

Abstract
Reverse transcription of retroviral RNA into linear double-stranded DNA and its integration into the host cell genome are essential steps in the retroviral life cycle. The nonhomologous end-joining (NHEJ) DNA repair pathway has been implicated in protecting cells from retrovirus-induced apoptosis caused by strand breaks in host cell DNA or unintegrated linear viral DNA. In eukaryotes, both the NHEJ and homologous recombination (HR) pathways play important roles in repairing DNA double-strand breaks. Here we show that the HR repair protein RAD52 modulates the outcome of recombinant HIV-l vector infection by markedly reducing the efficiency of productive integration events. Increased retroviral integration is the first major phenotype described for a RAD52 deficiency in mammalian cells. Mutations in other HR proteins (XRCC2, XRCC3 and BRCA2) do not markedly affect retroviral transduction rates, suggesting that the HR repair pathway per se does not influence retroviral infection. Instead, the mechanism of attenuation of retroviral infection by RAD52 appears to be based upon competition between the RAD52 protein and active integration complexes for the retroviral cDNA genome.
Keywords: DNA double-strand breaks, gene therapy, homologous recombination, RAD52, retrovirus
Introduction
The repair of DNA damage is an essential process by which cells maintain the integrity of their genome in order to prevent mutations, chromosomal aberrations and loss of genetic material. DNA double-strand breaks (DSBs) are particularly genotoxic forms of DNA damage because a single unrepaired DSB can lead to cell death via apoptosis (Huang et al, 1996; Rich et al, 2000). The repair of DNA DSBs can occur through either homologous recombination (HR) or nonhomologous end joining (NHEJ) (Van Gent et al, 2001; Jackson, 2002). Repair of DSBs by HR requires an undamaged homologous DNA sequence for use as a repair template to restore the integrity of the damaged DNA. Homology-directed DSB repair proceeds through either gene conversion or single-strand annealing (SSA) pathways (Paques and Haber, 1999; Johnson and Jasin, 2000). Proteins involved in HR pathways are the so-called RAD52 group of proteins that includes RAD52, RAD51 and its paralogues XRCC2, XRCC3, RAD51B, RAD51C and RAD51D (Thompson and Schild, 2001). In addition, the BRCA1 and BRCA2 proteins, encoded by genes associated with breast cancer predisposition, have also been implicated in HR-directed gene conversion pathways (Moynahan et al, 1999; 2001; Davies et al, 2001). Unlike HR, NHEJ does not require an undamaged DNA partner or extensive sequence homology but joins the broken ends of a DSB together. NHEJ repair requires the DNA-end-binding activity of the Ku70/80 heterodimer, its interacting partner—the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs)—and the complex between DNA ligase IV and XRCC4 (Smith and Jackson, 1999).
Reverse transcription and integration of viral double-stranded DNA into a host cell's genome are essential steps in the retroviral life cycle. These steps are mediated by the retroviral proteins reverse transcriptase (RT) and integrase (IN), respectively. After virus entry, the retroviral RNA is reverse transcribed by RT to produce a linear double-stranded DNA copy that includes directly repeated sequences at each end, known as long terminal repeats (LTRs). The linear retroviral cDNA is then 3′-recessed at both ends and joined to host cell chromosomal DNA via a direct trans-esterification reaction catalysed by IN. This strand transfer process results in short staggered DNA strand breaks in the host cell's DNA at the site of integration (Brown, 1997). Host cell DNA repair proteins must effectively repair these gapped DNA intermediates in order to complete the integration process. Several host cell DNA repair proteins and pathways, including the NHEJ DNA repair pathway (Daniel et al, 1999; Li et al, 2001; Jeanson et al, 2002), Poly(ADP-ribose)-polymerase (PARP; Gaken et al, 1996; Ha et al, 2001), ataxia telangiectasia mutated (ATM; Daniel et al, 2001), ATM-Rad3-related (ATR; Daniel et al, 2003) and RAD18 proteins (Mulder et al, 2002), have all been shown to be involved in the establishment of productive retroviral infections.
Loss of the NHEJ DNA repair pathway renders mammalian cells susceptible to apoptotic cell death following retroviral infection (Daniel et al, 1999; Li et al, 2001). Both host cell DNA damage caused by IN (Daniel et al, 1999) and excess retroviral double-stranded linear DNA resulting from RT activity (Li et al, 2001) may act as triggers for retrovirally induced apoptosis in NHEJ-deficient cells. Recent evidence also suggests that the NHEJ DNA repair pathway is responsible for the production of retroviral 2-LTR DNA circles that are generated from double-stranded linear cDNA (Li et al, 2001; Jeanson et al, 2002). Consistent with the idea that NHEJ plays an important role in retroviral infections is the finding that Ku, a key protein in initiating NHEJ repair, is physically associated with the Ty1 retrotransposon (Downs and Jackson, 1999) and retroviral preintegration complexes (Li et al, 2001; Lin and Engelman, 2003).
Although the involvement of the NHEJ DNA repair pathway in retroviral integration has been previously described, there is still little understanding of the role that might be played by HR-directed repair, the other major DNA DSB repair pathway. Here, we investigate both the role of the HR pathway in retroviral infection and the interplay between proteins associated with the HR and NHEJ pathways.
Results
Loss of RAD52 (but not other HR proteins) enhances retroviral infection
In mammalian cells, RAD51 is thought to be a critical component of the HR pathway and unlike the situation in yeast, disruption of RAD51 in mammalian cells results in cellular lethality (Lim and Hasty, 1996; Tsuzuki et al, 1996; Sonoda et al, 1998). This unfortunately complicates any detailed analysis of the potential role of RAD51 in retroviral integration. However, mammalian cells also contain several paralogues and regulators of RAD51 function. Paralogues of RAD51 include the XRCC2 and XRCC3 proteins (Thompson and Schild, 2001), while the breast cancer-associated protein BRCA2 has been implicated in mediating HR through RAD51 regulation (Davies et al, 2001; Moynahan et al, 2001). Unlike RAD51 deficiency, mammalian cells are available that are impaired in XRCC2, XRCC3 and BRCA2 function and these cells are viable but show significantly reduced HR efficiency, chromosomal instability and associated sensitivity to DNA-damaging agents (Takata et al, 2001; Thompson and Schild, 2001).
