

ABSTRACT
Double-stranded RNAs (dsRNA) produced during human cytomegalovirus (HCMV) infection activate the antiviral kinase protein kinase R (PKR), which potently inhibits virus replication. The HCMV pTRS1 and pIRS1 proteins antagonize PKR to promote HCMV protein synthesis and replication; however, the mechanism by which pTRS1 inhibits PKR is unclear. PKR activation occurs in a three-step cascade. First, binding to dsRNA triggers PKR homodimerizaton. PKR dimers then autophosphorylate, leading to a conformational shift that exposes the binding site for the PKR substrate eIF2α. Consistent with previous in vitro studies, we found that pTRS1 bound and inhibited PKR. pTRS1 binding to PKR was not mediated by an RNA intermediate, and mutations in the pTRS1 RNA binding domain did not affect PKR binding or inhibition. Rather, mutations that disrupted the pTRS1 interaction with PKR ablated the ability of pTRS1 to antagonize PKR activation by dsRNA. pTRS1 did not block PKR dimerization and could bind and inhibit a constitutively dimerized PKR kinase domain. In addition, pTRS1 binding to PKR inhibited PKR kinase activity. Single amino acid point mutations in the conserved eIF2α binding domain of PKR disrupted pTRS1 binding and rendered PKR resistant to inhibition by pTRS1. Consistent with a critical role for the conserved eIF2α contact site in PKR binding, pTRS1 bound an additional eIF2α kinase, heme-regulated inhibitor (HRI), and inhibited eIF2α phosphorylation in response to an HRI agonist. Together our data suggest that pTRS1 inhibits PKR by binding to conserved amino acids in the PKR eIF2α binding site and blocking PKR kinase activity.
IMPORTANCE The antiviral kinase PKR plays a critical role in controlling HCMV replication. This study furthered our understanding of how HCMV evades inhibition by PKR and identified new strategies for how PKR activity might be restored during infection to limit HCMV disease.
KEYWORDS: human herpesvirus, human cytomegalovirus, HCMV, gene expression, protein synthesis, mRNA translation, eIF2α, protein kinase R, PKR, innate immunity
INTRODUCTION
Protein kinase R (PKR) plays a critical role in the cellular intrinsic antiviral response (1). Double-stranded RNAs (dsRNAs) produced during viral infection activate PKR, leading to phosphorylation of its only known substrate, the α subunit of the eIF2 translation initiation factor (eIF2α) (2–4). Phosphorylation of eIF2α reduces formation of the ternary complex, which is composed of eIF2α, GTP, and a charged methionyl tRNA (tRNAMet) (5). Since the ternary complex is required for translation initiation, activation of PKR limits the synthesis of both cellular and viral proteins and inhibits virus replication (6, 7). Active PKR further limits virus replication by increasing type I interferon expression, stimulating apoptosis, inducing autophagy, and possibly activating inflammasomes (8–14). Together, these functions make PKR a potent antiviral effector capable of inhibiting the replication of many viruses.
Both RNA and DNA viruses generate dsRNA ligands that activate PKR during infection. Structured regions of RNA virus genomes and dsRNA replicative intermediates are the most likely triggers of PKR activation during RNA virus infection (15–17). dsRNAs produced by overlapping transcriptional units during DNA virus replication likely drive PKR activation (18, 19). While PKR exists as an inactive monomer in uninfected cells, binding to dsRNA, mediated by two amino-terminal dsRNA binding domains (dsRBDs), results in a conformational shift that allows for PKR dimerization. Dimerization results in PKR autophosphorylation on multiple serine and threonine residues; however, only phosphorylation on threonine 446 (Thr446) and 451 (Thr451) in the PKR kinase domain (KD) activation loop are necessary for PKR activation (16, 20–23). Following autophosphorylation, conserved residues within the αG helix in the C-lobe of the PKR kinase domain mediate its interaction with eIF2α (3, 4). PKR binding to dsRNA ligands thus leads to a catalytically active PKR capable of binding and phosphorylating eIF2α, resulting in inhibition of protein synthesis and decreased viral replication.
Almost every virus encodes a PKR inhibitor, underscoring the critical role of PKR in the antiviral response (1). In several cases a single virus employs multiple proteins to limit PKR activation or mitigate the effects of eIF2α phosphorylation. For example, herpes simplex virus (HSV) encodes three proteins that limit PKR activation and eIF2α phosphorylation to facilitate virus replication. The vhs protein degrades viral dsRNAs that otherwise activate PKR, while the US11 protein binds PKR and prevents PKR-dependent eIF2α phosphorylation (12, 24). In addition, the ICP34.5 protein recruits the cellular protein phosphatase 1 (PP1) to dephosphorylate eIF2α and ensure continued synthesis of viral proteins (25). Similarly, the murine cytomegalovirus (MCMV) m142 and m143 proteins and the vaccinia virus (VV) K3L and E3L proteins each function to limit PKR activation (26–33). Other viruses, including orthomyxoviruses, flaviviruses, and retroviruses, each encode at least one PKR antagonist, highlighting the important role of PKR antagonism for successful viral replication (15, 17, 34).
Like the above-mentioned viruses, human cytomegalovirus (HCMV) encodes PKR antagonists that are necessary for virus replication. The HCMV TRS1 and IRS1 proteins (pTRS1 and pIRS1, respectively) each inhibit PKR activation (7, 35–37). The amino-terminal two-thirds of both pTRS1 and pIRS1 are conserved and contain a noncanonical RNA binding domain that competitively inhibits PKR binding to dsRNA ligands in vitro (38, 39). While the carboxyl-terminal thirds of pTRS1 and pIRS1 differ in sequence, both proteins contain a C-terminal PKR binding domain and both can inhibit PKR activation (6, 7, 37). Expression of either pTRS1 or pIRS1 is sufficient to prevent PKR activation during HCMV infection of human fibroblasts, and viral protein synthesis and HCMV replication are dramatically reduced in the absence of both proteins (6, 7, 18). pTRS1 and pIRS1 have additional functions, including promoting mRNA translation and limiting autophagy (40–49). However, the viral replication defect observed in the absence of both pTRS1 and pIRS1 is largely reversed through inhibition of PKR expression (6, 7). Though the other functions of pTRS1 and pIRS1 may play important roles in other cell types, inhibiting PKR activation appears to be the predominant role for pTRS1 and pIRS1 during HCMV infection of primary human fibroblasts.
While the role of pTRS1 and pIRS1 as PKR antagonists is well established, questions remain concerning the mechanism by which they inhibit PKR. pTRS1 binds both dsRNA and PKR, but the relative importance of these two functions for PKR antagonism is unknown. In this study, we performed a series of molecular biology and biochemical experiments to further elucidate the molecular mechanism(s) by which pTRS1 prevents PKR activation. We found that pTRS1 binding to both dsRNA and PKR contributes to PKR antagonism, although pTRS1 binding to PKR is the predominant mechanism that mediates PKR inhibition by the full-length TRS1 protein. Our results suggest that pTRS1 does not block PKR dimerization but rather prevents activating autophosphorylation of PKR dimers. pTRS1 binding to PKR is mediated through interactions with conserved amino acids in the PKR eIF2α contact site and results in inhibition of PKR kinase activity. We also found that pTRS1 binds the heme-regulated inhibitor (HRI) kinase and inhibits eIF2α phosphorylation in response to the HRI agonist arsenite in PKR-deficient cells. Together, our results provide further insight into the mechanism of PKR antagonism by pTRS1.
RESULTS
pTRS1 inhibits PKR activation by dsRNA.
We began by examining the ability of pTRS1 to inhibit PKR activation in transfected cells. Increasing amounts of a synthetic dsRNA [poly(I·C)] were added to lysates of cells transfected with either a GFP or pTRS1 expression plasmid, and PKR activation was measured by Western blotting. PKR is activated by autophosphorylation on Thr446 (T446) and Thr451 (T451), although phosphorylation of T451 is dependent upon T446 phosphorylation (23, 50). We therefore used PKR T446 phosphorylation as a marker of PKR activation. Low levels of phosphorylated PKR were present in the absence of poly(I·C) in cells expressing GFP, likely due to stress from transfection. Poly(I·C) activated PKR in cells expressing GFP (Fig. 1A). In contrast, the presence of pTRS1 completely suppressed PKR activation, even at the highest doses of poly(I·C). Thus, pTRS1 is sufficient to prevent PKR activation by dsRNA in cell culture.
FIG 1.

