

Abstract
The initial characterization of the product of the vaccinia virus G5R gene, which is conserved in all poxviruses sequenced to date, is described. The G5 protein was detected in the core fraction of purified virions, and transcription and translation of the G5R open reading frame occurred early in infection, independently of DNA replication. Attempts to delete the G5R gene and isolate a replication-competent virus were unsuccessful, suggesting that G5R encodes an essential function. We engineered vaccinia virus mutants with clusters of charged amino acids changed to alanines and determined that several were unable to replicate at 40°C but grew well at 37°C. At the nonpermissive temperature, viral gene expression and DNA replication and processing were unperturbed. However, tyrosine phosphorylation and proteolytic cleavage of the A17 membrane protein and proteolytic cleavage of core proteins were inhibited at 40°C, suggesting an assembly defect. The cytoplasm of cells that had been infected at the nonpermissive temperature contained large granular areas devoid of cellular organelles or virus structures except for occasional short crescent-shaped membranes and electron-dense lacy structures. The temperature-sensitive phenotype of the G5R mutants closely resembled the phenotypes of vaccinia virus mutants carrying conditionally lethal F10R protein kinase and H5R mutations. F10, although required for phosphorylation of A17 and viral membrane formation, was synthesized by the G5R mutants under nonpermissive conditions. An intriguing possibility is that G5 participates in the formation of viral membranes, a poorly understood event in poxvirus assembly.
Vaccinia virus (VAC), the prototype of the poxvirus family of cytoplasmic DNA viruses, encodes proteins with functions in transcription, DNA replication, virion assembly, and host interactions (33). Of the nearly 200 VAC genes, about 50 have orthologues that have been recognized in all poxviruses sequenced to date, including those that infect insects (55). The functions of the putative proteins encoded by many of the highly conserved genes, however, remain unknown. Several reverse genetic methods have been developed to determine whether poxvirus genes are essential and to investigate their roles. One approach is to engineer inducer-dependent mutants (61). Another employs transient dominant selection to make temperature-sensitive or other mutations (18, 21). A third involves viral gene deletion and the propagation of the mutant in a complementing cell line (23).
Open reading frame (ORF) 082, referred to here as G5R, of the WR strain of VAC (accession number AY243312) is one of the approximately 50 ORFs conserved in all poxviruses. Although predicted to encode a protein of 434 amino acids lacking a signal peptide or transmembrane domain, expression of the gene has not been reported previously. Here, we show that the gene is transcribed early in infection and that the protein becomes incorporated into the core of mature virus particles. Following unsuccessful attempts to isolate a replication-competent VAC with a deletion of the G5R ORF, we engineered conditionally lethal temperature-sensitive mutations. At the nonpermissive temperature, the mutants exhibited no apparent defect in early functions such as gene expression and DNA replication. There were, however, protein-processing defects, and no recognizable viral membranes or other viral structures formed.
MATERIALS AND METHODS
Cells.
BS-C-1, BS-C-40, CV-1, and HeLa cells were grown in Earle's modified Eagle's medium (Quality Biological Inc.) supplemented with 10% fetal calf serum. Reduced serum (2.5%) was used for virus infections.
Plasmids.
PCR, carried out with vaccinia virus strain WR DNA as the template with the PCR Supermix High Fidelity (Invitrogen) reaction mixture, was used to copy the G5R ORF with an added V5 epitope tag at its 3′ end between an AscI and a BamHI site. The amplified product was inserted into AscI- and BamHI-cleaved pNEB193 (New England Biolabs). A 511-bp DNA segment, containing adjacent segments of the VAC J2R ORF, which encodes thymidine kinase (TK), and the J1R ORF flanked by SmaI and AscI sites was generated by PCR and inserted into the plasmid at one side of G5R. Next, a 536-bp DNA fragment, consisting of adjacent parts of the J2R and J3R ORFs, was amplified by PCR between a SalI and PstI site and inserted on the other side of G5R. The final plasmid, named pFFG5-V5, contained a V5-tagged copy of the G5R ORF flanked by portions of the VAC TK and neighboring genes. We confirmed the correct sequence of the cloned DNA segment of this plasmid and others constructed for this study.
A 569-bp DNA segment, consisting of a portion of the G4L ORF between a SmaI and a NotI site, was amplified by PCR and inserted into pZippy-NEO/GUS (15), which contains copies of the neomycin resistance (neo) and Escherichia coli β-glucuronidase (gus) genes under the control of VAC promoters. Since there is a small overlap of the G5R 5′ end and the G4L promoter, the amplified segment retained 50 bp of G5R. In addition, a 615-bp portion of the G5.5R gene between SalI and PstI restriction sites was amplified by PCR and inserted into pZippy-NEO/GUS. Because of a small overlap of the G5R 3′ end and the G5.5R promoter, another 60 bp of G5R were retained. The final plasmid, pFFG5RΔNG, contained copies of the neo and gus genes flanked by portions of the genes neighboring G5R.
A copy of the gene encoding enhanced green fluorescent protein (GFP), controlled by the VAC P11 late promoter, plus a copy of the E. coli xanthine-guanine phosphoribosyltransferase gene (gpt), controlled by the VAC P7.5 early and late promoter were inserted head to head in the pNEB193 plasmid. The latter DNA was then flanked by left and right segments of the TK and neighboring genes, as described above to form pFFNEBgptGFPTKr1. Charge-to-alanine cluster mutations in the G5R gene were made with the QuickChange site-directed mutagenesis kit (Stratagene). PCR was then used to place AscI and BamHI restriction sites at the 5′ and 3′ ends of the mutated G5R gene, respectively, and the purified DNA, which included the G5R promoter, was then inserted between the corresponding restriction sites in the GFP and gpt genes of pFFNEBgptGFPTKr1. The resulting plasmids, generically called pFFG5R-mut (in which the suffix mut is replaced by the number of the first amino acid mutated), contained a cassette consisting of a mutated G5R gene between the GFP and gpt genes, which was flanked by portions of the VAC TK and neighboring genes.
Recombinant viruses.
