

Abstract
Although human respiratory syncytial virus (RSV) is one of the most common viruses inducing respiratory tract infections in young children and the elderly, the genotype distribution and characteristics of RSV in northeastern China have not been investigated. Here, we identified 25 RSV‐A and 8 RSV‐B strains from 80 samples of patients with respiratory infections between February 2015 and May 2015. All 25 RSV‐A viruses were classified as the ON1 genotype, which rapidly spread and became the dominant genotype in the world since being identified in Ontario (Canada) in December 2010. All eight RSV‐B viruses belonged to the BA genotype with a 60‐nucleotide duplication, seven of which formed two new genotypes, BA‐CCA and BA‐CCB. The remaining RSV‐B virus clustered with one of the Hangzhou strains belonging to genotype BA11. Construction of a phylogenetic tree and amino acid substitution analysis showed that Changchun ON1 viruses exclusively constituted Lineages 3, 5 and 6, and contained several unique and newly identified amino acid substitutions, including E224G, R244K, L289I, Y297H, and L298P. Selective pressure was also evaluated, and various N and O‐glycosylation sites were predicted. This study provides the first genetic analysis of RSV in northeastern China and may facilitate a better understanding of the evolution of this virus locally and globally. J. Med. Virol. 89:222–233, 2017. © 2016 The Authors. Journal of Medical Virology Published by Wiley Periodicals, Inc.
Keywords: respiratory syncytial virus, genetic variability, mutation/mutation rate, glycoproteins
INTRODUCTION
Human respiratory syncytial virus (RSV) is the predominant cause of respiratory tract infections in children, the elderly and immunocompromised adults [Gonzalez et al., 2012; Avadhanula et al., 2014]. It causes severe bronchiolitis and pneumonia in infants under 2 years old and presents clinically with coughing and wheezing [Hirano et al., 2014; Halasa et al., 2015]. RSV, classified in the Pneumovirus genus of the Paramyxoviridae family, is an enveloped virus which contains a non‐segmented, single‐stranded, negative‐sense RNA genome of 15,200 nucleotides. Its genome encodes 11 proteins: NS1, NS2, N, P, M, SH, G, F, M2‐1, M2‐2, and L [Cane, 2001]. The attachment protein G is a type II surface glycoprotein which is comprised of an ectodomain, a transmembrane domain and a cytoplasmic domain [Roberts et al., 1994]. This glycoprotein varies in length, amino acid sequence, N‐linked and O‐linked glycosylation sites and is also the key determinant of the antigenicity of RSV [Palomo et al., 1991]. The rapid change of the glycoprotein may induce antigenic drift and, thus, enable RSV to escape from human immune responses. For this reason, humans are infected repeatedly, causing preventive vaccines to fail [Anderson et al., 1985].
Two highly variable regions (HVR1 and HVR2) are present in the ectodomain of the RSV G protein. Based on monoclonal antibody detection of the G protein together with the F protein, RSV can be divided into subgroups A and B. Furthermore, both subgroups can be subdivided into genotypes according to the HVR2 [Johnson et al., 1987; Galiano et al., 2005]. Subgroup A is categorized into 14 genotypes (NA1‐NA4, GA1‐GA7, SAA1, CB‐A, and ON1), while subgroup B includes 25 genotypes (BA1‐BA12, BA‐C, SAB1‐SAB4, GB1‐GB4,URU1‐2, CB‐B, CB1) [Cui et al., 2013b; Gimferrer et al., 2015; Ren et al., 2014]. ON1, a newly found genotype of subgroup A, was first identified in Ontario, Canada in 2010. This genotype has a characteristic 72‐nucleotide duplication in the HVR2 [Eshaghi et al., 2012]. This phenomenon resembles the BA genotype with a 60‐nucleotide duplication in the HVR2 of subgroup B [Trento et al., 2003]. Since its discovery, ON1 has spread globally and has been found mainly in Canada [Eshaghi et al., 2012], USA [Avadhanula et al., 2014], China [Cui et al., 2013a], Korea [Kim et al., 2014], Japan [Tsukagoshi et al., 2013], Thailand [Auksornkitti et al., 2013], India [Choudhary et al., 2013], Malaysia [Khor et al., 2013], Germany [Prifert et al., 2013], Spain [Trento et al., 2015], Italy [Pierangeli et al., 2014], Latvia [Balmaks et al., 2014], Croatia [Ivancic‐Jelecki et al., 2015], Kenya [Agoti et al., 2014], Panama [Trento et al., 2015], Philippines, Peru, and South Africa [Pretorius et al., 2013]. The distribution together with global and local transmission dynamics of ON1 has also been reported recently [Duvvuri et al., 2015].
As almost all children over 2 years of age have experienced at least one RSV infection, research on the molecular epidemiology of RSV, especially its G protein, would be of great clinical importance. Although the genetic features of RSV have been investigated in many countries, related data in China are relatively scarce. Our study is the first to investigate the genotype distribution and characteristics of RSV in northeastern China. In this study, we identified 25 RSV‐A positive and 8 RSV‐B positive samples. Phylogenetic and amino acid analyses were conducted in order to explore the molecular features and evolutionary relationships among different RSV isolates. Meanwhile, we carried out the most recent common ancestor (MRCA) analysis and predicted the glycosylation sites as well as the positive and negative selection sites. These findings may also provide information that will promote the development of RSV‐specific treatments and vaccines in the future.
MATERIALS AND METHODS
Collection of Clinical Samples
We collected nasal swabs from children under 5 years old with upper or lower respiratory tract infections from the Department of Pediatric Respiratory, First Hospital of Jilin University located in northeastern China. Eighty clinical samples were acquired during the spring from February to May in 2015. A nasal swab was obtained as soon as a child was diagnosed with a respiratory tract infection. The specimen was then transported directly to the laboratory and tested for the presence of RSV. Specimens were stored at −80°C if not examined immediately. All samples were collected after written informed consent was given by at least one parent for each child. This study was approved by the Ethics and Research Council of First Hospital of Jilin University.
