

Abstract
The National Institutes of Health's Mammalian Gene Collection (MGC) project was designed to generate and sequence a publicly accessible cDNA resource containing a complete open reading frame (ORF) for every human and mouse gene. The project initially used a random strategy to select clones from a large number of cDNA libraries from diverse tissues. Candidate clones were chosen based on 5′-EST sequences, and then fully sequenced to high accuracy and analyzed by algorithms developed for this project. Currently, more than 11,000 human and 10,000 mouse genes are represented in MGC by at least one clone with a full ORF. The random selection approach is now reaching a saturation point, and a transition to protocols targeted at the missing transcripts is now required to complete the mouse and human collections. Comparison of the sequence of the MGC clones to reference genome sequences reveals that most cDNA clones are of very high sequence quality, although it is likely that some cDNAs may carry missense variants as a consequence of experimental artifact, such as PCR, cloning, or reverse transcriptase errors. Recently, a rat cDNA component was added to the project, and ongoing frog (Xenopus) and zebrafish (Danio) cDNA projects were expanded to take advantage of the high-throughput MGC pipeline.
The Human Genome Project has produced several valuable resources for future scientific research. However, our understanding of biological systems function is still in its infancy. Even the availability of the complete human genome (Lander et al. 2001; Venter et al. 2001; The International Human Genome Sequencing Consortium, in prep.) and advanced drafts of the mouse (Waterston et al. 2002) and rat (Gibbs et al. 2004) genomes are not sufficient to define all of the transcribed and coding regions, given the current limitations of gene-prediction algorithms and the variable quality of the EST resources.
The NIH Mammalian Gene Collection (MGC; http://mgc.nci.nih.gov) program was established to provide a publicly accessible full-open reading frame (ORF) clone corresponding to each human and mouse protein-coding gene (Strausberg et al. 1999). The aim is to produce a community resource that consists of two components: (1) a set of clones that are publicly available without restriction and (2) the corresponding highly accurate cDNA sequence information submitted to the public nucleotide sequence databases. Other large-scale cDNA cloning efforts include two programs in Japan and one in Germany (Wiemann et al. 2001; Okazaki et al 2002; Ota et al 2004).
This paper provides an update on the current status of the MGC. Owing to the success of the project to date, the goals have been expanded to include the generation of a full-ORF clone collection for the rat. In addition, the MGC protocols are being applied to assist two other ongoing projects to generate full-ORF clones for two frog (Xenopus) species and the zebrafish (Danio rerio).
RESULTS AND DISCUSSION
Status of the Human and Mouse Full-ORF cDNA Collections
The MGC project initially took a random EST-based strategy to obtain full-ORF clones. The current MGC collection derives from >110 human and 80 mouse cDNA libraries made from a wide variety of tissues, cell lines, and development stages using different construction methods and vectors (Strausberg et al. 2002b; see http://mgc.nci.nih.gov for details). For each library, 5000-20,000 clones were sequenced at the 5′-end (5′-ESTs), and the sequences were clustered with all available data in dbEST using the UniGene algorithms (Pontius et al. 2002; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene). Candidate full-ORF clones were selected for full-insert sequencing if they fulfilled one of three criteria: (1) the insert included sequence that was 5′ to the starting methionine codon of a known gene; (2) the translated sequence was similar, but not identical to, the N terminus of a known protein; or (3) the 5′-sequence matched the statistical profile of the 5′-ends of known genes (Strausberg et al. 2002b). The candidate clone for each gene was sequenced to high quality (no uncertain base calls and an average estimated error rate of <1 in 50,000 nucleotides).
Fully sequenced clones were analyzed to determine whether they contained a complete coding sequence (CDS). Clones were then further analyzed with a combination of translating searches against the nonredundant protein database and a statistical assessment of the nucleotide sequence. This assessment involved the determination of the presence of an in-frame upstream stop codon and a sequence related to a Kozak consensus preceding the ORF, and an analysis of the properties of the 3′-untranslated region (Strausberg et al. 2002b). For known genes, that is, those in the RefSeq database (Pruitt et al. 2003; http://www.ncbi.nlm.nih.gov/RefSeq/) with a well-defined coding sequence, only clones in which the ORF comprised at least 50% or more of the longest known CDS were accepted into the MGC to avoid the preferential selection of splice forms corresponding to short products. Increasing the required ORF length to 80% of RefSeq would eliminate <2% of the clones (data not shown). The fully sequenced clones were also analyzed to eliminate those with potential frameshifts and chimeras. About 6% of the clones were found to have a frameshift, and 2% were chimeras. In instances of ambiguity and for genes without protein homologies, each clone was manually curated. These latter genes must have an ORF of at least 100 amino acids and cross at least one intron. These stringent requirements may mean that small, single-exon genes of unknown function were missed. If a clone failed any of the tests, another candidate was selected for sequencing. Only clones that were determined to be CDS-complete were submitted to GenBank with an MGC clone identifier (MGC:XXXXX) and the Entrez keyword “MGC,” whereas clones with frameshifts have a “frameshift” in the definition line and do not have the MGC clone identifier. All clones are available as a part of the MGC project through the I.M.A.G.E. distributors (Lennon et al. 1996).
As of March 2004, the MGC consisted of 11,298 human genes represented by 15,565 clones and 10,295 mouse genes represented by 12,974 clones (Table 1A). Thus, the size of the mouse clone collection is rapidly approaching that of the human collection, even though the initial emphasis of the project was on obtaining clones of human origin. In addition, 1383 clones of short variants from 1102 human genes are also available through the I.M.A.G.E. distributors.