To determine what role, if any, the HR pathway plays in regulating mammalian retroviral integration, we used cells impaired in XRCC2, XRCC3 or BRCA2 and infected them with HIV-1 luciferase reporter gene vectors before determining transduction efficiencies using luciferase activity as a readout (see Supplementary information). Figure 1A and B demonstrate little if any effect on retroviral transduction efficiencies with the loss of these key components of the mammalian HR pathway. By contrast, cells deficient in RAD52 show a 16-fold increase in transduction rates (Figure 1C). The HIV luciferase transduction assay results of isogenic mouse embryonic stem (ES) cells with targeted deletions of none (Rad52+/+), one (Rad52+/−) or both (Rad52−/−) Rad52 alleles also demonstrated that HIV-1 integration efficiency was sensitive to Rad52 gene dosage, since the deletion of one Rad52 allele also resulted in a 10-fold increase in transduction efficiency. Further evidence that RAD52 inhibits retroviral infection and that RAD52 deficiency leads to an increase in retroviral transduction was provided by RNA interference (RNAi) gene knockdown experiments using small interfering RNAs (siRNAs; Elbashir et al, 2001). A RAD52 siRNA was used to knock down RAD52 expression in HeLa cells (see Supplementary information) before HIV-1 luciferase transduction assays were performed. Figure 1D shows that RAD52 siRNA-transfected cells demonstrate an increase in HIV-1 vector transduction efficiency when compared to cells transfected with a nonspecific control siRNA. These data are consistent with those seen for Rad52 knockout mouse ES cells (Figure 1C), although the relative increase in HIV-1 transduction efficiency is much lower at only two- to three-fold over and above the control cells. This discrepancy could be due, in part, to the fact that siRNA knockdown is not 100% complete (see Supplementary information) with residual protein and activity remaining.
Figure 1.
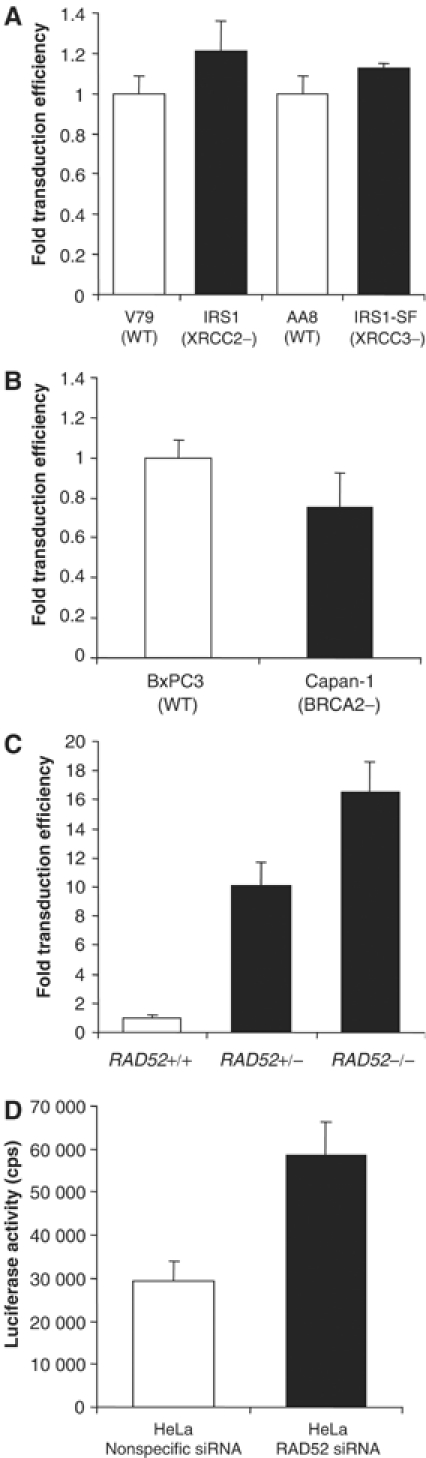
RAD52 (but not HR in general) is involved in modulating retroviral infection. (A) HIV-1 transduction assay results for XRCC2 (IRS1)- and XRCC3 (IRS1-SF)-defective hamster cell lines infected with HIV-1 luciferase retrovirus stocks. (B) HIV-1 luciferase transduction assays of BRCA2-deficient (Capan-1) human pancreatic cells. (C) HIV-1 luciferase transduction assay results for RAD52-deficient mouse ES cells. (D) HIV-1 luciferase transduction assay results for HeLa cells transfected with the siRNAs directed against RAD52. The assay itself is described in more detail in Supplementary information.
Overexpression of RAD52 inhibits HIV-1 infection and stable integration
Since the absence of RAD52 increases retroviral transduction, we wondered if its overexpression would have the converse effect. We generated a number of stably transfected HeLa cell lines containing either vector DNA or vector DNA expressing HA epitope-tagged RAD52. Overexpression of RAD52 was confirmed by immunoblot analysis of two independently isolated cell lines containing only vector DNA, and five cell lines containing the HA-RAD52-expressing vector. Both HA and RAD52 antibodies detected the overexpressed HA-RAD52 protein (Figure 2A; antibodies against Ku70, Ku80 and β-actin served as controls). The level of endogenous RAD52 protein is very low and could not be detected efficiently with our antibody. HIV-1 luciferase transduction assays revealed that retroviral transduction is equally efficient in HeLa and vector-only-transfected cell lines. However, in the HeLa cell lines overexpressing RAD52, transduction is reduced approximately five-fold (Figure 2A).
Figure 2.
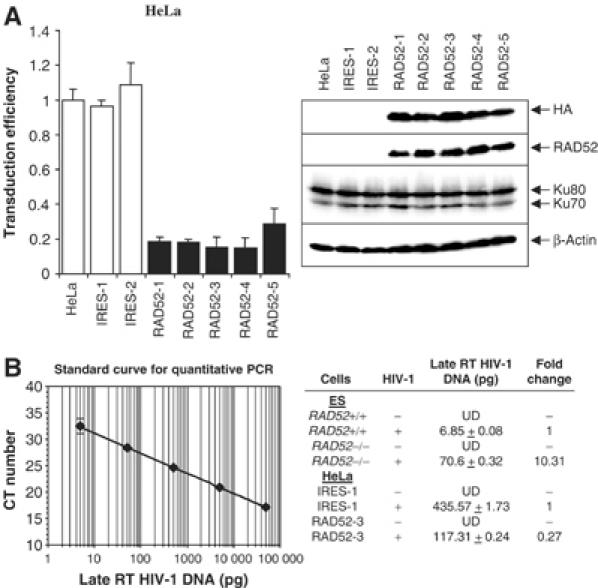
Overexpression of RAD52 impairs retroviral integration. (A) Parental HeLa cells (HeLa) and clones stably transfected with vector DNA only (IRES-1 and -2) or stably overexpressing HA-RAD52 (RAD52-1 through -5) infected with HIV-1 luciferase retroviral stocks. HIV-1 luciferase transduction assay results are shown in the left graph and immunoblot analysis of the HeLa cell clones are on the right. Luciferase transduction assay results are expressed as luciferase activity relative to parental HeLa cells. Western blots of HeLa cell clones were performed using antibodies raised against the HA tag epitope, RAD52, Ku70, Ku80 and β-actin (loading control). (B) Quantitative real-time PCR analysis of late RT HIV-1 DNA isolated from Rad52−/− ES cells and HeLa cell clones overexpressing HA-RAD52 at 21 days after infection. The standard curve for quantification was generated from HeLa cell DNA isolated 24 h after infection (left panel). The amount of late RT HIV-1 DNA in each cell line (pg per 50 ng genomic DNA), determined from the standard curve, is shown in the right panel. UD: undetectable.