Recombinant viruses were constructed and propagated as previously described (8, 17). CV-1 cells infected with the WR strain of VAC were transfected with pFFG5-V5 with Lipofectamine 2000 and Optimem reduced medium (Invitrogen) for 5 h. The cells were washed and incubated for an additional 48 h and then harvested. Serial dilutions of the lysates were used to infect A543 TK− cells in the presence of 25 μg of bromodeoxyuridine per ml. TK− recombinant viruses were selected and analyzed by PCR, and virus stocks were prepared. The TK− recombinant virus contained the G5R-V5 ORF under control of its natural promoter in addition to the original G5R gene. This virus was used to infect CV-1 cells that were later transfected with pFFG5RΔNG in order to replace the original G5R gene with the neo and gus genes by homologous recombination. Lysates were then used to infect BS-C-1 monolayers in the presence of 2 mg of geneticin (Gibco/Invitrogen) per ml and 100 μM 5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid (Clontech). The recombinant virus vG5R-V5, containing a single G5R ORF with a V5 tag under control of its original promoter, was selected, analyzed by PCR, and propagated.
CV-1 cells were infected with vG5R-V5 and transfected with individual pFFG5R-mut plasmids containing specific clusters of charged amino acids mutated to alanines. Recombinant viruses were generated as described above except that the incubation temperature was 31°C. Lysates were used to infect BS-C-1 cells at 31°C in the presence of mycophenolic acid, and plaques that were resistant to the drug and exhibited green fluorescence were picked and amplified.
Immunoblotting.
Cells were harvested, centrifuged, and suspended in gel loading buffer with added β-mercaptoethanol. Proteins were separated by electrophoresis on a sodium dodecyl sulfate (SDS)-4 to 20% polyacrylamide gel with Tris-glycine-SDS (Invitrogen) buffer and transferred to nitrocellulose membranes (Immobilon-P; Millipore). The membranes were blocked in 50 mM Tris-HCl (pH 7.5)-150 mM NaCl-0.05% Tween 20-2.5% nonfat dry milk as described (7). Mouse anti-V5 monoclonal antibody conjugated with horseradish peroxidase (Invitrogen) and Supersignal West Dura chemiluminescence substrates (Pierce) were used to detect V5-tagged proteins. Membranes were exposed to X-Omat film (Kodak).
Transcription analysis.
Cells were harvested, centrifuged, and resuspended in 0.2 ml of phosphate-buffered saline (PBS). Total RNA was extracted with the RNeasy RNA extraction kit (Qiagen). A 1-μg sample of total RNA was resolved in a denaturing 1% 3-(N-morpholino)propanesulfonic acid (MOPS)-agarose gel and transferred to a nylon membrane with the NorthernMax kit (Ambion). A PCR copy of the G5R ORF was labeled with [α-32P]dCTP by random priming with the Random Primers DNA labeling system (Invitrogen). Unincorporated nucleotides were removed by centrifugation through a G-50 column (Pharmacia). Hybridization was carried out at 68°C for 1 h in 10 ml of QuickHyb solution (Stratagene) in rolling tubes. Membranes were exposed to X-Omat film.
Measurement of VAC DNA accumulation and processing.
DNA was extracted from cells, immobilized on nylon membranes, and hybridized to PCR copies of the VAC I7L, H6R, and G7L ORFs that had been 32P labeled with the Random Primers DNA-labeling system (Invitrogen) essentially as described (19). DNA accumulation was determined with a phosphoimager.
To determine DNA size, infected cells were immobilized in agarose and incubated with proteinase K. The DNA was subjected to electrophoresis in a CHEF-DRII apparatus (Bio-Rad) and transferred to a nylon membrane essentially as described (19). DNA was cross-linked to the membrane and hybridized with a mixture of 32P-labeled probes as above.
DNA concatemer junctions were analyzed by restriction enzyme digestion and Southern blotting. Cells were harvested, centrifuged, washed, and suspended in 0.2 ml of PBS. DNA was extracted with the Qiamp blood kit (Qiagen). The DNA concentration of each sample was calculated by determining the absorbance at 260 nm, and 1 μg of total DNA was digested with BstEII. The digested DNA was resolved by electrophoresis in a 0.7% agarose gel and transferred to a positively charged nylon membrane. The membrane was hybridized to a 32P-labeled probe containing the first set of short direct repeats near the terminus of the VAC genome (31) and exposed to X-ray film.
Fluorescence microscopy.
Infected cells were washed once with PBS, fixed with 4% paraformaldehyde in PBS, and permeabilized with 0.05% saponin in PBS. Fixed cells were stained with anti-H3 antibody (7) followed by Texas Red-conjugated goat anti-rabbit immunoglobulin secondary antibody (Jackson ImmunoResearch Laboratories). Stained coverslips were mounted in Mowoil containing 1 μg of 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) (Molecular Probes) per ml and allowed to dry overnight. Images were collected on a Leica TCS-NT/SP inverted confocal microscope system (Coherent Radiation, Palo Alto, Calif.), and figures were prepared with the Adobe Photoshop 6.0 software.
RESULTS
G5R ORF is expressed early during VAC infection.
An A- and T-rich region, conforming to the consensus nucleotide sequence of an early poxvirus promoter (10), occurs upstream of the G5R initiation codon. A recombinant VAC with a V5 epitope-tagged copy of the G5R ORF was constructed in order to determine the time of expression experimentally. We did not modify the G5R gene in situ because of an overlap with the neighboring G4L and G5.5R genes and therefore constructed a recombinant virus with a C-terminal epitope-tagged copy of the G5R gene under control of its putative early promoter in the VAC TK locus. The selectable TK locus has been used repeatedly for making recombinant viruses, and gene insertions have little effect on virus replication in tissue culture cells. In the second step, the original G5R gene, except for the overlapping regions of G4L and G5.5 R, was replaced by the neo and gus genes for selection and screening purposes (Fig. 1A). The final construct, vG5R-V5, formed plaques similar to those of the parental VAC WR strain on BS-C-1 cells (Fig. 1B), indicating that neither the translocation of the gene nor the short C-terminal amino acid extension significantly affected virus replication.
FIG. 1.

Representation of portions of the VAC WR and vG5R-V5 genomes. (A) In the VAC WR genome, ORFs G4L, G5R, G5.5R, J1R, J2R (TK), and J3R are represented by boxes; the G4L, G5R, and G5.5 promoters (G4pr, G5pr, and G5.5pr, respectively) are indicated by arrowheads. In the VG5R-V5 genome, the G5R ORF was replaced by the neo gene (Neo) and the gus gene (Gus); a copy of the G5R gene containing a V5 epitope tag and under the control of its original promoter was inserted into the J2R (TK) gene site. (B) Plaque phenotypes of VAC WR and vG5R-V5. BS-C-1 cells were infected, incubated for 48 h, and then fixed and stained with crystal violet-ethanol solution.