RNA Extraction, Reverse Transcription, and Quantitative Real‐time PCR (qPCR) for Detection and Distinguishing of RSV Subgroups
TRIzol (Invitrogen, Carlsbad, CA) was used to extract viral RNA from 200 µl of each sample according to the manufacturer's instructions. The cDNA was generated using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA) and random primers according to the supplier's instructions. The qPCR was conducted on an Mx3005 P instrument (Agilent Technologies, Stratagene, La Jolla, CA) using the RealMaster Mix Kit (SYBR Green, Takara, Japan), and the following subgroup‐specific primers were designed using conserved sequences of the N gene of RSV: RSV‐A‐N‐F, 5′‐AGATCAACTTCTGTCATCCAGCAA‐3′; RSV‐A‐N‐R, 5′‐TTCTGCACATCATAATTAGGAG‐3′; RSV‐B‐N‐F, 5′‐AAGATGCAAATCATAAATTCACAGGA‐3′; RSV‐B‐N‐R, 5′‐TGATATCCAGCATCTTTAAGTA‐3′ [Gunson et al., 2005]. The qPCR assay was carried out in a 20‐µl volume consisting of 10 µl of 2.5 × RealMaster Mix/20 × SYBR Green solution containing HotMaster Taq DNA Polymerase, 0.5 µl of 10 ng/µl of each oligonucleotide primer, 3 µl of cDNA template and 6 µl of water. The target sequence amplification was conducted as follows: initial activation of HotMaster Taq DNA Polymerase at 95°C for 2 min, followed by 45 cycles of 95°C for 15 sec, 57°C for 15 sec and 68°C for 20 sec.
Nested PCR for G Gene Amplification and Sequencing of PCR Products
Nested PCR was performed on RSV positive samples detected by qPCR. The first round of amplification was carried out with the forward primer AG20, 5′‐GGGGCAAATGCAAACATGTCC‐3′, and the reverse primer F164, 5′‐GTTATGACACTGGTATACCAACC‐3′. The PCR parameters were as follows. After denaturation of cDNA samples at 94°C for 5 min, the amplification was performed in 40 cycles consisting of a denaturing step for 30 sec at 94°C, a primer annealing step for 30 sec at 54°C, and an elongation step for 1 min at 72°C, followed by extension at 72°C for 10 min. The first round PCR products were used as templates in the second round amplification with the primers BG10, 5′‐GCAATGATAATCTCAACCTC‐3′, and F1, 5′‐CAACTCCATTGTTATTTGCC‐3′. Thermocycling conditions were: 94°C for 5 min, followed by 30 cycles of 94°C for 45 sec, 54°C for 45 sec, and 72°C for 1 min, followed by a final extension at 72°C for 10 min [Agoti et al., 2012]. PCR products from positive reactions were confirmed on 1% agarose gels and were either sequenced directly or purified with an E.Z.N.A. Gel Extraction Kit (OMEGA, Norcross, GA) before being sequenced. All sequencing was performed by Comate Bioscience (Jilin Co., Ltd., Changchun, China) using the BigDye Terminatorv 3.1 kit and ABI‐PRISM 3730XL DNA sequencer (Applied Biosystems).
Phylogenetic Analysis
The second highly variable regions of the G gene of subgroups A and B were compared to reference strains available from GenBank. Reference strains representing the discovered genotypes were retrieved from GenBank (http://www.ncbi.nlm.nih.gov) as of August 11, 2015. All sequences were trimmed according to the HVR2. Multiple sequences were aligned using the ClustalW program of MEGA6 software [Tamura et al., 2013]. Identical sequences were identified by using ElimDupes (http://hcv.lanl.gov/content/sequence/ELIMDUPES/elimdupes.html).
Phylogenetic trees were constructed using the neighbor‐joining method, and the evolutionary distances were estimated using the p‐distance method. Analysis of complete gap deletion or missing data were also performed. The tree topologies were tested by 1,000 bootstrap replicates, and only bootstrap values >70% were shown in each unrooted tree. Lineages were also defined when the bootstrap value was more than 70%. The newly found RSV‐A and RSV‐B isolates in Changchun were deposited in GenBank under the accession numbers KU254612‐KU254644.
Deduced Amino Acid Analysis
Deduced amino acids were translated with MEGA6 software and edited using BioEdit software. RSV‐A isolates were compared to the prototype ON1, which was first found in Canada (GenBank accession no. JN257693), while RSV‐B isolates were contrasted with the prototype BA1 strain first identified in Buenos Aires, Argentina (GenBank accession no. AY333364). Sublineages were defined according to the amino acid substitutions within one lineage. The number of amino acid substitutions could be used to evaluate the genetic distance, and shared substitutions reflected a stable evolutionary trend within a sublineage.
Glycosylation Site Analysis
Putative N‐glycosylation sites were predicted if the amino acid sequence was Asn‐Xaa‐Thr/Ser, where Xaa was not a proline and accepted if the glycosylation potential was ≥0.5 using the NetNGlyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/) [Julenius, 2007]. Moreover, potential O‐glycosylation sites were determined by the NetOGly 3.1 server (http://www.cbs.dtu.dk/services/NetOGlyc-3.1/) and accepted if the G‐score was ≥0.5 [Julenius et al., 2005].
Evolutionary Analysis
The evolutionary rate and MRCA of the RSV isolates were estimated by using regression of the root‐to‐tip distance from the neighbor‐joining tree in Path‐O‐Gen software and by using the Bayesian Markov chain Monte Carlo approach in BEAST v1.6.1 software [Drummond and Rambaut, 2007]. In the Bayesian evolutionary analysis, sequences were analyzed with an uncorrelated lognormal relaxed clock using the GTR model, and the software was run through 50 million steps to reach convergence for local sequences, with sampling every 10,000 steps. In the analysis, the effective sample size (ESS) over 200 was ensured. Only RSV isolates with definite sampling dates were included in this study. For the phylogenetic tree, the 333‐nucleotide partial G genes of ON1 isolates identified in Changchun were used as the input.
Selective Pressure Analysis
Positively and negatively selected sites were calculated in order to evaluate the selective pressure on the HVR2 of the G gene. Datamonkey (http://www.datamonkey.org/) was used to calculate the synonymous (dS) and nonsynonymous (dN) substitution rates at each codon [Pond and Frost, 2005]. Three methods with an HKY 85‐nucleotide substitution bias model were applied to recognize the diversifying selection. The following three methods were used: single likelihood ancestor counting (SLAC), fixed effects likelihood (FEL) and internal fixed effects likelihood (IFEL). A P‐value <0.1 was used to define the positively (dN‐dS > 0) or negatively (dN‐dS < 0) selected sites.
RESULTS
RSV Distribution and Related Clinical Features
Using group‐specific primers for qPCR analysis, 25 RSV‐A and 8 RSV‐B positive samples were detected from the 80 clinical samples. The positive rates for RSV‐A and RSV‐B were 31.25% and 10%, respectively, and the total RSV infection rate was 41.25%. The clinical data showed that nearly all of the children had clinical manifestations of coughing and wheezing and were diagnosed with bronchiolitis or pneumonia. Children under 1‐year‐old accounted for the largest proportion of patients affected by RSV. Moreover, morbidity was not obviously different between males and females. Fever, nasal discharge and rale were not directly correlated with RSV infection.