To determine if the effect of RAD52 was due to the impairment of stable integration or through some other process, we transduced both Rad52−/− ES cells and RAD52-overexpressing HeLa clones and their control cells with the HIV-1 luciferase vectors and propagated the cells for 21 days before extracting high-molecular-weight genomic DNA and determining the level of proviral DNA by quantitative PCR (qPCR). Analysis of HIV-1 DNA content was carried out by qPCR of late HIV-1 RT products (Butler et al, 2001) in the transduced ES cells and shows that Rad52−/− cells contain approximately 10-fold more HIV-1 DNA than Rad52+/+ cells (Figure 2B). In contrast, the RAD52-overexpressing HeLa clone (RAD52-3) shows a four-fold reduction in HIV-1 DNA compared to control cells (IRES-1). qPCR was also performed using primers that detect unintegrated 2-LTR HIV-1 DNA (Butler et al, 2001). 2-LTR sequences were undetectable (see Supplementary information), indicating that the late HIV-1 RT DNA amplified in Figure 2B corresponds to integrated proviral DNA. Quantitative HIV-Alu PCR (Butler et al, 2001) was not used to determine proviral DNA content since mouse DNA does not contain Alu sequences. These qPCR results are in line with those seen for the HIV luciferase transduction assays (Figures 1C and 2A) and indicate that RAD52 acts primarily through impairing stable HIV-1 integration events.
The DNA-binding activity of RAD52 is required to inhibit HIV-1 transduction
The function of RAD52 in HR requires its interaction with both RPA and RAD51 (Paques and Haber, 1999). If the observed effect of RAD52 on retroviral transduction is indeed independent of its role in HR, then the domain of RAD52 required for interaction with RPA and RAD51 should be dispensable for the repression of transduction.
Two different internal deletion mutants of RAD52 were tested for their ability to modulate retroviral transduction. The first lacks amino-acid residues 43–177, which span the RAD52 multimerisation and DNA-binding domains (Figure 3A; Shen et al, 1996a; 1996b). The second mutant lacks residues 195–347, which encompass the RPA and RAD51 interaction domains (Park et al, 1996). Plasmids expressing full-length RAD52 or either of the two deletion mutants were transiently transfected in 293 cells and the expression of the RAD52 derivative was confirmed by immunoblotting (Figure 3B). HIV-1 luciferase transduction assays demonstrated that only the DNA-binding and self-interaction domains of RAD52, and not the RPA and RAD51 interaction domains, were required for the repression of retroviral transduction (Figure 3B). Since the loss of the RPA interaction domain has previously been shown to abolish the ability of RAD52 to stimulate HR (Park et al, 1996), this is consistent with data in Figure 1A and B, which suggest that HR repair activities do not directly influence the course of retroviral infections. However, the requirement for the DNA-binding activity of RAD52 suggests that a physical DNA interaction, possibly with HIV-1 DNA, is necessary to elicit the observed inhibitory effects of RAD52 on retroviral transduction.
Figure 3.
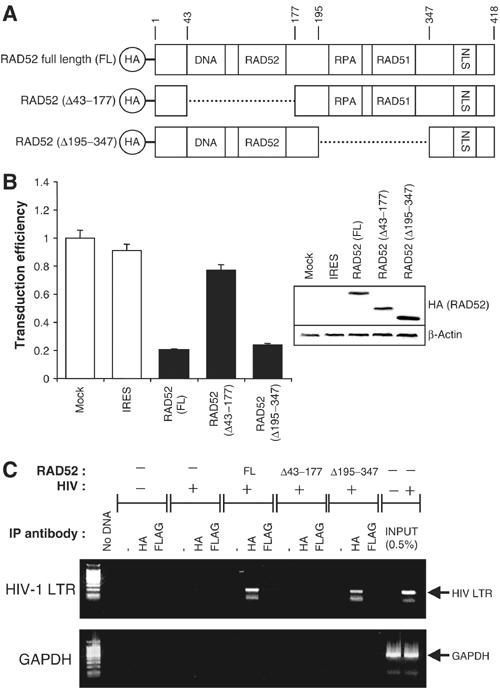
The DNA-binding domain of RAD52 is required for inhibition of retroviral infection. (A) Schematic of HA-tagged RAD52 and deletion mutants showing functional domains (DNA: DNA-binding domain; RAD52: RAD52 self-association domain; RPA: RPA-binding domain; RAD51: RAD51-binding domain; NLS: nuclear localisation signal, with amino-acid residues numbered). (B) 293 cells transiently transfected with full-length (FL) RAD52 and RAD52 deletion mutant expression plasmids subsequently infected with HIV-1 luciferase retroviral stocks. HIV-1 luciferase transduction results (left) and immunoblot analysis of cell lysates (right) are shown. Results are expressed as luciferase activity relative to untransfected 293 cells. Immunoblots were performed using anti-HA tag antibodies before re-probing for β-actin (loading control). (C) Chromatin immunoprecipitation (ChIP) analysis of 293 cells transiently transfected with FL-RAD52 or RAD52 deletion mutant expression plasmids and infected with HIV-1 luciferase retroviral stocks. Levels of HIV-1 DNA physically associated with HA-RAD52 were determined by immunoprecipitation with anti-HA antibodies and PCR performed using primers against HIV-1 LTR sequences (upper panel) or genomic GAPDH sequences (lower panel). Nonspecific control immunoprecipitations were performed using either no antibody (−) or an IgG1 isotype control anti-FLAG tag antibody. For reference, 0.5% of the total amount of HIV-1 LTR DNA formed during a typical infection is also shown (INPUT).
To test whether RAD52 can bind to HIV-1 DNA, chromatin immunoprecipitation (ChIP) assays were performed on 293 cells transiently transfected with RAD52 expression plasmids and transduced with the HIV-1 luciferase vector (Figure 3C). Immunoprecipitations using whole-cell extracts were carried out with antibodies against HA-RAD52 and with nonspecific controls (no antibody and isotype IgG1 anti-FLAG antibody). Associated DNA was then detected in a semiquantitative manner using PCR for HIV-1 LTR or genomic GAPDH gene sequences. Figure 3C demonstrates that full-length RAD52 is indeed associated with HIV-1 DNA, since HIV-1 LTR DNA sequences were readily amplified from HA ChIPs but not from nonspecific control ChIPs. As predicted, the RAD52 Δ43–177 DNA-binding mutant shows a greatly reduced affinity for HIV-1 DNA, whereas this is not the case for the Δ195–347 mutant. GAPDH DNA was not readily amplified in any ChIP, demonstrating the specificity of association of RAD52 with HIV-1 LTR DNA.
