in the absence or presence of 1 μM CP. After 24 h of incubation, conditioned media (CM) as well as cell pellets were collected (Supplemental Fig. 1A) and analyzed (Fig. 1). CM-RN were collected, speed-vacuum concentrated (10×), and used for treatment of microglia as well as VSC4.1 motoneurons.
FIGURE 1. Calpain inhibition prevents primary rat neuron death after IFN-γ stimulation.
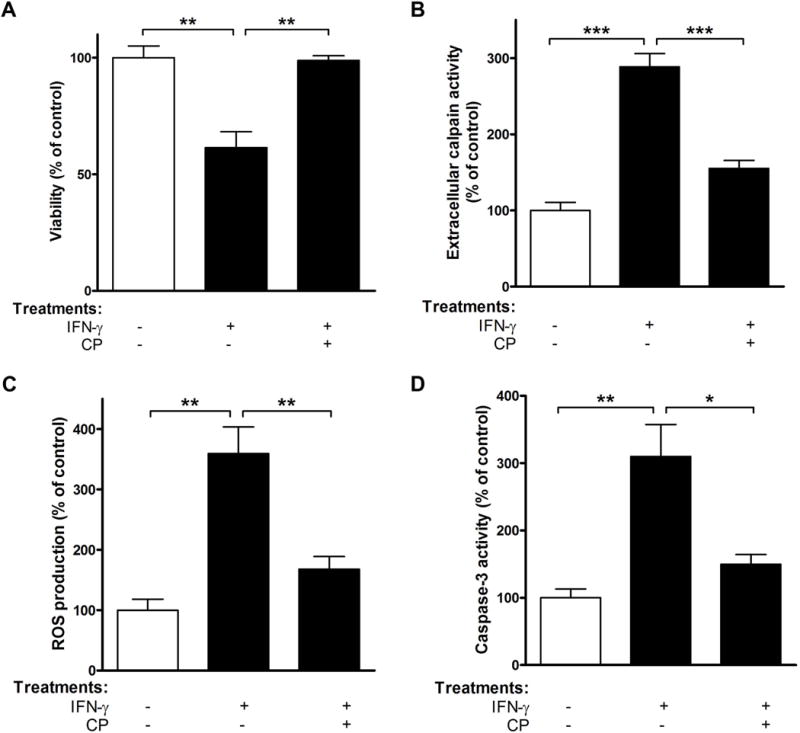
Primary RN from cortex were exposed to IFN-γ in the absence or presence of 1 μM CP for 24 h. CM and cells were then collected and subjected to (A) Trypan blue dye exclusion test; (B) extracellular calpain activity assay; (C) ROS production determination; (D) caspase-3 activity assay. *p<0.05; **p<0.01; ***p<0.001; N=3.
To determine if damaged neurons could activate microglia, primary rat microglia (RM) from cortex were pre-treated for one hour with CM-RN±CP followed by collection of CM and cells (Supplemental Fig. 1B) for subsequent analysis (Fig. 2). CM-RM were concentrated and used for further treatment.
FIGURE 2. Pre-treatment with calpeptin prevents death of rat primary cortical microglia by supernatant from IFN-γ-damaged neurons.
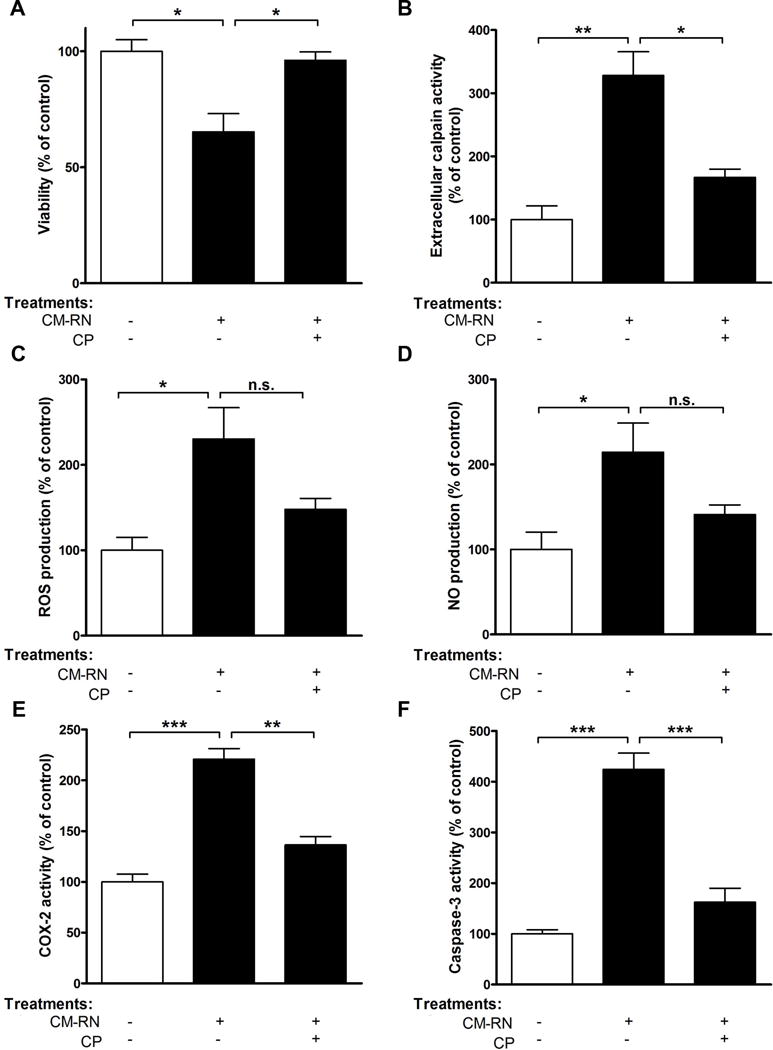
Primary RM from cortex exposed to CM-RN±CP. After 24 h of incubation, (A) viability, (B) extracellular calpain activity, (C) ROS production, (D) NO production, (E) COX-2 activity, and (F) caspase-3 activity were determined. *p<0.05; **p<0.01; ***p<0.001; n.s.-not significant; N=3.
Treatment of VSC4.1 Cells
VSC4.1 motoneurons were exposed to CM-RM, CM-RM±CP (1 h pre-treatment), CM-RM±SNJ-1945 (1 h pre-treatment), or CM-RM + siRNA for calpain 1 or CM-RM + siRNA for calpain 2 (1 h pre-treatment). After 24 hr of treatment, CM as well as cells were collected (Supplemental Fig. 1C) and analyzed (Fig. 3). The delivery of siRNA for calpain 1 (μcalpain) and calpain 2 (mcalpain) into VSC4.1 motoneurons was performed using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer’s instructions, as described earlier (Das et al. 2013b). Cell transfection was carried out 4–6 h, and the medium was then replaced with normal growth medium. Control cells were mock-transfected, and all experimental results were confirmed using scrambled siRNA. After 24 h cells were exposed to CM-RM.
FIGURE 3. Calpain inhibition/knockdown prevents VSC4.1 motoneuron cell death stimulated by condition media released from activated primary microglia.
VSC4.1 motoneurons were exposed to CM-RM; CM-RM+CP; CM-RM+SNJ-1945; CM-RM+μcalpain RNAi or CM-RM + mcalpain RNAi. After 24 h of treatment, (A) viability, (B) extracellular calpain activity, (C) ROS production, and (D) caspase-3 activity were determined. **p<0.01; ***p<0.001, N=3.