All RSV isolates were successfully sequenced. After sequence alignment and analysis, all RSV‐A isolates were found to possess the 72‐nucleotide duplication in the HVR2 of the G gene, which is characteristic of the genotype ON1. Moreover, all RSV‐B isolates were classified as the BA genotype based on the 60‐nucleotide duplication within the HVR2.
Phylogenetic Analysis of Genotype ON1 in Subgroup A
Of the total 87 ON1 isolates collected from China, only 56 were included in constructing the phylogenetic tree, while the remaining 31 were excluded due to duplication. All 25 Changchun ON1 viruses were included in order to evaluate the relative frequency within Changchun ON1 isolates. Among the 56 included ON1 isolates, 25 were from Changchun, 20 from Beijing, 6 from Hangzhou, 4 from Chongqing, and 1 from Shanghai. The phylogenetic analysis of these 56 ON1 viruses showed that the Chinese ON1 viruses were mainly divided into two branches, which could be further classified into seven lineages (Fig. 1). The larger branch with six lineages contained diversified clusters and most of the Chinese ON1 viruses. Thirty‐four isolates were genetically close to the Canadian ON1 prototypes and defined as Lineage 1. The remaining five lineages may have evolved from the original ON1 viruses, and each of them formed a well‐supported cluster with bootstrap values higher than 70%. Specifically, the ON1 isolates identified in Changchun could be classified into Lineages 1, 3, 5, and 6. While only ON1 isolates from Chongqing formed the smaller branch, the bootstrap value was greater than 90%, and these viruses were considered another lineage. Overall, we found that ON1 viruses isolated in Beijing, Hangzhou, Shanghai, and Changchun constituted Lineage 1. Lineages 2 and 4 only included ON1 viruses recognized in Beijing. Lineages 3, 5, and 6 were composed of Changchun ON1 viruses exclusively, while Lineage 7 included ON1 isolates found in Chongqing.
Figure 1.
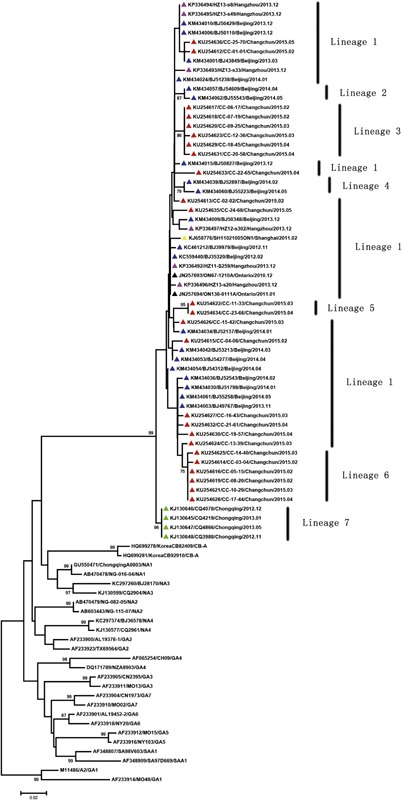
Phylogenetic tree of human RSV ON1 from China. The tree based on the HVR2 of the G protein was constructed with 333 nucleotides of the C‐terminal region of the G protein of ON1 viruses. The lengths of reference strains of other genotypes in subgroup A were trimmed according to the ON1 viruses. The tree was constructed using the neighbor‐joining method. ON1 viruses identified in Changchun, Beijing, Hangzhou, Chongqing, and Shanghai are preceded by a red, blue, pink, green, and yellow triangle, respectively. The two original ON1 prototypes discovered in Ontario, Canada are each labeled with a black triangle. The names of other genotypes in subgroup A are located at the end of each reference strain. Only bootstrap values greater than 70% are displaced at the branch nodes. Scale bars represent nucleotide substitutions per site.
Deduced Amino Acid Analysis of Genotype ON1
The ON1 viruses were characterized by a 24‐amino acid duplication in the HVR2 of the G protein. Moreover, from the alignment of the amino acid sequences (Fig. 2), we recognized two distinct amino acid substitutions, T253K and N273Y, in all the Chinese ON1 strains compared with non‐ON1 strains. These two substitutions also defined Lineage 1. Furthermore, other lineages carried one or several more amino acid substitutions compared with Lineage 1. Lineage 2 was composed of two Beijing ON1 viruses and characterized by a S299R substitution when compared with the Canadian prototype ON67‐1210A. Lineage 3 was composed of six Changchun ON1 viruses and featured a L298P substitution. Lineage 4 also included two Beijing ON1 viruses, and their defining substitution was V279I. Lineage 5 contained two Changchun ON1 viruses and three striking substitutions, E224G, R244K, and L274P. Lineage 6 included six Changchun ON1 viruses and even four notable substitutions, T249I, E262K, L289I, and Y297H. Lineage 7 was composed of four Chongqing ON1 viruses, and their characteristic substitutions were G232E and E262K. Geographically, Changchun ON1 viruses shared amino acid substitutions T249I and L274 P with those in Beijing and E262K with those in both Beijing and Chongqing. Furthermore, Changchun ON1 viruses also contained some defining amino acid substitutions, including E224G, R244K, L289I, Y297H, and L298P, which were unique and first identified in China.
Figure 2.
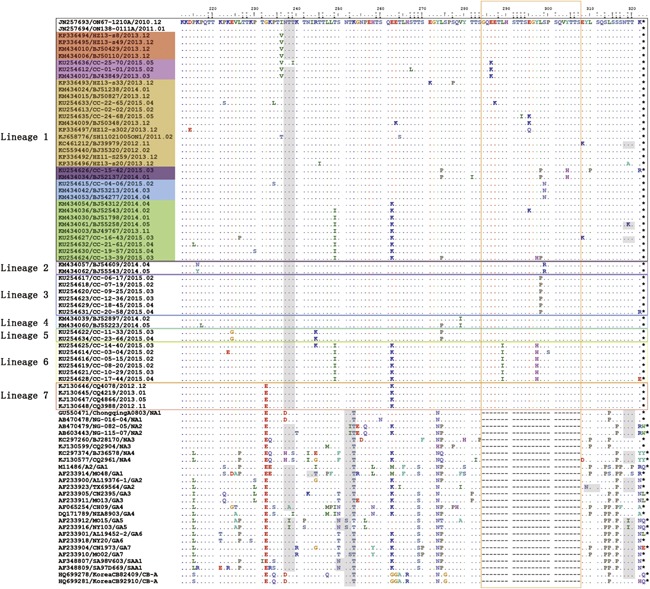
Alignments of deduced amino acids sequences of the G protein of ON1 viruses isolated in China. Alignments are shown relative to the prototype ON1 strain first detected in Ontario, Canada (GenBank accession number JN257693). Only the HVR2 of the G protein is included. Identical residues are indicated by dots, and stop codons are represented by asterisks. The horizontal boxes colored in black, purple, dark blue, light blue, green, yellow, and red indicate ON1 Lineage 1–7, respectively. Within Lineage 1, each sublineage is colored differently (red, pink, yellow, purple, light blue, and green). The vertical orange box marks the 24‐amino acid duplication in the G protein. Potential N‐glycosylation sites (NXT, where X is not proline) are highlighted in gray. The predicted O‐glycosylation sites of the prototype ON1 strain and the Chinese ON1 strains are indicated by black rectangular dots and gray rectangular dots, respectively. Reference strains with dashes are non‐ON1 genotypes in subgroup A and listed below the ON1 genotypes.
