

ABSTRACT
Chromatin is the nucleoprotein complex that protects and compacts eukaryotic genomes. It is responsible for a large part of the epigenetic control of transcription. The genomes of DNA viruses such as human cytomegalovirus (HCMV) are devoid of histones within virions but are chromatinized and epigenetically regulated following delivery to the host cell nucleus. How chromatin is initially assembled on viral genomes and which variant forms of the core histone proteins are deposited are incompletely understood. We monitored the deposition of both ectopically expressed and endogenous histones H3.1 and H3.2 (collectively, H3.1/2) and H3.3 during lytic and latent HCMV infections. Here, we show that they are deposited on HCMV genomes during lytic and latent infections, suggesting similar mechanisms of viral chromatin assembly during the different infection types and indicating that both canonical and variant core histones may be important modulators of infecting viral genomes. We further show that association of both H3.1/2 and H3.3 occurs independent of viral DNA synthesis or de novo viral gene expression, implicating cellular factors and/or virion components in the formation of chromatin on virion-delivered genomes during both lytic and latent infections.
IMPORTANCE It is well established that infecting herpesvirus genomes are chromatinized upon entry into the host cell nucleus. Why or how this occurs is a mystery. It is important to know why they are chromatinized in order to better understand cellular pathogen recognition (DNA sensing) pathways and viral fate determinations (lytic or latent) and to anticipate how artificially modulating chromatinization may impact viral infections. It is important to know how the genomes are chromatinized in order to potentially modulate the process for therapeutic effect. Our work showing that HCMV genomes are loaded with canonical and variant H3 histones during both lytic and latent infections strengthens the hypothesis that chromatinization pathways are similar between the two infection types, implicates virion or cellular factors in this process, and exposes the possibility that histone variants, in addition to posttranslational modification, may impact viral gene expression. These revelations are important to understanding and intelligently intervening in herpesvirus infections.
INTRODUCTION
The large genomes of eukaryotic cells must be highly compacted in order to fit within the restricted volume of the nucleus. To accomplish this, eukaryotic DNA is packaged into a repeating nucleoprotein structure known as chromatin, the basic subunit of which is the nucleosome (1, 2). The core nucleosome particle consists of approximately 146 bp of DNA wrapped nearly twice around a histone hetero-octamer consisting of two copies each of the four core histone proteins H2A, H2B, H3, and H4 (3). Nucleosomes are further compacted into higher-order chromatin fibers through the incorporation of the linker histone H1, other nonhistone proteins, and structural RNA components (4–6).
Wrapping DNA in nucleosomes compacts, protects, and organizes genomes. However, nucleosomes are intrinsically inhibitory to processes requiring access to the underlying DNA template such as transcription, replication, and repair (7–10). Therefore, chromatin and nucleosomes, in particular, must be highly dynamic (11). The dynamic nature of nucleosomes is achieved through chromatin-remodeling factors, the posttranslational modification (PTM) of histones, and the incorporation of histone variants (12, 13). ATP-dependent chromatin remodeling factors such as SWI/SNF, ISWI, and NuRD family complexes remove or reposition nucleosomes along DNA, allowing access to the underlying sequence (14). Histone PTMs include methylation, phosphorylation, acetylation, and monoubiquitination, among others (15, 16). Many histone PTMs correlate with different transcriptional states and define binding sites for chromatin-associated factors according to the histone code hypothesis (15, 17, 18).
In addition to positioning and PTMs, the incorporation of sequence variants of core histone proteins can have profound effects on chromatin structure and dynamics (12, 13, 19). Most histones in dividing cells are canonical histone proteins expressed from tandem gene arrays during S phase and are deposited onto chromatin concurrent with DNA synthesis (20–22). In contrast, nonallelic sequence variants of core histones are constitutively expressed throughout the cell cycle from single- or low-copy-number genes and are incorporated into chromatin largely through replication-independent mechanisms (20, 23–26). Major H2A variants include H2A.Z, H2A.X, and macroH2A that play specialized roles in transcription, the DNA damage response, and heterochromatin formation, respectively (20, 27). In addition to the canonical histones H3.1 and H3.2 (collectively designated H3.1/2), ubiquitously expressed H3 variants include centromere protein A (CENP-A) and H3.3 (26, 28). CENP-A marks centromeres, nucleates kinetochore formation, and is essential for proper chromosome segregation during mitosis (29). H3.3 is found at loci distinct from H3.1/2 and can mark either actively transcribed or transcriptionally repressed loci (30–32). In particular, H3.3 concentrated at telomeres, repetitive elements, endogenous retroviruses, and adjacent to centromeres appears to maintain the heterochromatin that silences transcription from these loci (31, 33–36).