Early transcription of the G5R gene under its natural promoter was demonstrated by Northern blotting. A transcript which migrated just above the 18S rRNA was detected at 2, 4, and 6 h after infection (Fig. 2A). Starting at 4 h and increasing at 6 and 12 h, the G5R probe reacted with heterogeneous species representing expected late readthrough transcripts. In the presence of AraC, which prevents the transcription of late genes, only the discrete band was detected (Fig. 2A).
FIG. 2.
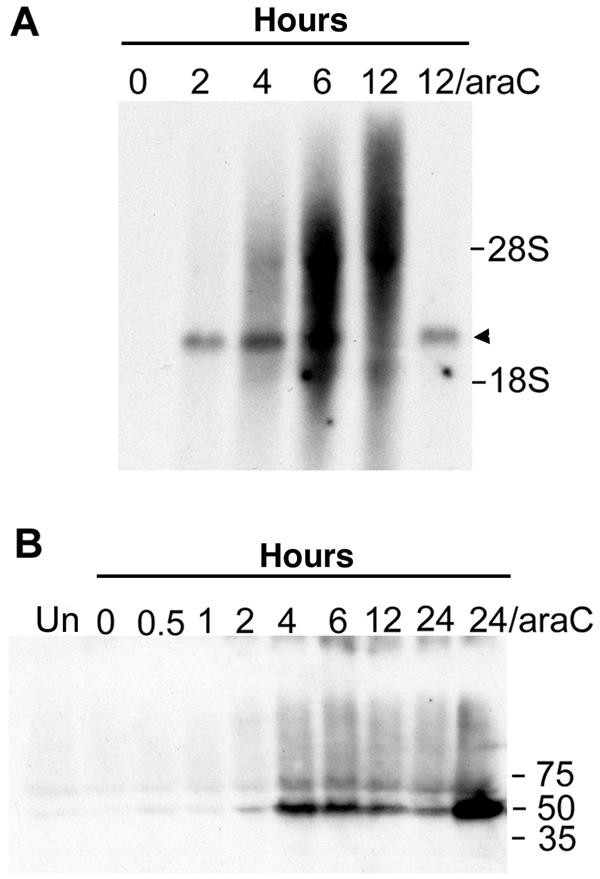
Temporal expression of the G5R gene. (A) Northern blot analysis. BS-C-1 cells were infected with vG5R-V5 in the absence or presence of 200 μM AraC and harvested at the indicated hours. Total RNA was extracted, resolved in a denaturing agarose gel, transferred to a nylon membrane, and hybridized to a 32P-labeled G5R gene probe. G5R transcripts are indicated by the arrowhead. The positions of 18S and 28S rRNAs are shown. (B) Western blot analysis. BS-C-1 cells that were uninfected (UN) or infected with vG5R-V5 in the presence or absence of 200 μM AraC were harvested at the indicated hours, and the lysates were analyzed by SDS-PAGE and Western blotting with a monoclonal antibody to the V5 epitope. The positions and masses (in kilodaltons) of marker proteins are indicated at the right.
Further evidence for early gene expression was obtained by protein analysis with SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting with monoclonal antibody to the V5 epitope. A protein with the expected mass of 50 kDa was detected at 2 h of infection, peaked at 4 h, and then appeared to decline (Fig. 2B). Larger amounts of G5 were detected in the presence of AraC than in its absence (Fig. 2B), again consistent with early gene expression.
Association of G5 with virus cores.
Virions comprised mainly viral late proteins, though there have been reports of early proteins associated with purified particles (2, 14). Intracellular mature virions were purified from cells infected with vG5R-V5 by sedimentation through a sucrose cushion followed by two successive sucrose gradient centrifugations. The association of G5 protein with purified virions was demonstrated by SDS-PAGE followed by immunoblotting with monoclonal antibody to V5. Detergent treatment was used to localize G5 within the particle. Purified virions were treated with solutions containing Tris-HCl alone or with NP-40 detergent or with NP-40 detergent and dithiothreitol reducing agent. Soluble proteins derived from membranes were separated from insoluble core-associated proteins by centrifugation, and both fractions were analyzed. The 50-kDa protein was detected exclusively in the pellet fraction regardless of the treatment (Fig. 3), indicating association with the virus core, consistent with the absence of predicted transmembrane regions.
FIG. 3.
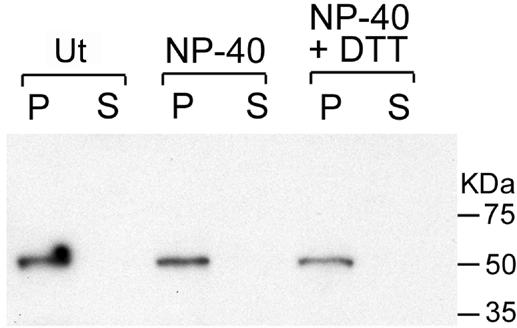
Association of G5 with cores of purified virions. Virions purified from cells infected with vG5R-V5 by sedimentation through a sucrose cushion and two sucrose gradients (16) were incubated in Tris buffer alone (Ut) with 0.5% NP-40, or with 0.5% NP-40 plus 50 mM dithiothreitol (DTT). After centrifugation, the supernatant (S) and pellet (P) fractions were analyzed by Western blotting with a monoclonal antibody to V5. The masses (in kilodaltons) and positions of protein markers are indicated at the right.
Clustered charge-to-alanine scanning mutagenesis.
Although conservation of the G5R ORF in all poxviruses suggested that it is essential for replication, we attempted to delete the gene from wild-type VAC by homologous recombination. The same neo and gus selection and screening system successfully employed as the second step in making vG5R-V5 was used, except that this time a copy of G5R-V5 was not present in the TK locus. Under these conditions, we failed to isolate even small plaques that expressed Gus.
With evidence that G5R is essential, we performed alanine-scanning mutagenesis (58) in order to make conditionally lethal mutations. In this procedure, charged amino acids in a charge cluster are converted to alanine, thereby weakening peptide interactions and resulting in proteins that are nonfunctional at elevated temperature. The G5R ORF has 11 charge clusters, defined as a group of at least three charged residues separated by no more than one neutral residue. Of those clusters, seven were chosen for mutagenesis (Fig. 4A). Although alanine-scanning mutagenesis has been applied to VAC successfully (12, 21, 25, 37), the procedure is laborious and involves extensive screening of plaques to distinguish mutants from wild-type virus. We devised an alternative procedure that requires an additional initial step but then saves considerable time by streamlining screening.
FIG. 4.