Moreover, Lineage 1 could be divided into several sublineages based on more specific and unique substitutions. Such substitutions were not sufficiently striking to separate their carriers into well‐supported clusters (bootstrap over 70%) in the phylogenetic tree and become a new lineage, but they indeed indicated subtle diversification. These stable amino acid substitutions included I236V, E286K, L274P, L298P, Y304H, S299N, E262K, and T249I.
Another important observation was that isolates of Lineages 2, 3, and 6 as well as some in Lineage 1 had amino acid substitutions in the duplication area. Seven substitution sites (E286K, L289I, Y297H, L298P, S299R, S299N, Y304H) out of the total 17 sites (E224G, G232E, T235K, I236V, R244K, T249I, E262K, N273Y, L274P, V279I, E286K, L289I, Y297H, L298P, S299R, S299N, Y304H) were located in this region, accounting for 41.2%.
Phylogenetic Analysis of Genotype BA in Subgroup B
The phylogenetic tree of Changchun RSV‐B isolates and their reference strains (Fig. 3) showed that the eight Changchun RSV‐B viruses could be classified into three clusters with bootstrap values of 90%, 97%, and 71%. Based on criteria previously put forward that bootstrap values of 70–100% and p‐distances of less than 0.07 can define a new genotype [Venter et al., 2001], two new genotypes were found to be circulating in Changchun and designated as BA‐CCA and BA‐CCB. These new genotypes were formed by five and two Changchun RSV‐B viruses, respectively. The phylogenetic tree also showed that the newly found genotype BA‐CCB may have evolved from the new genotype BA‐CCA, and one remaining Changchun RSV‐B virus was in the same cluster with the BA‐11 genotype reference virus. Furthermore, all of the eight RSV‐B viruses isolated in Changchun had a closer genetic relationship with the BA‐11 genotype reference strains acquired in Hangzhou, another city in China, since they formed a larger cluster. Therefore, the two new genotypes likely originated from the genotype BA‐11 that was also first identified in China [Yu et al., 2015].
Figure 3.
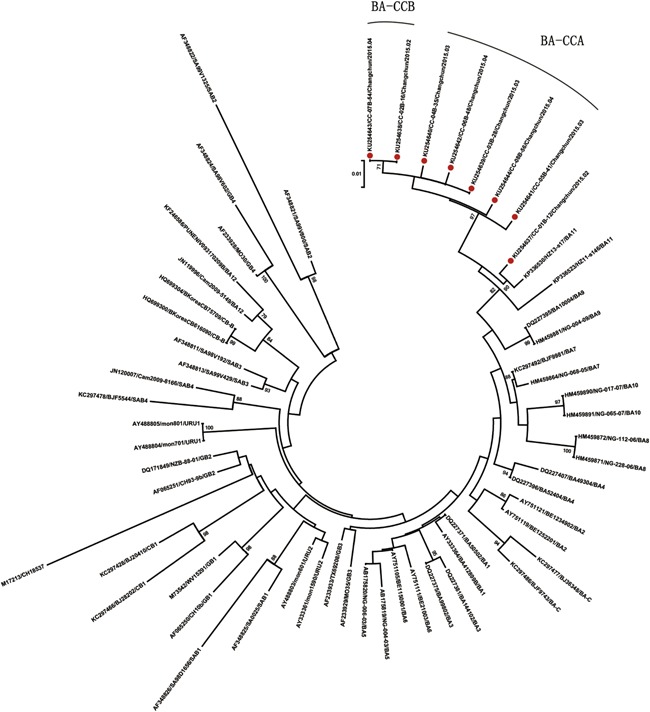
Phylogenetic tree of human RSV genotype BA found in Changchun. The 324 nucleotides of the HVR2 of the G protein were used to construct the neighbor‐joining tree. The reference strains in subgroup B were trimmed accordingly, and the corresponding genotype name is located at the end of each reference strain. Each BA virus detected in Changchun is preceded by a red circle. Only bootstrap values over 70% are shown on the branches. Scale bar indicates nucleotide substitutions per site.
Deduced Amino Acid Analysis of Genotype BA
The genotype BA was first recognized in Buenos Aires, Argentina with a 20‐amino acid duplication in the HVR2 of the G protein [Trento et al., 2003]. Since the genotype was discovered, it has been changing constantly and now can be divided into many more advanced genotypes. The eight BA viruses found in Changchun (Fig. 4) were found to share some common amino acid substitutions with other reference strains compared with the prototype BA1 strains, including K218T, L223P, S247P, T270I, V271A, and I281T. Moreover, seven of the eight viruses also contained a common H287Y substitution. However, unique substitutions were still found in the Changchun BA strains. Seven Changchun BA viruses showed a characteristic substitution of T254I. Five Changchun BA viruses possessed a unique T298M substitution, and two had a T290I substitution. The distinct changes in the Changchun BA strains were sufficient to form new clusters in the phylogenetic tree, BA‐CCA and BA‐CCB. Notably, none of the three unique substitutions of the Changchun BA viruses was within the duplication region.
Figure 4.

Alignments of deduced amino acid sequences of the G protein of the BA viruses isolated in China. Alignments are shown relative to the prototype BA1 strain first identified in Buenos Aires, Argentina (GenBank accession number AY333364). Only the HVR2 of the G protein is included. Identical residues are represented by dots, and stop codons are indicated by asterisks. The horizontal boxes of green and red show our newly found genotypes BA‐CCA and BA‐CCB. The vertical orange box represents the 20‐amino acid duplication in the G protein. Gray shading highlights potential N‐glycosylation sites (NXT, where X is not proline). The predicted O‐glycosylation sites of the prototype BA1 strain and the eight Changchun BA strains are indicated by black rectangular dots and gray rectangular dots, respectively. For each reference strain, the name of its genotype is located at the end.
