

ABSTRACT
PYRRO-C3D is a cephalosporin-3-diazeniumdiolate nitric oxide (NO) donor prodrug designed to selectively deliver NO to bacterial infection sites. The objective of this study was to assess the activity of PYRRO-C3D against nontypeable Haemophilus influenzae (NTHi) biofilms and examine the role of NO in reducing biofilm-associated antibiotic tolerance. The activity of PYRRO-C3D on in vitro NTHi biofilms was assessed through CFU enumeration and confocal microscopy. NO release measurements were performed using an ISO-NO probe. NTHi biofilms grown on primary ciliated respiratory epithelia at an air-liquid interface were used to investigate the effects of PYRRO-C3D in the presence of host tissue. Label-free liquid chromatography-mass spectrometry (LC/MS) proteomic analyses were performed to identify differentially expressed proteins following NO treatment. PYRRO-C3D specifically released NO in the presence of NTHi, while no evidence of spontaneous NO release was observed when the compound was exposed to primary epithelial cells. NTHi lacking β-lactamase activity failed to trigger NO release. Treatment significantly increased the susceptibility of in vitro NTHi biofilms to azithromycin, causing a log fold reduction (10-fold reduction or 1-log-unit reduction) in viability (P < 0.05) relative to azithromycin alone. The response was more pronounced for biofilms grown on primary respiratory epithelia, where a 2-log-unit reduction was observed (P < 0.01). Label-free proteomics showed that NO increased expression of 16 proteins involved in metabolic and transcriptional/translational functions. NO release from PYRRO-C3D enhances the efficacy of azithromycin against NTHi biofilms, putatively via modulation of NTHi metabolic activity. Adjunctive therapy with NO mediated through PYRRO-C3D represents a promising approach for reducing biofilm-associated antibiotic tolerance.
KEYWORDS: Haemophilus influenzae, biofilms, antibiotic resistance, nitric oxide, proteomics
INTRODUCTION
Nontypeable Haemophilus influenzae (NTHi) plays a major role in a number of chronic lung diseases, including chronic obstructive pulmonary disease (COPD), the fourth largest cause of mortality worldwide (1), and cystic fibrosis, where early childhood infection leads to an environment within the lung that is more susceptible to infection by Pseudomonas aeruginosa (2). NTHi is also the predominant early colonizer in primary ciliary dyskinesia (PCD), another genetic chronic lung disease characterized by a lack of mucociliary clearance, chronic lung infection, and lung function decline (3, 4).
Persistence of NTHi infection is often associated with biofilm formation, with NTHi biofilms being implicated in a number of clinical settings, including formation on middle ear epithelium during chronic otitis media (5), chronic rhinosinusitis (6), chronic obstructive pulmonary disease sputum (7), and lower respiratory tract diseases (8). The biofilm phenotype enables bacteria to evade the host immune response, benefit from increased antibiotic tolerance, and subsequently develop antibiotic resistance through horizontal gene transfer (9). Exopolysaccharide matrix formation by biofilm bacteria may also restrict the diffusion of antibiotics into biofilms and prevent ingress of immune cells (10). Increased expression of efflux pumps and β-lactamases have also been shown to contribute to increased tolerance. It is the change to a metabolically dormant phenotype, however, that potentially plays the most important role, rendering antibiotics that target cell division ineffective (11). Biofilm formation in NTHi has been associated with the reduced metabolic activity typical of that observed in other bacterial species, with preserved ability to respond to stress (12). As well as clinical isolates from a range of diseases having the ability to form in vitro biofilms (8), NTHi has also been shown to form biofilms on cultured respiratory epithelial surfaces with decreased antibiotic susceptibility (13, 14). NTHi biofilms also exhibit quorum signaling that is characteristic of other respiratory biofilm formers such as P. aeruginosa (15). While P. aeruginosa is the most widely studied biofilm-forming respiratory pathogen, many differences exist with NTHi. For example, P. aeruginosa biofilms form within mucus in vivo (16) rather than attached to the epithelial surface as NTHi biofilms do (5, 6, 13, 14). Also, cyclic di-GMP seems to play a pivotal role in the P. aeruginosa biofilm life cycle under the control of guanylate cyclases and phosphodiesterases (17); however, NTHi genome sequencing shows no domains coding for these enzymes (18).
Bacteria within biofilms can be triggered by various external factors to revert to a planktonic single-cell state, a process that not only facilitates propagation and recolonization elsewhere within the host but also renders the bacteria more susceptible to antibiotics (10). Development of a therapeutic approach that induces dispersal or reverses the metabolically dormant phenotype is therefore an attractive approach for improving antibiotic effectiveness in the treatment of biofilm-associated infections.
Nitric oxide (NO) is a ubiquitous signaling molecule that plays a wide range of biological roles in both prokaryotes and eukaryotes. Low-dose NO has been shown to signal a dispersal response in biofilms formed by a number of bacterial species, including P. aeruginosa, Escherichia coli, Serratia marcescens, Staphylococcus aureus, and also multispecies biofilms (19–21). However, NO also plays a number of important roles in the human host, meaning that administration of spontaneous NO donors as drugs would likely elicit undesirable side effects, particularly through alterations in circulatory dynamics (22–24). To address this, we have developed a novel class of targeted NO prodrugs (cephalosporin-3′-diazeniumdiolates) that are composed of a diazeniumdiolate (NONOate) NO donor attached to the 3′ position of first-generation cephalosporins. This innovative drug class was designed to selectively release the NONOate following cleavage of the β-lactam ring by bacterial β-lactamases, thereby targeting NO delivery directly to the site of infection (25) (Fig. 1a). We hypothesized that treatment of NTHi biofilms with a cephalosporin-3′-diazeniumdiolate (i.e., PYRRO-C3D K+ salt [Fig. 1a]) would signal a return to a planktonic phenotype, thereby increasing NTHi sensitivity toward conventional antibiotics. We investigated the activity of PYRRO-C3D, both alone and in combination with azithromycin, on biofilms formed in vitro and on primary respiratory epithelia grown at an air-liquid interface. High-throughput label-free proteomic analyses were performed to provide mechanistic insights into the role of NO in NTHi biofilms.
FIG 1.
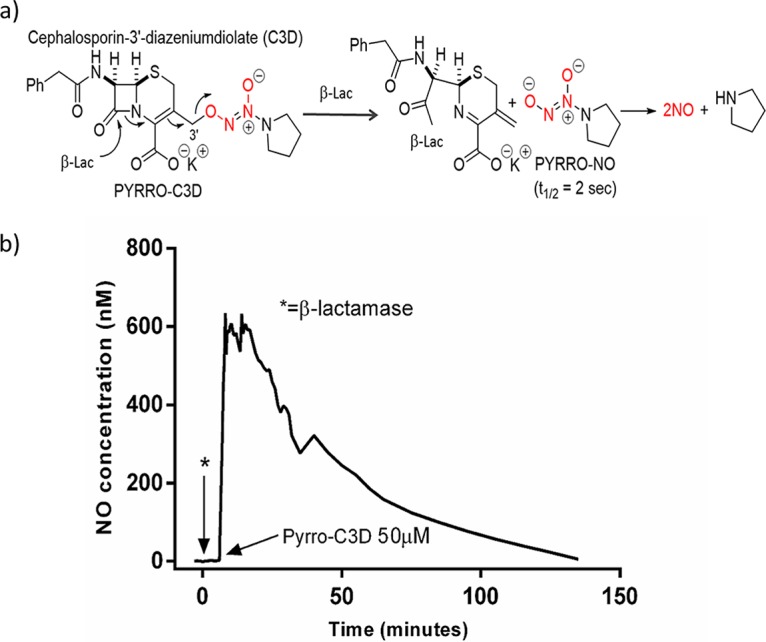
(a) Structure of PYRRO-C3D and NO release mechanism following β-lactam ring cleavage by β-lactamase. (b) NO release from PYRRO-C3D (50 μM in PBS) following activation with β-lactamase (penicillinase).