Table 1.
Project Summary
| Organism | Project start date | No. of ESTs generated | No. of genes targeted | No. of clones in the collectiona | No. of genes representeda (% of target) |
|---|---|---|---|---|---|
| A. Mammalian Gene Collection | |||||
| Homo sapiens | Summer 2000 | 1,470,000 | All | 15,565 | 11,289 |
| Mus musculus | Summer 2000 | 1,100,000 | All | 12,974 | 10,295 |
| Rattus norvegicus | Fall 2003 | 45,000 | 6200 | 658 | 641 (10) |
| B. Collaborating Projects | |||||
| Xenopus laevis | Fall 2002 | 194,000 | 9200 | 1981 | 1962 (22) |
| Xenopus tropicalis | Spring 2003 | 59,000 | 6500 | 553 | 550 (8) |
| Danio rerio | Fall 2002 | 138,000 | 10,000 | 3436 | 3011 (30) |
As of March 8, 2004.
Status of Other Organism Collections
Based on the utility of these well-characterized human and mouse full-ORF clones and the desire on the part of the scientific community for full-ORF clone sets from additional organisms, the MGC recently expanded its scope to include the rat. In addition, the MGC pipeline is being used to support the generation of such clones from frog (see http://xgc.nci.nih.gov/Info/) and zebrafish (see http://zgc.nci.nih.gov/Info/). The goals and progress of these projects are summarized in Table 1B. Although they use the MGC infrastructure, each of these projects is managed separately (Klein et al. 2002; Rasooly et al. 2003). The goals of all three differ from those of the human and mouse project inasmuch that they do not aim to capture clones representing all of the genes for these organisms. Based on the experience gained in generating the human and mouse collections, these more limited goals should be readily achievable by using the random selection protocols already developed and by the judicious use of 15-20 libraries derived from different developmental stages and tissues.
Analysis of the Human and Mouse Clones
The average ORF sizes in the collection are 1186 and 1299 nt for human and mouse, respectively. The sizes are smaller than the average RefSeq ORF sizes, which are 1607 nt for human and 1437 nt for mouse (L. Wagner, unpubl.). The size difference suggests that the large ORFs are currently underrepresented in the MGC. The size distribution of MGC clones as compared with the RefSeqs can be found in Supplemental Material #1; it should be noted that the collection does include several clones with large CDS. In addition, the MGC is underrepresented in rare transcripts (Supplemental Material #2) as would be expected in a random transcript sampling approach. Ohara et al. (1997) generated size-selected cDNA libraries of 3-10 kb in length to clone large transcripts. To date, they isolated and fully sequenced 1954 cDNAs with an average ORF of 2905 nt.
There are 8412 human genes with mouse orthologs and 7808 mouse genes with human orthologs. For 5351 genes, clones from both organisms were obtained (Fig. 1). As this overlap represents approximately two-thirds of each set, the clone selection protocol is apparently not biased in a major way toward selecting the same genes in both organisms. Because the mouse project included a fairly large number of early development cDNA libraries, which the human project did not, it could be expected that clones specific for embryonic stages would be enriched in the mouse collection. Interestingly, however, the genes for which clones have been obtained only in mouse but not human are neither more highly expressed in the embryo, nor enriched for embryo-specific expression (data not shown).
Figure 1.

Overlap of homologous human and mouse genes with representative MGC clones. Analyses were performed as described in Methods. There are 8412 human genes with mouse orthologs and 7808 mouse genes with human orthologs. The number of HomoloGene groups may include paralogous genes in addition to orthologous genes in 8% of the sets.
The set of human genes represented in the MGC collection was compared with the comprehensive, annotated, nonredundant set of genes in the RefSeq database (Pruitt et al. 2000). RefSeq contains 11,233 entries of human genes that are considered to be biologically significant transcripts on the basis of two or more independent publications. The MGC includes a candidate for 9081 of these genes. This high frequency indicates that the random EST-based strategy has been highly successful in identifying full-ORF clones of known genes. In addition, a large number of previously uncharacterized full-ORF sequences, whose function is still unknown, have also been recovered. However, this approach has now reached a point of diminishing return for the human, and is reaching saturation for the mouse collection (Fig. 2). Therefore, for these two organisms, the project must now shift to more directed strategies to obtain clones for the missing genes.
Figure 2.

Progress of gene capture over time. The number of (A) human and (B) mouse full-ORF MGC clones and the number of genes represented by these clones over the lifetime of the project are shown. (Inset) The number of ESTs sequenced as a function of time. ESTs from prior data sets, including human and mouse cDNA libraries from the CGAP (Schaefer et al. 2001; Strausberg 2001, Strausberg et al. 2002a), were used to jump start the MGC project.
Two directed strategies were evaluated. The first was based on the determination of tissue expression distribution of the missing genes (data not shown). A normalized and subtracted cDNA library was made from a tissue, placenta, in which many of the missing genes were expressed. However, the resulting yield of clones representing them was too low to make this a practical approach. Specifically, out of 16,800 EST reads, the project identified 101 full-length clones for known genes (0.6%) for full-length sequencing. In the second approach, gene-specific primers were used to amplify the coding region of the transcripts from tissues in which they are expressed, followed by cloning of the PCR product into a cDNA vector and sequencing multiple candidates for each gene. Pilot studies indicate that between 50% and 80% of the missing genes can be recovered by this method (Baross et al. 2004; Wu et al. 2004), although it is likely that a fraction will be difficult to obtain.