RAD52 competes with Ku for binding to HIV-1 LTR DNA
Since we had observed a gene dosage effect of RAD52 on retroviral transduction efficiency in the mouse ES cells (Figure 1C), we investigated whether the level of RAD52 protein could also influence the repression of transduction in human cells. 293 cells were transiently transfected with increasing amounts of RAD52 expression plasmid DNA, and immunoblot analysis of the cells demonstrated that RAD52 protein levels correlated with the amount of transfected plasmid (Figure 4A). In HIV luciferase transduction assays, we were able to demonstrate that the efficiency of retroviral transduction does indeed decrease in correlation with increasing amounts of RAD52 protein (Figure 4A). The gene dosage effect of RAD52 on retroviral transduction (Figures 1C and 4A), coupled with the observation that only RAD52 DNA binding is required to elicit this effect (Figure 3), suggests a possible mechanism of inhibition involving competition for the binding of HIV-1 cDNA intermediates between RAD52 and proteins involved in HIV-1 integration.
Figure 4.
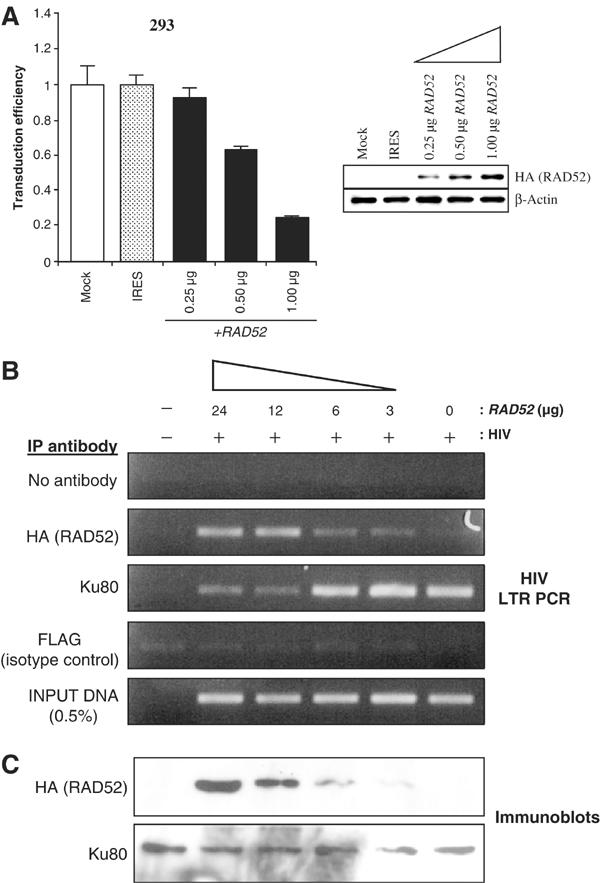
RAD52 can compete with Ku for binding to HIV-1. (A) 293 cells transiently transfected with an increasing amount of HA-RAD52 expression plasmid, then infected with HIV-1 luciferase retroviral stocks. HIV-1 luciferase transduction assay results are shown (left) along with immunoblot analysis of cell lysates (right). Results are expressed as luciferase activity relative to untransfected 293 cells. Immunoblots were performed using anti-HA tag antibodies before re-probing for β-actin (loading control). (B) Competitive PCR–ChIP analysis of 293 cells transiently transfected with increasing amounts of HA-RAD52 expression plasmid and infected with HIV-1 luciferase retroviral stocks. Results show HIV-1 LTR PCRs for ChIPs using antibodies against the HA-tag, Ku80 and nonspecific controls (no antibody (−) and IgG1 isotype control anti-FLAG tag antibody). PCR amplifications were normalised to the amount of HIV-1 LTR DNA used per ChIP (INPUT DNA). (C) Immunoblot analysis of input extracts used to perform the ChIP assays. The amount of HA-RAD52 or Ku80 protein used per ChIP analysis was determined by immunoblotting using anti-HA tag or anti-Ku80 antibodies.
One such competing protein might be Ku. A number of studies have shown that Ku and NHEJ DNA repair are required for efficient retroviral transduction (Daniel et al, 1999; Li et al, 2001; Jeanson et al, 2002) and that Ku is physically associated with retroviral preintegration complexes (Li et al, 2001; Lin and Engelman, 2003), presumably bound to cDNA ends. The ability of RAD52 to inhibit retroviral transduction could, therefore, be due to competition with Ku for viral cDNA.
To test this possibility, competitive PCR–ChIP assays were performed on 293 cells transfected with increasing amounts of RAD52 expression plasmid DNA and transduced with the HIV-1 luciferase vector. For all transfections, the total amount of DNA was kept constant (24 μg) by adding vector-only plasmid DNA. ChIPs were performed using antibodies against Ku80 and HA-tagged RAD52, and the amount of HIV-1 LTR DNA immunoprecipitated by each antibody was assessed by PCR (Figure 4B). This analysis demonstrates that in cells with high levels of RAD52 protein expression there is a much greater amount of RAD52 associated with HIV-1 LTR DNA (anti-HA ChIPs) compared with those with low levels of RAD52 protein expression. Importantly, anti-Ku80 ChIPs reveal the opposite finding, with cells expressing high levels of RAD52 demonstrating significantly reduced levels of Ku80-associated HIV-1 LTR DNA compared to cells with low RAD52 levels. The immunoblots of input extracts using HA and Ku80 antibodies, shown in Figure 4C, confirm that RAD52 protein expression levels correlate with the amount of expression plasmid transfected and that Ku80 expression remains unaffected. Together, these data support the hypothesis that RAD52 competes with Ku for binding to HIV-1 LTRs.
RAD52 suppresses 2-LTR circle DNA formation but does not enhance apoptosis
Previously, it has been established that proteins involved in NHEJ, including Ku, are required for the formation of retroviral 2-LTR circle DNA (Li et al, 2001; Jeanson et al, 2002). Since the absence of RAD52 and Ku has opposing effects on retroviral transduction efficiency, we assessed the efficiency of 2-LTR circle DNA formation (a possible indicator of NHEJ activity) in cells expressing different amounts of RAD52. The relative amounts of 2-LTR circle DNA were estimated by semiquantitative PCR analyses of DNA extracted from both Rad52+/+ or Rad52−/− ES cells infected with HIV-1 (Figure 5A) and on HIV-1-infected 293 cells transfected with expression plasmids for full-length RAD52 or RAD52 Δ43–177 DNA-binding-deficient mutant (Figure 5B). Analysis of mouse ES cells (Figure 5A) shows that 2-LTR circle DNA appeared at a similar rate in both Rad52+/+ and Rad52−/− cells at early times postinfection (4 h) but then accumulated to higher levels in Rad52−/− cells at later time points (particularly noticeable after 18 h). The 2-LTR circle DNA was also maintained at peak levels for longer (18–24 h) in Rad52−/− cells. By contrast, in 293 cells overexpressing full-length RAD52 (Figure 5B), 2-LTR circle DNA formation was significantly impaired, but not so in cells overexpressing the RAD52 Δ43–177 mutant that is incapable of repressing retroviral transduction (Figure 3). These data therefore indicate that the loss or gain of RAD52 can lead to the modulation of HIV-1 2-LTR circle DNA formation.
Figure 5.