Domains of pTRS1 necessary for PKR binding and inhibition. (A) Lysates from HEK293T cells transfected with either a GFP or pTRS1 expression vector were treated with poly(I·C) to induce PKR activation. Levels of phosphorylated PKR (T446) were measured by Western blotting. (B) Schematic showing location of pTRS1 functional domains and pTRS1 truncation mutants (RBD, dsRNA binding domain; PBD, PKR binding domain). Areas shaded gray are conserved between pTRS1 and pIRS1. (C) HEK293T cells were transfected with GFP, full-length TRS1, or the indicated TRS1 mutants. Lysates were treated with poly(I·C), and PKR phosphorylation (T446) was measured by Western blotting. (D) HEK293T cells were cotransfected with Myc-tagged kinase-dead PKR (K296R) and either full-length TRS1 or the indicated TRS1 mutants. Cell lysates were immunoprecipitated with anti-Myc (PKR) or IgG (specificity control) antibody. The presence of pTRS1 in the immune complexes was determined by Western blotting. Representative results from one of at least three independent experiments are shown for each panel.
We next examined the regions of pTRS1 necessary for PKR antagonism. In these experiments, we used a series of plasmids expressing amino- or carboxyl-terminal truncations of pTRS1, as they have previously been characterized in vitro (37, 39) (Fig. 1B and Table 1) and measured PKR T446 phosphorylation after poly(I·C) treatment (Fig. 1C). It is important to note that truncation mutants may unintentionally affect protein folding, and thus negative results using these mutants should be interpreted with caution. As before, full-length pTRS1 blocked activation of PKR by poly(I·C) treatment. However, pTRS1 lacking its amino-terminal 240 or 390 amino acids (pTRS1 240-795 or pTRS1 390-795, respectively), which contain the dsRNA binding domain, did not inhibit PKR activation. Similarly, a pTRS1 mutant lacking the final 116 amino acids (pTRS1 1-679), containing the PKR binding domain, also failed to antagonize PKR. Curiously, the removal of an additional 180 amino acids from the C terminus (pTRS1 1-550) restored PKR inhibition, but further deletion (pTRS1 1-370) again led to an inability to inhibit PKR activation.
TABLE 1.
pTRS1 and PKR mutations and functional descriptionsa
| Type of expression vector and plasmid name | Mutation | Description | Reference(s) |
|---|---|---|---|
| PKR expression vectors | |||
| PKR T446A | T446A | Mutation in activating phosphorylation site | 23 |
| PKR E375V | E375V | Confers resistance to inhibition by VV K3L | 27 |
| PKR A473T | A473T | ||
| PKR D486V | D486V | ||
| PKR T487A | T487A | Disrupts PKR phosphorylation of eIF2α | 3 |
| PKR E490A | F495R | ||
| PKR F495R | E490A | Mutation in conserved eIF2α αG helix residue | 4 |
| PKR K296R Flag | K296R | Kinase-dead PKR with N-terminal Flag tag | 3 |
| PKR K296R Myc | K296R | Kinase dead PKR with N-terminal Myc tag | |
| PKR GST-KD | NA | Doxycycline-inducible fusion of GST to the PKR kinase domain (aa 259–551) | |
| HRI-Myc | NA | HRI expression vector, C-terminal Myc tag | This study |
| pTRS1 expression vectors | |||
| WT | Full-length pTRS1 | Binds both dsRNA and PKR | 37, 39 |
| pTRS1 1-370 | C-terminal truncation mutant lacking final 425 aa | Contains RNA binding domain. Lacks PKR binding domain | |
| pTRS1 1-550 | C-terminal truncation mutant lacking final 245 aa | Contains RNA binding domain. Lacks PKR binding domain | |
| pTRS1 1-679 | C-terminal truncation mutant lacking final 116 aa | Contains RNA binding domain. Lacks PKR binding domain | |
| pTRS1 240-795 | N-terminal truncation mutant lacking the first 239 aa | Lacks RNA binding domain. Contains PKR binding domain | |
| pTRS1 390-795 | N-terminal truncation mutant lacking the first 299 aa | Lacks RNA binding domain. Contains PKR binding domain | |
| pTRS1triple | Full-length pTRS1 containing point mutations in the RNA binding domain (R121A, R124A, and R125A) | Lacks ability to bind dsRNA | 38 |
| pTRS1mut1 | Full-length pTRS1 containing point mutations in the PKR binding domain (R658A, D660A, D661A, and E662A) | Lacks ability to bind PKR | 7 |
| pTRS1mut1.triple | Full-length pTRS1 containing point mutations in the RNA (R121A, R124A, and R125A) and PKR (R658A, D660A, D661A, and E662A) binding domains | Lacks ability to bind both dsRNA and PKR | 7, 38 |
| pTRS1 1-550triple | pTRS1 deletion mutant lacking final 245 aa and containing point mutations in RNA binding domain (R658A, D660A, D661A, and E662A) | Lacks ability to bind both dsRNA and PKR | 38, 39 |
| pCW-pTRS1 | NA | Doxycycline-inducible full-length pTRS1 | This study |
WT, wild type; aa, amino acids; VV, vaccinia virus; NA, not applicable.
Purified pTRS1 and PKR directly interact in vitro, and deletion or mutation of the PKR binding domain ablates the ability of pTRS1 to inhibit PKR during HCMV infection (6, 7, 37). These results suggest that pTRS1 interaction with PKR is critical for PKR antagonism. We therefore tested the ability of each pTRS1 mutant to bind PKR in transfected cells (Fig. 1D). Cells were transfected with vectors expressing pTRS1 mutants together with kinase-dead PKR (K296R), and the presence of pTRS1 in PKR-specific immune complexes was determined by Western blotting. Only full-length pTRS1 bound to PKR. None of the pTRS1 truncation mutants copurified with PKR, even pTRS1 240-795 and pTRS1 390-795, which both contain the previously described PKR binding domain. Interestingly, pTRS1 1-550 inhibited PKR activation (Fig. 1C) but did not bind PKR. These data suggest that pTRS1 binding to PKR is sufficient, but not necessary, to prevent PKR activation by dsRNA. However, as noted above, negative results in this assay should be interpreted cautiously due to the potential effect of the truncations on protein folding.
Relative role of dsRNA and PKR binding by pTRS1 in PKR antagonism.
Amino acids 84 to 246 of pTRS1 contain a noncanonical double-stranded RNA binding domain (RBD) that allows purified pTRS1 to competitively inhibit the binding of purified PKR to dsRNA (39). This function suggests that in addition to inhibiting PKR via a direct interaction, pTRS1 may also inhibit PKR by preventing recognition of its dsRNA ligands (38, 39). Consistent with this idea, the pTRS1 1-550 mutant, which lacks the PKR binding domain, did not copurify with PKR but could still prevent PKR activation in response to dsRNA treatment. To determine if pTRS1 1-550 inhibits PKR by binding to dsRNA, we introduced three amino acid changes in the dsRBD of pTRS1 1-550 (pTRS1 1-550triple) that prevent binding of purified pTRS1 to dsRNA (38). We then measured the ability of this mutant to prevent poly(I·C)-induced PKR activation (Fig. 2A). pTRS1 1-550triple could not prevent PKR activation in response to poly(I·C) treatment. Thus, in the context of the truncated pTRS1 protein (pTRS1 1-550), RNA binding is necessary for PKR antagonism.
FIG 2.

PKR binding by full-length pTRS1 is necessary for inhibition of PKR activation. (A) Lysates from HEK293T cells transfected with full-length TRS1 (WT) or the indicated TRS1 mutants. pTRS1 1-550 (1-550) expresses only the first 550 amino acids of pTRS1. pTRS1triple (Triple) contains mutations that disrupt dsRNA binding. pTRS1 1-550triple (1-550 Triple) expresses the 1-550 truncation mutant containing mutations in the dsRNA binding domain. Lysates were incubated with poly(I·C) to activate PKR. PKR phosphorylation (T446) was examined by Western blotting. (B) Same as panel A, except with different TRS1 mutants. pTRS1mut1 (Mut1) contains mutations that disrupt PKR binding. (C) HEK293T cells were transfected with Myc-tagged kinase-dead PKR (K296R) together with full-length TRS1 or the indicated TRS1 mutants. pTRS1mut1.triple (Mut1-triple) contains point mutations in both the PKR and dsRNA binding domains. Cell lysates were immunoprecipitated with anti-Myc (PKR) or IgG (specificity control) antibody. The presence of pTRS1 in the immune complexes was determined by Western blotting. (D) Same as panel C, except that lysates were treated with micrococcal nuclease prior to immunoprecipitation. (E) MRC-5 fibroblasts were infected with HCMV at a multiplicity of infection (MOI) of 3 for 24 h. The presence of PKR in pTRS1 or IE1 specific immune complexes was determined by Western blotting. Representative results from one of at least three independent experiments are shown for each panel.