To determine whether conditioned medium (CM) from damaged neurons (CM-RN) could affect fresh naïve neuron viability and whether calpain inhibitor could prevent such damage, VSC4.1 motoneurons were treated with CM-RN±CP (1 h pre-treatment). Two types of negative controls were included: inactivation of IFN-γ by heating at 55°C for 30 min before primary rat culture stimulation and neutralization of IFN-γ by anti-IFN-γ antibody. VSC4.1 cells were either challenged with heat-inactivated IFN-γ (HI–IFN-γ) or with neutralizing antibody to IFN-γ in the absence or presence of 1 μM CP. CM and cells (Supplemental Fig. 1D) were collected and subjected to appropriate analysis (Fig. 4).
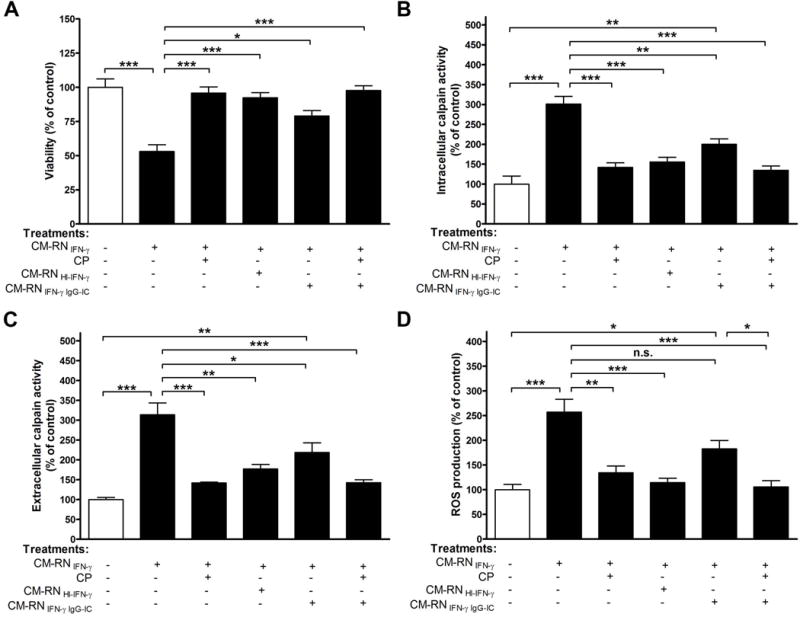
FIGURE 4. Pre-treatment with calpeptin prevents apoptotic death of VSC4.1 motoneurons by supernatant from IFN-γ damaged neurons.
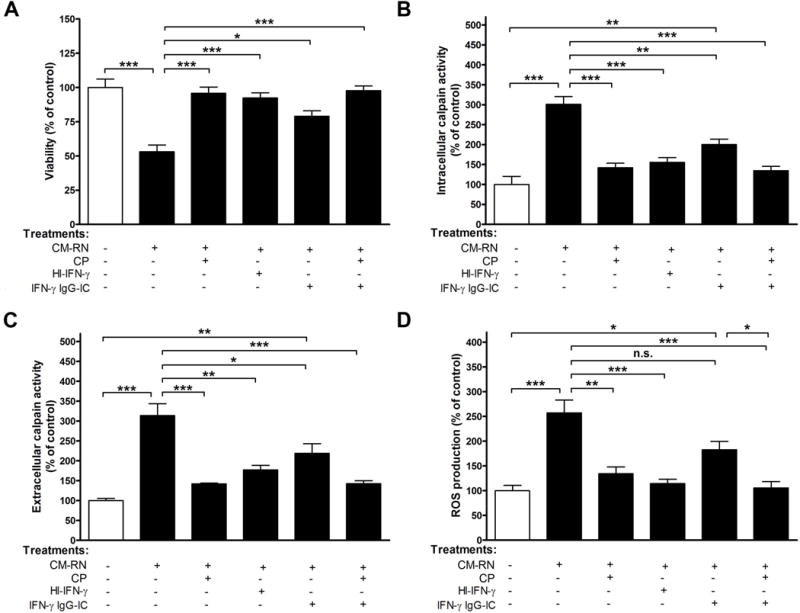
VSC 4.1 cells were exposed to CM-RN±CP; heat inactivated CM-RN; CM-RN treated with neutralizing antibody±CP. After specified treatment, (A) viability, (B) intracellular calpain activity, (C) extracellular calpain activity, and (D) ROS production were determined. p<0.05; **p<0.01; ***p<0.001; N=3.
Treatment of Human Primary Cells
Fetal human neurons (HN) were exposed to 50 μl of PBMCs, PBMCsAct, or PBMCsAct plus 1 μM or 5 μM CP (PBMCsAct+CP; 1 h pre-treatment) for 24 hr. After incubation, CM as well as cell pellets were collected (Supplemental Fig. 2A) and subjected to appropriate analysis (Fig. 5). PBMCsAct were concentrated and used for human microglia (HM) treatment.
FIGURE 5. Calpain inhibition prevents human neurons degeneration after their stimulation with PBMCsAct.
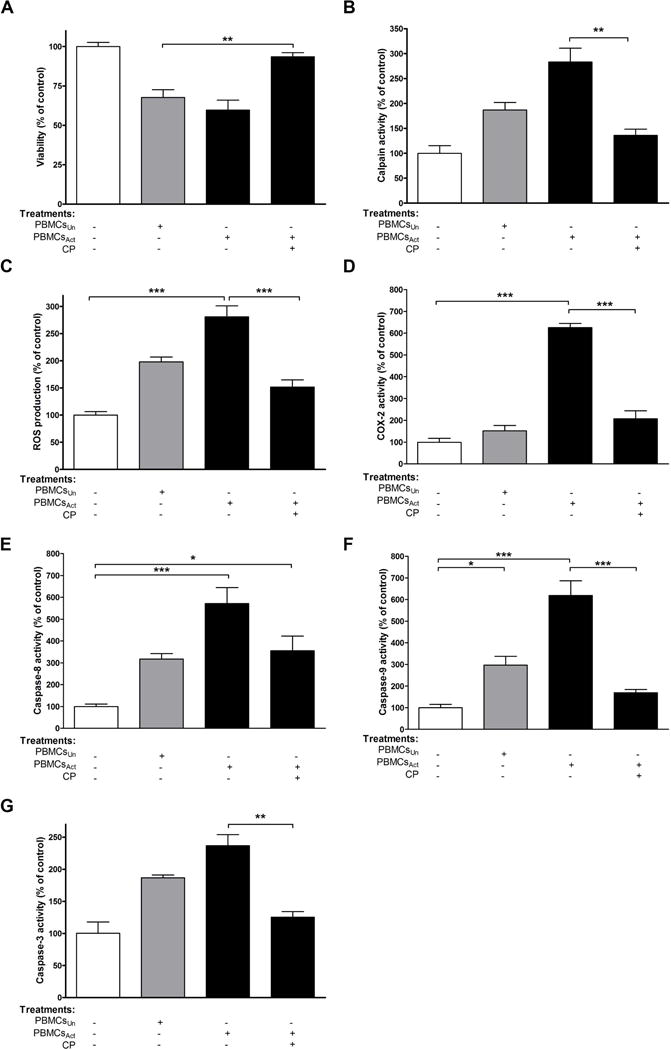
Fetal HN were exposed to PBMCs; PBMCsAct; or PBMCsAct + CP. After 24 h of incubation, (A) viability, (B) calpain activity, (C) ROS production, (D) COX-2 activity, (E) caspase-8 activity, (F) caspase-9 activity, and (G) caspase-3 activity were determined. *p<0.05; **p<0.001; ***p<0.001; N=3.
Primary HM isolated from undamaged cortex from unaffected brains of patients were treated with CM-HNPBMCsAct±CP. The CM from incubated microglia as well as the cell pellet were collected (Supplemental Fig. 2B) a (Fig. 6). CM-HM were concentrated and used for fresh or naïve HN treatment.