In addition to the well-characterized transcripts, the MGC project will pursue two additional classes of missing genes. The first includes putative, computer-predicted genes for which there is some experimental evidence for the transcript's existence (such as one or more ESTs, or an uncharacterized cDNA generated through a large-scale project). The second class consists of ab initio gene predictions based solely on computational methods.
Validation of the Human and Mouse MGC Clones
The availability of the reference human genome sequence (Lander et al. 2001; Venter et al. 2001; The International Human Genome Sequencing Consortium, in prep.), an advance draft of the mouse genome sequence (Waterston et al. 2002), and the rich reservoir of human ESTs and other clone sequences (Adams et al. 1991; Boguski et al. 1993; Williamson 1999; Brentani et al. 2003) provided an opportunity for more detailed analysis of the quality of the MGC clones. Comparison of the MGC clone sequences with the finished human genome sequence reveals differences at about 1 in every 1147 positions in the coding regions, a frequency of 0.00087. These differences could be due to biological reasons (e.g., natural variation, with an expected nucleotide sequence diversity of 0.00075; Bamshad and Wooding 2003), post-transcriptional mRNA editing (Parks 2000; Schaub and Keller 2002; Anant et al. 2003), or one or more experimental artifacts. Experimental artifacts could arise during growth of cells in tissue culture, RNA preparation, library generation (either by the lack of fidelity of the reverse transcriptase or the DNA polymerase), clone propagation, or sequencing of the cDNA or the genome. Because the sequence quality of the MGC clones is very high (a frequency of errors of <1 in 50,000 nt), sequence quality is not a dominant source of error. Of the observed differences with the human genome, 32% coincide with a variant recorded in the polymorphism database dbSNP (Sherry et al. 2001; http://www.ncbi.nlm.nih.gov/SNP). As not all human variation is currently represented in dbSNP, there are undoubtedly other variants in the MGC clones that represent bona fide biological variation. Hence, we conclude that a significant fraction of the observed sequence difference between the MGC clones and the human reference sequence represents natural variation in the human population.
Using alignment of the cDNA sequences to the genome, two special cases were analyzed in which essentially all of the possible sequences should be known and, therefore, a significant level of novel variation would not be expected. Analysis of these cases should provide independent estimates of clone quality. The first involved the clones for HLA genes; because of the extent to which this locus has been previously studied, few new alleles should be found in the MGC. The sequence of 24 of the 28 MGC HLA clones coded for an amino acid composition corresponding to known polymorphisms, suggesting that at least 85% of the HLA MGC clones correspond to variations known to exist in the human population.
The second case represented a situation in which population polymorphism should be reduced to a very low level. Mouse MGC clones isolated from a nonnormalized, high-quality cDNA library made from the inbred strain C57BL6/J were compared with finished sequence from the same mouse strain. Here, 97% of the MGC clones aligned perfectly, with an estimated error rate of 1 in 77,000 nt. As this is on the order of the sequence accuracy itself, it implies that there are very few other types of errors in these MGC clones. However, other C57BL6/J libraries did not match the genome sequence as well. The discrepancy rate in clones from normalized libraries was 1 in 650 nt, and several libraries made by a protocol aimed to enrich for long, full-length clones had a total discrepancy rate of 1 in 253 nt. These data suggest that cDNA library synthesis protocols substantially affect the error rate of the final product (see below).
Another approach to human clone validation involved analysis of the nature of observed coding changes. Selective pressures on coding regions will disfavor polymorphisms that change an amino acid (nonsynonymous, or NS, changes). Hence, biologically valid variation in the MGC would be expected to show fewer NS changes than would artifactual changes of individual clones caused by PCR, cloning, or reverse transcriptase errors. If the nucleotide differences in the MGC clones were completely random with a 4:1 transition:transversion ratio, the nonsynonymous fraction (fNS) would be 0.71 (D. Lipman and L. Wagner, unpubl.). In fact, however, the fNS is 0.52, indicating that missense changes are, indeed, selected against. This should be compared, however, with the observed fNS of 0.43 for all coding SNPs in dbSNP that have been validated by testing in a panel of genomic DNA samples (S. Sherry and L. Wagner, unpubl.). Assuming that the observed differences are a mixture of artifacts and polymorphisms leads to the simple formula:
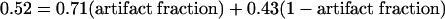 |
Therefore, ∼32% of the nonsynonymous variants in the overall MGC were experimentally introduced.
In yet another approach to assessing the possibility that some of the observed differences in sequence between MGC clones and the reference human genome are of artifactual origin, the MGC cDNA sequences have also been compared with the partially completed sequence of the chimpanzee (Pan troglodytes; http://www.ncbi.nlm.nih.gov/mapview; http://www.ebi.ac.uk/embl/indiex.html) genome to determine if any of the identified nucleotide differences match an ancestral allele. About 24% of the nonsynonymous cDNA alleles were found to match the Pan allele. In those instances, it can be concluded that the difference between the human cDNA and genomic sequence is likely to be a polymorphism (or, rarely, an error in the genome sequence), and therefore, those cDNAs are considered to be validated.
Sorting the human clones with NS coding changes by library provider identified one potential source of nucleotide differences. The results (Table 2) show that the library origin has a significant impact on the rate at which such differences were observed. Although this phenomenon was not investigated further, it is interesting to note that library maker #4, who had the highest sequence difference rate, used thermostabilized reverse transcriptase and performed subtraction and normalization protocols in which the second strand synthesis was done by a thermostable polymerase, whereas the other makers did not use either of these enzymes.