RESULTS
PYRRO-C3D elicits a direct antibacterial effect on planktonic, but not biofilm, NTHi.
Prior to treatment of NTHi, NO release from PYRRO-C3D was first confirmed following chemical activation by the β-lactamase enzyme penicillinase. Activation of 50 μM PYRRO-C3D resulted in rapid release of NO, reaching a maximum concentration of ∼600 nM over 14 min, which was followed by a gradual decline over a further 120 min (Fig. 1b). Treatment of planktonic NTHi cultures with increasing concentrations of PYRRO-C3D identified that concentrations of >50 μM inhibited growth (Fig. 2a). However, no reduction in biofilm viability was observed following PYRRO-C3D treatment at 10 nM to 100 μM over 2 h (Fig. 2b). Treatment of planktonic NTHi with 50 μM PYRRO-C3D released between 48 and 90 nM NO over 15 min, with the signal being quenched following the addition of the β-lactamase inhibitor clavulanate (Fig. 2c).
FIG 2.
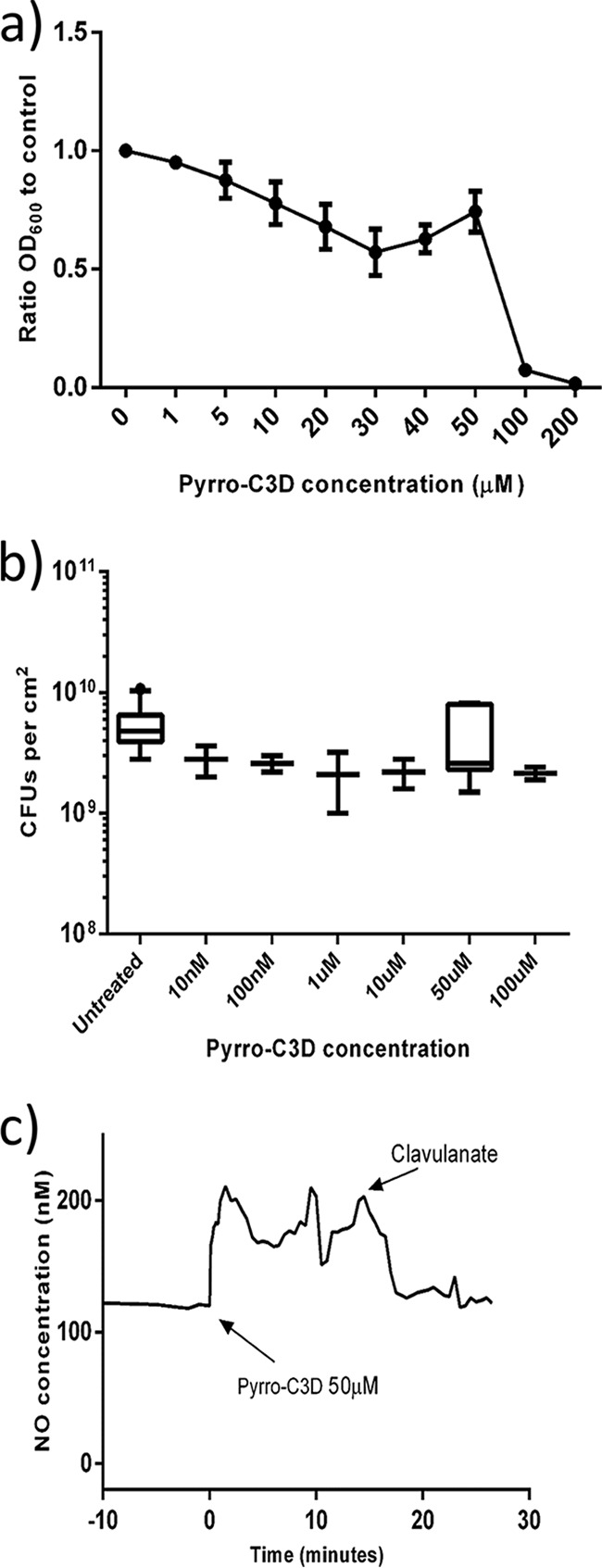
PYRRO-C3D elicits a direct antibacterial effect on planktonic NTHi. (a) Planktonic NTHi growth in the presence of PYRRO-C3D measured by absorbance (OD600; n = 4). (b) Viability of 72-h in vitro NTHi biofilm following 2-h treatment with PYRRO-C3D as measured by CFU enumeration (n = 4). (c) NO release from 50 μM PYRRO-C3D in the presence of planktonic NTHi. The signal was recorded over 15 min before quenching with the β-lactamase inhibitor clavulanate (n = 2).
PYRRO-C3D enhances NTHi biofilm susceptibility to azithromycin.
Previous research has shown that NO treatment of biofilms formed by several bacterial species reduces their tolerance toward antibiotics (26, 27). Treatment of established 72-h NTHi biofilms with 4 mg/ml azithromycin produced a slight reduction in viable NTHi within the biofilm (P = 0.0019; Fig. 3a). Complete killing, however, was not achieved despite the planktonic MIC for this strain being 0.001 mg/ml (data not shown). Combined treatment with 4 mg/ml azithromycin and 50 μM PYRRO-C3D resulted in a significant increase in bacterial killing, where a log fold reduction (10-fold reduction or 1-log-unit reduction) in viable cells was observed in the biofilm population (P = 0.0189; Fig. 3a). COMSTAT analysis of the live biofilm population following confocal imaging indicated that azithromycin alone had little impact on the viable biomass, while combined treatment with PYRRO-C3D produced a significant reduction (P = 0.0064; Fig. 3b). Notably, this reduction occurred despite a slight but significant increase in the live biofilm population occurring following treatment with PYRRO-C3D alone (Fig. 3b). This effect, however, does not appear to be mediated by biofilm dispersal, as a significant drop in the viable supernatant population with PYRRO-C3D alone was observed (P = 0.0061; Fig. 3c). No difference between azithromycin treatment alone and combined PYRRO-C3D–azithromycin was also observed (P = 0.0974; Fig. 3c). COMSTAT analysis also showed no significant difference in biofilm thickness across any of the treatments, suggesting a lack of biofilm dispersal (Fig. 3d). Measurement of biofilm density, assessed by average diffusion distance between live bacteria, revealed that PYRRO-C3D alone increased biofilm density, whereas treatment with azithromycin had no effect (Fig. 4). Combination treatment with azithromycin did, however, rescind the increase in biofilm density observed when treating with PYRRO-C3D alone (Fig. 4).
FIG 3.