FIGURE 6. Pre-treatment with calpeptin prevents apoptotic death of human primary microglia by supernatant from PBMC-damaged human neurons.
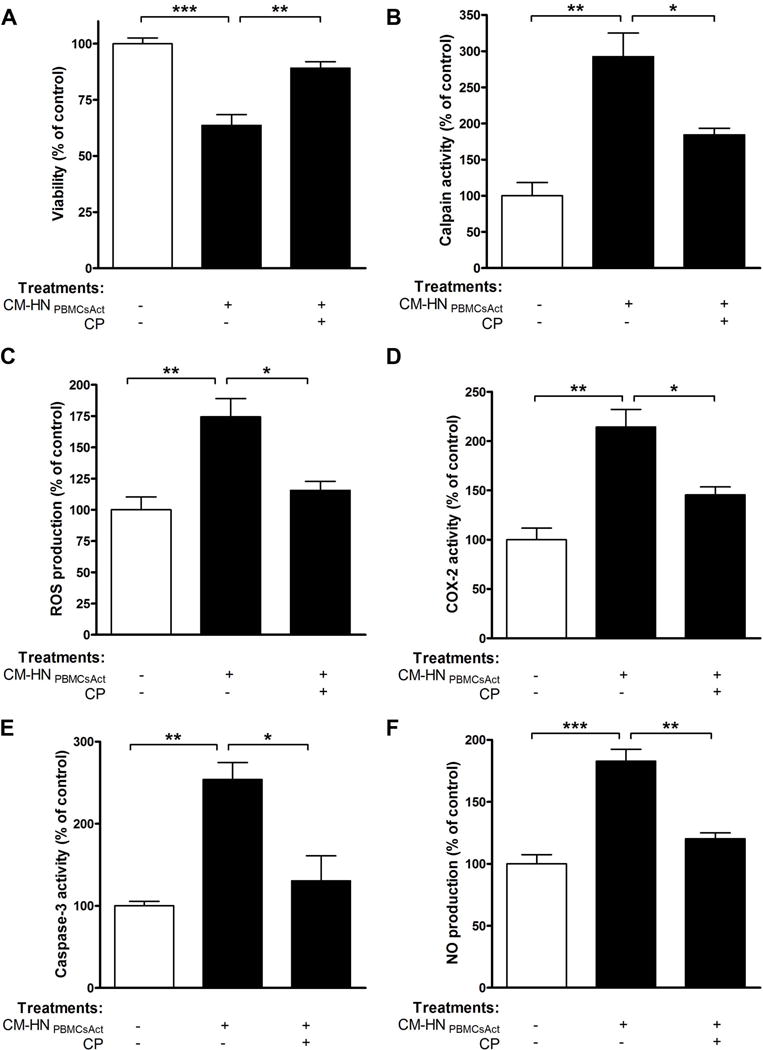
Primary HM from undamaged cortex were exposed to CM-HNPBMCsAct or CM-HNPBMCsAct+CP. After 24 h of incubation, (A) viability, (B) calpain activity, (C) ROS production, (D) COX-2 activity, (E) caspase-3 activity, and (F) NO production were determined. *p<0.05; **p<0.01; ***p<0.001; N=3.
To determine the effect of calpain upon microglia-mediated damage, fresh naive fetal HN were treated with CM-HM in the presence or absence of CP. CM and cells were collected after treatments (Supplemental Fig. 2C) and subjected to further analysis (Fig. 7). Following treatments, the mixture of attached and detached cell populations was used for estimation of the cell viability by the Trypan blue dye exclusion test (Das et al. 2004).
FIGURE 7. Calpain inhibition prevents microglia-mediated human neurons degeneration in vitro.
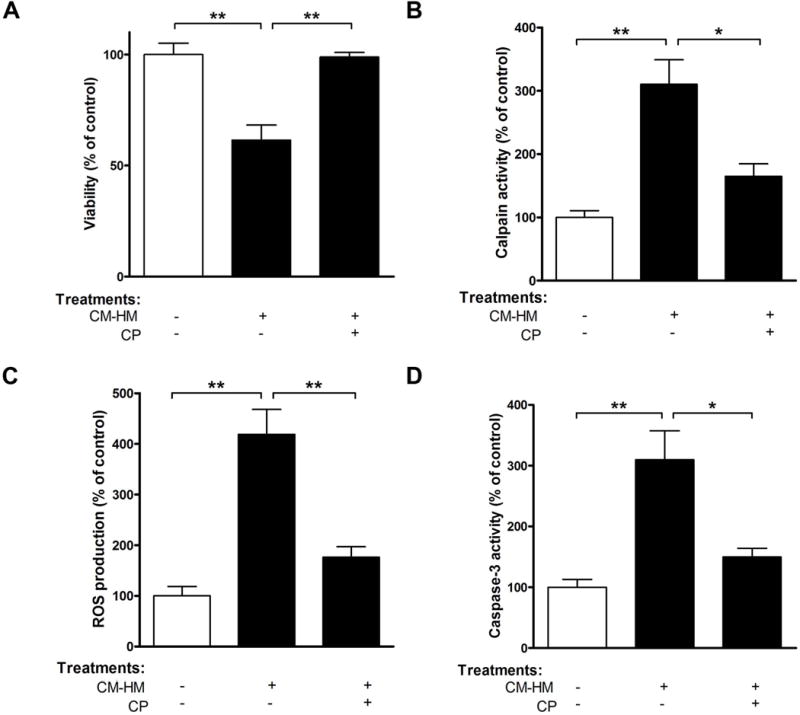
Fetal HN were exposed to CM-HM or CM-HM+CP. After 24 h of incubation, (A) viability, (B) calpain activity, (C) ROS production, and (D) caspase-3 activity were determined. *p<0.05; **p<0.01; ***p<0.001; N=3.
Calpain Activity Assay
Calpain activity in total cell lysates was determined based on the reaction with a calpain substrate, fluorogenic peptide (Ac-LLY-AFC), using a calpain activity assay kit (Abcam, Cambridge, MA). Briefly, cells were lysed then incubated with the substrate and reaction buffer for 1 h at 37°C in the dark. Proteolysis of the fluorescent probe (AFC) was monitored using a fluorimeter with filter settings of 400 nm for excitation and 505 nm for emission.
Measurement of Nitrite and ROS
The production of cytoactive molecules was measured as reported previously (Das et al. 2007, Davidge et al. 1995). Briefly, NO production was calculated from the amount of nitrite detected by Griess reaction. Absorbance was determined at 550 nm using a microplate reader. Net intracellular accumulation of ROS was detected using fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCF-DA), and the fluorescence was measured at 530 nm after excitation at 480 nm.
Assay for Cyclooxygenase-2 Activity
COX-2 activity was determined using a COX-2 activity assay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer’s protocol, as previously described (Sribnick et al. 2010). Briefly, cells (1×106) were homogenized in cold buffer 0.1 M Tris-HCl, pH 7.8 containing 1 mM EDTA, mixed with assay buffer, heme, and COX-1 inhibitor. Colorimetric substrate and arachidonic acid were added sequentially and incubated at 37°C for 10 min, and the absorbance was read at 590 nm.