Table 2.
Discrepancy Frequencies in Human cDNAs, Sorted by Library Synthesizers
| Library maker | Nucleotide difference rate vs. the genome | fNS | % of the time MGC allele is found in the chimpanzee |
|---|---|---|---|
| All MGC | 0.00087 | 0.52 | 0.238 |
| #1 | 0.00062 | 0.45 | 0.326 |
| #2 | 0.00074 | 0.50 | 0.243 |
| #3 | 0.00125 | 0.54 | 0.095 |
| #4 | 0.00144 | 0.61 | 0.042 |
The results from the aggregate collection are presented in row 1, and the results from the four major providers of human libraries are listed separately. fNS is the fraction of the discrepancies between the cDNA and the genomic sequence that are nonsynonymous at the amino acid level. The libraries are listed in the Supplemental Material (#3).
There are 1980 human genes that are represented only by a MGC clone that has at least one NS substitution compared with the reference genome, and the NS substitution is not present in dbSNP. The amino acid differences between the MGC-encoded proteins and the genome-encoded proteins can be assumed to be due to a mix of polymorphisms, cloning artifacts, and sequence errors. Actual sequencing errors are likely to be rare, and most of the cloning artifacts are likely to be in the MGC clones, because of the experimental parameters listed previously. When the differences to the human genome within the cDNA clones are compared with the corresponding Pan sequence, 20% of the MGC clones assessed fully agree with chimpanzee, and thus these are likely to represent true polymorphisms. But for a true polymorphism, the expectation is that the Pan sequence would match the cDNA and the genomic sequence with equal frequency. Therefore, by inference, an additional 20% of MGC cDNA clones probably carry NS changes that are biologically valid—although it is not possible to identify which clones these are. This leaves ∼1200 human genes in the MGC (10% of the total collection) for which the NS changes are likely due to experimental artifact. It should be noted that these same artifacts are likely to be found at some level in any collection of cDNAs, given the unavoidable nonzero error rates of thermostable polymerase, reverse transcriptase, and other components of the various protocols for generating full-length cDNA clones.
The results in this section exemplify the challenge in determining the extent of the various biological and experimental contributions to the nucleotide differences found in the MGC clones as compared with the reference genomes. Each analysis used a different subset of the data, and, therefore, it is not possible to compare the results directly. In addition, for the human, rare polymorphisms are hard to determine, and the comparison to the ancestral genome is limited by the gaps in the latter. In the case of the mouse clones, only 14 libraries were made from the same strain as the genome sequence, and, therefore, the number of clones used in the analyses were not very large.
The variations discovered during the clone characterization analyses have made it imperative to include detailed annotation in each GenBank record for each MGC clone. Accordingly, every human clone record now includes the following: the identification of every nucleotide difference with the reference human sequence, the inferred amino acid changes, if any, and a dbSNP reference number if the variant is already documented. In the future, additional properties of the differences, including changes within conserved motifs and the presence of ancestral alleles, will be added as will the annotation of mouse records.
Gaining Access to the MGC Resources
Since its inception, the MGC project has developed several independent Web sites as well as provided data to other public resources. Major project-relevant Web sites can be found in Table 3.
Table 3.
Web Sites Relevant to the Project
| Web site | Description |
|---|---|
| http://mgc.nci.nih.gov | Provides a list of genes and libraries, as well as information on library construction, vectors, and distribution resources for human, mouse, and rat. |
| http://xgc.nci.nih.gov | As above, but the Xenopus laevis and X. tropicalis. |
| http://zgc.nci.nih.gov | As above, but for Danio rerio. |
| http://image.llnl.gov | Provides information on cDNA clones from the MGC, XGC, and ZGC projects, including library and vector details, clone queries, links to full-ORF clone data files, and information on obtaining clones. |
| http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene | An experimental system for partitioning GenBank sequences into nonredundant sets of gene-oriented clusters for many organisms, updated periodically. In MGC, genes are defined on the basis of UniGene clustering. |
| http://genome.ucsc.edu/ | A genome browser that includes the MGC clones as part of their visualization options. |
The MGC Web site (http://mgc.nci.nih.gov) provides information about the full-ORF clone collections for human, mouse, and rat. The number of full-ORF clones and nonredundant genes for each species is listed on the home page and is updated weekly. The main page also provides links to lists of candidate clones awaiting full-insert sequencing, all of the EST sequences generated for the MGC project, and descriptions of the vectors and methods used to construct MGC libraries. Complete lists and sequences of full-ORF MGC clones can also be downloaded for each species from this Web site. A user can search for a full-ORF clone using either the gene symbol or a keyword search. All full-length clones are also available in NCBI's Entrez nucleotide database by searching for the keyword “MGC.” The result of such a query provides links to the other informatics resources, such as LocusLink (Pruitt and Maglott 2001; http://www.ncbi.nlm.nih.gov/LocusLink/), consolidated information about curated sequence and genetic loci), the GenBank record, and the I.M.A.G.E. clone information. The I.M.A.G.E. identifier links to the corresponding record in UniGene and NCI's Cancer Genome Anatomy Project (CGAP; http://cgap.nci.nih.gov). A modified version of BLAST was developed by MGC and supports alignment of a query sequence against all MGC full-length clone sequences. Similar full-length clone resources are available for the Xenopus Gene Collection (XGC; http://xgc.nci.nih.gov) and the Zebrafish Gene Collection (ZGC; http://zgc.nci.nih.gov).