Glycosylation Analysis
Almost all Chinese ON1 viruses had a putative N‐glycosylation site at amino acid position 237 (Fig. 2) except CC‐25‐70‐May‐2015 from Changchun in Lineage 1. Although position 318 fulfilled the typical N‐glycosylation site pattern, the glycosylation potential of most Chinese ON1 strains calculated by the NetNGlyc 1.0 server was below 0.5; however, these values were all above 0.4, and mostly above 0.49, near the threshold. In fact, three Chinese ON1 strains did have a predicted N‐glycosylation site at position 318. Among them, CC‐16‐43‐Mar‐2015 was from Changchun with the glycosylation potential of 0.5032. BJ/55258‐May‐2014 and BJ/39979‐Nov‐2012 were from Beijing with the glycosylation potential of 0.5566 and 0.5031, respectively (Fig. S1). All three viruses were classified into Lineage 1.
Thirty‐seven O‐glycosylation sites in the Canadian prototype ON1 strain were predicted, and seven of them were in the duplication region (Fig. 2). Overall, 44 O‐glycosylation sites in the Chinese ON1 strains were predicted, and 10 of them appeared in the duplication region. For each Chinese ON1 strain, the O‐glycosylation varied from 26 to 42. Specifically, those identified in Changchun had 28–42 O‐glycosylation sites.
Regarding the N‐glycosylation of Changchun BA viruses (Fig. 4), only three out of the eight isolates had a putative site at amino acid position 296. The other five isolates lost the N‐glycosylation site due to the substitution T298M. N‐glycosylation sites of other BA genotypes in subgroup B were also observed.
With the use of the NetOGly 3.1 server, we recognized 44 O‐glycosylation sites in the Argentinean prototype BA strain. Eleven sites were in the duplication region (Fig. 4). Overall, we also identified up to 44 O‐glycosylation sites in the Changchun BA strains, although they did not match with those of the prototype completely. Specifically, the number of O‐glycosylation sites of the Changchun BA viruses ranged from 40 to 44, and generally 10 of those sites were found in the duplication region for Changchun BA strains.
MRCA and Evolutionary Rate Analyses
The time of most recent common ancestor (TMRCA) of the 25 Changchun ON1 isolates from the root‐to‐tip linear regression analysis on the neighbor‐joining tree was June 2012 (Fig. 5), and the nucleotide substitution rate in the HVR2 of Changchun ON1 viruses was estimated to be 5.56 × 10‐3 substitutions per site per year. The alternative Bayesian method showed that the TMRCA of the Changchun ON1 viruses was March 2011 (95% highest probability density [HPD] interval, 2006.05–2014.97). The nucleotide substitution rate was 1.26 × 10−2 (95% HPD interval, 2.42 × 10−5 to 2.81 × 10−2).
Figure 5.
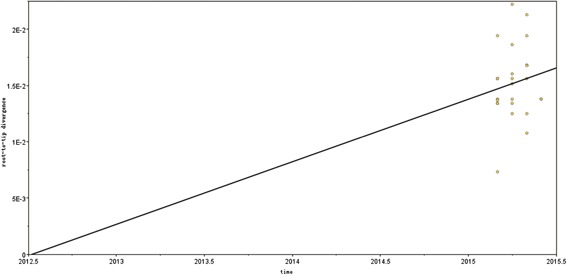
TMRCA and evolutionary rate of 25 ON1 viruses detected in Changchun. The analysis was performed by root‐to‐tip regression of the genetic distance from the neighbor‐joining tree. The evolutionary rate is represented by the nucleotide substitution rate. The plot shows the correlation between the branch lengths and the sampling dates of ON1 strains.
Selective Pressure Analysis of Global ON1 Viruses
We used 205 global ON1 nucleotide sequences to carry out the selective pressure analysis. Twenty‐eight sequences were identical even though they were recognized in different countries and were removed by Datamonkey. One German ON1 sequence contained a TAA stop codon in the middle of the whole sequence and was excluded as well. Thus, 176 unique global sequences were applied in this study. The positive and negative selection sites are listed in Tables I and II.
Table I.
Positive Selection Sites in the HVR2 of G Protein of ON1 Viruses
| Model | Positive selection sites a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SLAC | V225A | V225L | V225E | L247P | L274P | L274Q | L274R | |||
| FEL | L247P | N251D | N251Y | N251S | E262K | Y273H | L274P | L274Q | L274R | Y297H* |
| IFEL | Y273H | Y297H* | ||||||||
P‐value <0.1.
Sites in the 24‐amino acid duplication region are indicated by an asterisk.
Table II.
Negative Selection Sites in the HVR2 of G Protein of ON1 Viruses
| Model | Negative selection sites a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SLAC | P217 | Q218 | T227 | P230 | T239 | T245 | S277 | Q285* | S307* | ||||
| FEL | Q218 | T227 | P230 | T239 | I243 | T259 | S277 | Q278 | Q285* | S291* | S294* | S307* | T320 |
| IFEL | Q218 | T227 | P230 | S277 | S283 | Q285* | S291* | T292* | S307* | ||||
P‐value <0.1.
Sites in the 24‐amino acid duplication region are indicated by an asterisk.
For the positive selection sites, the mean dN/dS ratio was 0.785 according to the SLAC method. L247P, L274P, L274Q, and L274R were recognized as positive selection sites by both the SLAC and FEL methods. Y273H and Y297H* were identified by the FEL and IFEL methods.
The number of negative selection sites was larger than that of positive selection sites. Q218, T227, P230, S277, Q285*, and S307* were identified as negative selection sites by all three methods. T239 was recognized by the SLAC and FEL methods, while S291* was identified by the FEL and IFEL methods.
DISCUSSION
In our study, we conducted phylogenetic and amino acid analyses of RSV in Changchun, a northeastern city in China, to explore the molecular epidemiology of RSV transmission locally. Out of the total 80 samples, 25 RSV ON1 genotype isolates and 8 RSV BA genotype isolates were successfully sequenced. Sequence alignments showed at least one nucleotide variation in 19 ON1 isolates, and all of the 8 BA isolates were different. Therefore, we considered the local Changchun RSV isolates to be diversified, which was also reflected by the phylogenetic analyses. From the phylogenetic tree of RSV‐A ON1 viruses, four lineages were identified in Changchun. Lineages 3, 5, and 6 were found only in Changchun, while Lineage 1 was recognized in not only Changchun but also other cities in China. On the whole country scale, ON1 had formed seven lineages in 4 years. Lineage 1 was still the main lineage circulating in China, since most ON1 strains isolated fell into this lineage.