PYRRO-C3D increases NTHi in vitro biofilm susceptibility toward azithromycin treatment. (a and b) NTHi biofilms grown in vitro for 72 h and treated with 50 μM PYRRO-C3D and 4 mg/ml azithromycin (Azithro), both individually and in combination, for 2 h were assessed for viability through CFU enumeration (a) and COMSTAT analysis (b) of live stained bacteria (n = 5). (c and d) Viability of the supernatant population following treatment measured by CFU enumeration (c) and maximum biofilm thickness measured by confocal microscopy (d). Values that were statistically significantly different are indicated by bars and asterisks as follows: *, P ≤ 0.05; **, P < 0.01; ***, P ≤ 0.001. Values were not statistically significantly different (ns) are also indicated.
FIG 4.
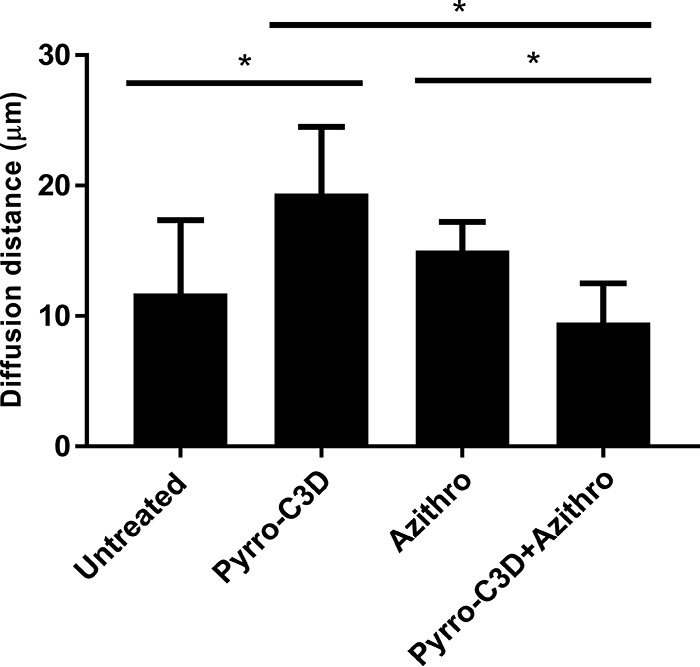
PYRRO-C3D treatment increases NTHi biofilm density. Confocal images of NTHi biofilms treated with 50 μM PYRRO-C3D and 4 mg/ml azithromycin, both individually and in combination, for 2 h were processed using COMSTAT software to calculate the average diffusion distance between live bacteria within biofilms. *, P ≤ 0.05.
Response of NTHi biofilms to PYRRO-C3D is NO mediated.
Having established that PYRRO-C3D potentiates the activity of azithromycin against NTHi biofilms, experiments were next performed to examine whether the effect was NO mediated. Treatment of biofilms with an equivalent concentration (50 μM) of the spontaneous NO donor diethylamine NONOate (DEA/NO) alone had no effect on biofilm viability (P = 1.08; Fig. 5a). In contrast to the PYRRO-C3D–azithromycin combination, no increase in antibiotic efficacy was observed when DEA/NO was coadministered with azithromycin (P = 0.56; Fig. 5a). Treatment with the NO scavenger carboxy-2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (cPTIO) nullified the increase in azithromycin susceptibility observed in the presence of PYRRO-C3D (P = 0.0001), suggesting that the potentiation effect is NO mediated but requires slower, more controlled release of NO than is achievable with DEA/NO (Fig. 5b). Treatment with cephaloram, the parent first-generation cephalosporin from which PYRRO-C3D is derived (but lacking an NO donor), also showed no effect on biofilm viability in the presence or absence of azithromycin, suggesting that the potentiation response with PYRRO-C3D does not arise from β-lactam-mediated antibacterial activity (Fig. 5a). The β-lactamase inhibitor clavulanate abrogated the potentiation response, consistent with NO release from PYRRO-C3D requiring β-lactamases (Fig. 5b). This finding was corroborated by the absence of potentiation observed when treating a non-β-lactamase-producing strain (HI5) with PYRRO-C3D and azithromycin (P = 0.24), while observing a strong effect with an alternative β-lactamase-producing strain (HI6) (P < 0.0001; Fig. 6).
FIG 5.

Response of NTHi biofilms to PYRRO-C3D is NO mediated. (a and b) Viability of 72-h in vitro NTHi biofilms following 2 h treatment with 50 μM DEA/NO, cephaloram, and PYRRO-C3D (a) and 50 μM cPTIO and clavulanate (Clav) (b), both individually and in combination with 4 mg/ml azithromycin for 2 h. Viability was measured by CFU enumeration. *, P ≤ 0.05.
FIG 6.
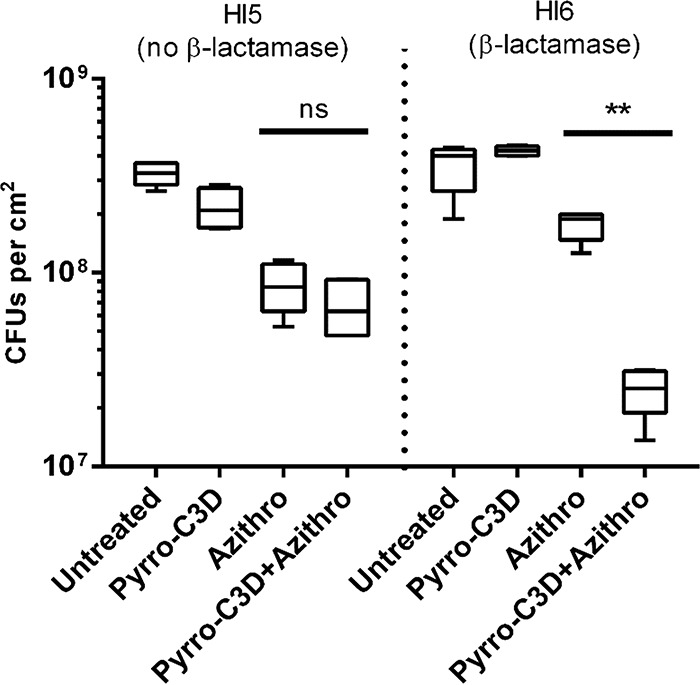
NO release from PYRRO-C3D is dependent on NTHi β-lactamase production. Viability of 72-h in vitro biofilms formed by β-lactamase-producing (HI6) and non-β-lactamase-producing (HI5) NTHi isolates following treatment with 50 μM PYRRO-C3D and 4 mg/ml azithromycin, both individually and in combination for 2 h, as assessed by CFU enumeration (n = 5). **, P ≤ 0.01; ns, not significant.
PYRRO-C3D increases azithromycin susceptibility of NTHi biofilms grown on primary respiratory epithelial cells.
PYRRO-C3D treatment of NTHi biofilms formed on primary respiratory ciliated epithelial cells at an air-liquid interface (ALI) was used to investigate whether the presence of human host cells affected the activity of the compound. Lack of NO release from PYRRO-C3D in the absence of NTHi cells was first confirmed, where NO release was detected only after introduction of β-lactamase (Fig. 7a). Scanning electron microscopy (SEM) was used to confirm NTHi biofilm formation following 72-h coculture before proceeding with compound treatments (Fig. 7c). As observed in the in vitro NTHi-only biofilm model, treatment of the cocultures with PYRRO-C3D alone had no effect on viability (P = 0.41), and treatment with azithromycin alone resulted in a log fold reduction (10-fold reduction or 1-log-unit reduction) (P = 0.0007). When used in combination, azithromycin and PYRRO-C3D produced a significant 2-log-unit reduction in viability relative to controls (P = 0.0026; Fig. 7b).