All MGC full-ORF clones are available to all researchers for unencumbered use and can be purchased from several commercial I.M.A.G.E. distributors. The “where to buy” link on the MGC Web site allows users to directly access the Web sites of U.S. and worldwide distributors. In addition, most of the clones that were sequenced in the project, whether representing a full ORF or not, and that can be identified by their I.M.A.G.E. identifier, can be obtained from many of the same clone distributors.
Information about MGC clones in the context of the reference genome sequence can be obtained at the UCSC (University of California, Santa Cruz) Genome Browser (Kent et al. 2002; http://genome.ucsc.edu). On the browser, a specific MGC track can be activated to visualize the location of MGC clones in the reference genome, and links from this track can show the alignment of the cDNA with the reference sequence. All of the search and visualization functionality of this browser can be used to identify genes with MGC clones and to provide additional information about the corresponding genes, including the gene's position in the genome, sequence variation, and sequence conservation with other genomes.
In conclusion, the MGC project has already generated a large, well-documented, and increasing useful collection of cDNA clones containing full-ORFs for human and mouse genes. Targeted methods to recover the missing cDNAs are under way. The project has recently been expanded include clones for another species, Rattus norvegicus, and comparable methods are being used to generate collections of genes for Xenopus laevis, Xenopus tropicalis, and Danio rerio.
METHODS
cDNA Library Production
Descriptions of methods for the library construction can be found at the Web sites http://mgc.nci.nih.gov/Info/ (for human, mouse and rat), http://xgc.nci.nih.gov/Info/ (for X. laevis and X. tropicalis) and http://zgc.nci.nih.gov/Info/ (for D. rerio). The complete sequence for each of the MGC vectors is found at http://image.llnl.gov/image/html/vectors.shtml. The catalog of the cDNA libraries resulting in full-length MGC clones can also be found at the Web sites.
Library Characterization, Screening, Selection of Full-ORF Candidate Clones, and Sequencing
The core methods have been previously described (Strausberg et al. 2002b). Recent modifications to the pipeline include: (1) 3′-sequences are not generated, except in the Xenopus project, where ∼40% of the clones have 3′-reads; (2) 2000-5000 clones from each library are sequenced at the 5′-end, and if a library is deemed to be of a high quality, 5000 clones are added. The identification of clones for full-insert sequencing for Xenopus and Danio are based on one of the following criteria: a BLAST search against the well-characterized, complete mRNAs; the presence of a starting methionine and an alignment of at least 95% identity over at least 100 nt; or a translating BLAST search against proteins from organisms whose genomes have been sequenced requiring both a starting methionine and an alignment of the sequences with an e-value of at most 10-6, except for the region at the protein's N terminus. This region is excepted because many distant homologs do not have well-conserved N termini and a clone may be a candidate for full-insert sequencing if the length of nonalignment is the same in X. laevis, X. tropicalis, or D. rerio cDNA and the orthologous protein(s). The selection of Rattus clones is the same as described for Xenopus and Danio, but requires the homology to the 5′-end as the rat and mouse have at least 93% homology (Makalowski and Boguski 1998). The full-length sequencing is performed by one of three methods as described (Strausberg et al. 2002b) to an accuracy of less than 1 error per 50,000 nt. The GenBank accession nos. generated by the MGC project can be found in Supplemental material #4.
Determination of Nucleotide Differences
The sequences of all human and mouse MGC clones were aligned to the genome sequence of the organism of origin (NCBI build 34 for human and NCBI build 31 for mouse), and all discrepancies between MGC clones and genomic sequence were recorded. The best placement of each clone on the genomic sequence with canonical splice-site recognition was used. Within the ORF, all differences were identified as either synonymous or nonsynonymous by using the protein-coding sequence of the MGC clones. The differences, which correspond to known mRNA variations recorded in dbSNP v. 117 (http://www.ncbi.nlm.nih.gov/SNP/), were identified. Finally, alignments of human MGC clones to the P. troglodytes genome were generated to identify the ancestral alleles. The nonoverlapping local alignments of at least 98% identity to chimpanzee genomic contigs were used, because of the evolutionary distance and the unfinished status of the Pan genome.
Determination of Human and Mouse Overlap
Homology relationships were taken from the HomoloGene resource (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=homologene), which is constructed by automated comparison of gene sets from more than a dozen complete eukaryotic genomes. Comparisons are performed in a progressive manner using the taxonomic tree to guide the process. Paralogous genes from the same species may be included in a HomoloGene group if they are closer to each other than to an outgroup species. The number of HomoloGene groups may include paralogous genes in addition to orthologous genes in 8% of the sets.