However, the local evolutionary pattern was more obvious. Apart from the exclusive ON1 lineages in Changchun, this pattern was also reflected by Lineages 2 and 4, which were found only in Beijing, and Lineage 7, which was identified only in Chongqing. In terms of the relationship between different lineages, we can infer that Lineages 5 and 6 evolved from two clusters of Lineage 1 viruses. Moreover, Changchun ON1 viruses recognized in 2015 may have originated from Beijing ON1 viruses isolated in 2014, potentially from two separate sources. On the other hand, in the deduced amino acid alignment, we could also divide Lineage 1 viruses into sublineages. Characteristic substitutions distinguished the genetic relationship between different lineages and sublineages more clearly. In particular, the reverse substitutions of E262K and G232E may help RSV evade the human immune system or improve the probability of RSV survival. This type of mutation has been called a flip‐flop pattern [Botosso et al., 2009]. For Changchun RSV‐B BA viruses, unique cluster and amino acid substitutions were exhibited as well. Five Changchun RSV‐B BA viruses formed a new genotype BA‐CCA, and the remaining three formed the new genotype BA‐CCB. Three characteristic substitutions, T254I, T290I, and T298M, were exhibited by only Changchun BA isolates.
The genetic diversifications of Changchun ON1 and BA viruses revealed that RSV may be able to adapt to the local environment and change frequently. Furthermore, the regional pattern of RSV distribution was indicated by the newly found genotypes CB1 and NA3, and RSV‐B in particular was reported to spread less effectively with a higher variation specific to the local populations in Kenya and England [Cui et al., 2013b; Agoti et al., 2014, 2015]. Here, we further observed a rapid local variation in RSV‐A ON1, which has been noted previously to spread rapidly.
All of the Changchun RSV‐A viruses belonged to genotype ON1 viruses, and all of the Changchun RSV‐B viruses belonged to genotype BA. These findings confirmed the rapid global dissemination of RSV with nucleotide duplication and demonstrated their strong ability to survive and replace other RSV genotypes, as was also shown in Italy and Germany [Pierangeli et al., 2014; Tabatabai et al., 2014]. The relatively high rate of amino acid substitutions in the duplication region may account for this phenomenon. Moreover, we can see that the N‐glycosylation sites were relatively fewer and more conserved than the O‐glycosylation sites, which varied in number and position for different viruses. The amino acid substitutions were closely connected to the glycosylation. The T298M substitution caused two BA‐CCB and three BA‐CCA viruses to lose the only N‐glycosylation site in the HVR2. In this manner, the amino acid substitutions and changes of glycosylation sites can enable viruses to alter their antigenic characteristics and evade host immunity [Cane et al., 1991; Palomo et al., 2000].
We calculated the TMRCA of ON1 viruses from Changchun using both the linear regression method and Bayesian Markov chain Monte Carlo approach. For Changchun ON1 viruses, the TMRCA was around 2011–2012, which was approximately 1 or 2 years after the first ON1 was detected in Canada. However, the nucleotide substitution rates varied between the two methods. The rates obtained by the Bayesian approach were about twice as much as those of the linear regression method. We considered the results obtained by the Bayesian approach more convincing since the 95% HPD intervals were relatively narrow, and the ESS was over 200. The low R square value also decreased the reliability of the linear regression method. The substitution rate of Changchun ON1 viruses in our study was 1.26 × 10‐2, higher than that of viruses from other places. This finding may be attributed to the rapidly changing and diversified ON1 viruses within the local environment.
Selective pressure is closely connected to the antigenic evolution of RSV and other respiratory viruses. Positively selected sites can change the amino acid sequence and benefit the survival of viruses, while negatively selected sites can further sustain functions of viruses [Botosso et al., 2009]. In our study, we used three methods to predict the selection sites [Kushibuchi et al., 2013]. SLAC is a conservative approach but more suitable for a large number of sequences, while FEL and IFEL are considered more effective and can provide similar results [Kosakovsky Pond and Frost, 2005]. The positive selection analyses showed that some sites in our results were the same as those identified in Japan, but they still revealed unique sites recognized only in China [Hirano et al., 2014]. For the SLAC method, V225A, V225L, V225E, L247P, and L274Q were exclusive. For the FEL method, L247P, E262K, Y273H, and L274Q were unique. However, S260N was only found in Japan by the IFEL method. The negative selection sites predicted by the Japanese researchers and Chinese researchers were also not completely identical. P217, Q218, S277, Q285*, and S307* were only identified in our study, while T231 and S291* were only found in a Japanese study using the SLAC method. Moreover, by the FEL method, Q218, T227, T239, Q278, S294*, and T320 were found in China, while P222, E224, T231, and T245 were found in Japan. Last but not least, Q218, T227, P230, S283, T292*, and S307* were included in our study, and only T231 was unique in the study conducted in Japan by the IFEL method. Those differences may have resulted from differences in the number and diversity of the global ON1 sequences selected between the studies. The research carried out in Japan only included one ON1 virus isolated in Beijing. However, during 2014 and 2015, ON1 appeared in many cities in China, and these ON1 viruses needed to be analyzed nationally and together with previously isolated global ON1 viruses. Our report is not only the first to study the molecular and genetic characteristics of RSV ON1 genotype in northeastern China, but it is also the first to summarize the ON1 viruses isolated throughout China.
We only acquired 33 positive samples from the spring of 2015 and focused on the current distribution of RSV. Due to the low sample size, we may have missed some genotypes, and the degree of diversity may be limited. However, we did find some meaningful local evolutionary characteristics of these viruses in northeastern China, which were quite distinct from isolates in other regions. The phylogenetic trees and amino acid alignments showed that RSV isolates in Changchun were diversified and evolved rapidly even in such a short period. Further studies on the yearly distribution and molecular epidemiology of RSV genotypes will need to be conducted in the following years.
In conclusion, this report of human RSV in Changchun not only provided evidence of ON1 rapidly spreading across the world, but it also emphasized the significance of studying this virus at the local level. Local evolution was exemplified by the characteristic lineages for RSV‐A and new genotypes, BA‐CCA and BA‐CCB, since RSV‐B was found specifically in Changchun. Moreover, the TMRCA and nucleotide substitutions should be researched in order to determine the pace of RSV evolution geographically. Amino acid changes, glycosylation sites and selective pressure all contribute to antigenic features of RSV, and their analyses may explain the cause of these frequent viral re‐infections. Such information also helps to determine the feasibility of developing RSV vaccines. Further ongoing molecular epidemiology studies, as well as whole genome analyses, also should be conducted, for example, to find associations between infection with RSV isolates and the severity of clinical symptoms.
AUTHORS’ CONTRIBUTIONS
Conceived and designed the experiments: WYZ, SCH, and XFY. Performed the experiments: YXZ, LL, ZLL, and MH. Analyzed the data: SCH, WYZ, YXZ, SHW, and ZLL. Contributed reagents/materials/analysis tools: WYZ, XFY, YXZ, SHW, ZLL, and JLL. Contributed to the writing of the manuscript: WYZ, SCH, and YXZ.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. The N‐glycosylation potential of (A) CC‐16‐43‐Mar‐2015, (B) BJ/55258‐May‐2014 and (C) BJ/39979‐Nov‐2012.