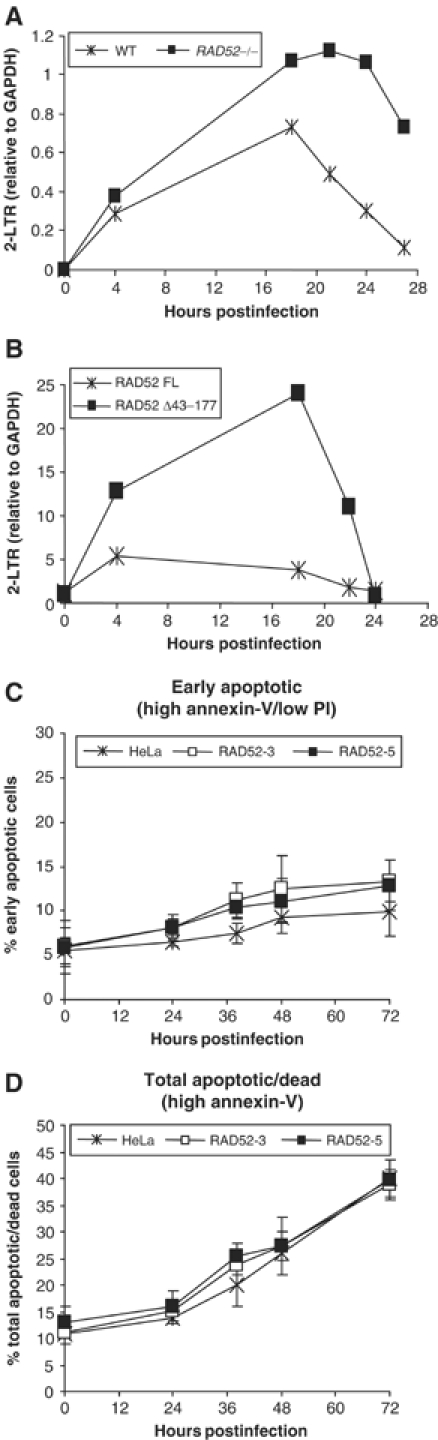
2-LTR circle DNA formation but not apoptosis is modulated by RAD52 expression. Semiquantitative PCR analysis of DNA extracted from HIV-1-infected cells with different levels of RAD52 expression. (A) 2-LTR PCR analysis of wild-type (Rad52+/+; 1B10) and Rad52−/− mouse ES cells. (B) 2-LTR PCR analysis of 293 cells transfected with either full-length HA-RAD52 (RAD52 FL) or DNA-binding deletion mutant HA-RAD52 (RAD52 Δ43–177) expression plasmids. All PCR quantification results are expressed as a normalised ratio of 2-LTR DNA:GAPDH control DNA. HeLa and two RAD52-overexpressing stable cells clones (RAD52-3 and RAD52-5) were infected with HIV-1 and apoptosis was quantified by annexin-V staining and flow cytometry. Live/dead cell discrimination was also performed by counter-staining with propidium iodide (PI). (C) The percentage of cells undergoing the early stages of apoptosis at increasing time points after infection with HIV-1 luciferase retrovirus stocks. (D) The percentage of both dead and apoptotic cells (all high annexin-V-stained cells) at increasing time points after HIV-1 luciferase retrovirus infection.
In addition to affecting HIV-1 2-LTR circle formation, Ku and the NHEJ apparatus have been implicated in protecting cells from retrovirus-induced apoptosis (Daniel et al, 1999; Li et al, 2001). In the study by Li et al (2001) it was suggested that 2-LTR circle DNA formation mediated by NHEJ could be directly responsible for protection from apoptosis, since it may remove potentially proapoptotic retroviral cDNA ends formed after reverse transcription. If this model is correct, then we might expect a correlation between RAD52 inhibition of 2-LTR circle DNA formation and increased apoptosis of HIV-1-infected cells. HIV-1 vector-induced apoptosis was assessed for both HeLa cells and RAD52-overexpressing HeLa clones RAD52-3 and -5. The proportion of cells undergoing apoptosis was determined by annexin-V staining and the proportion of live/dead cells was determined by propidium iodide (PI) counter-staining. Our results show that the extent of apoptosis between HeLa and the RAD52-overexpressing clones was not significantly different (Figure 5C and D). Both early-stage apoptosis (high annexin-V but low PI staining) and the total apoptotic/dead (all annexin-V-positive cells) populations were not increased in either of the two RAD52-overexpressing HeLa clones. To confirm these results, apoptosis/cell death was also assessed using caspase-activation assays and trypan blue staining. Equivalent results were obtained (data not shown) indicating no demonstrable differences between the cell lines. Similar apoptosis analyses using 293 cells transiently transfected with the RAD52 expression plasmid or with Rad52−/− mouse ES cells also failed to show any major differences in HIV-1 vector-induced apoptosis when compared to control cells (data not shown). These results therefore indicate that despite the effect on retroviral transduction efficiency and NHEJ-mediated 2-LTR circle DNA formation, RAD52 does not appear to influence HIV-1-induced apoptosis.
Discussion
The data presented have shown that the HR protein RAD52 suppresses retroviral infection in mammalian cells. Retroviral transduction can be increased by as much as 16-fold in the absence of RAD52. Previously, no overt phenotype resulting from the lack of RAD52 had been detected in mammalian cells and the results presented here represent the most dramatic in vivo effect of RAD52 inactivation to date. The absence of Rad52 in mice, for example, does not result in DNA damage sensitisation, cancer predisposition or defects associated with meiotic recombination (Rijkers et al, 1998). In fact, the only HR defect detected in Rad52−/− mouse ES cells was a modest (1.5-fold) reduction in homologous gene targeting. Although no overt DNA repair phenotypes are associated with RAD52 deficiency in mammalian cells, genetic and biochemical experiments have revealed that RAD52 plays a major role in HR (Kanaar and Hoeijmakers, 1998). Our results indicate, however, that the HR function of RAD52 is not involved in attenuating retroviral infection because other proteins required for HR, such as XRCC2, XRCC3 and BRCA2, do not greatly influence the outcome of HIV-1 infections, either positively or negatively (Figure 1). Thus, the HR DSB repair pathway is superfluous with respect to establishing productive retroviral integration. Two other observations further support this notion. Firstly, the domain of RAD52 responsible for interaction with other HR proteins is not required for the suppression of retroviral infection. Instead, this activity resides in the DNA-binding and RAD52 self-interaction domain (Figure 3). Secondly, suppression can be achieved by the overexpression of RAD52 alone and occurs in a dose-dependent manner (Figure 4).
Analysis of 2-LTR circle DNA during retroviral infection, a process that requires Ku and NHEJ repair (Li et al, 2001; Jeanson et al, 2002), shows that its formation can be modulated by the levels of RAD52 expression (Figure 5). These observations, coupled with the demonstration that RAD52 can compete with and effectively displace Ku from the ends of HIV-1 cDNA (Figure 4B), indicate that RAD52 can interfere with Ku-directed NHEJ repair activity. The exact mechanism of this competition, however, is far from clear. Although it has been proposed that RAD52 may directly compete with Ku for DNA ends (Van Dyck et al, 1999), it is also possible that single-stranded DNA character at the ends of the retroviral LTRs could mean that RAD52 has an indirect effect on Ku binding. Without knowing the exact nature of retroviral LTR DNA in infected cells and the way in which RAD52 and Ku proteins bind to the LTRs, it is difficult to distinguish between these possibilities.