Alanine-scanning mutagenesis. (A) The translated sequence of the G5R ORF is shown, with clusters of charged amino acids in light gray. The amino acid number of the first charged amino acid of each underlined cluster that was mutated is indicated. (B) Representation of portions of the genome of vG5R mutants (vG5R-Mut). Labeling is similar to that in Fig. 1A. The mutated G5R gene is flanked by the GFP and gpt genes. A hypothetical charge-to-alanine mutation is represented by the bar labeled mut within G5R.
Because the neighboring G4L and G5.5R genes overlap the two ends of the G5R gene, vG5R-V5 (Fig. 1A) was used as the parental virus. A plasmid containing the GFP and gpt genes controlled by VAC promoters and separated by AscI and BamHI sites between the left and right segments of the TK gene was designed. A copy of the G5R ORF containing charged amino acid codons of a cluster mutated to alanines was then inserted between the AscI and BamHI sites of the above plasmid. The resulting plasmid was transfected into cells infected with vG5R-V5, and virus from plaques that formed in the presence of mycophenolic acid and expressed GFP at 31°C were isolated and analyzed. DNA sequencing demonstrated that the frequency of picking desired recombinants was 100%. We named the recombinant viruses, depicted schematically in Fig. 4B, according to the first mutated charged amino acid in the cluster as follows: vG5R79, vG5R85, vG5R96, vG5R118, vG5R167, vG5R325, and vG5R350. An additional mutant virus, vG5R167/325, with two charge clusters mutated, and one with one wild-type G5R, vG5Rwt, were also made.
Plaque phenotypes of G5R mutants.
The size of wild-type VAC strain WR plaques in BS-C-40 cells varied with temperature in the following manner: 37°C > 40°C > 31°C (Fig. 5). A similar relationship between temperature and plaque size was found for vG5R-V5, the parental TK− strain WR virus used for mutant isolation (not shown). The alanine substitution mutants produced plaques slightly smaller in size than those of WR at 31 and 37°C but, with the exception of vG5R96 and to a lesser extent vGFR325, exhibited severe temperature sensitivity at 40°C (Fig. 5). In contrast, vG5Rwt, containing the authentic G5R ORF, made distinctly larger plaques at 40°C than any of the mutants (Fig. 5). We chose the stringent mutants vG5R167 and vG5R350 as well as the double mutant vG5R167/325 for further analysis.
FIG. 5.

Plaque formation at 31, 37, and 40°C. Confluent BS-C-40 cells were infected with VAC WR or G5R mutants generated through clustered charge-to-alanine mutagenesis. Cells were incubated for 48 h, fixed, and stained with crystal violet. The mutants are named according to the first charged amino acid of the mutated cluster, as shown in Fig. 4A. vG5Rwt contains an unmutated G5 ORF.
Yields of G5R mutants under one-step growth conditions.
Failure to form plaques can be due to reduced virus replication or cell-to-cell spread. These two phenotypes can be distinguished by one-step virus growth experiments. VAC WR reproduced nearly as well at 40°C as at 31 or 37°C (Fig. 6). In contrast, growth of the three mutant viruses was severely inhibited at 40°C but not at 31 or 37°C (Fig. 6). Because the mutants were able to grow well at 37°C, this temperature was used as the permissive temperature in subsequent experiments.
FIG. 6.

One-step virus growth. BS-C-40 monolayers in six-well plates were infected with 10 PFU of VAC WR, vG5R167, vG5R350, or vG5R167/325 per cell at 31, 37, or 40°C. After 1 h, the inocula were removed, the cells were washed twice with warm PBS, and fresh medium at the appropriate temperature was added. At various times after infection, cells were harvested, washed, lysed by freezing and thawing, and sonicated. Virus yields were then determined by plaque assay in BS-C-1 cells at 37°C.
Viral DNA synthesis and processing under nonpermissive conditions.
The possibility that the G5 protein is required for viral DNA replication was investigated. Cells were infected with VAC WR, vG5R167, vG5R350, or vG5R167/325 at 37 or 40°C, and the accumulation of viral DNA was determined. In cells infected with VAC WR, DNA accumulation was similar at 37 and 40°C for the first 6 h (Fig. 7). At later times, viral DNA accumulation was reduced at the high temperature. A similar pattern was found for the three temperature-sensitive mutants analyzed (Fig. 7), indicating that they did not have a specific block in viral DNA synthesis.
FIG. 7.

Viral DNA synthesis. BS-C-40 cells were infected at a multiplicity of 10 PFU per cell with VAC WR, vG5R167, vG5R167/325, or vG5R350 and incubated at 37 or 40°C. At the indicated times, total DNA was extracted, blotted onto a membrane, and hybridized to a 32P-labeled VAC DNA probe. Radioactivity was measured with a phosphorimager.
VAC DNA replicates in the form of large concatemers, which are rapidly resolved into unit-length genomes (3, 36). Mutants that have blocks in viral late protein synthesis (11, 32) or repress expression of the virus-encoded Holiday junction endonuclease (19), however, accumulate concatemers. Concatemer formation and resolution can be assessed by pulsed-field gel electrophoresis of DNA from infected cells. Alternatively, resolution of DNA concatemer junctions can be analyzed by restriction enzyme digestion. Each concatemer junction consists of an imperfect palindrome that is resolved into two hairpin termini. When viral DNA is digested with the restriction enzyme BstEII, fragments of 2.6 and 1.3 kbp, corresponding to the concatemer junction and hairpin terminus, respectively, are formed and can be detected with a labeled DNA probe corresponding to sequences near the termini of the genome (3).
We used the two techniques to investigate viral DNA processing at the permissive and nonpermissive temperatures. In cells infected with VAC WR, the resolved hairpin termini accumulated at 37 and 40°C, whereas the junction fragment remained at a low level throughout (Fig. 8A). Similar results were observed for vG5R350 (Fig. 8A). In keeping with these results, mostly monomeric genomes were detected upon pulsed-field gel electrophoresis of DNA isolated from cells infected with either VAC WR or vG5R350 at 37 and 40°C (Fig. 8B). Similar results were also obtained with vG5R167 and vG5R167/325 (not shown). Taken together, our results suggested that the temperature-sensitive phenotype of the mutants did not result from a defect in DNA synthesis or processing.
FIG. 8.

Processing of viral DNA into unit-length genomes. BS-C-40 cells were infected with 10 PFU of WR or vG5R350 per cell and incubated at 37 or 40°C for 2, 4, 6, 12, or 24 h. (A) Concatemer junction analysis. Cells were harvested, and total DNA was isolated, digested with BstEII, resolved in agarose gels, transferred to membranes, and hybridized to a 32P-labeled probe consisting of the linearized hairpin terminus of the VAC genome. Processed telomere fragments of 1.3 kbp (T) and concatemer junction fragments of 2.6 kbp (J) are indicated. (B) Pulsed-field analysis. Viral DNA was analyzed by pulsed-field gel electrophoresis, transferred to a membrane, and hybridized to a 32P-labeled VAC DNA probe. Gel wells (W) and the monomeric (M) and dimeric (D) forms of the genome are indicated.