Colorimetric Assays for the Measurement of Caspase-8, Caspase-9, and Caspase-3 Activities
Measurements of caspase activities in cells were performed using commercially available caspase-8, caspase-9, and caspase-3 assay kits (Sigma), as previously reported (Das et al. 2006a). Briefly, activities were assayed based on hydrolysis of the Ac-IETD p-nitroaniline (pNA) by caspase-8, Ac-LEHD pNA by caspase-9, and Ac-DEVD pNA by caspase-3. The absorbance read at 405 nm. Caspase activities in μmol of pNA released from the substrate per min per ml of cell lysate were calculated based on the formula: OD × d/εmM × t × v, where d was dilution factor, t was reaction time, v was volume of sample and the constant value εmM=10.5. Data were expressed as % of change compared to control ± SEM, where control was set as 100%.
Statistical Analysis
Results of at least 3 different experiments were expressed as the percentage of control where control values were set to 100%. Significant differences between groups were determined by one-way analysis of variance ANOVA followed by Tukey’s multiple comparison test using GraphPad PRISM 5.0. Significant differences were indicated by *p<0.05, **p<0.01, or ***p<0.001.
RESULTS
IFN-γ Induced Cell Death in Primary Rat Neurons
Primary RN culture was used as an in vitro model to investigate the cytotoxic action of the Th1 pro-inflammatory cytokine IFN-γ, a potent autoimmune inducer known to be upregulated in MS. As expected, neurons were affected by IFN-γ treatment, as indicated by a 2.6 fold reduction of viability after 24 h of treatment (Fig. 1A) and 3-fold increased (p<0.001) calpain activity (Fig. 1B). The increased vulnerability of neurons to IFN-γ injury was almost completely abolished by the calpain inhibitor CP.
Next, the mechanisms involved in cell death were investigated, and whether calpain inhibition could block neuron injury was determined. Neurons exposed to IFN-γ demonstrated a significant increase in ROS production (3.6 fold, p<0.01) at 24 h post-exposure (Fig. 1C). Activation of caspase-3, considered a central regulator of apoptosis, consistently increased by 3.1 fold (p<0.01), confirming that cells were already committed to apoptosis (Fig. 1D). Treatment with CP attenuated both ROS generation and caspase-3 activity following Th1 cytokine insult (Fig. 1, C–D). This suggests that calpain acts to increase the vulnerability of neurons to IFN-γ by affecting both ROS generation and caspase-3 activity.
Injured Primary Rat Neurons Secrete Factors that Activated Rat Microglia In Vitro
Deleterious microglial activation may be triggered by mediators released from damaged neurons causing a self-propelling cycle, a mechanism for chronic neuronal loss proposed in diverse neurodegenerative diseases (Block & Hong 2005, Gao et al. 2003, Levesque et al. 2010). In line with this, CM-RN stimulated with IFN-γ was used to activate primary RM. Microglial cells were vulnerable to the toxic mediators that were released into the CM after neuronal damage, as indicated by reduced cell viability (2.9 fold, p<0.05, Fig. 2A). The significant increases were observed in a variety of pro-inflammatory and apoptotic mediators such as calpain activity (3.3 fold, p<0.01, Fig. 2B), ROS generation (2.3 fold, p<0.05, Fig. 2C), NO production (2.1 fold, p<0.05, Fig. 2D), COX-2 activity (3.2 fold, p<0.001, Fig. 2E), and caspase-3 activity (4.2 fold, p<0.001, Fig. 2F). Calpain inhibition suppressed the effects of these mediators with caspase 3 activity being considered the most significant (Fig. 2F). This suggests that secretory factors, especially calpain, from damaged RNs activated microglial cells in vitro.
Calpain Inhibition/Knockdown Prevented VSC 4.1 Cell Death Stimulated by Secreted Factors Released From Damaged Primary Neurons
In parallel studies using VSC4.1 motoneurons, treatment with CM-RM was shown to decrease cell viability (Fig. 3A) while calpain activity (Fig. 3B), ROS production (Fig. 3C), and caspase-3 activity (Fig. 3D) were significantly increased (p<0.001). Calpain inhibition by cell-permeable inhibitor CP or the novel water-soluble calpain inhibitor SNJ-1945 significantly suppressed these effects (p<0.001; Fig. 3, B–D).
A similar effect was observed when a specific siRNA-oligonucleotide for calpain 1 or calpain 2 was transfected into VSC4.1 cells as revealed by decreased calpain activity (Fig. 3B). The microglia-induced damage indicated by ROS production (Fig. 3C), and caspase 3-activity (Fig. 3D) was significantly reduced (p<0.001) whereas non-specific siRNA had no inhibitory effect (data not shown). This is indicative that inhibition of calpain secreted from damaged primary neurons may prevent cell death.
Secreted Factors Released From IFN-γ Damaged Primary Neurons Induced VSC 4.1 Motoneuron Cell Death
To investigate whether damaged neurons could affect fresh naïve neuron viability, VSC4.1 cells were treated with CM-RN±CP (1 h pre-treatment). CM-RN were incubated CP and SNJ for 1 h prior to incubation with VSC4.1 cells. In a parallel experiment, primary microglia were also challenged with heat inactivated IFN-γ or neutralizing antibody treated CM-RN±CP. Treatment of CM-RN with CP was done 1 h prior to incubation with VSC4.1. As expected, VSC 4.1 cells showed increased sensitivity to CM-RN (Fig. 4A). No significant change in viability was seen after treatment with CM-RN+CP or comparable sets of cells treated with heat inactivated and neutralizing antibody treated CM-RN±CP (Fig. 4A). As demonstrated, the CM-RN treatment not only decreased cell viability but also increased calpain activity (Fig. 4, B–C) and elevated oxidative stress response, as revealed by ROS production (Fig. 4D). CP treatment inhibited calpain activity (Fig. 4, panels B–C) as well as production of ROS (Fig. 4D), and this inhibitory effect was closely linked to IFN-γ activity being dramatically decreased after heat inactivation of IFN-γ (Fig. 4, B–D). Calpain activity and production of ROS were also evaluated after treatment of CM-RN with anti-IFN-γ neutralizing antibody (Fig. 4, B–D), and they were significantly decreased. Therefore, secretory factors from damaged primary neuron both increased the activity of cell death markers and decreased cell viability.
Activated PBMCs from MS Patients Secreted Factors that Induced Damage to Human Neurons in vitro and Calpain Inhibition Prevented Damage
The second model used activated PBMCs as insulting stimulus in human primary cultures. Previously, activated PBMCs were shown to be a source of auto-reactive T cells releasing Th1/Th17 pro-inflammatory cytokines (Imam et al. 2007, Smith et al. 2011). This model was chosen based on the general consensus supporting a role of T cells subsets and their contribution to the disease initiation and perpetuation in EAE and MS. First, the major player and the mechanism that contributed to IFN-γ–initiated activation of neuron-microglia associated with calpain-mediated bi-directional cytotoxicity in rat cultures were determined. Secondly, whether the same or a similar mechanism contributing to bi-directional cytotoxicity is elicited by activated PBMCs from MS patients was explored.
Neurons treated with PBMCsAct showed reduced viability that was reversed by CP treatment (Fig. 5A). Calpain activity of neurons treated with PBMCs vs. PBMCsAct was also monitored. As shown in Fig. 5B, calpain activity was increased by 86.77 ± 15.26% and 183.1 ± 27.89% in neurons treated with PBMCs and PBMCsAct, respectively, suggesting that PBMCs were already secreting calpain even without CD3/CD28 in vitro activation. Calpain activity was decreased when HN were treated with PBMCsAct +CP (2.1 fold, p<0.01, Fig. 5B).
Our results on HN indicate that treatment with PBMCsAct significantly increased ROS production (2.8 fold, p<0.001) while CP treatment significantly decreased ROS generation (1.9 fold, p<0.001, Fig. 5C). As expected, COX-2 activity was greatly increased (6.3 fold, p<0.001), and CP treatment reduced COX-2 activity (3 fold, p<0.001) in exposed neurons (Fig. 5D).