Previous studies have shown that Ku deficiency is associated with both the loss of 2-LTR circle DNA formation (Li et al, 2001; Jeanson et al, 2002) and increased apoptosis (Daniel et al, 1999; Li et al, 2001) following retroviral infection. It has been speculated that the formation of 2-LTR circle DNA and the induction of apoptosis are directly linked. One NHEJ-dependent cell survival model (Li et al, 2001) argues for a direct physical elimination mechanism whereby linear retroviral cDNA ends that represent proapoptotic signals are removed through circularisation by the Ku-dependent NHEJ pathway, thereby protecting the host cell from apoptosis. In a simple extension of this model, the prevention of Ku binding to the retroviral LTRs, either through the absence of Ku or alternatively when RAD52 competes with Ku for retroviral LTR DNA, should result in an abrogation of NHEJ activity, a decrease in the number of 2-LTR DNA circles observed and apoptosis of the host cell resulting in a nonproductive infection. This simple RAD52–Ku competition model for retroviral suppression, while attractive, is not consistent with our experimental observations. Significantly, we have been unable to demonstrate any substantial differences in the level of HIV-1-induced cell killing in either Rad52−/− or in RAD52-overexpressing cells (Figure 5), as might be predicted. This means that although RAD52 affects the end-joining activity of Ku, it does not interfere with Ku-dependent protection from retrovirus cDNA-induced apoptosis. This disparity can only be explained if either the removal of the proapoptotic linear retroviral cDNA through LTR circle formation is not required for apoptosis suppression or linear retroviral DNA can be removed by alternative pathways. The possibility that LTR circle DNA formation and apoptosis suppression are not directly linked has been suggested as an alternative model in which to explain earlier (Li et al, 2001) and more recent (Kilzer et al, 2003) observations. Certainly, the data presented here suggest that the role of Ku and the NHEJ pathway in preventing HIV-1-induced apoptosis is, in fact, more complex than was initially envisaged.
The findings presented here therefore require the invocation of an alternative hypothesis in order to explain the RAD52 suppression of retroviral integration, because the inhibitory activity of RAD52 on HIV-1 infection cannot be explained through the modulation of cell survival. Our data suggest that HIV-1 transduction is influenced as a direct consequence of the binding of RAD52 to HIV-1 cDNA and that this binding correlates with a decrease in the number of stable integration events. The simplest explanation therefore is that RAD52 can suppress HIV-1 integration. Although beyond the scope of this study, it could be envisaged that RAD52 may prevent the association of other retroviral cDNA-binding proteins, in addition to Ku, that form part of the preintegration complex (Bowerman et al, 1989). Candidate proteins that might be affected include integration cofactors such as Ini1 (Kalpana et al, 1994), HMG I(Y) (Farnet and Bushman, 1997), BAF (Lee and Craigie, 1998) as well as the viral IN or RT proteins themselves. In addition, the postreplication/translesion DNA repair protein RAD18 has been shown to interact with HIV-1 IN (Mulder et al, 2002) and studies in yeast have shown that DNA repair pathway switching involving the Srs2 helicase can be modulated by both RAD18 and RAD52 (Broomfield et al, 2001). These observations therefore provide a possible link between RAD52, RAD18 and HIV-1 integration.
Based on our data, we propose a model in which suppression of retroviral infection by RAD52 may be due to the effective capping of retroviral cDNA ends by RAD52 such that the loading or recruitment of other proteins or complexes required for efficient reverse transcription or integration is blocked. In this model, the major influence of RAD52 is through inhibition of integration and not through the enhancement of apoptosis. This model is only likely to be valid, however, if there are differences between the binding characteristics of Ku and RAD52, such that Ku can bind to the viral cDNA while still accommodating (or perhaps enhancing) the binding of RT or IN and its associated cellular cofactors, while RAD52 inhibits cofactor binding. In this regard, it is notable that multiple Ku subunits can load onto and translocate along the DNA strand (Yoo and Dynan, 1999), which would be potentially compatible with the recruitment of additional proteins, such as IN, to the ends of the viral cDNA. Also consistent with this idea are data demonstrating that Ku does not affect the cleavage or strand-transfer activities of the IN protein in vitro (Li et al, 2001). The competitive involvement of Ku and RAD52 proteins in retrovirus transduction suggests that DNA repair pathways may represent another cellular process that is hijacked by viruses to complete their infectious cycles. Modulation of NHEJ or HR DNA repair proteins therefore provides the potential to regulate viral infection. For example, by targeting the NHEJ pathway and its associated signalling pathways, it may be possible to repress retroviral infections. Alternatively, inhibition of RAD52 gene expression could lead to a considerable enhancement of retroviral vector-based gene transduction, significantly increasing the potential of retroviral-based gene therapy.
Materials and methods
Expression vectors
RAD52 cDNA was cloned in-frame with an HA epitope in pIRESneo2 (Clontech) to yield the expression plasmid pIREsneo2-HA-RAD52. RAD52 deletion mutants Δ43–177 and Δ195–347 were made by digestion of RAD52 cDNA with Bsu36I–HinDIII and BglII–XbaI respectively, then blunt-ending with Klenow DNA polymerase before re-ligation. These deletions maintained the reading frame of RAD52.
Antibodies
RAD52 antibody (H-300) was obtained from Santa Cruz Biotechnology; HA antibodies from Boehringer Mannheim (12CA5) and Sigma (HA7); the Ku70 (S5C11) and Ku80 (111) antibodies from NeoMarkers-Labvision Corp.; and the FLAG (M2) and β-actin (AC-15) antibodies from Sigma.
Cell lines
HeLa and 293 cells were grown in DMEM with 10% FBS (Invitrogen). Mouse ES cell line 1B10 (Rad52+/+) and its Rad52+/− and Rad52−/− derivatives (Rijkers et al, 1998) were grown on gelatinised dishes in DMEM with 15% FBS and 1000 U/ml leukaemia inhibitory factor (ESGRO, Chemicon International Inc.). V79 (XRCC2+) and the XRCC2-defective derivative IRS1 (Jones et al, 1995; Thacker et al, 1995), AA8 (XRCC3+) and its XRCC3-defective derivative IRS1-SF (Tebbs et al, 1995) hamster cell lines and the BRCA2-defective cell line Capan-1 (Abbott et al, 1998) and BxPC3 (BRCA2+) human pancreatic cell lines have been described previously. HeLa-IRES and HeLa-RAD52 stable clones were made by transfection of HeLa cells with pIRESneo2HA and pIRESneo2-HA-RAD52 plasmids, respectively. Individual clones were expanded after selection with 500 μg/ml G418 (Invitrogen). RAD52-overexpressing clones were identified by immunoblot analysis with HA and RAD52 antibodies.