FIG 7.
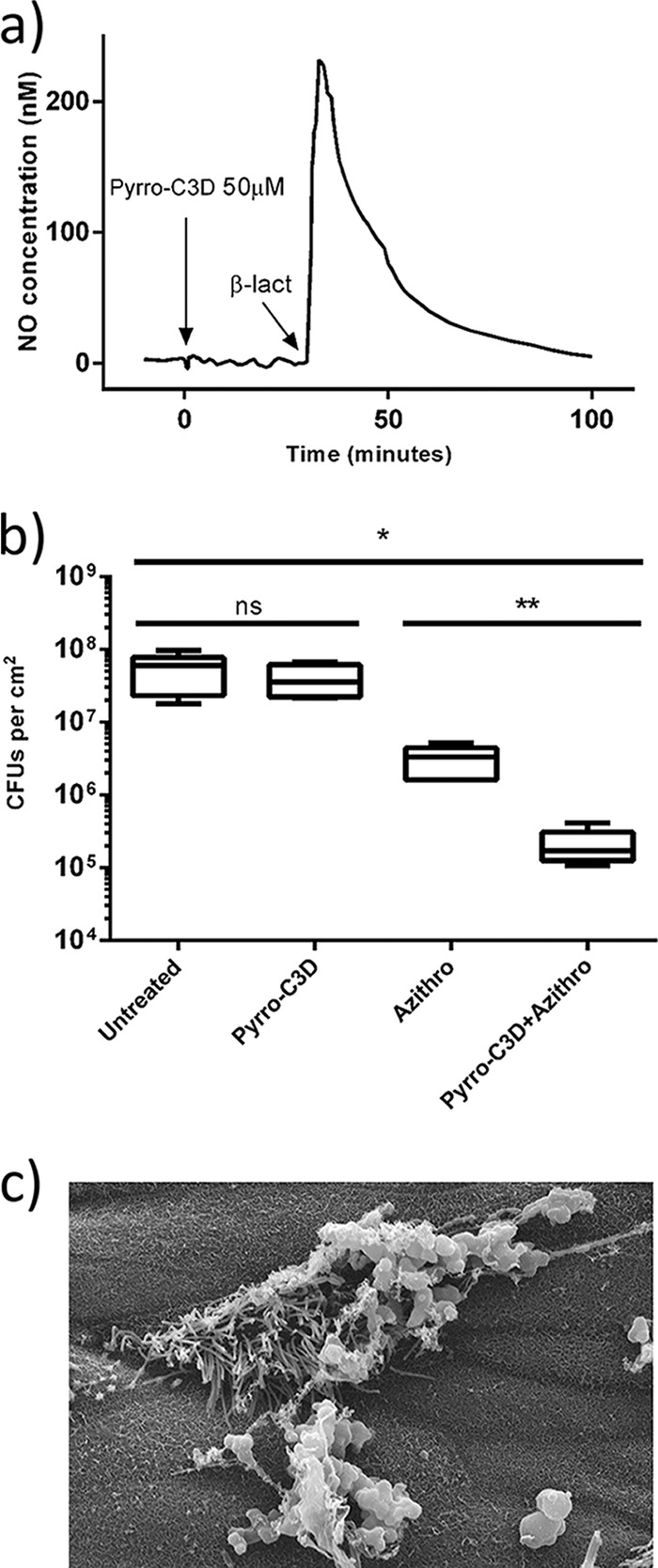
PYRRO-C3D treatment increases azithromycin susceptibility of NTHi biofilms grown on primary respiratory epithelial cells. (a) Measurement of NO release from 50 μM PYRRO-C3D in the presence of primary respiratory epithelial cells isolated from the air-liquid interface (ALI) before and after activation with 10 U of β-lactamase (β-lact) (penicillinase). (b) Viability of 72-h NTHi biofilms grown at the ALI on primary respiratory epithelial cells following 2-h treatment with 50 μM PYRRO-C3D and 4 mg/ml azithromycin, both alone and in combination, as assessed by CFU enumeration (n = 5). *, P ≤ 0.05; **, P ≤ 0.01. (c) SEM image of a 72-h NTHi biofilm formed at an ALI on primary respiratory epithelial cells. Magnification of ×5,000.
Nitric oxide treatment regulates protein expression in NTHi biofilms.
Similar to Streptococcus pneumoniae, NTHi lacks proteins possessing the GGDEF, EAL, and HD-GYP domains, which are important in the turnover of the secondary messenger cyclic di-GMP (c-di-GMP) in response to NO signals (17). Label-free proteomic analyses were therefore performed to probe the mechanism by which NO affects NTHi biofilms. In total, 277 proteins were identified, and 127 were expressed in both untreated biofilms and biofilms treated with 50 μM sodium dinitroprusside (SNP). Of these proteins, 16 showed significantly increased expression (5% false-discovery rate [FDR]) following SNP treatment and were primarily involved in either metabolic or transcriptional/translational processes (Table 1). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of all proteins showing increased expression (>1.5-fold change) identified significant enrichment of ribosome pathways (34 proteins; FDR of 1.2 × 10−28) and glycolysis/gluconeogenesis (9 proteins; FDR of 1.55 × 10−5).
TABLE 1.
Differentially expressed proteins in in vitro NTHi biofilms following SNP (50 μM) treatmenta
| UniProt code or accession no. | Protein nameb | Gene | Ratio of treated/untreated NTHi biofilmsc |
|---|---|---|---|
| A0A0H3PBJ4 | Glucose-specific PTS enzyme IIA component* | crr | 24.15 |
| A0A0H3PK54 | Lipoprotein (d-methionine uptake)* | metQ | 21.52 |
| A0A0H3PBW8 | Phosphoglycerate kinase* | pgk | 21.00 |
| A0A0H3PFB4 | DNA-directed RNA polymerase subunit alpha* | rpoA | 16.33 |
| A0A0H3PJ51 | 2,3-Bisphosphoglycerate-dependent phosphoglycerate mutase* | gpmA | 13.37 |
| A0A0H3PG63 | Elongation factor G* | fusA | 12.96 |
| A0A0H3PMV3 | Pyruvate kinase* | pykA | 11.85 |
| A0A0H3PI75 | Inosine-5′-monophosphate dehydrogenase* | guaB | 11.41 |
| A0A0H3PCZ6 | l-Lactate dehydrogenase* | lldD | 10.50 |
| A0A0H3PF36 | ATP synthase subunit β | atpD | 9.97 |
| A0A0H3PLN7 | Pyridoxal 5′-phosphate synthase subunit* | pdxS | 9.15 |
| A0A0H3PLM6 | Pyruvate dehydrogenase E1 component* | aceE | 8.25 |
| A0A0H3PG47 | Chaperone protein ClpB | clpB | 7.89 |
| A0A0H3PC20 | NAD nucleotidase | nucA | 6.99 |
| A0A0H3PFF9 | Long-chain fatty acid transport protein | ompP1 | 6.09 |
| A0A0H3PF51 | CTP synthetase | pyrG | 5.75 |
The 16 proteins that showed significantly increased expression in in vitro NTHi biofilms following treatment with 50 μM sodium dinitroprusside (SNP) with a 5% false-discovery rate (FDR) are listed.