Complete List of Authors
MGC Program Team
Daniela S. Gerhard,4 Lukas Wagner,5 Elise A. Feingold,6 Carolyn M. Shenmen,5 Lynette H. Grouse,4 Greg Schuler,5 Steven L. Klein,7 Susan Old,8 Rebekah Rasooly,9 Peter Good,6 Mark Guyer,6 Allison M. Peck,6 Jeffery G. Derge,10 David Lipman,5 and Francis S. Collins6
Additional Bioinformatics and MGC Web Site
Wonhee Jang,5 Steven Sherry,5 Mike Feolo,5 Leonie Misquitta,5 Eduardo Lee,5 Kirill Rotmistrovsky,5 Susan F. Greenhut,4 Carl F. Schaefer,11 Kenneth H. Buetow,11 Tom I. Bonner,17 David Haussler,12 Jim Kent,12 Mark Diekhans,12 Terry Furey,12 and Michael Brent13
cDNA Clone Management
mRNA Preparation
cDNA Library Preparation
Florence Hsie,15 Tom Driscoll,15 M. Bento Soares,16 Maria F. Bonaldo,16 Tom L. Casavant,16 Todd E. Scheetz,16 Michael J. Brown-stein,17 Ted B. Usdin,17 Shiraki Toshiyuki,18 Piero Carninci,18 Yulan Piao,19 Dawood B. Dudekula,19 Minoru S.H. Ko,19 Koichi Kawakami,32 Yutaka Suzuki,20 Sumio Sugano,20 C.E. Gruber,21 M.R. Smith,21 Blake Simmons,22 Troy Moore,22 Richard Waterman,23 Stephen L. Johnson,23 Yijun Ruan,24 Chia Lin Wei,24 and S. Mathavan24
cDNA Full-Insert Sequencing
Baylor College of Medicine Human Genome Sequencing Center
Preethi H. Gunaratne,25 Jiaqian Wu,25 Angela M. Garcia,25 Stephen W. Hulyk,25 Edwin Fuh,25 Ye Yuan,25 Anna Sneed,25 Carla Kowis,25 Anne Hodgson,25 Donna M. Muzny,25 John McPherson,25 and Richard A. Gibbs25
Institute for Systems Biology
Jessica Fahey,3,26 Erin Helton,26 Mark Ketteman,26 Anuradha Madan,3,26 Stephanie Rodrigues,3,26 Amy Sanchez,26 Michelle Whiting,26 and Anup Madan3,26
NIH Intramural Sequencing Center
Alice C. Young,27 Keith D. Wetherby,27 Steven J. Granite,27 Peggy N. Kwong,27 Charles P. Brinkley,27 Russell L. Pearson,27 Gerard G. Bouffard,27 Robert W. Blakesly,27 and Eric D. Green27
Stanford Human Genome Center
Mark C. Dickson,28 Alex C. Rodriguez,28 Jane Grimwood,28 Jeremy Schmutz,28 and Richard M. Myers28
British Columbia Cancer Agency Genome Sciences Centre
Yaron S.N. Butterfield,29 Malachi Griffith,29 Obi L. Griffith,29 Martin I. Krzywinski,29 Nancy Liao,29 Ryan Morrin,29 Diana Palmquist,29 Anca S. Petrescu,29 Ursula Skalska,29 Duane E. Smailus,29 Jeff M. Stott,29 Angelique Schnerch,29 Jacqueline E. Schein,29 Steven J.M. Jones,29 Robert A. Holt,29 Agnes Baross,29 and Marco A. Marra29
Department of Genetics and Genome Sequencing Center, Washington University Medical School
Sandra Clifton30
EST Sequencing
Agencourt Bioscience Corporation
Acknowledgments
The Mammalian Gene Collection Program is an NIH interinstitute effort receiving financial and scientific support from many individual institutes and centers within the NIH. A complete list of these institutes is available on the MGC Web site. Special thanks go to Robert L. Strausberg and Richard D. Klausner, who were involved in initiating the project. The MGC Program has received excellent guidance from members of the External Scientific Committee: Barbara Wold, Philip Sharp, Geoffrey Duyk, Connie Cepko, Stewart Scherer, Lincoln Stein, Ronald Davis, Richard Klausner, and Edward Harlow. The XGC Program has received excellent guidance from Aaron Zorn, Ken Cho, Bruce Blumberg, Enrique Amaya, Nancy Papalopulu, and Jane Rogers. The ZGC Program has received excellent guidance from members of their advisory committee: Bruce Birren, Jane Rogers, Will Talbot, Monte Westerfield, and Len Zon. Additional contributions have been made by Judy Mietz (NCI), Michael Chang (NCRR), Adam Felsenfeld (NHGRI), Tyl Hewitt (NICHD), Deborah Henken (NICHD), Nancy Freeman (NIDCD), Rochelle Small (NIDCR), Danilo Tagle (NIND), and Lynn Schriml (NCBI). The rat program has benefited greatly from the advice and involvement of Howard Jacob. D.S.G. would like to acknowledge the valuable assistance of Cyndy Izadi in the preparation of this manuscript. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. N01-C0-12400. Members of the Baylor College of Medicine Human Genome Sequencing Center are thanked for support on this project. Rachel Dickhoff and Julia Greene of the Institute for Systems Biology provided helpful discussions and excellent assistance. Keith Wetherby, Russell Pearson, Nicole Dietrich, Peggy Kwong, and Stephen Granite of the NIH Intramural Sequencing Center provided superb technical and computational assistance. Special thanks are extended to the many contributing members of the Stanford Human Genome Center for support on this project. The following members of the University of British Columbia Genome Sciences Centre are thanked for their valuable contributions in cDNA sequencing and helpful discussions: J. Asano, S. Chan, N. Girn, R. Guin, R. Kustsche, S. Lee, K. MacDonald, C. Mathewson, T. Olson, P. Pandoh, A.-L. Prabhu, L. Spence, J. Stott, S. Taylor, K. Teague, M. Tsai, G. Yang, and S. Zuyderduyn.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data for the full-length clones from this study have been submitted to GenBank under accession nos. BC000001-BC077073.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2596504.
National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
National Center for Biotechnology Information, National Library of Medicine, Bethesda, Maryland 20894, USA.