Synthesis of viral proteins under nonpermissive conditions.
We examined viral protein synthesis under permissive and nonpermissive conditions. Because host cell protein synthesis is repressed during the first 6 h of infection, viral proteins can be selectively labeled with radioactive amino acids. Accordingly, cells were infected with VAC WR, vG5R167, vG5R167/325, or vG5R350 at 37 or 40°C and at various times were metabolically labeled for 30 min with a mixture of [35S]methionine and [35S]cysteine. The cell extracts were analyzed by SDS-PAGE followed by autoradiography. Data for VAC WR and vG5R350 are shown in Fig. 9. The shutoff of host protein synthesis and the onset of viral protein synthesis were similar under all conditions. At the higher temperature, however, there was a slightly faster progression, so that the 12-h patterns at 40°C looked similar to the 24-h patterns at 37°C for both WR and vG5R350. In addition, labeling declined from 12 to 24 h at 40°C for both viruses. The results for vG5R167 and vG5R167/325 were similar to those for vG5R350 (not shown).
FIG. 9.

Metabolic labeling of infected cells. BS-C-40 cells were infected with 10 PFU per cell of VAC WR or vG5R350 at 37 or 40°C and incubated for 2, 6, 12, or 24 h. At each time point, monolayers were washed with methionine-, cysteine-, and serum-deficient medium for 15 min. Cells were then pulse labeled for 30 min with 100 μCi of [35S]methionine and [35S]cysteine per ml, harvested, and analyzed by SDS-PAGE followed by autoradiography. The masses and positions of protein markers are indicated at the center of figure.
Analysis of virus assembly under nonpermissive conditions.
We used confocal and electron microscopy to examine virus assembly in cells infected with mutant viruses. Cells infected with VAC WR or vG5R350 at 37 or 40°C were fixed and stained with DAPI to visualize DNA. Images collected with a confocal microscope showed stained nuclei as well as cytoplasmic DNA in viral factories, recognized as discrete juxtanuclear sites, in both WR- and vG5R350-infected cells at either the permissive or nonpermissive temperature (Fig. 10). The cells were also stained with antibody to the VAC H3 protein, which has been shown to localize primarily in the viral factories (7). At each temperature, the H3 protein had a punctate appearance within the factory areas (Fig. 10). Similar results were obtained when cells were infected with vG5R167 or vG5R167/325 (data not shown). These observations indicated that viral factories containing DNA and viral proteins formed at the nonpermissive temperature.
FIG. 10.

Localization of DNA and viral protein in viral factories. BS-C-40 cells were infected with 5 PFU per cell of VAC WR or vG5R350 and incubated at 37 or 40°C for 12 h. Cells were then fixed, permeabilized, and labeled with the DNA-specific dye DAPI and anti-H3 antibody (α-H3) followed by Texas Red-conjugated goat anti-rabbit immunoglobulin antibody. Arrowheads indicate representative cytoplasmic DNA factories adjacent to the larger nuclei. In some cases the factories overlap the nuclei and are not clearly resolved from them.
Since the viral factory areas appeared normal, we examined cells infected with wild-type and temperature-sensitive mutant viruses at 37 and 40°C by transmission electron microscopy. At 37°C, virus morphogenesis was similar in both WR- and mutant virus-infected cells. Crescents and immature virions in factory areas of cells infected with vG5R350 at 37°C are shown in Fig. 11A. Other fields contained intracellular mature virions as well as enveloped forms (not shown). However, in cells infected with vG5R350 at 40°C, there were large granular regions devoid of cellular organelles and viral structures, except for an occasional small crescent (Fig. 11B and C). A large lacy structure was frequently present near the periphery of the granular regions (Fig. 11C). Cells infected with vG5R167 or vG5R167/325 at 40°C were indistinguishable from those infected with vG5R350 (not shown). In contrast, virus formation was normal in cells infected with VAC WR at 40°C (Fig. 11D). The microscopic appearance of cells infected with the G5R temperature-sensitive mutants at the nonpermissive temperature is unusual and was only described previously for conditionally lethal mutations that mapped to the F10L (47, 53) and H5R (12) ORFs. Thus, an early and virtually complete block in morphogenesis occurred in cells infected with G5R mutants at the nonpermissive temperature.
FIG. 11.

Electron microscopy of infected cells. BS-C-40 cells were infected with 5 PFU per cell of vG5R350 at 37°C (A) or 40°C (B and C) or with VAC WR at 40°C (D). At 12 h after infection, the cells were fixed and prepared for transmission electron microscopy (8). Electron micrographs are shown, with the scale indicated by the bars. Abbreviations; N, nucleus, M, mitochondrion; c, viral crescent; IV, immature virions; n, nucleoid within an immature virion; CEV, cell-associated enveloped virion; V, viroplasm. The arrow in panel C points to the lacy structure at the edge of the granular region.
Protein-processing defects under nonpermissive conditions.
Several of the major proteins of the VAC core are cleaved from precursors during a process that is coupled to virion morphogenesis (26). Thus, the drug rifampin and numerous mutations that block morphogenesis inhibit cleavage of core proteins. Based on the electron microscopy results described above, we anticipated that there would be defects in protein processing at the nonpermissive temperature. Although cleavage of the core protein precursor p4b to 4b occurred normally in cells infected with VAC WR at 37 or 40°C, this step was greatly inhibited at the higher temperature in cells infected with vG5R350 (Fig. 12A) and the other temperature-sensitive mutants (data not shown). We did not carry out pulse-chase experiments because of the difficulty in strictly maintaining the 40°C temperature during the required manipulations.
FIG. 12.

Cleavage and phosphorylation of viral proteins. (A) Cells were infected with 10 PFU per cell of WR or vG5R350 at 37 or 40°C for 8, 12, and 24 h. At each time point, cells were harvested, and lysates were prepared for SDS-PAGE and Western blotting with antiserum to the VAC p4b/4b protein. Arrowheads indicate the positions of p4b and 4b. The masses (in kilodaltons) and positions of protein markers are indicated at the center of figure. (B) Cells were infected with 10 PFU per cell of WR, vG5R167 (lane 1), vG5R167/325 (lane 2), or vG5R350 (lane 3) at 37 or 40°C. After 16 h, the cells were harvested,and lysates were analyzed by SDS-PAGE and Western blotting with antibody to A17. The positions and masses (in kilodaltons) of marker proteins are indicated at the right. The membrane used for panel B was stripped and reprobed with an antiphosphotyrosine antibody that recognizes phosphorylated A17 (C) and with F10 antibody (D).