In contrast, we found that pTRS1 could still inhibit PKR activation when the same dsRBD mutations were introduced into full-length pTRS1 (pTRS1triple) (Fig. 2A). We reasoned this could be due to pTRS1 binding to PKR through the previously defined PKR binding domain (PBD). We therefore mutated amino acids in pTRS1 necessary for binding to PKR (7) either alone (pTRS1mut1) or in combination with the dsRBD mutations (pTRS1mut1.triple). Mutation of the pTRS1 PBD ablated both binding to PKR (Fig. 2C) and the ability to inhibit poly(I·C)-induced PKR activation, even when the dsRBD was intact (Fig. 2B), suggesting that the primary mechanism of PKR antagonism by full-length pTRS1 is mediated through interactions with PKR. To further explore this hypothesis, we tested if pTRS1 associated with PKR via an RNA intermediate (Fig. 2D). Micrococcal nuclease treatment had no effect on pTRS1 binding to PKR. We also demonstrated that pTRS1 interacts with PKR in HCMV-infected cells, which had not been previously demonstrated (Fig. 2E). Together, these data suggest that pTRS1 has two distinct mechanisms to inhibit PKR activation. In some circumstances, pTRS1 may prevent PKR activation by sequestering dsRNA from PKR, independent of binding to PKR. However, at least in this system, the primary mechanism of PKR inhibition by full-length pTRS1 requires an RNA-independent interaction with PKR.
pTRS1 blocks PKR activation at a step after PKR dimerization.
Binding to dsRNA induces PKR dimerization, which is necessary for PKR activation (3, 51). We thus hypothesized that pTRS1 might block PKR activation by preventing PKR dimerization. To test this hypothesis, cells expressing pTRS1 were cotransfected with plasmids encoding Myc- and Flag-tagged PKR K296R. Catalytically inactive PKR was used in these experiments to limit toxicity. Poly(I·C) was added to cell lysates to induce PKR dimerization, and the ability of Flag-tagged PKR to be copurified with Myc-tagged PKR was measured by immunoprecipitation followed by Western blotting (Fig. 3). As expected, Flag-tagged PKR was copurified with Myc-tagged PKR in the absence of pTRS1. Similar amounts of Flag-PKR associated with Myc-PKR in the presence of pTRS1, suggesting that pTRS1 does not affect PKR homodimer formation. In addition, we detected pTRS1 in the Myc-PKR immune complexes, suggesting that pTRS1 might bind to PKR dimers. These data show that pTRS1 does not inhibit PKR activation by preventing PKR dimerization and suggest that pTRS1 may in fact bind to PKR dimers.
FIG 3.
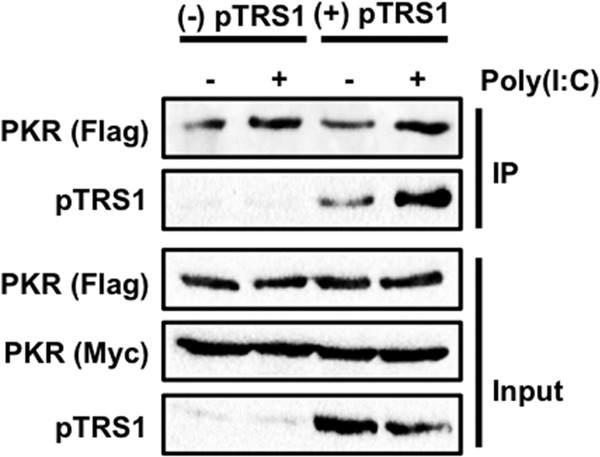
pTRS1 does not prevent PKR dimerization. HEK293T cells expressing pTRS1 from an inducible promoter (293T-pTRS1i) were cotransfected with Flag- and Myc-tagged PKR K296R expression vectors. pTRS1 expression was induced with doxycycline treatment. Cell lysates were treated with poly(I·C), and PKR was immunoprecipitated with anti-Myc antibody. The interaction of Flag- and Myc-tagged PKR was examined by Western blotting. Representative results from one of at least three independent experiments are shown.
To further test if pTRS1 prevents PKR activation at a step after dimerization, we determined if pTRS1 could bind and inhibit a constitutively dimerized, active PKR. The glutathione S-transferase (GST) protein constitutively forms dimers; replacing the amino-terminal dsRBD of PKR with GST results in a constitutively dimerized, and thus constitutively active, PKR kinase domain (GST-KD) (52). We asked if pTRS1 copurified with GST-KD to determine if pTRS1 could bind the dimerized PKR kinase domain (Fig. 4A). As long-term expression of GST-KD is toxic, a doxycycline-inducible promoter was used to regulate GST-KD expression. Wild-type pTRS1 was copurified with glutathione resin from cells expressing GST-KD, while the PKR binding domain mutant (pTRS1mut1) was not. We next determined if pTRS1 could inhibit the activity of a constitutively dimerized PKR KD by measuring the effect of pTRS1 expression on GST-KD autophosphorylation (Fig. 4B). pTRS1 expression decreased GST-KD autophosphorylation compared to those of GFP and pTRS1mut1 controls. Together, these results suggest that pTRS1 binds and inhibits dimerized PKR and that pTRS1 binds PKR through an interaction with the PKR KD.
FIG 4.

pTRS1 binds and inhibits a constitutively active PKR kinase domain. (A) HEK293T cells were cotransfected with a doxycycline-inducible GST-tagged PKR kinase domain (GST-KD) expression vector and an expression vector encoding either pTRS1 or pTRS1mut1 (Mut1). GST-KD expression was induced with doxycycline treatment. Glutathione resin was used to capture the GST-KD, and the copurification of pTRS1 or pTRS1mut1 was examined by Western blotting. (B) Cells were transfected as in panel A, with the addition of cells cotransfected with a GFP expression vector and the GST-KD expression vector. GST-KD expression was induced with doxycycline treatment, and levels of GST-KD autophosphorylation (T446) were determined by Western blotting. Representative results from one of at least three independent experiments are shown for each panel.
Amino acids in the PKR eIF2α contact site are important for pTRS1 binding.
Previous studies identified key amino acids in the αG helix of the PKR kinase domain that are necessary for binding of PKR to its substrate eIF2α (Fig. 5A) (3, 4). To determine if pTRS1 interacted with the PKR eIF2α binding site, we examined the ability of pTRS1 to bind and inhibit previously characterized PKR mutants with single amino acid changes in the αG helix (Table 1). Mutation of PKR residues Thr487 and Phe495 to alanine and arginine (T487A and F495R, respectively) ablate PKR phosphorylation of eIF2α but do not affect PKR catalytic activity or autophosphorylation (3). Two amino acids within the PKR αG helix, T487 and Glu490 (E490), are conserved in all eIF2α kinases (4). Therefore, we generated plasmids expressing PKR T487A, F495R, or E490A mutants and measured their binding to pTRS1 (Fig. 5B). While pTRS1 was efficiently copurified with wild-type PKR, very low levels of pTRS1 were recovered with the T487A and E490A PKR mutants, which were visible only after overexposure of the blot. pTRS1 was copurified with the PKR F495R mutant, but at reduced levels. pTRS1 also maintained the ability to bind a PKR mutant in which the activating phosphorylation site had been mutated to alanine (T446A) (Fig. 5B). These data suggest that amino acids in the PKR eIF2α contact site are important for pTRS1 binding and that PKR activation is not necessary for its interaction with pTRS1.
FIG 5.

pTRS1 interacts with the PKR kinase domain to prevent PKR autophosphorylation. (A) PyMOL image of the PKR kinase domain and eIF2a (PDB code 2A1A). The PKR aG helix is represented in cyan. Amino acids required for eIF2a binding are shown in magenta, while mutations that confer resistance to inhibition by K3L are shown in blue. (B) 293T-pTRS1i cells were transfected with Myc-tagged PKR K296R or the indicated Myc-tagged PKR mutants. pTRS1 expression was induced with doxycycline treatment before lysates were immunoprecipitated with anti-Myc (PKR) or IgG (specificity control) antibody. The presence of pTRS1 in the immune complexes was determined by Western blotting. (C) PKR-deficient HeLa cells (PKR KO) were transfected with the indicated PKR mutants together with vectors expressing either GFP or TRS1. Cells were transfected with poly(I·C) to activate PKR and PKR autophosphorylation (T446) was measured by Western blotting. (D and E) Same as panels B and C, except that cells were transfected with the indicated PKR mutants. Representative results from one of at least three independent experiments are shown for each panel.