National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892, USA.
National Institute of Heart Lung and Blood, National Institutes of Health, Bethesda, Maryland 20892, USA.
National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, USA.
SAIC-Frederick, Inc., National Cancer Institute at Frederick, Frederick, Maryland 21702, USA.
National Cancer Institute, Center for Bioinformatics, Rockville, Maryland 20852, USA.
Laboratory of Cell Biology, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, USA.
Center for Biomolecular Science & Engineering, University of California, Santa Cruz, Santa Cruz, California 95064, USA.
Laboratory for Computational Genomics, Washington University, St. Louis, Missouri 63130, USA.
The I.M.A.G.E. Consortium, Biology and Biotechnology Research Program, Lawrence Livermore National Laboratory, Livermore, California 94550, USA.
BD Biosciences Clontech, Palo Alto, California 94303, USA.
Department of Pediatrics, University of Iowa Health Care, Iowa City, Iowa 52242, USA.
Genome Science Laboratory, RIKEN Genomic Science Laboratory, Saitama 351-0198, Japan.
National Institute on Aging, NIH, Baltimore, Maryland 21224, USA.
National Institute of Genetics, Mishima 411-8540, Japan.
Department of Medical Genome Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Tokyo 108-8639, Japan.
Express Genomics, Frederick, Maryland 21701, USA.
Open Biosystems, Huntsville, Alabama 35806, USA.
Department of Genetics, Washington University School of Medicine, St. Louis, Missouri 63130, USA.
Genome Institute of Singapore, Singapore 138672.
Baylor College of Medicine Human Genome Sequencing Center, Baylor College of Medicine, Houston, Texas 77030, USA.
Present address: University of Iowa Hospitals and Clinics, Iowa City, IA 52242, USA.
The Institute for Systems Biology, Seattle, Washington 98103, USA.
NIH Intramural Sequencing Center, Gaithersburg, Maryland 20877, USA.
Stanford Human Genome Center, Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA.
University of British Columbia Genome Sciences Centre, BC Cancer Agency, Vancouver BC, V5Z 4S6 Canada.
Department of Genetics and Genome Sequencing Center, Washington University Medical School, St. Louis, Missouri 63130, USA.
Agencourt Bioscience Corporation, Beverly, Massachusetts 01915, USA.
References
- Adams, M.D., Kelley, J.M., Gocayne, J.D., Dubnick, M., Polymeropoulos, M.H., Xiao, H., Merril, C.R., Wu, A., Olde, B., Moreno, R.F., et al. 1991. Complementary DNA sequencing: Expressed sequence tags and Human Genome Project. Science 252: 1651-1656. [DOI] [PubMed] [Google Scholar]
- Anant, S., Blanc, V., and Davidson, N.O. 2003. Molecular regulation, evolutionary, and functional adaptations associated with C to U editing of mammalian apolipoproteinB mRNA. Prog. Nucleic Acid Res. Mol. Biol. 75: 1-41. [DOI] [PubMed] [Google Scholar]
- Bamshad, M. and Wooding, S.P. 2003. Signatures of natural selection in the human genome. Nat. Rev. Genet. 4: 99-111. [DOI] [PubMed] [Google Scholar]
- Baross, Á, Butterfield, T.S.N., Coughlin, S.M., Zeng, T., Griffith, M., Griffith, O.L., Petrescu, A.S., Smailus, D.E., Khattra, J., McDonald, H.L., et al. 2004. Systematic recovery and analysis of full-ORF human cDNA clones. Genome Res. (in press). [DOI] [PMC free article] [PubMed]
- Boguski, M.S., Lowe, T.M., and Tolstoshev, C.M. 1993. dbEST—Database for “expressed sequence tags.” Nat. Genet. 4: 332-333. [DOI] [PubMed] [Google Scholar]
- Brentani, H., Caballero, O.L., Camargo, A.A., da Silva, A.M., da Silva Jr., W.A., Dias Neto, E., Grivet, M., Gruber, A., Guimaraes, P.E., Hide, W., et al. 2003. The generation and utilization of a cancer-oriented representation of the human transcriptome by using expressed sequence tags. Proc. Natl. Acad. Sci. 100: 13418-13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs, R.A., Weinstock, G.M., Metzker, M.L., Muzny, D.M., Sodergren, E.J., Scherer, S., Scott, G., Steffen, D., Worley, K.C., Burch, P.E., et al. 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493-521. [DOI] [PubMed] [Google Scholar]
- Kent, W.J., Sugnet, C.W., Furey, T.S., Roskin, K.M., Pringle, T.H., Zahler, A.M., and Haussler, D. 2002. The human genome browser at UCSC. Genome Res. 12: 996-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, S.L., Strausberg, R.L., Wagner, L., Pontius, J., Clifton, S.W., and Richardson, P. 2002. Genetic and genomic tools for Xenopus research: The NIH Xenopus initiative. Dev. Dyn. 225: 384-391. [DOI] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Lennon, G., Auffray, C., Polymeropoulos, M., and Soares, M.B. 1996. The I.M.A.G.E. Consortium: An integrated molecular analysis of genomes and their expression. Genomics 33: 151-152. [DOI] [PubMed] [Google Scholar]
- Makalowski, W. and Boguski, M.S. 1998. Synonymous and nonsynonymous substitution distances are correlated in mouse and rat genes. J. Mol. Evol. 47: 119-121. [DOI] [PubMed] [Google Scholar]
- Ohara, O., Nagase, T., Ishikawa, K., Nakajima, D., Ohira, M., Seki, N., and Nomura, N. 1997. Construction and characterization of human brain cDNA libraries suitable for analysis of cDNA clones encoding relatively large proteins. DNA Res. 4: 53-59. [DOI] [PubMed] [Google Scholar]
- Okazaki, Y., Furuno, M., Kasukawa, T., Adachi, J., Bono, H., Kondo, S., Nikaido, I., Osato, N., Saito, R., Suzuki, H., et al. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563-573. [DOI] [PubMed] [Google Scholar]
- Ota, T., Suzuki, Y., Nishikawa, T., Otsuki, T., Sugiyama, T., Irie, R., Wakamatsu, A., Hayashi, K., Sato, H., Nagai, K., et al. 2004. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat. Genet. 36: 40-45. [DOI] [PubMed] [Google Scholar]
- Parks, T.N. 2000. The AMPA receptors of auditory neurons. Hear. Res. 147: 77-91. [DOI] [PubMed] [Google Scholar]
- Pontius, J.U., Wagner, L., and Schuler, G.D. 2002. Part 3. Querying and linking the data, 21. UniGene: A unified view of the transcriptome. In The NCBI Handbook (internet; ed. J. McEntyre), National Library of Medicine, Bethesda, MD.