Although cleavages of the A17 membrane protein and core proteins are mediated by the same protease (1), they occur at different stages of morphogenesis. Cleavage of A17 is inhibited by conditionally lethal mutations that map to the F10L ORF (4, 47) and prevent viral membrane formation but not by rifampin, which interferes with the maturation of viral membranes into crescents. We found that cleavage of A17 occurred in cells infected with VAC WR, vG5R350, vG5R167, or vG5R167/325 at 37°C (Fig. 12B). At 40°C, however, A17 was only cleaved in cells infected with VAC WR (Fig. 12B). Similarly, tyrosine phosphorylation of A17 was also greatly inhibited in cells infected with any of the temperature-sensitive mutants under nonpermissive conditions (Fig. 12C). Because phosphorylation of A17 is dependent on the F10 kinase (4, 13, 47), the blots were stripped and reprobed with antibody to F10. Surprisingly, the F10 bands were even more intense in samples from cells infected with the temperature-sensitive mutant at 40°C than at 37°C or from cells infected with VAC WR at either temperature (Fig. 12D), suggesting enhanced stability under nonpermissive conditions. Previous studies indicated that the stability of F10 was decreased under other conditions (48), indicating that the activity of this protein may be partly regulated by turnover.
DISCUSSION
Although orthologues of the VAC G5R ORF are present in all poxviruses, no information was available about their expression or role. In the first part of this study, we showed that G5 is expressed early in VAC infection and is associated with the cores of purified virions, which are assembled at late times. Although virions contain mainly the products of late genes that require viral DNA replication for their expression, G5 is not the only early protein that has been reported to be associated with virions. The B1 (2, 29, 52) and E8 (14) proteins as well as enzymes involved in transcription (34) are expressed early and are present in cores. B1 is a serine/threonine protein kinase that is required for DNA replication (38), though it may have additional functions. E8 has been proposed to have a function related to the enclosure of replication sites by the endoplasmic reticulum (51).
Reverse genetics was used to gain insight into the role of G5. Our attempt to delete the G5R ORF was unsuccessful, suggesting that the gene is essential and encouraging us to engineer conditionally lethal mutations. Because stringent inducer-dependent mutations that regulate early genes have not been described, we chose to make temperature-sensitive mutants by alanine-scanning mutagenesis (58). Although this approach has been applied successfully to VAC (12, 21, 25, 37), the transient dominant selection procedure (18) can be tedious when large numbers of mutants need to be screened. Therefore, we devised a more efficient method in which the essential step was to flank the mutated ORF with the gpt and GFP genes so that recombinant viruses could be selected with an antibiotic and the plaques could be screened under a fluorescence microscope. Each virus clone isolated in this way encoded a mutated G5R allele. With the previous method, numerous clones had to be screened by PCR, and only a small number of them had mutations.
We mutated 7 of the 11 charge clusters in the G5R ORF and tested the recombinant viruses for temperature sensitivity at 31, 37, and 40°C. Several of the mutants exhibited stringent temperature sensitivity at 40°C but still formed plaques of nearly normal size at 37°C. We chose 37 and 40°C as the permissive and nonpermissive temperatures, respectively, and great care was taken to avoid lowering the latter during experiments. The 37°C permissive temperature was preferred over 31°C because the virus cycle was considerably delayed and the virus yield was decreased at the lower temperature.
The inability to isolate a deletion mutant and the temperature sensitivity of the alanine-scanning mutants indicated that the G5R gene product has an essential role in virus reproduction. Given the early expression of G5R, we anticipated that this protein would affect gene expression or DNA replication. However, that was not the case, as the SDS-PAGE profiles of metabolically labeled viral proteins at the nonpermissive temperature were similar to that of the wild-type virus. Although viral DNA replication declined earlier at 40°C than at 37°C, this was true for the wild-type virus as well as the mutants. In addition, we found that resolution of genome concatemers occurred under nonpermissive conditions. Surprisingly, the only defect discovered was related to virus assembly, which is a late step in the virus life cycle.
During VAC reproduction, viral DNA and proteins accumulate in distinct areas of the cytoplasm called virus factories. The first discernible virus structures are crescent membranes with granular material in their concavities (9). Neither the structure nor the origin of the membrane forming the crescent is well understood (20, 22, 24, 39, 45). Association of the membranes with the granular material is dependent on several viral proteins that form a complex (46, 48, 50). The crescents extend to form spherical immature virions, which undergo a condensation process involving cleavage of core proteins at AG/X sites by a viral protease (1, 6, 26, 35, 56) and the formation of disulfide bonds in the cytoplasmic domains of membrane proteins by a novel virus-encoded redox pathway (43, 44, 59) to form infectious intracellular mature virions.
Cells infected with the G5R temperature-sensitive mutants at the nonpermissive temperature formed virus factories containing DNA and viral proteins, which were seen by confocal microscopy. At a higher resolution of the electron microscope, however, neither immature nor mature virions were discerned. Instead, there were large cytoplasmic areas devoid of cellular organelles, with only an occasional small viral crescent. Although this phenotype is uncommon, similar phenotypes were found previously in cells infected with mutants carrying conditionally lethal mutations that mapped to the H5R (12) and F10L (47, 53, 57) genes. The H5R gene is transcribed at early and late stages of infection, and the protein appears to function in transcription (5, 27, 42) as well as morphogenesis. F10 is a dual-specificity protein kinase (13, 28, 53, 57). Although F10 expression is required for phosphorylation of the A17 and A14 membrane components (4, 13, 30) and the A30 core protein (48), the phenotypes of mutants carrying conditionally lethal A17L, A14L, and A30L mutations can be distinguished from those of F10L, H5R, and G5R mutations. In cells infected with A30L mutants under nonpermissive conditions, empty immature virions form (49, 50). The numerous membrane vesicles that appear when expression of A17L (40, 60) or A14L (41, 54) is repressed were not seen with the F10L, H5R, and G5R mutants. The most intriguing possibility is that F10, H5, and G5 proteins are involved in the formation of viral membranes, one of the least-understood events in poxvirus replication.
Acknowledgments
We thank Eugene Koonin for computer-based gene and protein analyses, Norman Cooper for cells and viruses, and Brian Ward, Frank De Silva, and Patricia Szajner for helpful discussions and advice on protocols.