We next tested the ability of pTRS1 to prevent activation of each of the above-mentioned PKR mutants. pTRS1 and the PKR mutants were expressed in PKR-deficient cells, and PKR autophosphorylation was measured after poly(I·C) transfection (Fig. 5C). Consistent with previous results, poly(I·C) induced the autophosphorylation of each PKR mutant in control cells expressing GFP, demonstrating that each PKR mutant was catalytically active (3, 4, 27). Though the PKR F495 mutant was activated to lower levels than wild-type PKR in control cells, pTRS1 prevented autophosphorylation of the PKR F495R mutant. In contrast, the PKR T487A and E490A mutants were resistant to inhibition by pTRS1, consistent with the inability of pTRS1 to bind these mutants. From these data, we conclude that binding of pTRS1 to the PKR αG helix is necessary for inhibition of PKR activation.
The vaccinia virus PKR antagonist K3L also binds PKR at an eIF2α contact residue within the αG helix, Asp486 (D486). Recently, PKR mutants resistant to K3L binding and inhibition were identified (Fig. 5A) (27). To determine if pTRS1 bound PKR in a manner structurally similar to that of K3L, we tested the ability of pTRS1 to bind and inhibit three PKR mutants (the E375V, A473T, and D486V mutants) that escape inhibition by K3L (Fig. 5D). We found that pTRS1 efficiently bound both the PKR E375V and A473T mutants but showed decreased binding to the PKR D486V mutant. pTRS1 prevented autophosphorylation of the PKR E375V and A473T mutants and reduced autophosphorylation of the PKR D486V mutant (Fig. 5E). These data suggest that while pTRS1 and K3L interact with the same region of PKR, these interactions occur in a structurally distinct manner.
pTRS1 inhibits PKR kinase activity.
The inhibition of PKR autophosphorylation by pTRS1 at a step after PKR dimerization suggested that pTRS1 might inhibit PKR kinase activity. To test this hypothesis, we measured PKR kinase activity in the presence of pTRS1 (Fig. 6). Cells were transfected with a Myc-PKR expression plasmid together with plasmids expressing the control protein GFP, wild-type pTRS1, or pTRS1mut1, which cannot bind PKR. We then measured PKR kinase activity in Myc-specific immune complexes using a peptide from histone H2A as a substrate. The histone H2A peptide has previously been shown to be a substrate for PKR in vitro (3). PKR kinase activity was significantly lower in pTRS1-expressing cells than in control cells expressing GFP or cells expressing kinase-dead PKR K296R. The pTRS1mut1 protein did not inhibit PKR kinase activity, consistent with its inability to bind PKR and prevent PKR autophosphorylation. In fact, in some experiments slightly elevated PKR kinase activity was found in cells expressing pTRS1mut1, though the difference was not statistically significant. Together with the above-described data, these results suggest that pTRS1 binding to the PKR kinase domain inhibits PKR kinase activity, preventing activating autophosphorylation of PKR and subsequent phosphorylation of its substrate eIF2α.
FIG 6.
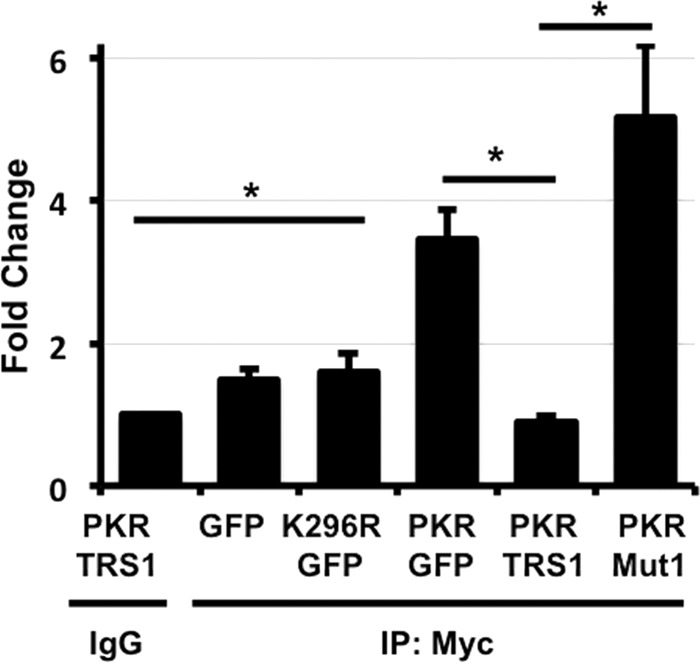
pTRS1 inhibits PKR kinase activity. HEK293T cells were transfected with the indicated expression vectors (PKR K296R, kinase-dead PKR; Mut1, pTRS1 containing PKR binding domain mutations). PKR was immunoprecipitated with anti-Myc antibody and resuspended in kinase buffer. The kinase reaction was performed “on bead” as described in Materials and Methods, using histone H2A peptide as a substrate. PKR kinase activity was measured using the ADP-Glo kinase assay. The amount of kinase activity is normalized to the background activity associated with nonspecific (IgG) immunoprecipitates, which was set to 1. Statistical significance was determined using an unpaired Student t test (n = 3; *, P < 0.05).
pTRS1 binds and inhibits an additional eIF2α kinase.
Our results show that pTRS1 binds and inhibits PKR through interactions with amino acids in the PKR eIF2α contact site. Interestingly, two of the amino acids in the PKR αG helix important for pTRS1 binding are conserved in other eIF2α kinases. To further test the hypothesis that interaction with the PKR eIF2α contact site is critical for pTRS1 binding, we determined if pTRS1 could also bind an additional eIF2α kinase, HRI. We found that HRI was copurified with pTRS1 in transfected 293T cells (Fig. 7A) and in HCMV-infected fibroblasts (Fig. 7B). Treating cells with arsenite leads to an accumulation of reactive oxygen species (ROS), which activates the HRI kinase, resulting in eIF2α phosphorylation (53). To determine if pTRS1 could prevent HRI-dependent eIF2α phosphorylation, we measured eIF2α phosphorylation after arsenite treatment in PKR-deficient cells (Fig. 7C). Arsenite induced a dose-dependent increase in eIF2α phosphorylation, which was decreased in cells expressing pTRS1. This result is consistent with our previous study showing that pTRS1 inhibits stress granule formation in PKR-deficient cells treated with arsenite (6). As HRI activation does not require an interaction with dsRNA, these data further confirm that pTRS1 can inhibit eIF2α kinases independent of its ability to bind RNA. In addition, these data further support the conclusion that residues in the eIF2α contact site are critical for pTRS1 interaction with PKR and may mediate interactions with additional eIF2α kinases.
FIG 7.

pTRS1 binds HRI and limits its activation. (A) 293T-pTRS1i cells were transfected with Myc-tagged HRI, and pTRS1 expression was induced with doxycycline. Lysates were immunoprecipitated with anti-Myc (PKR) or control antibody (αTubulin), and the presence of HRI the immune complexes was determined by Western blotting. (B) MRC-5 fibroblasts were infected with HCMV at an MOI of 3 for 72 h. The amount of HRI that was copurified with pTRS1 specific immune complexes or with beads alone (control) was determined by Western blotting. (C) PKR KO-pTRS1i cells were treated with doxycycline to induce pTRS1 expression and subsequently treated with increasing concentrations of arsenite (ARS) to activate HRI. Levels of eIF2α phosphorylation were measured by Western blotting. The ratio of phosphorylated eF2α to total eIF2α levels (P/total) was calculated by densitometry. Representative results from one of at least three independent experiments are shown for each panel.