The proteins that showed significantly expressed expression in in vitro NTHi biofilms following treatment with 50 μM SNP with a 1% FDR are indicated by an asterisk. PTS, phosphotransferase system.
Ratio of CFU in SNP-treated biofilms/untreated biofilms (in vitro NTHi biofilms).
DISCUSSION
NTHi, a common commensal in the upper respiratory tract, is an opportunistic pathogen responsible for localized and chronic lung infections associated with lung diseases. Biofilm formation by NTHi has been identified as playing a key role in both colonization and disease, and it contributes to ineffective antibiotic treatment (28). NO has been shown to signal a dispersal response in several biofilm-forming species, rendering the released bacteria susceptible to antibiotics (19–21). The ubiquitous nature of this molecule in human physiology, however, means that clinical implementation of nonspecific NO treatments with, for example, gaseous NO or spontaneous NO donors, could result in many side effects (22–24). Cephalosporin-3′-diazeniumdiolates like PYRRO-C3D, designed to target NO release specifically to bacterial infection sites, present a promising solution to this problem. Our findings indicate that PYRRO-C3D released low concentrations of NO upon contact with NTHi and that this release is specifically triggered by β-lactamases, as evidenced by the cessation of NO release in the presence of the β-lactamase inhibitor clavulanate. These data, as well as the lack of NO release when PYRRO-C3D was applied to respiratory epithelial cells in the absence of NTHi, demonstrate the specificity of the prodrug activation by bacterial cells, an attribute that would likely reduce the risk of NO-mediated side effects in vivo.
Regulation of the intracellular secondary messenger c-di-GMP plays a pivotal role in controlling both biofilm formation and dispersal, with increased levels promoting formation through increased aggregation, extracellular matrix and adhesin production, and reduced levels signaling a dispersal response (17, 29, 30). c-di-GMP levels are regulated by diguanylate cyclases (containing GGDEF domains) that are responsible for its synthesis and phosphodiesterases (containing HD-GYP and EAL domains) that catalyze degradation (30). Increased activity of these phosphodiesterases has been linked to specific external triggers such as nutrient deprivation, hypoxia, and NO (19, 31–33). A putative NO-sensing domain linked to both GGDEF and EAL domains, termed the NO-induced biofilm dispersal locus A (NbdA) (33), could be responsible for these downstream effects. However, it is unlikely that these mechanisms operate in Haemophilus spp., as genome-wide sequencing of NTHi (Rd KW20), Haemophilus ducreyi, Haemophilus parainfluenzae, and Haemophilus parasuis indicates a lack of proteins possessing the GGDEF, EAL, or HD-GYP domain (18). Our data support this, since low concentrations of NO did not appear to reduce the number of viable cells remaining in biofilms following PYRRO-C3D treatment. However, our proteomic data suggest that an alternative signaling pathway may be involved.
Proteomic analyses comparing untreated and NO-treated NTHi biofilms demonstrated increased expression of 16 proteins involved in metabolic or transcriptional/translational processes. This was confirmed by KEGG pathway analysis of all differentially expressed proteins, which showed overrepresentation of ribosome and glycolysis/gluconeogenesis pathways. A similar response was recently observed in Streptococcus pneumoniae biofilms, where low-dose NO modulated both translation and metabolism (26). This is particularly interesting given that S. pneumoniae also lacks the GGDEF, EAL, and HD-GYP domain-containing proteins associated with the c-di-GMP pathway. It is possible that this is because both species inhabit the same nasopharyngeal niche, which not only provides an environment with limited nutrient availability but also one with low levels of NO produced by epithelial cells (34). Of particular interest was the increased expression of the d-methionine binding lipoprotein MetQ following NO treatment, an amino acid that has previously been shown to play a role in dispersal of P. aeruginosa, S. aureus, and Staphylococcus epidermidis biofilms (35, 36). Interestingly, MetQ is also linked to a number of iron chelation/transporter proteins. It is known that iron can interfere with P. aeruginosa biofilm formation by inhibiting genes associated with biofilm formation, while SapF mediates heme utilization and is involved in both biofilm persistence and coordination (37, 38). NO treatment of E. coli has also been shown to inhibit a global regulator (Fur) that uses iron as a cofactor, affecting a wide range of metabolic processes, such as the stress response and iron metabolism (39), and has been implicated in NTHi virulence (40).
While several studies have shown that NO-mediated dispersal of biofilms reduces antibiotic tolerance (19–21), the regulation of pneumococcal biofilm metabolism was recently shown to provide an alternative mechanism for reducing tolerance (26). As PYRRO-C3D increases the susceptibility of NTHi biofilms to treatment with azithromycin, it is possible that a mechanism similar to that observed in pneumococcus is also responsible for reduction in tolerance observed here. Abrogation of the potentiation effect in the presence of the β-lactamase inhibitor clavulanate and the NO scavenger cPTIO, in addition to the lack of response with cephaloram, confirmed that the response to PYRRO-C3D was indeed NO mediated. It is worth noting that treatment of a strain lacking β-lactamase activity failed to potentiate the activity of azithromycin, suggesting that PYRRO-C3D would likely be effective only against NTHi biofilms capable of β-lactamase production.
Observing a significant improvement in azithromycin efficacy when used alongside PYRRO-C3D but not in the presence of an equivalent concentration of the spontaneous NO donor DEA/NO suggests that a bacterium-targeted NO donor, such as PYRRO-C3D, would be more effective in the treatment of biofilm-associated infections. While the half-life of DEA/NO is around 90 s, the release of NO from PYRRO-C3D continues for up to 120 min, suggesting that slow but sustained release is beneficial. Moreover, it was particularly noteworthy that NTHi biofilm susceptibility to combined PYRRO-C3D and azithromycin treatment was even more pronounced when the biofilms were grown on primary epithelial cells—a more physiologically relevant model of biofilm infections in the respiratory tract.
Conclusions.
In conclusion, this study has shown that the novel NO donor prodrug PYRRO-C3D is effective in specifically targeting NO release to β-lactamase-producing NTHi biofilms and that through modulation of metabolic activity, the compound potentiates the antibacterial activity of azithromycin. This effect is not seen in the absence of β-lactamase production. PYRRO-C3D used in combination with azithromycin thus warrants further investigation as a potential treatment for chronic, biofilm-based NTHi infections.
MATERIALS AND METHODS
Ethics.
Local and national research and development (R&D) and ethical approvals were obtained (Southampton and South West Hampshire Research Ethics 06/Q1704/105 and 07/Q1702/109).
Bacterial strains and growth conditions.
NTHi strain HI4 was isolated from the sputum of a primary ciliary dyskinesia (PCD) patient. Strains HI5 and HI6 were isolated from nasal swabs of healthy children participating in a nasal carriage study. All experiments were performed using strain HI4 unless stated otherwise. HI4 and HI6 were β-lactamase-producing strains, while HI5 lacked β-lactamase activity. Strains were subcultured onto Columbia agar with chocolate horse blood (Columbia blood agar base [CBA]; Oxoid, U.K.) and grown at 37°C and 5% CO2. Colonies were resuspended in brain heart infusion (BHI) broth (Oxoid, U.K.) supplemented with 10 μg/ml hemin and 2 μg/ml NAD.