Within virions, the DNA genomes of herpesviruses are not bound to histones (37–41). However, DNA viral genomes that enter the nucleus upon infection, including those of the herpesviruses, rapidly become associated with histones (38, 42, 43). The chromatin assembled when cellular histones associate with viral DNA appears to differ in some respects from cellular chromatin. For example, viral chromatin generally has lower histone occupancy and perhaps more unstable nucleosomes than cellular chromatin (42–45). However, transcriptional states of viral chromatin during either lytic replication or latency are, in all known cases, regulated by the same epigenetic mechanisms that control cellular transcription (46–49).
Specific histone PTMs associated with lytic and latent herpesvirus genomes have been identified, and discovering the mechanisms through which they are written, read, and erased has received significant effort. Less attention has been paid to the chromatin assembly process itself. Initial chromatinization occurs prior to viral genome replication and is not restricted to S phase (42, 43, 45, 50). Therefore, constitutively expressed histones deposited in a replication-independent manner are the prime candidates to first decorate infecting viral genomes. Indeed, a histone with these characteristics, H3.3, is found associated with lytic herpes simplex virus 1 (HSV-1) and latent Epstein-Barr virus (EBV) genomes (50, 51).
For human cytomegalovirus (HCMV), histones of the H2A, H2B, H3, and H4 classes all are found associated with viral genomes (42), but whether these are canonical or variant histones (or both) has not been examined. Furthermore, most of the analysis of HCMV chromatinization has been performed on lytic genomes, whereas only a few PTMs on latent genomes have been interrogated. Here, we assessed the deposition of canonical histones H3.1/2 and variant histone H3.3 onto HCMV lytic and latent genomes. We found both the canonical and variant histones incorporated into lytic and latent viral chromatin. Furthermore, their deposition required neither transcription from nor replication of the viral genome. As histone variants can play significant roles in regulating transcription (52), understanding which histones are deposited onto viral genomes increases our ability to decipher how viral chromatin interfaces with cellular defenses and directs viral transcription, replication, and packaging.
MATERIALS AND METHODS
Cells and infections.
TERT (telomerase reverse transcriptase)-immortalized human fibroblasts (TERT-HFs) and normal human dermal fibroblasts (NHDFs; Clonetics) were maintained in Dulbecco's modified Eagle medium (DMEM; Sigma). THP-1 cells (TIB-202; ATCC) were maintained in RPMI 1640 medium (Invitrogen). Medium was supplemented with 10% (vol/vol) fetal bovine serum (FBS; Sigma) and 1× penicillin-streptomycin with l-glutamine (G1146; Sigma). Primary CD34+ cells derived from cord blood (2C-101; Lonza) were maintained in hematopoietic progenitor growth medium (HPGM; Lonza) supplemented with recombinant human thrombopoietin (TPO), recombinant human stem cell factor (SCF), and recombinant human Flt3 ligand (all from Peprotech), as described previously (53). Cells were infected with HCMV in minimal volume for 60 min, followed by addition of medium to normal culture volumes. For UV inactivation, virus stocks were exposed to a 245-nm light source at 0.12 J/cm3 for 2 min on ice using a UV Stratalinker 2400 (Stratagene) prior to infection, as previously described (54).
Inhibitors and antibodies.
Where indicated in the figure legends and on the figures, valproic acid (VPA) (1 mM; Sigma) was added 3 h prior to infection of THP-1 cells, and phosphonoacetic acid (PAA) (100 μg/ml; Sigma) was added 24 h postinfection of NHDFs. The following antibodies were from commercial sources: anti-FLAG M2 (F1804; Sigma), anti-histone H3 (pan-H3) (ab1791; Abcam), anti-beta-actin (ab8226; Abcam), anti-tubulin (DM 1A; Sigma), anti-histone H2B (07-371; Millipore), anti-lamin A/C (sc-7292; Santa Cruz Biotechnology), anti-UL44 (CA006-100; Virusys), anti-hemagglutinin (HA) (HA.11; Covance), anti-histone H3.1/2 (ABE154; Millipore), anti-histone H3.3 (09-838; Millipore), and anti-Daxx (D7810; Sigma). Monoclonal antibodies against IE1 (1B12), pp28 (CMV157), and pp71 (2H10-9) have been described previously (55–57).