DISCUSSION
In this study, we sought to examine the mechanism by which HCMV pTRS1 inhibits the antiviral kinase PKR. Our results suggest that pTRS1 employs two mechanisms to block PKR activation. In isolation, the amino-terminal 550 amino acids of pTRS1, which contain the RNA binding domain, can prevent PKR activation independent of binding to PKR (Fig. 1). However, in the context of full-length pTRS1, mutations that disrupt pTRS1 RNA binding do not affect its ability to prevent PKR activation (Fig. 2). Rather, binding of pTRS1 to PKR appears to be essential for PKR antagonism. pTRS1 did not block PKR dimerization (Fig. 3) but instead could bind and inhibit a constitutively dimerized PKR kinase domain (Fig. 4). pTRS1 binding to PKR inhibits PKR kinase activity (Fig. 6) and is mediated through interactions with conserved amino acids in the PKR eIF2α contact site. Mutation of these residues rendered PKR resistant to pTRS1 antagonism (Fig. 5). Consistent with this finding, pTRS1 interacted with an additional eIF2α kinase, HRI, and limited HRI activation in PKR-deficient cells (Fig. 7). Together, our data suggest that inhibition of PKR kinase activity, mediated by pTRS1 binding to conserved residues within the PKR eIF2α contact site, is the dominant mechanism of PKR antagonism.
Both pTRS1 and pIRS1 contain a noncanonical RNA binding domain in their conserved N termini. Although the affinity of pTRS1 for dsRNA is lower than that of PKR, the higher abundance of pTRS1 and pIRS1 in relation to PKR during infection may allow for competition for dsRNA binding (38, 39). Our data show that in some contexts, dsRNA binding by pTRS1 contributes to PKR antagonism. pTRS1 1-550 did not bind PKR (Fig. 1D) but maintained the ability to inhibit PKR activation in response to poly(I·C) treatment (Fig. 1C). Mutations within pTRS1 1-550 that disrupt dsRNA binding (1-550triple) ablated the ability of pTRS1 1-550 to inhibit PKR (Fig. 2A), suggesting that in the context of the truncated protein, dsRNA binding is necessary for PKR inhibition. Interestingly, this truncation mutant comprises the conserved region between pTRS1 and pIRS1, suggesting that dsRNA binding by both proteins may contribute to PKR antagonism during infection, although the function of dsRNA binding by pTRS1/pIRS1 during infection has not been examined. It is also possible that pTRS1 1-550 has a higher binding affinity for dsRNA than full-length pTRS1, allowing for efficient binding and sequestration of poly(I·C) in vitro. Nevertheless, these data suggest that in some contexts, dsRNA binding by pTRS1 contributes to PKR antagonism.
While dsRNA binding was necessary for the pTRS1 1-550 mutant to inhibit PKR, the ability of full-length pTRS1 to bind dsRNA was dispensable for PKR antagonism (Fig. 2B). Consistent with this result, pTRS1 binding to PKR was not dependent on an RNA intermediate (Fig. 2D) and pTRS1 inhibited the activation of a constitutively active PKR kinase domain lacking its dsRNA binding domains (Fig. 4B). Instead, we found that an interaction between PKR and full-length pTRS1 was necessary for PKR antagonism (Fig. 2). Thus, at least in this system, pTRS1 binding to PKR, rather than competition for dsRNA ligands, is the primary mechanism by which pTRS1 prevents PKR activation. This is consistent with previous studies showing that the pTRS1 PKR binding domain is necessary for PKR inhibition during HCMV infection in primary human fibroblasts (7, 35). However, as discussed above, our results suggest that dsRNA binding by pTRS1 does play a role in PKR inhibition. Previous studies have shown that a pTRS1 mutant incapable of binding dsRNA (pTRS1triple) did not rescue replication of a vaccinia virus lacking its PKR antagonist, even though pTRS1triple maintained the ability to bind PKR (38). While dsRNA accumulates in HCMV-infected cells (18), the signals driving PKR activation during HCMV infection are unknown. Perhaps these two functions of pTRS1, binding to dsRNA and PKR, are most important for PKR inhibition in different settings or in response to different PKR-activating signals.
We also identified the domain of PKR necessary for its association with pTRS1. Our finding that pTRS1 could bind and inhibit a constitutively active PKR kinase domain (GST-KD) suggested that an interaction between pTRS1 and the PKR kinase domain is necessary for PKR antagonism (Fig. 4A and B). Using a series of previously described PKR point mutants that retain PKR catalytic activity (3, 4, 27), we found that pTRS1 interacts with specific amino acids in the αG helix of the PKR kinase domain that are necessary for binding to its substrate, eIF2α (Fig. 5). pTRS1 was not copurified with PKR mutants containing αG helix mutations (PKR T487A or E490A mutant) and weakly interacted with an additional αG helix mutant, the PKR D486V mutant (Fig. 5B and D). The loss of binding correlated with a decrease in the ability of pTRS1 to inhibit activation of these PKR mutants (Fig. 5C and E). The PKR T487A and E490A mutants were completely resistant to inhibition by pTRS1, while the D486V mutant was partially resistant. In contrast, mutations in PKR residues more distal to the eIF2α contact site (E375V and A473T) had a minimal effect on pTRS1 binding, and pTRS1 prevented activation of both mutants in response to poly(I·C) treatment (Fig. 5D and E). Thus, our data suggest that inhibition of PKR by pTRS1 requires an interaction with the eIF2α contact site in the PKR kinase domain.
These results were reminiscent of PKR inhibition by the vaccinia virus K3L protein. K3L binds the PKR αG helix and functions as a PKR pseudosubstrate (26, 27, 54). Mutation of PKR E375 or A473 to valine and threonine, respectively, confers resistance of PKR to K3L inhibition (27). We found that pTRS1 bound both the E375 and A473 PKR mutants (Fig. 5D) and prevented their autophosphorylation in response to poly(I·C) treatment (Fig. 5E). These results are again consistent with a model in which pTRS1 binding mediates PKR antagonism. In addition, these data suggest that pTRS1 inhibition of PKR activation occurs in a manner structurally distinct from that of other viral PKR antagonists.
In addition to a distinct binding modality, we found that pTRS1 also differs from K3L in its mode of PKR inhibition. K3L does not inhibit PKR catalytic activity but rather prevents phosphorylation of eIF2α by acting as a PKR pseudosubstrate (26, 54). Our data show that pTRS1 binds PKR and inhibits PKR kinase activity (Fig. 6). Importantly, the kinase assay used measures the conversion of ATP to AMP as a result of kinase activity. In the presence of pTRS1, AMP accumulation is reduced, indicating a loss of PKR catalytic activity. Thus, unlike K3L, pTRS1 does not act as a PKR pseudosubstrate but rather inhibits PKR kinase activity toward the histone H2A substrate. Together, these results suggest that pTRS1 binds the PKR kinase domain and inhibits its catalytic activity.
When considered in sum, our data suggest that pTRS1 inhibits PKR activation and subsequent eIF2α phosphorylation at multiple levels. In some scenarios, pTRS1 may sequester dsRNA ligands to prevent PKR dimerization and activation. In addition, we found that pTRS1 inhibits PKR at a step after dimerization through an interaction with the αG helix of the PKR kinase domain. pTRS1 binding to the kinase domain inhibits PKR catalytic activity, thus preventing autophosphorylation of dimerized PKR and subsequent phosphorylation of eIF2α. While not addressed herein, the fact that pTRS1 binds to PKR residues necessary for its interaction with eIF2α suggests that pTRS1 could also prevent eIF2α recognition by PKR. Thus, inhibition of PKR kinase activity by pTRS1 ensures that eIF2α is not phosphorylated, even when PKR has bound activating dsRNA ligands.
As the two residues in PKR recognized by pTRS1 are conserved across all eIF2α kinases, we reasoned that pTRS1 might bind and inhibit additional eIF2α kinases. We previously found that pTRS1 can prevent stress granule formation in response to arsenite treatment in PKR-deficient cells, suggesting that pTRS1 has the ability to block HRI-dependent eIF2α phosphorylation (53). In this study, we found that pTRS1 binds HRI (Fig. 7A and B) and limits eIF2α phosphorylation in response to arsenite treatment in PKR-deficient cells (Fig. 7C). These data further support the conclusion that residues in the PKR αG helix necessary for eIF2α binding are critical for interaction of pTRS1 with PKR. In addition, these data suggest that pTRS1 could have a more general role in suppressing cellular stress responses during HCMV infection, allowing for efficient cellular and viral protein synthesis in the face of robust cellular stress induced by infection.
MATERIALS AND METHODS
Cells and viruses.