REFERENCES
- 1.Ansarah-Sobrinho, C., and B. Moss. 2004. Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J. Virol. 78:6335-6343. [DOI] [PMC free article] [PubMed]
- 2.Banham, A., and G. L. Smith. 1992. Vaccinia virus gene B1R encodes a 34-kDa serine/threonine protein kinase that localizes in cytoplasmic factories and is packaged into virions. Virology 191:803-812. [DOI] [PubMed] [Google Scholar]
- 3.Baroudy, B. M., S. Venkatesan, and B. Moss. 1982. Structure and replication of vaccinia virus telomeres. Cold Spring Harbor Symp. Quant. Biol. 47:723-729. [DOI] [PubMed] [Google Scholar]
- 4.Betakova, T., E. J. Wolffe, and B. Moss. 1999. Regulation of vaccinia virus morphogenesis: phosphorylation of the A14L and A17L membrane proteins and C-terminal truncation of the A17L protein are dependent on the F10L protein kinase. J. Virol. 73:3534-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Black, E. P., N. Moussatche, and R. C. Condit. 1998. Characterization of the interactions among vaccinia virus transcription factors G2R, A18R, and H5R. Virology 245:313-322. [DOI] [PubMed] [Google Scholar]
- 6.Byrd, C. M., T. C. Bolken, and D. E. Hruby. 2002. The vaccinia virus I7L gene product is the core protein proteinase. J. Virol. 76:8973-8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.da Fonseca, F. G., A. Weisberg, E. J. Wolffe, and B. Moss. 2000. Characterization of the vaccinia virus H3L envelope protein: topology and post-translational membrane insertion via the C-terminal hydrophobic tail. J. Virol. 74:7508-7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.da Fonseca, F. G., E. J. Wolffe, A. Weisberg, and B. Moss. 2000. Effects of deletion or stringent repression of the H3L envelope gene on vaccinia virus replication. J. Virol. 74:7518-7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dales, S., and L. Siminovitch. 1961. The development of vaccinia virus in Earle's L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 10:475-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davison, A. J., and B. Moss. 1989. The structure of vaccinia virus early promoters. J. Mol. Biol. 210:749-769. [DOI] [PubMed] [Google Scholar]
- 11.DeLange, A. M. 1989. Identification of temperature-sensitive mutants of vaccinia virus that are defective in conversion of concatemeric replicative intermediates to the mature linear DNA genome. J. Virol. 63:2437-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMasi, J., and P. Traktman. 2000. Clustered charge-to-alanine mutagenesis of the vaccinia virus H5 gene: isolation of a dominant, temperature-sensitive mutant with a profound defect in morphogenesis. J. Virol. 74:2393-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derrien, M., A. Punjabi, R. Khanna, O. Grubisha, and P. Traktman. 1999. Tyrosine phosphorylation of A17 during vaccinia virus infection: Involvement of the H1 phosphatase and the F10 kinase. J. Virol. 73:7287-7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doglio, L., M. A. De, S. Schleich, N. Roos, and J. K. Locker. 2002. The Vaccinia virus E8R gene product: a viral membrane protein that is made early in infection and packaged into the virions' core. J. Virol. 76:9773-9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dvoracek, B., and T. Shors. 2003. Construction of a novel set of transfer vectors to study vaccinia virus replication and foreign gene expression. Plasmid 49:9-17. [DOI] [PubMed] [Google Scholar]
- 16.Earl, P. L., N. Cooper, S. Wyatt, B. Moss, and M. W. Carroll. 1998. Preparation of cell cultures and vaccinia virus stocks, p. 16.16.1-16.16.3. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. John Wiley and Sons, New York, N.Y.
- 17.Earl, P. L., B. Moss, L. S. Wyatt, and M. W. Carroll. 1998. Generation of recombinant vaccinia viruses, p. 16.17.1-16.17.19. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. Wiley Interscience, New York, N.Y.
- 18.Falkner, F. G., and B. Moss. 1990. Transient dominant selection of recombinant vaccinia viruses. J. Virol. 64:3108-3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia, A. D., and B. Moss. 2001. Repression of vaccinia virus Holliday junction resolvase inhibits processing of viral DNA into unit-length genomes. J. Virol. 75:6460-6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimley, P. M., E. N. Rosenblum, S. J. Mims, and B. Moss. 1970. Interruption by rifampin of an early stage in vaccinia virus morphogenesis: Accumulation of membranes which are precursors of virus envelopes. J. Virol. 6:519-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hassett, D. E., and D. E. Condit. 1994. Targeted construction of temperature-sensitive mutations in vaccinia virus by replacing clustered charged residues with alanine. Proc. Natl. Acad. Sci. USA 91:4554-4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollinshead, M., A. Vanderplasschen, G. L. Smith, and D. J. Vaux. 1999. Vaccinia virus intracellular mature virions contain only one lipid membrane. J. Virol. 73:1503-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holzer, G., and F. G. Falkner. 1997. Construction of a vaccinia virus deficient in the essential DNA repair enzyme uracil DNA glycosylase by a complementing cell line. J. Virol. 71:4997-5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Husain, M., and B. Moss. 2003. Evidence against an essential role of COPII-mediated cargo transport to the endoplasmic reticulum-Golgi intermediate compartment in the formation of the primary membrane of vaccinia virus. J. Virol. 77:11754-11766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishii, K., and B. Moss. 2001. Role of vaccinia virus A20R protein in DNA replication: construction and characterization of temperature-sensitive mutants. J. Virol. 75:1656-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katz, E., and B. Moss. 1970. Formation of a vaccinia virus structural polypeptide from a higher molecular weight precursor: inhibition by rifampicin. Proc. Natl. Acad. Sci. USA 6:677-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovacs, G. R., and B. Moss. 1996. The vaccinia virus H5R gene encodes late gene transcription factor 4: purification, cloning and overexpression. J. Virol. 70:6796-6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin, S., and S. S. Broyles. 1994. Vaccinia protein kinase 2: a second essential serine/threonine protein kinase encoded by vaccinia virus. Proc. Natl. Acad. Sci. USA 91:7653-7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin, S., W. Chen, and S. S. Broyles. 1992. The vaccinia virus B1R gene product is a serine/threonine protein kinase. J. Virol. 66:2717-2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mercer, J., and P. Traktman. 2003. Investigation of structural and functional motifs within the vaccinia virus A14 phosphoprotein, an essential component of the virion membrane. J. Virol. 77:8857-8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merchlinsky, M., C. Garon, and B. Moss. 1988. Molecular cloning and sequence of the concatemer junction from vaccinia virus replicative DNA: Viral nuclease cleavage sites in cruciform structures. J. Mol. Biol. 199:399-413. [DOI] [PubMed] [Google Scholar]
- 32.Merchlinsky, M., and B. Moss. 1989. Resolution of vaccinia virus DNA concatemer junctions requires late gene expression. J. Virol. 63:1595-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss, B. 1996. Poxviridae: the viruses and their replication, p. 2637-2671. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed., vol. 2. Lippincott-Raven Publishers, Philadelphia, Pa.