Recombinant HIV-1 vectors and retrovirus production
Recombinant HIV-1 retroviral stocks were produced by transient cotransfection of the HIV-1 gag/pol-packaging constructs LΔP2GPH (Haselhorst et al, 1998), HIV-1 luciferase transfer vector pHR'Luc and VSVG envelope expression plasmids (Naldini et al, 1996). HIV-1 viral titres were estimated using the HIV-1 p24 gag antigen ELISA kit (Beckman-Coulter).
Retroviral infection and luciferase assays
For HIV-1 luciferase transduction assays, cells were seeded in white 96-well plates and exposed to retroviral stocks at an MOI of 0.5. Luciferase activity, which will only occur after successful transduction (see Supplementary information), was quantified 48–72 h postaddition of virus on a Packard TopCount-NXT microplate scintillation counter using Bright-Glo luciferase assay reagent (Promega Corporation). For coupled, transient RAD52 transfection–retrovirus transduction assays, 293 cells were transfected with expression plasmid DNA using Lipofectamine-2000 reagent (Invitrogen). At 24 h post-transfection, cells were exposed to retroviral stocks at an MOI of 0.5. At 48 h postaddition of virus, cells were harvested in passive lysis buffer (PLB; Promega Corporation) and samples split for analysis of protein expression by immunoblot and HIV-1 infectivity by luciferase activity.
RAD52 siRNA knockdown experiments
RAD52 siRNA (target sequence: AAAGACUACCUGAGAUCACUA) and nonspecific control siRNA (AAATTCTATCACTAGCGTGAC) were synthesised and preduplexed by Dharmacon Inc. HeLa cells were transfected with 100 nM siRNA duplexes (see Supplementary information) before performing HIV-1 luciferase assays.
PCR analysis of HIV-1 cDNA forms
Infected cells were harvested in PBS with 100 μg/ml RNase A (Sigma) at room temperature for 10 min. Samples were heated at 95°C for 10 min and then digested with 300 μg/ml proteinase K at 56°C for 4 h. DNA was isolated by ethanol precipitation in the presence of yeast tRNA carrier. PCR analysis of HIV-1 cDNA forms using primers for 2-LTR circle DNA (primers LTR8 and LTR9) was performed as described by Naldini et al (1996). GAPDH PCR was performed with primer pairs GA3 (AGGTCCACCACCCTGTTGC) and GA5 (AGAAGACTGTGGATGGCCCC). All PCR reactions were limited to 20–26 cycles to ensure linearity of amplification. Bands were detected by Southern blot and hybridisation to 32P-labelled HIV-LTR or GAPDH cDNA probes (Rediprime kit, Amersham-Pharmacia). Bands were quantified by densitometry using the Cyclone phosphoimaging system (Packard). For some experiments, quantitative real-time PCR analysis of late HIV-1 DNA was performed as described by Butler et al (2001) using an ABI Prism 7000 Sequence Detection System. Here, late RT HIV-1 DNA was quantified from 50 ng of total genomic DNA in 25 μl reactions. PCR standards were obtained from serially diluted genomic DNA isolated from HeLa cells 24 h after transduction with retroviral stocks and made up to 50 ng in uninfected genomic DNA. Control qPCRs for unintegrated 2-LTR HIV-1 DNA and GAPDH (total genomic) DNA were also performed for each sample (see Supplementary information).
HIV-1 chromatin immunoprecipitations
293 cells were seeded in multiple 10-cm dishes and transfected with plasmid DNA using Lipofectamine-2000 reagent (Invitrogen). At 24 h post-transfection, cells were exposed to retroviral stocks at an MOI of 0.25. DNA–protein interactions were then fixed with formaldehyde (final concentration 1%) at 37°C for 10 min. Cells were sonicated four times in 10 s pulses in sonication buffer (50 mM Tris pH 8.0, 1% SDS, 10 mM EDTA, protease inhibitors). Immunoprecipitation of protein–DNA complexes was performed using 2–4 mg of cell supernatants per antibody that were diluted in IP buffer (20 nM Tris pH 8.0, 0.1% SDS, 2 mM EDTA, 1% Triton X-100, 50 mM NaCl, protease inhibitors) and precleared for 1 h with protein-G–Sepharose beads (Amersham-Pharmacia). Small aliquots for protein and DNA input samples were taken at this point. Antibodies were added (2–4 μg per mg of precleared cell supernatant) with 4°C overnight incubation. Protein-G–Sepharose beads were added and incubated for 2 h. Beads were washed twice in low-salt buffer (IP buffer+150 mM NaCl), twice in high-salt buffer (IP buffer+500 mM NaCl), once in LiCl wash buffer (10 mM Tris pH 8.0, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate, 250 mM LiCl) and twice in TE. Protein–DNA complexes were extracted in elution buffer (50 mM Tris pH 8.0, 1% SDS, 100 mM NaHCO3, 10 mM EDTA) and crosslinks reversed by adding 200 mM NaCl and heating at 65°C for 4 h. DNA was purified after proteinase K digestion, phenol:chloroform:IAA extraction and ethanol precipitation. HIV-1 DNA was detected by PCR using primers LTR5 and LTR6 (Naldini et al, 1996) as described.
Cell viability and apoptosis assays
Cells were exposed to retroviral stocks at an MOI of 10. Cells were harvested and apoptotic/dead cells were stained by incubation with annexin-V and PI (BD Pharmingen). The numbers of live (annexin-V/PI negative), early apoptotic (annexin-V positive/PI negative) and late apoptotic/dead (annexin-V/PI positive) cells were quantified by flow cytometric analysis using a BD FACScalibur and CellQuest software.
Supplementary Material
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3
Acknowledgments
We thank Albert Pastink for the Rad52+/− and Rad52−/− ES cells, Andrew Lever for the recombinant HIV-1 expression vectors, Fabrizio d'Adda di Fagagna for help with the ChIP assays, Sylvain Daujat and Tony Kouzarides for use of their real-time PCR machine and Mark Albertella for the pIRESneo2-HA expression vector.