HEK293T cells and MRC-5 fibroblasts were cultured in Dulbecco's modified Eagle medium (Sigma) supplemented with 10% fetal bovine serum (Sigma) and 100 U/ml of penicillin-streptomycin (Sigma) at 37°C and 5% CO2. PKR-deficient HeLa cells (PKR KO), HEK293T cells containing an inducible TRS1 expression cassette (293T-pTRS1i), and PKR-deficient cells containing an inducible TRS1 expression cassette (PKR KO-pTRS1i) were maintained in 1 μg/ml of puromycin in normal growth media. PKR-deficient HeLa cells have been previously described (35). The HCMV AD169inGFP strain was used for all infections. This strain is derived from a bacterial artificial chromosome (BAC) and contains a green fluorescent protein (GFP) reporter driven by the simian virus 40 (SV40) promoter.
Generation of recombinant plasmids.
Vectors expressing full-length TRS1 or TRS1 truncation mutants fused to a carboxyl-terminal 6×His epitope were a kind gift from Adam Geballe (University of Washington [39]). To generate full-length TRS1 containing arginine-to-alanine mutations that disrupt dsRNA binding (R121A, R124A, and R125A [38]), the full-length TRS1 expression plasmid was amplified with primers Triple F and Triple R (Table 2), and recircularized using Gibson cloning (New England BioLabs [NEB]). The R121A, R124A, and R125A point mutations were introduced into the 1-550 TRS1 expression vector, as described above, using the pcDNATRS1 1-550 plasmid as a template. Point mutations within the pTRS1 PKR binding domain that disrupt PKR binding (R658A, D660A D,661A, and E662A [7]) were incorporated into full-length TRS1 and TRS1triple using primers Mut1 F and Mut1 R (Table 1) followed by recircularization using a Gibson reaction (New England BioLabs), in which a mixture of DNA exonuclease, DNA polymerase, and DNA ligase anneal overlapping cDNA ends into a contiguous double-stranded DNA molecule. The portion of each clone containing the TRS1 open reading frame (ORF) was sequenced to ensure the absence of unintended mutations.
TABLE 2.
Primer and gBlock gene fragment sequencesa

Lowercase sequences represent Kozak sequences.
The human protein kinase R (PKR) ORF containing homology arms to the pcDNA3.1 V5 His expression vector was ordered as a gBlock gene fragment (IDT; Table 2). The pcDNA3.1 vector was digested with BamHI and XhoI, and the full-length PKR ORF was inserted using a Gibson cloning reaction according to the manufacturer's directions. The kinase-dead PKR (K296R) ORF containing homology arms to the pcDNA3.1 expression vector was also ordered as a gBlock gene fragment (Table 2) and introduced into pcDNA3.1 by Gibson cloning as described above. A Flag epitope was fused to the N terminus of PKR K296R by amplification of the PKR ORF with primers PKRflag F and PKR R (Table 2) followed by Gibson cloning into the pcDNA3.1 vector digested with BamHI and XhoI as described above. An N-terminal Myc tag was added to both wild-type PKR and PKR K296R as described above using the primers PKRmyc F and PKR R. The following point mutations were introduced into PKR: E375V, T446A, A473T, D486V, T487A, E490A, and F495R using primer pairs PKR_E375V F and PKR_E375V R, PKR_A446T F and PKR_A446T R, PKR_T473A F and PKR_T473A R, PKR_D486V F and PKR_D486V R, PKR_T487A F and PKR_T487A R, PKR_E490A F and PKR_E490A R, and PKR_F495R F and PKR_F495R R, respectively. All primer sequences are listed in Table 2. The full-length PKR expression plasmid was amplified from the Myc-tagged wild-type PKR and the Myc-tagged PKR K296R expression plasmids with the above-listed primer pairs. The PCR products were then gel extracted and circularized using Gibson cloning. For each PKR mutant, the portion of each vector containing the PKR ORF was sequenced to confirm the absence of additional mutations.
The PKR kinase domain (KD) fused to glutathione S-transferase (GST) was created by first amplifying GST from pET-41 b(+) using primers GST-pCW F and GST R (Table 2). The PKR KD (amino acids 259 to 551) was then amplified from the full-length PKR expression vector using primers KDGST F and KDGSTpCW R (Table 2). The plasmid pCW-Cas9 (Addgene; number 50661) was digested with NheI and BamHI to remove the Cas9 open reading frame. The GST ORF and PKR KD were then cloned into the digested pCW vector using a three-part Gibson reaction. Full-length TRS1 was introduced into the pCW plasmid by amplifying the TRS1 ORF using primers TRS1ind F and TRS1ind R (Table 2), followed by Gibson assembly into pCW-Cas9 previously digested with NheI and BamHI. Sanger sequencing was used to confirm the absence of unintended mutations.
The heme-regulated inhibitor (HRI) ORF was amplified from the pJP1563 vector (DNASU; number 38827) using primers HRImyc F and HRImyc R (Table 2). The HRI ORF was then cloned into the pcDNA3.1 vector digested with BamHI and XhoI via Gibson reaction. The complete HRI ORF was sequenced to ensure the absence of unintended mutations.
Production of lentivirus stocks and pTRS1-inducible cell lines.
Lentivirus stocks were generated by transfecting HEK293T cells with the pCW TRS1 expression plasmid (described above) and lentivirus packaging mix (Sigma) using polyethylenimine (PEI; Polysciences). The medium was collected at 48 h and 72 h after transfection and clarified by passage through a 0.22-μm filter. The virus was then stored at −80°C until use. 293T-pTRS1i and PKR KO-pTRS1i cells were generated by transduction with the pCW TRS1 lentivirus in the presence of 8 μg/ml of Polybrene. After bulk selection in 1 μg/ml of puromycin for 48 h, clonal cell lines were isolated by limiting dilution in media containing puromycin. Clonal cell lines were assayed for minimal basal expression of pTRS1 and maximal induction of pTRS1 expression after treatment with 1 μg/ml of doxycycline by Western blotting.
PKR and HRI activation assays.
The ability of pTRS1 or pTRS1 mutants to inhibit PKR was measured in HEK293T cells. Cells were transfected with 1 μg of either a GFP or TRS1 expression plasmid using PEI and harvested at 36 h after transfection. Cells were washed with phosphate-buffered saline (PBS), pelleted, and lysed in 120 μl of PKR lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% NP-40, 10% glycerol, and 1 mM EDTA with protease and phosphatase inhibitors [Roche]) on ice for 15 min. Insoluble debris was removed by centrifugation at 21,000 × g for 10 min at 4°C. Unless otherwise indicated, 1 mg/ml of poly(I·C) (LMW; Invivogen) was added to 100 μl of supernatant for 30 min at 30°C to activate PKR. To examine PKR KD activation, HEK293T cells were transfected with 500 ng of GST-KD expression plasmid together with 500 ng of either GFP, TRS1 or TRS1mut1 expression plasmid for 36 h. Doxycycline (1 μg/ml) was added to cells overnight prior to harvest to induce GST-KD expression. Cells were washed once with PBS, pelleted, and stored at −80°C until processed. To measure activation of PKR mutants in PKR KO cells, PKR KO-pTRS1i cells were cotransfected with 500 ng of PKR expression plasmid and 500 ng of either GFP or TRS1 expression plasmid. Twenty hours after DNA transfection, cells were transfected with 20 μg of poly(I·C) using Lipofectamine 2000 (Thermo Fisher) for 4 h. Cells were washed once in PBS, pelleted, and stored at −80°C until processed.
PKR KO-pTRS1i cells were used to measure the effect of pTRS1 on arsenite-induced HRI activation. Cells were treated overnight with doxycycline (1 μg/ml) to induce pTRS1 expression. The next day, cells were treated with 0.05, 0.25, or 0.5 mM sodium arsenite (Sigma) for 2 h to activate HRI (53). Cells were then washed once with PBS, pelleted, and stored at −80°C until processed.
Analysis of protein expression.