- Pruitt, K.D. and Maglott, D.R. 2001. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 29: 137-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt, K.D., Katz, K.S., Sicotte, H., and Maglott, D.R. 2000. Introducing RefSeq and LocusLink: Curated human genome resources at the NCBI. Trends Genet. 16: 44-47. [DOI] [PubMed] [Google Scholar]
- Pruitt, K.D., Tatusova, T., and Maglott, D.R. 2003. NCBI Reference Sequence project: Update and current status. Nucleic Acids Res. 31: 34-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasooly, R.S., Henken, D., Freeman, N., Tompkins, L., Badman, D., Briggs, J., and Hewitt, A.T. 2003. Genetic and genomic tools for zebrafish research: The NIH zebrafish initiative. Dev. Dyn. 228: 490-496. [DOI] [PubMed] [Google Scholar]
- Schaefer, C., Grouse, L., Buetow, K., and Strausberg, R.L. 2001. A new cancer genome anatomy project web resource for the community. Cancer J. 7: 52-60. [PubMed] [Google Scholar]
- Schaub, M. and Keller, W. 2002. RNA editing by adenosine deaminases generates RNA and protein diversity. Biochimie 84: 791-803. [DOI] [PubMed] [Google Scholar]
- Sherry, S.T., Ward, M.H., Kholodov, M., Baker, J., Phan, L., Smigielski, E.M., and Sirotkin, K. 2001. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 29: 308-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strausberg, R.L. 2001. The Cancer Genome Anatomy Project: New resources for reading the molecular signatures of cancer. J. Pathol. 195: 31-40. [DOI] [PubMed] [Google Scholar]
- Strausberg, R.L., Feingold, E.A., Klausner, R.D., and Collins, F.S. 1999. The mammalian gene collection. Science 286: 455-457. [DOI] [PubMed] [Google Scholar]
- Strausberg, R.L., Buetow, K.H., Greenhut, S.F., Grouse, L.H., and Schaefer, C.F. 2002a. The cancer genome anatomy project: Online resources to reveal the molecular signatures of cancer. Cancer Invest. 20: 1038-1050. [DOI] [PubMed] [Google Scholar]
- Strausberg, R.L., Feingold, E.A., Grouse, L.H., Derge, J.G., Klausner, R.D., Collins, F.S., Wagner, L., Shenmen, C.M., Schuler, G.D., Altschul, S.F., et al. 2002b. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. 99: 16899-16903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter, J., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- Wiemann, S., Weil, B., Wellenreuther, R., Gassenhuber, J., Glassl, S., Ansorge, W., Bocher, M., Blocker, H., Bauersachs, S., Blum, H., et al. 2001. Toward a catalog of human genes and proteins: Sequencing and analysis of 500 novel complete protein coding human cDNAs. Genome Res. 11: 422-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, A.R. 1999. The Merck Gene Index project. Drug Discov. Today 4: 115-122. [DOI] [PubMed] [Google Scholar]
- Wu, J.Q., Garcia, A.M., Hulyk, S., Sneed, A., Kowis, C., Yuan, Y., Steffen, D., McPherson, J.D., Gunaratne, P.H., and Gibbs, R.A. 2004. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques 36: 698-700. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://cgap.nci.nih.gov; Cancer Genome Anatomy Project.
- http://genome.ucsc.edu; UCSC Genome Browser.
- http://image.llnl.gov/image/html/vectors.shtml; complete sequence for each of the MGC vectors.
- http://mgc.nci.nih.gov; MGC.
- http://www.ebi.ac.uk/embl/indiex.html; chimpanzee.
- http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=homologene; HomoloGene resource.
- http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene; Unigene.
- http://www.ncbi.nlm.nih.gov/LocusLink/; LocusLink.
- http://www.ncbi.nlm.nih.gov/mapview; chimpanzee.
- http://www.ncbi.nlm.nih.gov/RefSeq/; RefSeq database.
- http://www.ncbi.nlm.nih.gov/SNP; the polymorphism database dbSNP.
- http://xgc.nci.nih.gov/Info/; Xenopus gene collection.
- http://zgc.nci.nih.gov/Info/; zebrafish gene collection.