Whether PBMCsAct could stimulate caspase-8, an apoptotic initiator in the extrinsic pathway, was also determined. While a marked increase in caspase-8 activity occurred after 24 h of stimulation (5.7 fold, p<0.001), this effect was abolished by 1.6 fold following CP treatment (Fig. 5E). Our data demonstrate that the loss of HN viability was associated with the consequent activation of the intrinsic pathway, caspase-9 (6.2 fold, p<0.001, Fig. 5F), and the final executioner, caspase-3. Caspase-3 was increased by 86.8% and 136.3% in HN treated with PBMCs and PBMCsAct, respectively (Fig. 5G). In contrast, treatment of HN with PBMCsAct+CP decreased caspase-3 activity by 1.9 fold (p<0.001), as shown in Fig. 5G. Thus, treatment with activated PBMCs increased calpain activation and HN cell death via the intrinsic pathway in vitro.
Calpain Inhibition Prevented Human Microglia Damage in vitro
Whether toxic mediators released from damaged HN could cause microglia injury was also investigated. Our results indicate that treatment with CM-HN reduced microglia viability (p<0.001, Fig. 6A) and demonstrated increases in calpain activity (2.9 fold, p<0.01, Fig. 6B), ROS production (p<0.01, Fig. 6C), COX-2 activity (p<0.01, Fig. 6D), and caspase-3 activity (p<0.01, Fig. 6D). The increase in ROS generation was accompanied by a significant increase in NO production (1.8 fold, p<0.001, Fig. 6F). Furthermore, calpain inhibition suppressed the effects of these toxic mediators and restored microglial viability (Fig. 6, A–F), which suggests that calpain secreted by HNs induced microglial damage.
Activated Microglia Released Factors that Induced Apoptosis in Naïve Human Neurons: the Effect Reversed by Calpain Inhibition
To explore the role of activated human microglia on cell viability, the effect of CM-HM on neurons was examined. Treatment of HN with CM-HM produced significant injury, causing decreased cell viability to 61.5% (p<0.01, Fig. 7A). The neuronal toxicity of CM-HM was attenuated by pretreatment with CP (p<0.01, Fig. 7A). Our study using human primary cultures confirmed the role of calpain as a key signal molecule released from activated neurons, as increased extracellular calpain activity was observed upon exposure of naïve HN to CM-HM (3.1 fold, p<0.01, Fig. 7B). To further reinforce the role of calpain, released by activated microglial cells into CM, parallel neuronal cultures were co-incubated with CP. As shown in Fig. 7A, CP reduced percentage of apoptotic cells and neuronal viability was almost completely restored.
Decreased cell viability was also associated with a concomitant increase in ROS production (4.2 fold, p<0.01, Fig. 7C), suggesting the involvement of oxidative damage in this process. Consistent with this result, activity of caspase-3, a critical player in apoptosis, was also increased (3.1 fold, p<0.01, Fig. 7D). In contrast to these apoptotic features, pretreatment of HN with CP followed by treatment with CM-HM suppressed the effects of these neurotoxic mediators (Fig. 7, panels B–D). Therefore, calpain secretion by microglia induces apoptosis in HN and the viability of HN could be restored following treatment with CP.
Discussion
Over the years many ideas have been proposed to explain the pathophysiology of neurodegenerative processes in progressive AD, MS, and PD (Correale 2014, Frischer et al. 2009, Kassmann et al. 2007, Meuth et al. 2008). The triggering mechanisms by which this progression occurs remain unclear. Since neuronal dysfunction and injury are thought to be present at the onset of these disease, damaged neurons may release factors that can not only bring their own demise, but also are likely to promote microglial and PBMC activation, contributing to progression of the degenerative process (Block & Hong 2005, Kim et al. 2005, Peterson et al. 2001). In MS, cortical lesions exhibit neuronal injury, including neuritic swellings and axonal transection (Peterson et al. 2001). Studies have confirmed the concept that axonal transection begins at the disease onset, and the cumulative axonal loss provides the pathologic substrate for the progressive disability experienced by most long-term MS patients. The transition from relapsing-remitting MS subtype to its secondary progressive stage and the subsequent development of progressive permanent motor disability occurs when a threshold of neuronal or axonal loss is reached or when adaptive immune response of the central nervous system (CNS) is exhausted (Reddy et al. 2000). Current therapeutic options for progressive MS are disappointing and remain challenging, reflecting a lack of understanding of pathogenic microglial mechanisms driving this MS subtype. Microglial activation correlates well with the presence of cortical lesions, alteration of synaptic function, and axonal transport, each indicative of neuronal dysfunction (Rasmussen et al. 2007). T cell infiltrates into CNS were found in mice with experimental autoimmune encephalomyelitis (EAE) at disease onset and through the acute phase whereas microglial activation was sustained during the chronic phases (Murphy et al. 2010, Rasmussen et al. 2007, Vainchtein et al. 2014). However, at present, the mode of persistent microglial activation in MS/EAE is unknown. Using both rat and human primary cultures as in vitro models, the mechanism of chronic neuronal cell death was studied following exposure to two different insulting stimuli: IFN-γ and PBMCsAct from MS patients. The exposure of RN to IFN-γ, which mimics that occurring in MS inflammation, caused their injury and functional impairment (Fig. 1).
One of the most important cell death initiators in this process was the increased intracellular Ca2+ concentration that activated calpain and implicated in related disturbances. Whether treatment of retinal ganglion cells (RGCs) with IFN-γ could increase Ca2+ and trigger calpain activation, which might lead to cell death, was previously examined (Das et al. 2006b), and degeneration of RGCs has been implicated in loss of visual function in demyelinating and other diseases (Das et al. 2013a, Shields & Banik 1998, Smith et al. 2011). In addition, elevated production of ROS, NO, and COX-2 are also factors involved in chronic neuronal loss (McTigue & Tripathi 2008). Another player involved in neuronal injury by IFN-γ in this in vitro model, as demonstrated here, was caspase-3. It should be emphasized that increased calpain activity is an important factor required to cleave inactive caspase-3 to its active form, as caspase-3 is one of the calpain substrates (Blomgren et al. 2001). Prompt activation of this caspase was also observed in microglia treated with CM-RN (Fig. 2), indicating that caspase-3 activation is a characteristic hallmark of IFN-γ–induced apoptotic machinery.
While intracellular calpain is involved in physiological function, over-activation of calpain is found in pathologic conditions and has been implicated in cell injury and degeneration in many neurodegenerative diseases, including MS (Crocker et al. 2003, Ray & Banik 2003, Shields et al. 1999, Shields et al. 1998, Trager et al. 2014, Samantaray et al. 2015). Pathologic calpain has been found to activate microglia and to be involved in activation of T cells and their migration in demyelinating conditions (Guyton et al. 2010, Smith et al. 2011, Trager et al. 2014). Degenerated neurons release several signaling molecules, including nucleotides, cytokines, and chemokines, to recruit microglia and enhance their activities (Biber et al. 2007). The death of naïve neurons by CM-RN, as shown, indicates that normal neurons are susceptible to factors that are released from damaged neurons, including calpain, which acts as a signaling protease, thus propelling their own demise and confirming earlier findings (Levesque et al. 2010, Siman et al. 2005, Smith et al. 2012). In addition, activation of microglia by released calpain and possibly other factors complete the cycle and perpetuate the progressive degeneration process by reactive gliosis (Levesque et al. 2010, Samantaray et al. 2015, Smith et al. 2012) (Fig. 4). In line with this, a recent report demonstrated the signaling protease matrix metalloprotease-3 was released from damaged neurons and activated microglia (Kim et al. 2005). Thus, the bi-directional interaction between neurons and microglia is important for understanding of chronic neuroinflammation or degeneration and gives us clues for future therapeutic strategy against neurodegenerative disorders. One strategy may be to block neuron damage with the use of calpain inhibitor, which will prevent subsequent microglial activation and disease progression. Calpain inhibition by CP or SNJ-1945 suppressed this process, which was further confirmed by knockdown of calpain using siRNA (Fig. 3). Such inhibition of microglia and T cell activation by calpain inhibitors has been found to block neuron and axon damage with reduction of the degenerative processes in demyelinating and other conditions (Crocker et al. 2003, Guyton et al. 2010, Levesque et al. 2010, Samantaray et al. 2015, Trager et al. 2014).