Unless otherwise stated, cells were lysed in RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.25% deoxycholate, and 1 mM EDTA containing protease and phosphatase inhibitors; Roche). Protein concentration was determined using a Bradford assay. Equal amounts of protein were resolved on 10% SDS-PAGE gels, transferred to nitrocellulose membranes (Amersham), and blocked in 5% nonfat milk in TBS-T (50 mM Tris [pH 7.4], 150 mM NaCl, 0.1% Tween 20) for 1 h at room temperature (RT) or overnight at 4°C. For mouse monoclonal antibodies, membranes were incubated with primary antibodies for either 1 h at RT or overnight at 4°C in 1% bovine serum albumin (BSA) in TBS-T. For rabbit polyclonal antibodies, membranes were blocked as described above and incubated at 4°C overnight in 5% BSA in TBS-T, with the exception of the anti-His antibody, which was diluted in 1% BSA in TBST-T. Membranes were incubated in appropriate secondary antibodies (KPL) at a 1:10,000 dilution in 1% BSA in TBS-T for 1 h at RT. A modified protocol was used for total and phosphorylated eIF2α blots. Membranes probed for total eIF2α were blocked and incubated in primary antibody as described above and then incubated in secondary antibody (1:10,000) in 5% milk in TBS-T for 1 h at RT before being developed. Membranes probed for phospho-eIF2α (Ser51) were blocked in 5% BSA in TBS-T overnight at 4°C prior to an overnight incubation in primary antibody at 4°C. Membranes were then incubated in secondary antibody (1:10,000) in 1% BSA in TBS-T for 1 h at RT. Antibodies against the following were used: pTRS1 (1:100 [43]), His (1:1,000; CST; number 2365S), total PKR (1:1,000; Santa Cruz; sc-707), phospho-PKR (Thr446) (1:1,000; Abcam; ab32036), Myc (1:1,000; CST; number 2276S), Flag (1:1,000; Sigma; F3165), eIF2α (1:1,000; CST; number 9722S), phospho-eIF2α (Ser51) (1:1,000; CST; number 3398S), HRI (1:1,000; Abcam; ab28530), and tubulin (1:20,000; Sigma; T6199).
Immunoprecipitation.
HEK293T cells were transfected with 5 μg of the desired plasmid for a total of 36 h. Where indicated, doxycycline (1 μg/ml) was added 18 h prior to harvest to induce pTRS1 expression. Cells were lysed in 1 ml of PKR lysis buffer for 15 min on ice. Insoluble debris was removed by centrifugation for 10 min at 21,000 × g at 4°C. One hundred microliters of protein A/G Sepharose beads (Santa Cruz) in PKR lysis buffer (20-μl packed-bead volume) was added to each sample and nutated at 4°C for 30 min to preclear the lysate. Where indicated, 2,000 U of micrococcal nuclease (NEB) and 5 μl of 0.5 M CaCl2 were added to each sample prior to the preclearing step and incubated at 37°C for 30 min to digest nucleic acid species. The beads were then removed and the lysate was transferred to a new tube containing 1 μg of the desired antibody, or 1 μg of either mouse IgG or tubulin antibody as a negative control. Samples were nutated with antibody at 4°C for 4 h, after which 100 μl of protein A/G beads in PKR lysis buffer (20-μl packed-bead volume) was added to each sample. For the anti-TRS1 immunoprecipitation, antibodies were preconjugated to protein A/G beads overnight before addition to sample lysates. Samples were nutated for 90 min after the addition of the beads. The beads were collected by brief centrifugation and washed three times with 1 ml of PKR lysis buffer before addition of 30 μl of 1× sample buffer (0.1 M Tris-HCl [pH 6.8], 6% glycerol, 2% SDS, 0.1 M dithiothreitol [DTT], 0.002% bromophenol blue). The samples were boiled at 95°C for 10 min, briefly centrifuged at 21,000 × g, and stored at −20°C prior to analysis by Western blotting.
For the glutathione Sepharose pulldown, HEK293T cells were cotransfected with 2.5 μg of inducible GST-PKR kinase domain (GST-KD) expression plasmid and 2.5 μg of either TRS1 or TRS1mut1 expression plasmid for 36 h. GST-KD expression was induced by addition of 1 μg/ml of doxycycline 18 h before harvest. Upon harvest, cells were washed once with PBS, pelleted, and lysed in 500 μl of wash buffer (20 mM HEPES [pH 7.0], 100 mM NaCl, 1 mM DTT, 1% glycerol) for 15 min on ice. Insoluble debris was removed by centrifugation at 21,000 × g for 10 min at 4°C. One hundred microliters of washed glutathione Sepharose 4B (GE) (15-μl packed-Sepharose volume) was added to each sample. Samples were nutated at RT for 1 h and then washed three times with 1 ml of wash buffer. Thirty microliters of 1× sample buffer was added to the beads, after which samples were boiled, briefly centrifuged, and stored at −20°C until analysis by Western blotting.
PKR kinase assay.
HEK293T cells were transfected with 5 μg of the desired plasmids for 36 h. For cotransfections, 2.5 μg of each plasmid was transfected. Immunoprecipitation using anti-Myc or mouse IgG was performed as described above. After a washing in PKR lysis buffer, beads were resuspended in 25 μl of 1× kinase reaction buffer (40 mM Tris [pH 7.5], 20 mM MgCl2, 0.1 mg/ml of BSA, 500 μM ATP [Promega]) containing 7.5 μg of histone H2A peptide (NEB) as a substrate. The kinase reaction was run for 30 min at RT. The ADP-Glo kinase assay (Promega) was then used to determine PKR kinase activity. Briefly, 25 μl of ADP-Glo reagent was added to each kinase reaction and incubated at RT for 40 min. Fifty microliters of kinase detection reagent was added to each sample and incubated at RT for an additional 40 min, after which the luminescence of each sample was read using a luminometer (Molecular Devices). Fold change in luminescence compared to the IgG control immunoprecipitation was calculated, and statistical significance was determined using an unpaired Student t test.
ACKNOWLEDGMENTS
We thank members of the Moorman, Heise, de Silva, and de Paris labs as well as the members of the UNC Virology community for helpful discussions and input. We also thank Westefson Sanders for continued support and Anne Beall for her valuable feedback.
This work was supported by NIH grant AI03311 to N.J.M. Additional support was provided by the North Carolina University Cancer Research Fund and an award from the UNC Virology Training grant (T32 AI07419 to B.Z.).
REFERENCES
- 1.García MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barber GN, Wambach M, Wong ML, Dever TE, Hinnebusch AG, Katze MG. 1993. Translational regulation by the interferon-induced double-stranded-RNA-activated 68-kDa protein kinase. Proc Natl Acad Sci U S A 90:4621–4625. doi: 10.1073/pnas.90.10.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE. 2005. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2α substrate recognition. Cell 122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 4.Dar AC, Dever TE, Sicheri F. 2005. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 5.Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, Hinnebusch AG. 2001. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol 21:5018–5030. doi: 10.1128/MCB.21.15.5018-5030.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziehr B, Vincent HA, Moorman NJ. 2016. Human cytomegalovirus pTRS1 and pIRS1 antagonize protein kinase R to facilitate virus replication. J Virol 90:3839–3848. doi: 10.1128/JVI.02714-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braggin JE, Child SJ, Geballe AP. 2016. Essential role of protein kinase R antagonism by TRS1 in human cytomegalovirus replication. Virology 489:75–85. doi: 10.1016/j.virol.2015.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balachandran S, Kim CN, Yeh WC, Mak TW, Bhalla K, Barber GN. 1998. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J 17:6888–6902. doi: 10.1093/emboj/17.23.6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Der SD, Yang Y-L, Weissmann C, Williams BRG. 1997. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc Natl Acad Sci U S A 94:3279–3283. doi: 10.1073/pnas.94.7.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perry AK, Chen G, Zheng D, Tang H, Cheng G. 2005. The host type I interferon response to viral and bacterial infections. Cell Res 15:407–422. doi: 10.1038/sj.cr.7290309. [DOI] [PubMed] [Google Scholar]
- 11.Tallóczy Z, Jiang W, Virgin HW, Leib DA, Scheuner D, Kaufman RJ, Eskelinen E-L, Levine B. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc Natl Acad Sci U S A 99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lussignol M, Queval C, Bernet-Camard M-F, Cotte-Laffitte J, Beau I, Codogno P, Esclatine A. 2013. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol 87:859–871. doi: 10.1128/JVI.01158-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting JP-Y, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ. 2012. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yim HC, Wang D, Yu L, White CL, Faber PW, Williams BR, Sadler AJ. 2016. The kinase activity of PKR represses inflammasome activity. Cell Res 26:367–379. doi: 10.1038/cr.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tu Y-C, Yu C-Y, Liang J-J, Lin E, Liao C-L, Lin Y-L. 2012. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J Virol 86:10347–10358. doi: 10.1128/JVI.00525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinicke LA, Wong CJ, Lary J, Nallagatla SR, Diegelman-Parente A, Zheng X, Cole JL, Bevilacqua PC. 2009. RNA dimerization promotes PKR dimerization and activation. J Mol Biol 390:319–338. doi: 10.1016/j.jmb.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brand SR, Kobayashi R, Mathews MB. 1997. The Tat protein of human immunodeficiency virus type 1 is a substrate and inhibitor of the interferon-induced, virally activated protein kinase, PKR. J Biol Chem 272:8388–8395. doi: 10.1074/jbc.272.13.8388. [DOI] [PubMed] [Google Scholar]