References
- Abbott DW, Freeman ML, Holt JT (1998) Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst 90: 978–985 [DOI] [PubMed] [Google Scholar]
- Bowerman B, Brown PO, Bishop JM, Varmus HE (1989) A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev 3: 469–478 [DOI] [PubMed] [Google Scholar]
- Broomfield S, Hryciw T, Xiao W (2001) DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae. Mutat Res 486: 167–184 [DOI] [PubMed] [Google Scholar]
- Brown PO (1997) Integration. In Retroviruses, Coffin JM, Hughes SH, Varmus HE (eds) pp 161–203. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [PubMed] [Google Scholar]
- Butler SL, Hansen MS, Bushman FD (2001) A quantitative assay for HIV DNA integration in vivo. Nat Med 7: 631–634 [DOI] [PubMed] [Google Scholar]
- Daniel R, Kao G, Taganov K, Greger JG, Favorova O, Merkel G, Yen TJ, Katz RA, Skalka AM (2003) Evidence that the retroviral DNA integration process triggers an ATR-dependent DNA damage response. Proc Natl Acad Sci USA 100: 4778–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel R, Katz RA, Merkel G, Hittle JC, Yen TJ, Skalka AM (2001) Wortmannin potentiates integrase-mediated killing of lymphocytes and reduces the efficiency of stable transduction by retroviruses. Mol Cell Biol 21: 1164–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel R, Katz RA, Skalka AM (1999) A role for DNA-PK in retroviral DNA integration. Science 284: 644–647 [DOI] [PubMed] [Google Scholar]
- Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC (2001) Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell 7: 273–282 [DOI] [PubMed] [Google Scholar]
- Downs JA, Jackson SP (1999) Involvement of DNA end-binding protein Ku in Ty element retrotransposition. Mol Cell Biol 19: 6260–6268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Farnet CM, Bushman FD (1997) HIV-1 cDNA integration: requirement of HMG I(Y) protein for function of preintegration complexes in vitro. Cell 88: 483–492 [DOI] [PubMed] [Google Scholar]
- Gaken JA, Tavassoli M, Gan SU, Vallian S, Giddings I, Darling DC, Galea-Lauri J, Thomas MG, Abedi H, Schreiber V, Menissier-de Murcia J, Collins MK, Shall S, Farzaneh F (1996) Efficient retroviral infection of mammalian cells is blocked by inhibition of poly(ADP-ribose) polymerase activity. J Virol 70: 3992–4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha HC, Juluri K, Zhou Y, Leung S, Hermankova M, Snyder SH (2001) Poly(ADP-ribose) polymerase-1 is required for efficient HIV-1 integration. Proc Natl Acad Sci USA 98: 3364–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselhorst D, Kaye JF, Lever AM (1998) Development of cell lines stably expressing human immunodeficiency virus type 1 proteins for studies in encapsidation and gene transfer. J Gen Virol 79: 231–237 [DOI] [PubMed] [Google Scholar]
- Huang LC, Clarkin KC, Wahl GM (1996) Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc Natl Acad Sci USA 93: 4827–4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP (2002) Sensing and repairing DNA double-strand breaks. Carcinogenesis 23: 687–696 [DOI] [PubMed] [Google Scholar]
- Jeanson L, Subra F, Vaganay S, Hervy M, Marangoni E, Bourhis J, Mouscadet JF (2002) Effect of Ku80 depletion on the preintegrative steps of HIV-1 replication in human cells. Virology 300: 100–108 [DOI] [PubMed] [Google Scholar]
- Johnson RD, Jasin M (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 19: 3398–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NJ, Zhao Y, Siciliano MJ, Thompson LH (1995) Assignment of the XRCC2 human DNA repair gene to chromosome 7q36 by complementation analysis. Genomics 26: 619–622 [DOI] [PubMed] [Google Scholar]
- Kalpana GV, Marmon S, Wang W, Crabtree GR, Goff SP (1994) Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science 266: 2002–2006 [DOI] [PubMed] [Google Scholar]
- Kanaar R, Hoeijmakers JH (1998) Genetic recombination: from competition to collaboration. Nature 391: 335–338 [DOI] [PubMed] [Google Scholar]
- Kilzer JM, Stracker T, Beitzel B, Meek K, Weitzman M, Bushman FD (2003) Roles of host cell factors in circularization of retroviral DNA. Virology 314: 460–467 [DOI] [PubMed] [Google Scholar]
- Lee MS, Craigie R (1998) A previously unidentified host protein protects retroviral DNA from autointegration. Proc Natl Acad Sci USA 95: 1528–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Olvera JM, Yoder KE, Mitchell RS, Butler SL, Lieber M, Martin SL, Bushman FD (2001) Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J 20: 3272–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DS, Hasty P (1996) A mutation in mouse Rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol 16: 7133–7143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CW, Engelman A (2003) The barrier-to-autointegration factor is a component of functional human immunodeficiency virus type 1 preintegration complexes. J Virol 77: 5030–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M (1999) Brca1 controls homology-directed DNA repair. Mol Cell 4: 511–518 [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Pierce AJ, Jasin M (2001) BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell 7: 263–272 [DOI] [PubMed] [Google Scholar]
- Mulder LC, Chakrabarti LA, Muesing MA (2002) Interaction of HIV-1 integrase with DNA repair protein hRad18. J Biol Chem 277: 27489–27493 [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Paques F, Haber JE (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63: 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MS, Ludwig DL, Stigger E, Lee SH (1996) Physical interaction between human RAD52 and RPA is required for homologous recombination in mammalian cells. J Biol Chem 271: 18996–19000 [DOI] [PubMed] [Google Scholar]
- Rich T, Allen RL, Wyllie AH (2000) Defying death after DNA damage. Nature 407: 777–783 [DOI] [PubMed] [Google Scholar]
- Rijkers T, Van Den Ouweland J, Morolli B, Rolink AG, Baarends WM, Van Sloun PP, Lohman PH, Pastink A (1998) Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol Cell Biol 18: 6423–6429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Cloud KG, Chen DJ, Park MS (1996a) Specific interactions between the human RAD51 and RAD52 proteins. J Biol Chem 271: 148–152 [DOI] [PubMed] [Google Scholar]
- Shen Z, Peterson SR, Comeaux JC, Zastrow D, Moyzis RK, Bradbury EM, Chen DJ (1996b) Self-association of human RAD52 protein. Mutat Res 364: 81–89 [DOI] [PubMed] [Google Scholar]
- Smith GC, Jackson SP (1999) The DNA-dependent protein kinase. Genes Dev 13: 916–934 [DOI] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S (1998) Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 17: 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S (2001) Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 21: 2858–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbs RS, Zhao Y, Tucker JD, Scheerer JB, Siciliano MJ, Hwang M, Liu N, Legerski RJ, Thompson LH (1995) Correction of chromosomal instability and sensitivity to diverse mutagens by a cloned cDNA of the XRCC3 DNA repair gene. Proc Natl Acad Sci USA 92: 6354–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker J, Tambini CE, Simpson PJ, Tsui LC, Scherer SW (1995) Localization to chromosome 7q36.1 of the human XRCC2 gene, determining sensitivity to DNA-damaging agents. Hum Mol Genet 4: 113–120 [DOI] [PubMed] [Google Scholar]
- Thompson LH, Schild D (2001) Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat Res 477: 131–153 [DOI] [PubMed] [Google Scholar]
- Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T (1996) Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci USA 93: 6236–6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck E, Stasiak AZ, Stasiak A, West SC (1999) Binding of double-strand breaks in DNA by human Rad52 protein. Nature 398: 728–731 [DOI] [PubMed] [Google Scholar]
- van Gent DC, Hoeijmakers JH, Kanaar R (2001) Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2: 196–206 [DOI] [PubMed] [Google Scholar]
- Yoo S, Dynan WS (1999) Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res 27: 4679–4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3