Our results indicating IFN-γ-induced apoptosis corroborate similar findings in human primary cultures. Neurons treated with PBMCsAct+CP reduced the secretion of inflammatory mediators, including ROS production, COX-2 activity, and activity of caspases, which in turn reduced neuronal cell death (Fig. 5). Apoptosis, a fundamental process involved in the death and degeneration of cells that occurs in progressive MS, is triggered in the “execution” phase by activation of caspases – a family of cysteine proteases that cleaves critical intracellular substrates. As reported here, exposure of HN to PBMCsAct induced apoptosis via activation of caspases-8, -9, and -3 (Fig. 6). Activation of caspase-8 indicated involvement of the extrinsic (death receptor-mediated) pathway of apoptosis while activation of caspase-9 followed by activation of caspase-3 suggested participation of the intrinsic (mitochondria-mediated) pathway as well. Treatment with CP partially blocked activation of caspases-8, -9, and-3, supporting the notion that neuroprotection is, in part, due to inhibition of both the receptor-mediated and the mitochondrial apoptotic pathway, and both involve activities of calpain consistent with the action of calpain inhibitors.
Beneficial or destructive outcomes in the CNS may be determined by a dialogue between innate and adoptive immune responses that are in most cases T cells and microglia. Our data support the hypothesis that T cells and microglia are the main drivers of MS pathology, and both cell types have been found to overexpress pathogenic calpain in this disease (Friese & Fugger 2007, Shields & Banik 1998, Shields et al. 1999). Based on our data, we propose a cascade of events that culminates in progressive MS (Fig. 8). Thus, calpain released from damaged neurons not only kills surrounding normal neurons and activates T cells, it also promotes activation of microglia contributing to different functions. Microglia are not likely to be committed to one function (Hu et al. 2015, Jeong et al. 2015) and conversion between different phenotypes seems now critical next step to clarify the factors that initiate or promote such changes. Thus, inhibition of microglia and T cell activation by calpain inhibitor as a therapeutic agent may block neurons and axon damage and reduce progression of the degenerative process.
FIGURE 8. Main mechanisms involved in progressive nature of MS driven by neuron-microglia interaction.
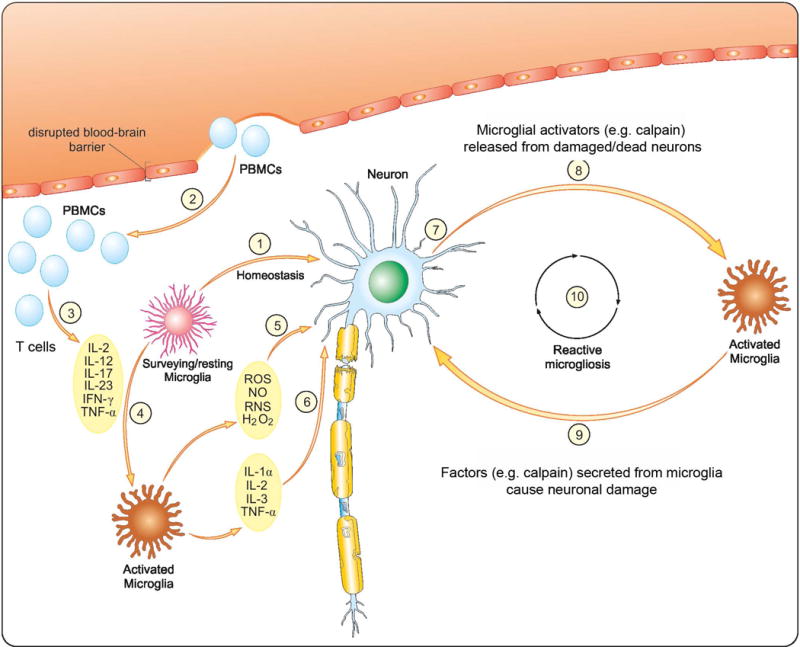
(1) In the surveillance state microglia monitor brain parenchyma and detect danger signals. (2) As a consequence of brain injury an activated peripheral blood mononuclear cells (PBMCs) infiltrate CNS through disrupted the blood-brain barrier. (3) The activated PBMCs release pro-inflammatory cytokines Th1 and Th17 (Imam et al. 2007, Smith et al. 2011). (4) Homeostasis is lost and resident microglia change phenotype to an activated state. Activation of microglia is associated with production of pro-inflammatory cytokines (5) as well as toxic metabolites (6) (McDowell et al. 2011). Supernatant from activated microglia containing released cytokines causes neurons injury (7) (McDowell et al. 2011). Increased production of different cytotoxic substances, including calpain and cytokines, induces important alterations in cortical neurons and axons. Released from dying/damaged neurons calpain-containing conditioned media activates microglial cells (8) while conditioned media from activated microglia causes injury to surrounding naïve neurons (9). Activation of microglia and neurons induce calpain-mediated bi-directional cytotoxicity to initiate a self-propelling cycle of neurons and microglia damage/death with pathology of reactive microgliosis (10). Calpain inhibitors (e.g., calpeptin) modulate calpain-mediated bi-directional cytotoxicity and could halt progressive MS.
Our study should lead to further understanding of the mechanisms underlying the progressive nature of MS and other neurodegenerative diseases and particularly the nature, activation, and role of microglia. Although the goal is to attenuate neuronal damage, our results indicate that communication between neurons and microglia is bi-directional, involving several modulatory factors, including calpain. Modulation of microglial activation for therapeutic purposes – suppressing deleterious actions while simultaneously preserving their protective functions – appears well worth pursuing. Since microglia participation is critical in many neurodegenerative diseases, especially those with an inflammatory component, ongoing research upon microglia modulation could provide greater insight necessary before any potential strategies for targeting microglia to attenuate inflammation and modify disease course is feasible. Targeting the interaction between neurons, microglia, and T cells is a potential approach to constrain the chronic inflammation and curb the progressive form of MS.
Supplementary Material
Acknowledgments
We gratefully acknowledge the funding support in part by the R01 grants from the National Institute of Neurological Disorders – National Institutes of Health, Bethesda, MD, USA (NS-41088, NS-56176 and NS-65456); non-HHS Research Project (I01) award from Veterans Affairs (5I01BX002349–02) for the work to NLB. Special thanks are to the Kosciuszko Foundation for the fellowship 2014/15 and to the Polish-U.S Fulbright Commission (2015/16 Senior Award Grant) to MP. The authors also thank Mrs. Katarzyna Izydorczyk for her excellent graphic skills in rendering the diagrams of the cell treatments and the Figure depicting the mechanisms proposed for microglia-neuron interactions driving progressive MS. We also thank Ms. Denise Matzelle for editorial assistance of the manuscript.