- 18.Marshall EE, Bierle CJ, Brune W, Geballe AP. 2009. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J Virol 83:4112–4120. doi: 10.1128/JVI.02489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacquemont B, Roizman B. 1975. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and of double-stranded RNA prepared from them. J Virol 15:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson E, Quartararo C, Brown RS, Shi Y, Yao X, Cole JL. 2010. Analysis of monomeric and dimeric phosphorylated forms of protein kinase R. Biochemistry 49:1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Husain B, Hesler S, Cole JL. 2015. Regulation of PKR by RNA: formation of active and inactive dimers. Biochemistry 54:6663–6672. doi: 10.1021/acs.biochem.5b01046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemaire PA, Anderson E, Lary J, Cole JL. 2008. Mechanism of PKR activation by dsRNA. J Mol Biol 381:351–360. doi: 10.1016/j.jmb.2008.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romano PR, Garcia-Barrio MT, Zhang X, Wang Q, Taylor DR, Zhang F, Herring C, Mathews MB, Qin J, Hinnebusch AG. 1998. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2α kinases PKR and GCN2. Mol Cell Biol 18:2282–2297. doi: 10.1128/MCB.18.4.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sciortino MT, Parisi T, Siracusano G, Mastino A, Taddeo B, Roizman B. 2013. The virion host shutoff RNase plays a key role in blocking the activation of protein kinase R in cells infected with herpes simplex virus 1. J Virol 87:3271–3276. doi: 10.1128/JVI.03049-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Zhang C, Chen X, Yu J, Wang Y, Yang Y, Du M, Jin H, Ma Y, He B, Cao Y. 2011. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. The J Biol Chem 286:24785–24792. doi: 10.1074/jbc.M111.232439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawagishi-Kobayashi M, Silverman JB, Ung TL, Dever TE. 1997. Regulation of the protein kinase PKR by the vaccinia virus pseudosubstrate inhibitor K3L is dependent on residues conserved between the K3L protein and the PKR substrate eIF2alpha. Mol Cell Biol 17:4146–4158. doi: 10.1128/MCB.17.7.4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seo EJ, Liu F, Kawagishi-Kobayashi M, Ung TL, Cao C, Dar AC, Sicheri F, Dever TE. 2008. Protein kinase PKR mutants resistant to the poxvirus pseudosubstrate K3L protein. Proc Natl Acad Sci U S A 105:16894–16899. doi: 10.1073/pnas.0805524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romano PR, Zhang F, Tan SL, Garcia-Barrio MT, Katze MG, Dever TE, Hinnebusch AG. 1998. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol Cell Biol 18:7304–7316. doi: 10.1128/MCB.18.12.7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thakur M, Seo EJ, Dever TE. 2014. Variola virus E3L Zα domain, but not its Z-DNA binding activity, is required for PKR inhibition. RNA 20:214–227. doi: 10.1261/rna.042341.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Budt M, Niederstadt L, Valchanova RS, Jonjić S, Brune W. 2009. Specific inhibition of the PKR-mediated antiviral response by the murine cytomegalovirus proteins m142 and m143. J Virol 83:1260–1270. doi: 10.1128/JVI.01558-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Child SJ, Geballe AP. 2009. Binding and relocalization of protein kinase R by murine cytomegalovirus. J Virol 83:1790–1799. doi: 10.1128/JVI.01484-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostermann E, Warnecke G, Waibler Z, Brune W. 28 October 2015. Knockout of the host resistance gene Pkr fully restores replication of murine cytomegalovirus (MCMV) m142 and m143 mutants in vivo. J Virol doi: 10.1128/JVI.02003-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valchanova RS, Picard-Maureau M, Budt M, Brune W. 2006. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J Virol 80:10181–10190. doi: 10.1128/JVI.00908-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li S, Min J-Y, Krug RM, Sen GC. 2006. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349:13–21. doi: 10.1016/j.virol.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Ziehr B, Vincent HA, Moorman NJ. 2016. Human cytomegalovirus pTRS1 and pIRS1 antagonize PKR to facilitate virus replication. J Virol doi: 10.1128/JVI.02714-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Child SJ, Hakki M, De Niro KL, Geballe AP. 2004. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol 78:197–205. doi: 10.1128/JVI.78.1.197-205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hakki M, Marshall EE, De Niro KL, Geballe AP. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J Virol 80:11817–11826. doi: 10.1128/JVI.00957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bierle CJ, Semmens KM, Geballe AP. 2013. Double-stranded RNA binding by the human cytomegalovirus PKR antagonist TRS1. Virology 442:28–37. doi: 10.1016/j.virol.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hakki M, Geballe AP. 2005. Double-stranded RNA binding by human cytomegalovirus pTRS1. J Virol 79:7311–7318. doi: 10.1128/JVI.79.12.7311-7318.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ziehr B, Lenarcic E, Vincent HA, Cecil C, Garcia B, Shenk T, Moorman NJ. 2015. Human cytomegalovirus TRS1 protein associates with the 7-methylguanosine mRNA cap and facilitates translation. Proteomics 15:1983–1994. doi: 10.1002/pmic.201400616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, Esclatine A. 2012. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J Virol 86:2571–2584. doi: 10.1128/JVI.05746-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mouna L, Hernandez E, Bonte D, Brost R, Amazit L, Delgui LR, Brune W, Geballe AP, Beau I, Esclatine A. 2016. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy 12:327–342. doi: 10.1080/15548627.2015.1125071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romanowski MJ, Shenk T. 1997. Characterization of the human cytomegalovirus irs1 and trs1 genes: a second immediate-early transcription unit within irs1 whose product antagonizes transcriptional activation. J Virol 71:1485–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adamo JE, Schröer J, Shenk T. 2004. Human cytomegalovirus TRS1 protein is required for efficient assembly of DNA-containing capsids. J Virol 78:10221–10229. doi: 10.1128/JVI.78.19.10221-10229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iskenderian AC, Huang L, Reilly A, Stenberg RM, Anders DG. 1996. Four of eleven loci required for transient complementation of human cytomegalovirus DNA replication cooperate to activate expression of replication genes. J Virol 70:383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pari GS, Anders DG. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J Virol 67:6979–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pari GS, Kacica MA, Anders DG. 1993. Open reading frames UL44, IRS1/TRS1, and UL36-38 are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA synthesis. J Virol 67:2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strang BL, Geballe AP, Coen DM. 2010. Association of human cytomegalovirus proteins IRS1 and TRS1 with the viral DNA polymerase accessory subunit UL44. J Gen Virol 91:2167–2175. doi: 10.1099/vir.0.022640-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strang BL, Bender BJ, Sharma M, Pesola JM, Sanders RL, Spector DH, Coen DM. 2012. A mutation deleting sequences encoding the amino terminus of human cytomegalovirus UL84 impairs interaction with UL44 and capsid localization. J Virol 86:11066–11077. doi: 10.1128/JVI.01379-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang F, Romano PR, Nagamura-Inoue T, Tian B, Dever TE, Mathews MB, Ozato K, Hinnebusch AG. 2001. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem 276:24946–24958. doi: 10.1074/jbc.M102108200. [DOI] [PubMed] [Google Scholar]
- 51.Dey M, Mann BR, Anshu A, Mannan MA-u. 2014. Activation of protein kinase PKR requires dimerization-induced cis-phosphorylation within the activation loop. J Biol Chem 289:5747–5757. doi: 10.1074/jbc.M113.527796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ung TL, Cao C, Lu J, Ozato K, Dever TE. 2001. Heterologous dimerization domains functionally substitute for the double-stranded RNA binding domains of the kinase PKR. EMBO J 20:3728–3737. doi: 10.1093/emboj/20.14.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McEwen E, Kedersha N, Song B, Scheuner D, Gilks N, Han A, Chen J-J, Anderson P, Kaufman RJ. 2005. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem 280:16925–16933. doi: 10.1074/jbc.M412882200. [DOI] [PubMed] [Google Scholar]
- 54.Dar AC, Sicheri F. 2002. X-ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol Cell 10:295–305. doi: 10.1016/S1097-2765(02)00590-7. [DOI] [PubMed] [Google Scholar]