Planktonic experiments.
Flat-bottomed 96-well culture plates (Fisher Scientific, U.K.) were inoculated with ∼1.0 × 107 mid-exponential NTHi grown in supplemented BHI. PYRRO-C3D (10 mM) (in dimethyl sulfoxide [DMSO]) was diluted in supplemented BHI and added to wells at final concentrations ranging from 1 to 200 μM, with supplemented BHI used as an untreated control. Cultures were incubated at 37°C and 5% CO2 for 24 h. Absorbance (optical density at 595 nm [OD595]) was measured using an EZ Read 400 spectrophotometer (Biochrom) (n = 3).
In vitro biofilm experiments.
Untreated polystyrene six-well tissue culture plates (Corning Incorporated, USA) were inoculated with ∼2.0 × 108 mid-exponential NTHi grown in supplemented BHI. Plates were incubated at 37°C and 5% CO2 for 72 h and media replaced with fresh supplemented BHI daily. Biofilms were then washed twice with Hanks' balanced salt solution (HBSS) to remove unattached cells before being treated with 10 nM to 100 μM PYRRO-C3D, 50 μM (each) carboxy-2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (cPTIO), diethylamine (DEA) or NO (DEA/NO), clavulanate and cephaloram, and 4 mg/ml azithromycin for 2 h at 37°C and 5% CO2. Biofilms were washed twice, resuspended in 1 ml HBSS, vortexed, and then serially diluted before being spot plated onto CBA and incubated at 37°C and 5% CO2. For confocal imaging, biofilms were grown on 35-mm untreated glass bottom CELLview culture dishes (Greiner Bio One, U.K.) and prepared as described above. Following treatment, biofilms were stained with a LIVE/DEAD BacLight bacterial viability kit (Life Technologies, USA) per the manufacturer's instructions and examined using a Leica SP8 laser confocal scanning microscope (LCSM) with an inverted stand and using a 63× oil immersion lens. Sequential scanning was performed using 1-μm sections, and the images were analyzed using COMSTAT 2.0 software (41).
Epithelial cell coculture experiments.
Nasal epithelial cells were obtained from healthy volunteers, cultured through two passages, and then placed on 12-mm transwells (0.4-μm pore size) as previously described (42). Once the cells were confluent, the apical medium was removed, and the cells were fed at the basolateral surface every 48 h. A minimum of 4 weeks after ciliation, transepithelial resistance was measured to confirm an intact epithelial surface (43). NTHi in minimum essential medium (MEM) containing HEPES without glutamine (ThermoFisher, U.K.) was applied to the apical surfaces of the epithelial cells at a multiplicity of infection (MOI) of 100:1. Cocultures were grown for 72 h at 37°C and 5% CO2 with the medium changed every 24 h. Both the apical and basolateral surfaces were washed with Hanks balanced salt solution (HBSS) prior to treatment. Cocultures were treated with compounds and processed for CFU enumeration as described before. Transwell membranes were removed and processed as previously described (44), and the remaining biofilms were imaged using an FEI Quanta 250 scanning electron microscope.
Nitric oxide measurements.
Nitric oxide release from 50 μM PYRRO-C3D in phosphate-buffered saline (PBS) was measured using an ISO-NO probe (World Precision Instruments, USA) per the manufacturer's instructions. PYRRO-C3D was activated through the addition of Bacillus cereus penicillinase (10 U; Sigma, U.K.), and NO release was recorded over 130 min. NO release from PYRRO-C3D in the presence of mid-exponential NTHi cells was measured for 15 min before quenching the reaction with 50 μM clavulanate (β-lactamase inhibitor). For epithelial cell coculture measurements, 750 μl of PBS was added to the apical surface, and the probe was inserted. Baseline NO release was measured for 30 min before activating PYRRO-C3D through the addition of 10 U of B. cereus penicillinase.
Proteomic analysis.
An alternative NO donor (sodium dinitroprusside [SNP]) was used in place of PYRRO-C3D to characterize the response of NTHi biofilms to NO, without being confounded by any activity arising from the β-lactam component of PYRRO-C3D (28). Untreated and 50 μM SNP-treated in vitro NTHi biofilms were resuspended in 1 ml of HBSS and washed twice by centrifugation at 10,000 × g for 5 min at 4°C. The supernatant was discarded, the pellet was resuspended in 1 M triethylammonium bicarbonate containing 4 M guanidine hydrochloride, 10 mg/ml lysozyme, and 100 mM triethylammonium bicarbonate (TEAB) prepared in HBSS, and incubated at 37°C for 30 min. Samples were bead beaten with 0.1-mm zirconium oxide beads at 50 Hz for 5 min and centrifuged at 3,000 × g for 2 min at room temperature, and the supernatants were filter sterilized through 0.22-μm polyethersulfone membranes to remove any remaining intact cells. Samples were precipitated overnight in 100% ethanol at −20°C, centrifuged at 12,000 × g for 5 min at 4°C, and resuspended in 100 mM TEAB with 0.1% Rapigest SF surfactant (Waters, U.K.). Protein solutions were heat treated at 80°C for 10 min and then briefly vortexed. Dithiothreitol (DTT) (in 100 mM TEAB) was added to a final concentration of 2.5 nM and then heat treated at 60°C for 10 min. After the solution was allowed to cool, it was spun at 10,000 × g, and iodoacetamide was added at a final concentration of 7.5 mM before the solution was incubated at room temperature for 30 min in the dark. Protein samples were digested in sequencing-grade modified porcine trypsin solution (Promega, Southampton, United Kingdom) at final concentration of 1.3 μg/ml overnight at 37°C. Trifluoroacetic acid was added to a final concentration of 0.5%, and the mixtures were incubated for 30 min at 37°C before they were centrifuged at 13,000 × g for 10 min. The supernatant was lyophilized and resuspended in 200 mM ammonium formate with 100 fmol of enolase as an internal standard.
(i) Mass spectrometry of NTHi biofilm protein samples.
Peptide separations were performed using a nanoAcquity UPLC system (Waters, U.K.). For the first dimension separation, 1.0 μl of the peptide digest was injected onto a Symmetry C18 trapping cartridge (180 μm by 20 mm) (Waters, U.K.). After 5-min washing of the trap column, peptides were separated on a BEH130 C18 column (75-μm inner diameter [i.d.] by 250 mm; 1.7 μm) (Waters, U.K.) using a linear gradient of 5 to 40% buffer B (buffer A is 0.1% formic acid in H2O, and buffer B is 0.1% formic acid in acetonitrile) over 90 min with a washing step to 85% buffer B at a flow rate of 300 nl/min. All separations were automated and performed online, and the solutions were sprayed directly into the nanospray source of the mass spectrometer. Mass spectrometric (MS) experiments were all performed using a Waters G2-S Synapt HDMS (high-definition mass spectrometry) mass spectrometer operating in elevated-energy mass spectrometry (MSe) mode. Data were acquired from 50 to 2000 m/z with ion mobility enabled using alternate low- and high-collision energy (CE) scans. Low CE was 5 V and elevated, ramped from 20 to 40V. The lock mass [Glu-fibrinopeptide, (M + 2H)+2, m/z = 785.8426] was infused at a concentration of 100 fmol/μl at 300 nl/min, and spectra were acquired every 13 s.