ACKNOWLEDGMENTS
We thank Chunyan Dai for reagents of RNA extraction, reverse transcription, qPCR, and nested PCR. We also thank Phuong Thi Sarkis for editorial assistance.
Contributor Information
Wenyan Zhang, Email: zhangwenyan@jlu.edu.cn.
Shucheng Hua, Email: shuchenghuajdyy@126.com.
REFERENCES
- Agoti CN, Mwihuri AG, Sande CJ, Onyango CO, Medley GF, Cane PA, Nokes DJ. 2012. Genetic relatedness of infecting and reinfecting respiratory syncytial virus strains identified in a birth cohort from rural Kenya. J Infect Dis 206:1532–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoti CN, Otieno JR, Gitahi CW, Cane PA, Nokes DJ. 2014. Rapid spread and diversification of respiratory syncytial virus genotype ON1, Kenya. Emerg Infect Dis 20:950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoti CN, Otieno JR, Munywoki PK, Mwihuri AG, Cane PA, Nokes DJ, Kellam P, Cotten M. 2015. Local evolutionary patterns of human respiratory syncytial virus derived from whole‐genome sequencing. J Virol 89:3444–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LJ, Hierholzer JC, Tsou C, Hendry RM, Fernie BF, Stone Y, McIntosh K. 1985. Antigenic characterization of respiratory syncytial virus strains with monoclonal antibodies. J Infect Dis 151:626–633. [DOI] [PubMed] [Google Scholar]
- Auksornkitti V, Kamprasert N, Thongkomplew S, Suwannakarn K, Theamboonlers A, Samransamruajkij R, Poovorawan Y. 2013. Molecular characterization of human respiratory syncytial virus, 2010‐2011: Identification of genotype ON1 and a new subgroup B genotype in Thailand. Arch Virol 159:499–507. [DOI] [PubMed] [Google Scholar]
- Avadhanula V, Chemaly RF, Shah DP, Ghantoji SS, Azzi JM, Aideyan LO, Mei M, Piedra PA. 2014. Infection with novel respiratory syncytial virus genotype Ontario (ON1) in adult hematopoietic cell transplant recipients, texas, 2011‐2013. J Infect Dis 211:582–589. [DOI] [PubMed] [Google Scholar]
- Balmaks R, Ribakova I, Gardovska D, Kazaks A. 2014. Molecular epidemiology of human respiratory syncytial virus over three consecutive seasons in Latvia. J Med Virol 86:1971–1982. [DOI] [PubMed] [Google Scholar]
- Botosso VF, Zanotto PM, Ueda M, Arruda E, Gilio AE, Vieira SE, Stewien KE, Peret TC, Jamal LF, Pardini MI, Pinho JR, Massad E, Sant'anna OA, Holmes EC, Durigon EL. 2009. Positive selection results in frequent reversible amino acid replacements in the G protein gene of human respiratory syncytial virus. PLoS Pathog 5:e1000254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane PA. 2001. Molecular epidemiology of respiratory syncytial virus. Rev Med Virol 11:103–116. [DOI] [PubMed] [Google Scholar]
- Cane PA, Matthews DA, Pringle CR. 1991. Identification of variable domains of the attachment (G) protein of subgroup A respiratory syncytial viruses. J Gen Virol 72:2091–2096. [DOI] [PubMed] [Google Scholar]
- Choudhary ML, Anand SP, Wadhwa BS, Chadha MS. 2013. Genetic variability of human respiratory syncytial virus in Pune, Western India. Infect Genet Evol 20:369–377. [DOI] [PubMed] [Google Scholar]
- Cui G, Qian Y, Zhu R, Deng J, Zhao L, Sun Y, Wang F. 2013a. Emerging human respiratory syncytial virus genotype ON1 found in infants with pneumonia in Beijing, China. Emerg Microbes Infect 2:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Zhu R, Qian Y, Deng J, Zhao L, Sun Y, Wang F. 2013b. Genetic variation in attachment glycoprotein genes of human respiratory syncytial virus subgroups a and B in children in recent five consecutive years. PLoS ONE 8:e75020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvvuri VR, Granados A, Rosenfeld P, Bahl J, Eshaghi A, Gubbay JB. 2015. Genetic diversity and evolutionary insights of respiratory syncytial virus A ON1 genotype: Global and local transmission dynamics. Sci Rep 5:14268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshaghi A, Duvvuri VR, Lai R, Nadarajah JT, Li A, Patel SN, Low DE, Gubbay JB. 2012. Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: A novel genotype with a 72 nucleotide G gene duplication. PLoS ONE 7:e32807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiano MC, Luchsinger V, Videla CM, De Souza L, Puch SS, Palomo C, Ricarte C, Ebekian B, Avendano L, Carballal G. 2005. Intragroup antigenic diversity of human respiratory syncytial virus (group A) isolated in Argentina and Chile. J Med Virol 77:311–316. [DOI] [PubMed] [Google Scholar]
- Gimferrer L, Campins M, Codina MG, Martin Mdel C, Fuentes F, Esperalba J, Bruguera A, Vilca LM, Armadans L, Pumarola T, Anton A. 2015. Molecular epidemiology and molecular characterization of respiratory syncytial viruses at a tertiary care university hospital in Catalonia (Spain) during the 2013‐2014 season. J Clin Virol 66:27–32. [DOI] [PubMed] [Google Scholar]
- Gonzalez PA, Bueno SM, Carreno LJ, Riedel CA, Kalergis AM. 2012. Respiratory syncytial virus infection and immunity. Rev Med Virol 22:230–244. [DOI] [PubMed] [Google Scholar]
- Gunson RN, Collins TC, Carman WF. 2005. Real‐time RT‐PCR detection of 12 respiratory viral infections in four triplex reactions. J Clin Virol 33:341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasa N, Williams J, Faouri S, Shehabi A, Vermund SH, Wang L, Fonnesbeck C, Khuri‐Bulos N. 2015. Natural history and epidemiology of respiratory syncytial virus infection in the Middle East: Hospital surveillance for children under age two in Jordan. Vaccine 33:6479 –6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano E, Kobayashi M, Tsukagoshi H, Yoshida LM, Kuroda M, Noda M, Ishioka T, Kozawa K, Ishii H, Yoshida A, Oishi K, Ryo A, Kimura H. 2014. Molecular evolution of human respiratory syncytial virus attachment glycoprotein (G) gene of new genotype ON1 and ancestor NA1. Infect Genet Evol 28:183–191. [DOI] [PubMed] [Google Scholar]