Abbreviations used
- CM
conditioned media
- CNS
central nervous system
- COX-2
cyclooxygenase-2
- CP
calpeptin
- HI-IFN-γ
heat inactivated IFN-γ
- HM
human microglia
- HN
human neurons
- MS
multiple sclerosis
- NO
nitric oxide
- PBMCsAct
activated peripheral blood mononuclear cells
- PBMCs
unactivated peripheral blood mononuclear cells
- RM
rat microglia
- RN
rat neurons
- CM-RN
CM from RN stimulated with IFN-γ
- CM-RN±CP
CM from RN stimulated with IFN-γ in the presence or absence of CP
- CM-RM
CM from RM treated with CM-RN
- CM-RM±CP
CM from RM treated with CM-RN in the presence or absence of CP
- CM-RNHI-IFN-γ
CM from RN treated with heat inactivated IFN-γ
- CM-HNPBMCs
CM from HN treated with PBMCs
- CM-HNPBMCsAct
CM from HN treated with activated PBMCs (PBMCsAct)
- CM-HNPBMCsAct±CP
CM from HN treated with PBMCsAct in the presence or absence of CP
- CM-HM±CP
CM from HM treated with CM-HNPBMCsAct in the presence or absence of CP
- CM-HN±CP
CM from HN treated with CM-HM in the presence or absence of CP
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- VSC4.1
ventral spinal cord 4.1
Footnotes
Involves human subjects: Yes
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if No, skip complete sentence
=> if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.”
Conflicts of interest: none
=> if ‘none’, insert “The authors have no conflict of interest to declare.”
=> otherwise insert info unless it is already included
Conflict of interest statement
The authors have no conflicting financial interests. Calpain inhibitor SNJ 1945 was obtained from Senju Pharmaceuticals, Kobe, Japan.
References
- Balasubramaniam B, Carter DA, Mayer EJ, Dick AD. Microglia derived IL-6 suppresses neurosphere generation from adult human retinal cell suspensions. Experimental eye research. 2009;89:757–766. doi: 10.1016/j.exer.2009.06.019. [DOI] [PubMed] [Google Scholar]
- Banati RB, Newcombe J, Gunn RN, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123(Pt 11):2321–2337. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- Banik NL, McAlhaney WW, Hogan EL. Calcium-stimulated proteolysis in myelin: evidence for a Ca2+-activated neutral proteinase associated with purified myelin of rat CNS. J Neurochem. 1985;45:581–588. doi: 10.1111/j.1471-4159.1985.tb04026.x. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K. Developments in neuronal cell culture. Nature. 1988;336:185–186. doi: 10.1038/336185a0. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Hauser KF, Kindy MS. Cell proliferation and protooncogene induction in oligodendroglial progenitors. J Neurosci Res. 1992;32:340–349. doi: 10.1002/jnr.490320306. [DOI] [PubMed] [Google Scholar]
- Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends in neurosciences. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123(Pt 6):1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol. 2001;14:271–278. doi: 10.1097/00019052-200106000-00003. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Progress in neurobiology. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature reviews Neuroscience. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Chauhan A, Turchan J, Pocernich C, Bruce-Keller A, Roth S, Butterfield DA, Major EO, Nath A. Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J Biol Chem. 2003;278:13512–13519. doi: 10.1074/jbc.M209381200. [DOI] [PubMed] [Google Scholar]
- Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS letters. 1987;223:284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- Correale J. The role of microglial activation in disease progression. Mult Scler. 2014;20:1288–1295. doi: 10.1177/1352458514533230. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Smith PD, Jackson-Lewis V, et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:4081–4091. doi: 10.1523/JNEUROSCI.23-10-04081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Banik NL, Patel SJ, Ray SK. Dexamethasone protected human glioblastoma U87MG cells from temozolomide induced apoptosis by maintaining Bax:Bcl-2 ratio and preventing proteolytic activities. Molecular cancer. 2004;3:36. doi: 10.1186/1476-4598-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Banik NL, Ray SK. Mechanism of apoptosis with the involvement of calpain and caspase cascades in human malignant neuroblastoma SH-SY5Y cells exposed to flavonoids. Int J Cancer. 2006a;119:2575–2585. doi: 10.1002/ijc.22228. [DOI] [PubMed] [Google Scholar]
- Das A, Banik NL, Ray SK. Garlic compounds generate reactive oxygen species leading to activation of stress kinases and cysteine proteases for apoptosis in human glioblastoma T98G and U87MG cells. Cancer. 2007;110:1083–1095. doi: 10.1002/cncr.22888. [DOI] [PubMed] [Google Scholar]
- Das A, Garner DP, Del Re AM, Woodward JJ, Kumar DM, Agarwal N, Banik NL, Ray SK. Calpeptin provides functional neuroprotection to rat retinal ganglion cells following Ca2+ influx. Brain Res. 2006b;1084:146–157. doi: 10.1016/j.brainres.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Das A, Guyton MK, Smith A, Wallace Gt, McDowell ML, Matzelle DD, Ray SK, Banik NL. Calpain inhibitor attenuated optic nerve damage in acute optic neuritis in rats. J Neurochem. 2013a;124:133–146. doi: 10.1111/jnc.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Sribnick EA, Wingrave JM, Del Re AM, Woodward JJ, Appel SH, Banik NL, Ray SK. Calpain activation in apoptosis of ventral spinal cord 4.1 (VSC4.1) motoneurons exposed to glutamate: calpain inhibition provides functional neuroprotection. J Neurosci Res. 2005;81:551–562. doi: 10.1002/jnr.20581. [DOI] [PubMed] [Google Scholar]
- Das A, Wallace Gt, Reiter RJ, Varma AK, Ray SK, Banik NL. Overexpression of melatonin membrane receptors increases calcium-binding proteins and protects VSC4.1 motoneurons from glutamate toxicity through multiple mechanisms. Journal of pineal research. 2013b;54:58–68. doi: 10.1111/j.1600-079X.2012.01022.x. [DOI] [PubMed] [Google Scholar]
- Davidge ST, Baker PN, Laughlin MK, Roberts JM. Nitric oxide produced by endothelial cells increases production of eicosanoids through activation of prostaglandin H synthase. Circulation research. 1995;77:274–283. doi: 10.1161/01.res.77.2.274. [DOI] [PubMed] [Google Scholar]
- De Groot CJ, Montagne L, Janssen I, Ravid R, Van Der Valk P, Veerhuis R. Isolation and characterization of adult microglial cells and oligodendrocytes derived from postmortem human brain tissue. Brain research Brain research protocols. 2000;5:85–94. doi: 10.1016/s1385-299x(99)00059-8. [DOI] [PubMed] [Google Scholar]
- Dheen ST, Jun Y, Yan Z, Tay SS, Ling EA. Retinoic acid inhibits expression of TNF-alpha and iNOS in activated rat microglia. Glia. 2005;50:21–31. doi: 10.1002/glia.20153. [DOI] [PubMed] [Google Scholar]
- Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Current medicinal chemistry. 2007;14:1189–1197. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- Friese MA, Fugger L. T cells and microglia as drivers of multiple sclerosis pathology. Brain. 2007;130:2755–2757. doi: 10.1093/brain/awm246. [DOI] [PubMed] [Google Scholar]
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. FASEB J. 2003;17:1954–1956. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Das A, Samantaray S, Wallace GCt, Butler JT, Ray SK, Banik NL. Calpeptin attenuated inflammation, cell death, and axonal damage in animal model of multiple sclerosis. J Neurosci Res. 2010;88:2398–2408. doi: 10.1002/jnr.22408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton MK, Wingrave JM, Yallapragada AV, Wilford GG, Sribnick EA, Matzelle DD, Tyor WR, Ray SK, Banik NL. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J Neurosci Res. 2005;81:53–61. doi: 10.1002/jnr.20470. [DOI] [PubMed] [Google Scholar]
- Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11:56–64. doi: 10.1038/nrneurol.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam SA, Guyton MK, Haque A, Vandenbark A, Tyor WR, Ray SK, Banik NL. Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. J Neuroimmunol. 2007;190:139–145. doi: 10.1016/j.jneuroim.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta neuropathologica. 2003;106:518–526. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- Jana M, Jana A, Pal U, Pahan K. A simplified method for isolating highly purified neurons, oligodendrocytes, astrocytes, and microglia from the same human fetal brain tissue. Neurochemical research. 2007;32:2015–2022. doi: 10.1007/s11064-007-9340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SY, Jeon R, Choi YK, Jung JE, Liang A, Xing C, Wang X, Lo EH, Song YS. Activation of microglial TLR3 promotes neuronal survival against cerebral ischemia. J Neurochem. 2015 doi: 10.1111/jnc.13441. [DOI] [PubMed] [Google Scholar]
- Kassmann CM, Lappe-Siefke C, Baes M, et al. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet. 2007;39:969–976. doi: 10.1038/ng2070. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kim SS, Cho JJ, Choi DH, Hwang O, Shin DH, Chun HS, Beal MF, Joh TH. Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:3701–3711. doi: 10.1523/JNEUROSCI.4346-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque S, Wilson B, Gregoria V, Thorpe LB, Dallas S, Polikov VS, Hong JS, Block ML. Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain. 2010;133:808–821. doi: 10.1093/brain/awp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Gao HM, Wang JY, Jeohn GH, Cooper CL, Hong JS. Role of nitric oxide in inflammation-mediated neurodegeneration. Annals of the New York Academy of Sciences. 2002;962:318–331. doi: 10.1111/j.1749-6632.2002.tb04077.x. [DOI] [PubMed] [Google Scholar]
- Long-Smith CM, Sullivan AM, Nolan YM. The influence of microglia on the pathogenesis of Parkinson’s disease. Progress in neurobiology. 2009;89:277–287. doi: 10.1016/j.pneurobio.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2010;7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell ML, Das A, Smith JA, Varma AK, Ray SK, Banik NL. Neuroprotective effects of genistein in VSC4.1 motoneurons exposed to activated microglial cytokines. Neurochem Int. 2011;59:175–184. doi: 10.1016/j.neuint.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem. 2008;107:1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Mehla R, Chauhan A. HIV-1 differentially modulates autophagy in neurons and astrocytes. J Neuroimmunol. 2015;285:106–118. doi: 10.1016/j.jneuroim.2015.06.001. [DOI] [PubMed] [Google Scholar]
- Meuth SG, Simon OJ, Grimm A, Melzer N, Herrmann AM, Spitzer P, Landgraf P, Wiendl H. CNS inflammation and neuronal degeneration is aggravated by impaired CD200-CD200R-mediated macrophage silencing. J Neuroimmunol. 2008;194:62–69. doi: 10.1016/j.jneuroim.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Moss DW, Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. The European journal of neuroscience. 2001;13:529–538. doi: 10.1046/j.1460-9568.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Annals of neurology. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- Polazzi E, Contestabile A. Reciprocal interactions between microglia and neurons: from survival to neuropathology. Reviews in the neurosciences. 2002;13:221–242. doi: 10.1515/revneuro.2002.13.3.221. [DOI] [PubMed] [Google Scholar]
- Rasmussen S, Wang Y, Kivisakk P, Bronson RT, Meyer M, Imitola J, Khoury SJ. Persistent activation of microglia is associated with neuronal dysfunction of callosal projecting pathways and multiple sclerosis-like lesions in relapsing–remitting experimental autoimmune encephalomyelitis. Brain. 2007;130:2816–2829. doi: 10.1093/brain/awm219. [DOI] [PubMed] [Google Scholar]
- Ray SK, Banik NL. Calpain and its involvement in the pathophysiology of CNS injuries and diseases: therapeutic potential of calpain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:173–189. doi: 10.2174/1568007033482887. [DOI] [PubMed] [Google Scholar]
- Reddy H, Narayanan S, Arnoutelis R, Jenkinson M, Antel J, Matthews PM, Arnold DL. Evidence for adaptive functional changes in the cerebral cortex with axonal injury from multiple sclerosis. Brain. 2000;123(Pt 11):2314–2320. doi: 10.1093/brain/123.11.2314. [DOI] [PubMed] [Google Scholar]
- Samantaray S, Knaryan VH, Shields DC, Cox AA, Haque A, Banik NL. Inhibition of Calpain Activation Protects MPTP-Induced Nigral and Spinal Cord Neurodegeneration, Reduces Inflammation, and Improves Gait Dynamics in Mice. Mol Neurobiol. 2015;52:1054–1066. doi: 10.1007/s12035-015-9255-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Putative role of calpain in the pathophysiology of experimental optic neuritis. Experimental eye research. 1998;67:403–410. doi: 10.1006/exer.1998.0537. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Saido TC, Banik NL. A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc Natl Acad Sci U S A. 1999;96:11486–11491. doi: 10.1073/pnas.96.20.11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Tyor WR, Deibler GE, Hogan EL, Banik NL. Increased calpain expression in activated glial and inflammatory cells in experimental allergic encephalomyelitis. Proc Natl Acad Sci U S A. 1998;95:5768–5772. doi: 10.1073/pnas.95.10.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Zhang C, Roberts VL, Pitts-Kiefer A, Neumar RW. Novel surrogate markers for acute brain damage: cerebrospinal fluid levels corrrelate with severity of ischemic neurodegeneration in the rat. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2005;25:1433–1444. doi: 10.1038/sj.jcbfm.9600138. [DOI] [PubMed] [Google Scholar]
- Smith AW, Doonan BP, Tyor WR, Abou-Fayssal N, Haque A, Banik NL. Regulation of Th1/Th17 cytokines and IDO gene expression by inhibition of calpain in PBMCs from MS patients. J Neuroimmunol. 2011;232:179–185. doi: 10.1016/j.jneuroim.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Klaver AC, Coffey MP, Dang L, Loeffler DA. Effects of intravenous immunoglobulin on alpha synuclein aggregation and neurotoxicity. Int Immunopharmacol. 2012;14:550–557. doi: 10.1016/j.intimp.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Sribnick EA, Samantaray S, Das A, Smith J, Matzelle DD, Ray SK, Banik NL. Postinjury estrogen treatment of chronic spinal cord injury improves locomotor function in rats. J Neurosci Res. 2010;88:1738–1750. doi: 10.1002/jnr.22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Progress in neurobiology. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006a;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Wang J, Kawanokuchi J, Mitsuma N, Mizuno T, Suzumura A. Interferon-gamma induces microglial-activation-induced cell death: a hypothetical mechanism of relapse and remission in multiple sclerosis. Neurobiology of disease. 2006b;22:33–39. doi: 10.1016/j.nbd.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Trager N, Smith A, Wallace IvG, Azuma M, Inoue J, Beeson C, Haque A, Banik NL. Effects of a novel orally administered calpain inhibitor SNJ-1945 on immunomodulation and neurodegeneration in a murine model of multiple sclerosis. J Neurochem. 2014;130:268–279. doi: 10.1111/jnc.12659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchtein ID, Vinet J, Brouwer N, Brendecke S, Biagini G, Biber K, Boddeke HW, Eggen BJ. In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia. 2014;62:1724–1735. doi: 10.1002/glia.22711. [DOI] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney NP, Eidem TM, Peng H, Huang Y, Zheng JC. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J Neurochem. 2009;108:1343–1359. doi: 10.1111/j.1471-4159.2009.05886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatz M, Starling A. Calpains and disease. The New England journal of medicine. 2005;352:2413–2423. doi: 10.1056/NEJMra043361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.