(ii) Identification of proteins from MS spectra.
The raw mass spectra were processed using ProteinLynx Global Server version 3.0 (enabled by Symphony pipeline software; Waters, U.K.) to generate a reduced charge state and deisotoped precursor lists, with associated product ion mass lists. These mass lists were searched against the H. influenzae strain 3655 UniProt protein sequence (downloaded June 2016). A maximum of one missed cleavage was allowed for tryptic digestion, and the allowed variable modification was set to contain oxidation of methionine. Carboxyamidomethylation of cysteine was set at a fixed modification.
Statistical analysis.
Statistical analyses were performed using GraphPad version 6.04 and unpaired t tests. Data reported with a significance of ≤0.05 were considered statistically different. Analysis of identified proteins was corrected for multiple analysis using a false-discovery rate (FDR) of 5%. Proteins that were changed either >1.5-fold or <0.7-fold following NO treatment were analyzed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (45, 46) to identify overrepresented biological pathways.
ACKNOWLEDGMENTS
We thank the Southampton Biomedical Imaging Unit for their support in biofilm imaging and Paul Skipp at the Southampton Centre for Proteomic Research for his support in the proteomic analyses. We also thank Stuart Clarke at the University of Southampton, Department of Clinical & Experimental Sciences, for provision of the nontypeable Haemophilus influenzae strains used in this study.
M.J.K. acknowledges funding from Australian Cystic Fibrosis Research Trust (ACFRT, 2014).
We declare that we have no conflicts of interest.
REFERENCES
- 1.Halbert RJ, Natoli JL, Gano A, Badamgarav E, Buist AS, Mannino DM. 2006. Global burden of COPD: systematic review and meta-analysis. Eur Respir J 28:523–532. doi: 10.1183/09031936.06.00124605. [DOI] [PubMed] [Google Scholar]
- 2.Ramsey KA, Ranganathan S, Park J, Skoric B, Adams AM, Simpson SJ, Robins-Browne RM, Franklin PJ, de Klerk NH, Sly PD, Stick SM, Hall GL, Arest CF. 2014. Early respiratory infection is associated with reduced spirometry in children with cystic fibrosis. Am J Respir Crit Care Med 190:1111–1116. doi: 10.1164/rccm.201407-1277OC. [DOI] [PubMed] [Google Scholar]
- 3.Alanin MC, Nielsen KG, von Buchwald C, Skov M, Aanaes K, Hoiby N, Johansen HK. 2015. A longitudinal study of lung bacterial pathogens in patients with primary ciliary dyskinesia. Clin Microbiol Infect 21:1093.e1–1093.e7. doi: 10.1016/j.cmi.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 4.Rogers GB, Carroll MP, Zain NMM, Bruce KD, Lock K, Walker W, Jones G, Daniels TWV, Lucas JS. 2013. Complexity, temporal stability, and clinical correlates of airway bacterial community composition in primary ciliary dyskinesia. J Clin Microbiol 51:4029–4035. doi: 10.1128/JCM.02164-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall-Stoodley L, Hu FZ, Gieseke A, Nistico L, Nguyen D, Hayes J, Forbes M, Greenberg DP, Dice B, Burrows A, Wackym PA, Stoodley P, Post JC, Ehrlich GD, Kerschner JE.. 2006. Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. JAMA 296:202–211. doi: 10.1001/jama.296.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swords WE. 2012. Nontypeable Haemophilus influenzae biofilms: role in chronic airway infections. Front Cell Infect Microbiol 2:97. doi: 10.3389/fcimb.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy TF, Kirkham C, Sethi S, Lesse AJ. 2005. Expression of a peroxiredoxin-glutaredoxin by Haemophilus influenzae in biofilms and during human respiratory tract infection. FEMS Immunol Med Microbiol 44:81–89. doi: 10.1016/j.femsim.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 8.Obaid NA, Jacobson GA, Tristram S. 2015. Relationship between clinical site of isolation and ability to form biofilms in vitro in nontypeable Haemophilus influenzae. Can J Microbiol 61:243–245. doi: 10.1139/cjm-2014-0763. [DOI] [PubMed] [Google Scholar]
- 9.Fux CA, Costerton JW, Stewart PS, Stoodley P. 2005. Survival strategies of infectious biofilms. Trends Microbiol 13:34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Hall-Stoodley L, Stoodley P. 2009. Evolving concepts in biofilm infections. Cell Microbiol 11:1034–1043. doi: 10.1111/j.1462-5822.2009.01323.x. [DOI] [PubMed] [Google Scholar]
- 11.Hoiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. 2010. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents 35:322–332. doi: 10.1016/j.ijantimicag.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 12.Post D, Held JM, Ketterer MR, Phillips NG, Sahu A, Apicella MA, Gibson BW. 2014. Comparative analyses of proteins from Haemophilus influenzae biofilm and planktonic populations using metabolic labeling and mass spectrometry. BMC Microbiol 14:329. doi: 10.1186/s12866-014-0329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starner TD, Zhang N, Kim G, Apicella MA, McCray PB. 2006. Haemophilus influenzae forms biofilms on airway epithelia: implications in cystic fibrosis. Am J Respir Crit Care Med 174:213–220. doi: 10.1164/rccm.200509-1459OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker WT, Jackson CL, Coles J, Lackie PM, Faust SN, Hall-Stoodley L, Lucas JS. 2014. Ciliated cultures from patients with primary ciliary dyskinesia produce nitric oxide in response to Haemophilus influenzae infection and proinflammatory cytokines. Chest 145:668–669. doi: 10.1378/chest.13-2398. [DOI] [PubMed] [Google Scholar]
- 15.Unal CM, Singh B, Fleury C, Singh K, Chavez de Paz L, Svensater G, Riesbeck K. 2012. QseC controls biofilm formation of non-typeable Haemophilus influenzae in addition to an AI-2-dependent mechanism. Int J Med Microbiol 302:261–269. doi: 10.1016/j.ijmm.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 16.Sriramulu DD, Lünsdorf H, Lam JS, Römling U. 2005. Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J Med Microbiol 54:667–676. doi: 10.1099/jmm.0.45969-0. [DOI] [PubMed] [Google Scholar]
- 17.Hickman JW, Tifrea DF, Harwood CS. 2005. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A 102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou S-H, Galperin MY. 2016. Diversity of cyclic di-GMP-binding proteins and mechanisms. J Bacteriol 198:32–46. doi: 10.1128/JB.00333-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barraud N, Hassett DJ, Hwang S-H, Rice SA, Kjelleberg S, Webb JS. 2006. Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J Bacteriol 188:7344–7353. doi: 10.1128/JB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barraud N, Storey MV, Moore ZP, Webb JS, Rice SA, Kjelleberg S. 2009. Nitric oxide-mediated dispersal in single- and multi-species biofilms of clinically and industrially relevant microorganisms. Microb Biotechnol 2:370–378. doi: 10.1111/j.1751-7915.2009.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlag S, Nerz C, Birkenstock TA, Altenberend F, Götz F. 2007. Inhibition of staphylococcal biofilm formation by nitrite. J Bacteriol 189:7911–7919. doi: 10.1128/JB.00598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, Feelisch M, Fukuto J, Wink DA. 2004. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem 385:1–10. [DOI] [PubMed] [Google Scholar]