- 34.Moss, B., B. Y. Ahn, B. Amegadzie, P. D. Gershon, and J. G. Keck. 1991. Cytoplasmic transcription system encoded by vaccinia virus. J. Biol. Chem. 266:1355-1358. [PubMed] [Google Scholar]
- 35.Moss, B., and E. N. Rosenblum. 1973. Protein cleavage and poxvirus morphogenesis: Tryptic peptide analysis of core precursors accumulated by blocking assembly with rifampicin. J. Mol. Biol. 81:267-269. [DOI] [PubMed] [Google Scholar]
- 36.Moyer, R. W., and R. L. Graves. 1981. The mechanism of cytoplasmic orthopoxvirus DNA replication. Cell 27:391-401. [DOI] [PubMed] [Google Scholar]
- 37.Punjabi, A., K. Boyle, J. DeMasi, O. Grubisha, B. Unger, M. Khanna, and P. Traktman. 2001. Clustered charge-to-alanine mutagenesis of the vaccinia virus A20 gene: temperature-sensitive mutants have a DNA-minus phenotype and are defective in the production of processive DNA polymerase activity. J. Virol. 75:12308-12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rempel, R. E., and P. Traktman. 1992. Vaccinia virus-B1 kinase-phenotypic analysis of temperature-sensitive mutants and enzymatic characterization of recombinant proteins. J. Virol. 66:4413-4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Risco, C., J. R. Rodriguez, C. Lopez-Iglesias, J. L. Carrascosa, M. Esteban, and D. Rodriguez. 2002. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 76:1839-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodríguez, D., C. Risco, J. R. Rodríguez, J. L. Carrascosa, and M. Esteban. 1996. Inducible expression of the vaccinia virus A17L gene provides a synchronized system to monitor sorting of viral proteins during morphogenesis. J. Virol. 70:7641-7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez, J. R., C. Risco, J. L. Carrascosa, M. Esteban, and D. Rodriguez. 1998. Vaccinia virus 15-kilodalton (A14L) protein is essential for assembly and attachment of viral crescents to virosomes. J. Virol. 72:1287-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosel, J. L., P. L. Earl, J. P. Weir, and B. Moss. 1986. Conserved TAAATG sequence at the transcriptional and translational initiation sites of vaccinia virus late genes deduced by structural and functional analysis of the HindIII H genome fragment. J. Virol. 60:436-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Senkevich, T. G., C. L. White, E. V. Koonin, and B. Moss. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc. Natl. Acad. Sci. USA 99:6667-6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Senkevich, T. G., C. L. White, E. V. Koonin, and B. Moss. 2000. A viral member of the ERV1/ALR protein family participates in a cytoplasmic pathway of disulfide bond formation. Proc. Natl. Acad. Sci. USA 97:12068-12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sodeik, B., R. W. Doms, M. Ericsson, G. Hiller, C. E. Machamer, W. van't Hof, G. van Meer, B. Moss, and G. Griffiths. 1993. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J. Cell Biol. 121:521-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szajner, P., H. Jaffe, A. S. Weisberg, and B. Moss. 2003. Vaccinia virus G7L protein interacts with the A30L protein and is required for association of viral membranes with dense viroplasm to form immature virions. J. Virol. 77:3418-3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szajner, P., A. S. Weisberg, and B. Moss. 2004. Evidence for an essential catalytic role of the F10 protein kinase in vaccinia virus morphogenesis. J. Virol. 78:257-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szajner, P., A. S. Weisberg, and B. Moss. 2004. Physical and functional interactions between vaccinia virus F10 protein kinase and virion assembly proteins A30 and G7. J. Virol. 78:266-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szajner, P., A. S. Weisberg, and B. Moss. 2001. Unique temperature-sensitive defect in vaccinia virus morphogenesis maps to a single nucleotide substitution in the A30L gene. J. Virol. 75:11222-11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szajner, P., A. S. Weisberg, E. J. Wolffe, and B. Moss. 2001. Vaccinia virus A30L protein is required for association of viral membranes with dense viroplasm to form immature virions. J. Virol. 75:5752-5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tolonen, N., L. Doglio, S. Schleich, and J. Krijnse Locker. 2001. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol. Biol. Cell 12:2031-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Traktman, P., M. K. Anderson, and R. E. Rempel. 1989. Vaccinia virus encodes an essential gene with strong homology to protein kinases. J. Biol. Chem. 264:21458-21461. [PubMed] [Google Scholar]
- 53.Traktman, P., A. Caligiuri, S. A. Jesty, and U. Sankar. 1995. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of morphogenesis. J. Virol. 69:6581-6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Traktman, P., K. Liu, J. DeMasi, R. Rollins, S. Jesty, and B. Unger. 2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: Construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74:3682-3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Upton, C., S. Slack, A. L. Hunter, A. Ehlers, and R. L. Roper. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J. Virol. 77:7590-7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vanslyke, J. K., C. A. Franke, and D. E. Hruby. 1991. Proteolytic maturation of vaccinia virus core proteins-identification of a conserved motif at the N termini of the 4b and 25K virion proteins. J. Gen. Virol. 72:411-416. [DOI] [PubMed] [Google Scholar]
- 57.Wang, S., and S. Shuman. 1995. Vaccinia virus morphogenesis is blocked by temperature-sensitive mutations in the F10 gene, which encodes protein kinase 2. J. Virol. 69:6376-6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wertman, K. F., D. G. Drubin, and D. Botstein. 1992. Syst. mutational analysis of the yeast ACT1 gene. Genetics 132:337-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.White, C. L., T. G. Senkevich, and B. Moss. 2002. Vaccinia virus G4L glutaredoxin is an essential intermediate of a cytoplasmic disulfide bond pathway required for virion assembly. J. Virol. 76:467-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolffe, E. J., D. M. Moore, P. J. Peters, and B. Moss. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang, Y., and B. Moss. 1991. Inducer-dependent conditional-lethal mutant animal viruses. Proc. Natl. Acad. Sci. USA 88:1511-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]