- Ivancic‐Jelecki J, Forcic D, Mlinaric‐Galinovic G, Tesovic G, Nikic Hecer A. 2015. Early evolution of human respiratory syncytial virus ON1 strains: Analysis of the diversity in the C‐Terminal hypervariable region of glycoprotein gene within the first 3.5 years since their detection. Intervirology 58:172–180. [DOI] [PubMed] [Google Scholar]
- Johnson PR, Spriggs MK, Olmsted RA, Collins PL. 1987. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: Extensive sequence divergence between antigenically related proteins. Proc Natl Acad Sci USA 84:5625–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julenius K. 2007. NetCGlyc 1.0: Prediction of mammalian C‐mannosylation sites. Glycobiology 17:868–876. [DOI] [PubMed] [Google Scholar]
- Julenius K, Molgaard A, Gupta R, Brunak S. 2005. Prediction, conservation analysis, and structural characterization of mammalian mucin‐type O‐glycosylation sites. Glycobiology 15:153–164. [DOI] [PubMed] [Google Scholar]
- Khor CS, Sam IC, Hooi PS, Chan YF. 2013. Displacement of predominant respiratory syncytial virus genotypes in Malaysia between 1989 and 2011. Infect Genet Evol 14:357–360. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Kim DW, Lee WJ, Yun MR, Lee HY, Lee HS, Jung HD, Kim K. 2014. Rapid replacement of human respiratory syncytial virus A with the ON1 genotype having 72 nucleotide duplication in G gene. Infect Genet Evol 26:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SD. 2005. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222. [DOI] [PubMed] [Google Scholar]
- Kushibuchi I, Kobayashi M, Kusaka T, Tsukagoshi H, Ryo A, Yoshida A, Ishii H, Saraya T, Kurai D, Yamamoto N, Kanou K, Saitoh M, Noda M, Kuroda M, Morita Y, Kozawa K, Oishi K, Tashiro M, Kimura H. 2013. Molecular evolution of attachment glycoprotein (G) gene in human respiratory syncytial virus detected in Japan 2008–2011. Infect Genet Evol 18:168–173. [DOI] [PubMed] [Google Scholar]
- Palomo C, Cane PA, Melero JA. 2000. Evaluation of the antibody specificities of human convalescent‐phase sera against the attachment (G) protein of human respiratory syncytial virus: Influence of strain variation and carbohydrate side chains. J Med Virol 60:468–474. [PubMed] [Google Scholar]
- Palomo C, Garcia‐Barreno B, Penas C, Melero JA. 1991. The G protein of human respiratory syncytial virus: Significance of carbohydrate side‐chains and the C‐terminal end to its antigenicity. J Gen Virol 72:669–675. [DOI] [PubMed] [Google Scholar]
- Pierangeli A, Trotta D, Scagnolari C, Ferreri ML, Nicolai A, Midulla F, Marinelli K, Antonelli G, Bagnarelli P. 2014. Rapid spread of the novel respiratory syncytial virus A ON1 genotype, central Italy, 2011 to 2013. Euro Surveill 19:pii: 20843. [DOI] [PubMed] [Google Scholar]
- Pond SL, Frost SD. 2005. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533. [DOI] [PubMed] [Google Scholar]
- Pretorius MA, van Niekerk S, Tempia S, Moyes J, Cohen C, Madhi SA, Venter M, Group SS. 2013. Replacement and positive evolution of subtype A and B respiratory syncytial virus G‐protein genotypes from 1997–2012 in South Africa. J Infect Dis 208:S227–S237. [DOI] [PubMed] [Google Scholar]
- Prifert C, Streng A, Krempl CD, Liese J, Weissbrich B. 2013. Novel re spiratory syncytial virus a genotype, Germany, 2011‐2012. Emerg Infect Dis 19:1029–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren L, Xia Q, Xiao Q, Zhou L, Zang N, Long X, Xie X, Deng Y, Wang L, Fu Z, Tian D, Zhao Y, Zhao X, Li T, Huang A, Liu E. 2014. The genetic variability of glycoproteins among respiratory syncytial virus subtype A in China between 2009 and 2013. Infect Genet Evol 27:339–347. [DOI] [PubMed] [Google Scholar]
- Roberts SR, Lichtenstein D, Ball LA, Wertz GW. 1994. The membrane‐associated and secreted forms of the respiratory syncytial virus attachment glycoprotein G are synthesized from alternative initiation codons. J Virol 68:4538–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabatabai J, Prifert C, Pfeil J, Grulich‐Henn J, Schnitzler P. 2014. Novel respiratory syncytial virus (RSV) genotype ON1 predominates in Germany during winter season 2012–13. PLoS ONE 9:e109191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trento A, Abrego L, Rodriguez‐Fernandez R, Gonzalez‐Sanchez MI, Gonzalez‐Martinez F, Delfraro A, Pascale JM, Arbiza J, Melero JA. 2015. Conservation of G‐protein epitopes in respiratory syncytial virus (Group A) despite broad genetic diversity: Is antibody selection involved in virus evolution? J Virol 89:7776–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trento A, Galiano M, Videla C, Carballal G, Garcia‐Barreno B, Melero JA, Palomo C. 2003. Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J Gen Virol 84:3115–3120. [DOI] [PubMed] [Google Scholar]
- Tsukagoshi H, Yokoi H, Kobayashi M, Kushibuchi I, Okamoto‐Nakagawa R, Yoshida A, Morita Y, Noda M, Yamamoto N, Sugai K, Oishi K, Kozawa K, Kuroda M, Shirabe K, Kimura H. 2013. Genetic analysis of attachment glycoprotein (G) gene in new genotype ON1 of human respiratory syncytial virus detected in Japan. Microbiol Immunol 57:655–659. [DOI] [PubMed] [Google Scholar]
- Venter M, Madhi SA, Tiemessen CT, Schoub BD. 2001. Genetic diversity and molecular epidemiology of respiratory syncytial virus over four consecutive seasons in South Africa: Identification of new subgroup A and B genotypes. J Gen Virol 82:2117–2124. [DOI] [PubMed] [Google Scholar]
- Yu X, Kou Y, Xia D, Li J, Yang X, Zhou Y, He X. 2015. Human respiratory syncytial virus in children with lower respiratory tract infections or influenza‐like illness and its co‐infection characteristics with viruses and atypical bacteria in Hangzhou, China. J Clin Virol 69:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. The N‐glycosylation potential of (A) CC‐16‐43‐Mar‐2015, (B) BJ/55258‐May‐2014 and (C) BJ/39979‐Nov‐2012.