- 23.Clutton-Brock J. 1967. Two cases of poisoning by contamination of nitrous oxide with higher oxides of nitrogen during anaesthesia. Br J Anaesth 39:388–392. doi: 10.1093/bja/39.5.388. [DOI] [PubMed] [Google Scholar]
- 24.Jardeleza C, Thierry B, Rao S, Rajiv S, Driling A, Miljkovic D, Paramasavian S, James C, Dong D, Thomas N, Vreugde S, Prestidge CA, Wormald PJ. 2015. An in vivo safety and efficacy demonstration of a topical liposomal nitric oxide donor treatment for Staphylococcus aureus biofilm-associated rhinosinusitis. Transl Res 166:683–692. doi: 10.1016/j.trsl.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 25.Barraud N, Kardak BG, Yepuri NR, Howlin RP, Webb JS, Faust SN, Kjelleberg S, Rice SA, Kelso MJ. 2012. Cephalosporin-3′-diazeniumdiolates: targeted NO-donor prodrugs for dispersing bacterial biofilms. Angew Chem Int Ed Engl 51:9057–9060. doi: 10.1002/anie.201202414. [DOI] [PubMed] [Google Scholar]
- 26.Allan RN, Morgan S, Brito-Mutunayagam S, Skipp P, Feelisch M, Hayes SM, Hellier W, Clarke SC, Stoodley P, Burgess A, Ismail-Koch H, Salib RJ, Webb JS, Faust SN, Hall-Stoodley L. 2016. Low concentrations of nitric oxide modulate Streptococcus pneumoniae biofilm metabolism and antibiotic tolerance. Antimicrob Agents Chemother 60:2456–2466. doi: 10.1128/AAC.02432-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barraud N, Kelso MJ, Rice SA, Kjelleberg S. 2015. Nitric oxide: a mediator of biofilm dispersal with applications in infectious diseases. Curr Pharm Des 21:31–42. [DOI] [PubMed] [Google Scholar]
- 28.Wu S, Li X, Gunawardana M, Maguire K, Guerrero-Given D, Schaudinn C, Wang C, Baum MM, Webster P. 2014. Beta-lactam antibiotics stimulate biofilm formation in non-typeable Haemophilus influenzae by up-regulating carbohydrate metabolism. PLoS One 9:e99204. doi: 10.1371/journal.pone.0099204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee VT, Matewish JM, Kessler JL, Hyodo M, Hayakawa Y, Lory S. 2007. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol 65:1474–1484. doi: 10.1111/j.1365-2958.2007.05879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caly D, Bellini D. 2015. Targeting cyclic di-GMP signalling: a strategy to control biofilm formation? Curr Pharm Des 21:12–24. [DOI] [PubMed] [Google Scholar]
- 31.Rao F, Yang Y, Qi Y, Liang Z-X. 2008. Catalytic mechanism of cyclic di-GMP-specific phosphodiesterase: a study of the EAL domain-containing RocR from Pseudomonas aeruginosa. J Bacteriol 190:3622–3631. doi: 10.1128/JB.00165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.An S, Wu J, Zhang L-H. 2010. Modulation of Pseudomonas aeruginosa biofilm dispersal by a cyclic-di-GMP phosphodiesterase with a putative hypoxia-sensing domain. Appl Environ Microbiol 76:8160–8173. doi: 10.1128/AEM.01233-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Heine S, Entian M, Sauer K, Frankenberg-Dinkel N. 2013. NO-induced biofilm dispersion in Pseudomonas aeruginosa is mediated by an MHYT domain-coupled phosphodiesterase. J Bacteriol 195:3531–3542. doi: 10.1128/JB.01156-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collins SA, Gove K, Walker W, Lucas JSA. 2014. Nasal nitric oxide screening for primary ciliary dyskinesia: systematic review and meta-analysis. Eur Respir J 44:1589–1599. doi: 10.1183/09031936.00088614. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez CJ, Akers KS, Romano DR, Woodbury RL, Hardy SK, Murray CK, Wenke JC. 2014. d-Amino acids enhance the activity of antimicrobials against biofilms of clinical wound isolates of Staphylococcus aureus and Pseudomonas aeruginosa. Antimicrob Agents Chemother 58:4353–4361. doi: 10.1128/AAC.02468-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramon-Perez ML, Diaz-Cedillo F, Ibarra JA, Torales-Cardena A, Rodriquez-Martinez S, Jan-Roblero J, Cancino-Diaz ME, Cancino-Diaz JC. 2014. d-Amino acids inhibit biofilm formation in Staphylococcus epidermidis strains from ocular infections. J Med Microbiol 63:1369–1376. doi: 10.1099/jmm.0.075796-0. [DOI] [PubMed] [Google Scholar]
- 37.Musk DJ, Banko DA, Hergenrother PJ. 2005. Iron salts perturb biofilm formation and disrupt existing biofilms of Pseudomonas aeruginosa. Chem Biol 12:789–796. doi: 10.1016/j.chembiol.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Vogel AR, Szelestey BR, Raffel FK, Sharpe SW, Gearinger RL, Justice SS, Mason KM. 2012. SapF-mediated heme-iron utilization enhances persistence and coordinates biofilm architecture of Haemophilus. Front Cell Infect Microbiol 2:42. doi: 10.3389/fcimb.2012.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D'Autreaux B, Touati D, Bersch B, Latour J-M, Michaud-Soret I. 2002. Direct inhibition by nitric oxide of the transcriptional ferric uptake regulation protein via nitrosylation of the iron. Proc Natl Acad Sci 99:16619–16624. doi: 10.1073/pnas.252591299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrison A, Santana EA, Szelestey BR, Newsom DE, White P, Mason KM. 2013. Ferric uptake regulator and its role in the pathogenesis of nontypeable Haemophilus influenzae. Infect Immun 81:1221–1233. doi: 10.1128/IAI.01227-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, Ersboll BK, Molin S. 2000. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146:2395–2407. doi: 10.1099/00221287-146-10-2395. [DOI] [PubMed] [Google Scholar]
- 42.Hirst RA, Jackson CL, Coles JL, Williams G, Rutman A, Goggin PM, Adam EC, Pahe A, Evans HJ, Lackie PM, O'Callaghan C, Lucas JS. 2014. Culture of primary ciliary dyskinesia epithelial cells at air-liquid interface can alter ciliary phenotype but remains a robust and informative diagnostic aid. PLoS One 9:e89675. doi: 10.1371/journal.pone.0089675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denker BM, Nigam SK. 1998. Molecular structure and assembly of the tight junction. Am J Physiol 274:F1–F9. [DOI] [PubMed] [Google Scholar]
- 44.Allan RN, Skipp P, Jefferies J, Clarke SC, Faust SN, Hall-Stoodley L, Webb J. 2014. Pronounced metabolic changes in adaptation to biofilm growth by Streptococcus pneumoniae. PLoS One 9:e107015. doi: 10.1371/journal.pone.0107015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. 2016. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]