Epitope-tagged cell lines.
Fibroblast and THP-1 cell lines expressing either histone H3.1 or H3.3 with C-terminal Flag and HA tags from the elongation factor 1α (EF1α) promoter were generated by lentiviral transduction. Tagged cDNAs for H3.1 and H3.3 were amplified by PCR from pOZ-H3.1 and pOZ-H3.3 (58) (a gift from Yoshihiro Nakatani, Harvard Medical School) and cloned into the EcoRI/BamHI sites of pSIN-EF2-puro (Addgene plasmid 16580) (59). Recombinant lentivirus was produced by cotransfection of 293Y cells with pSIN-EF2-puro-eH3, pMD-VSV-G (where VSV-G is vesicular stomatitis virus G protein), and pΔR8.91 (a gift from Nathan Sherer, University of Wisconsin—Madison). TERT-HFs and THP-1 cells were transduced with recombinant lentivirus in the presence of 8 μg/ml Polybrene, and populations of stably transduced cells were selected with puromycin.
Western blotting.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer with protease and phosphatase inhibitors or 1% SDS containing 2% β-mercaptoethanol, and equal amounts of lysates were separated by SDS-PAGE, transferred to Optitran membranes (GE Healthcare), and analyzed by Western blotting as previously described (60).
Subnuclear fractionations.
Cells were collected and washed once in cold phosphate-buffered saline (PBS). An aliquot was lysed in 1% SDS containing 2% β-mercaptoethanol as total lysate, and the remaining cells were pelleted and resuspended in cold CSK buffer (10 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], pH 6.8, 100 mM NaCl, 300 mM sucrose, 2 mM MgCl2, 1 mM EGTA, and 1 mM dithiothreitol [DTT]) containing 0.5% Triton X-100. Samples were incubated on ice for 5 min and then pelleted at 5,000 × g for 3 min at 4°C. Supernatant was collected as the soluble fraction, and the pellets were washed once in cold CSK buffer and then resuspended in CSK buffer. RNase-free DNase (M6101; Promega) was added to 100 units/ml, and samples were incubated at 37°C for 2 h. Ammonium sulfate was added to a final concentration of 0.25 M, and samples were incubated on ice 5 min prior to centrifugation at 5,000 × g for 3 min at 4°C. Supernatant was collected as the chromatin fraction, and pellets were washed once in CSK buffer containing 2 M NaCl and resuspended in urea buffer (8 M urea, 10 mM Tris, pH 8.0) as the nuclear matrix fraction. All buffers were supplemented with protease and phosphatase inhibitors just prior to use.
Immunoprecipitations.
Immunoprecipitations were performed as previously described (61) with minor modifications. Briefly, cells were collected and washed once with PBS and once with hypotonic buffer (15 mM HEPES, pH 7.5, 20 mM KCl, 5 mM MgCl2). Cell pellets were resuspended in hypotonic buffer supplemented with 0.02% NP-40, 10 μg/ml RNase A, and protease and phosphatase inhibitors and incubated at 4°C with rotation for 15 min. NaCl was added to a final concentration of 400 mM with agitation to extract nuclear proteins, and extracts were incubated at 4°C with rotation for 15 min prior to centrifugation at 15,000 × g for 30 min at 4°C. Supernatant was incubated with 2 μg of antibody overnight at 4°C with rotation. Antibody complexes were collected with protein A+G magnetic beads (88802; Thermo) and washed for 5 min three times with hypotonic buffer containing 400 mM NaCl. Bound proteins were eluted in 1× SDS loading buffer containing β-mercaptoethanol and boiled before being run on SDS-PAGE gels.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (54). Briefly, infected cells were collected, washed once with PBS, and fixed with 1% formaldehyde for 8 min at room temperature, followed by addition of glycine to 125 mM to quench the reaction. Nuclei were then isolated, and chromatin was sheared by sonication. Chromatin from approximately 1 million cells per reaction mixture was incubated with 2 to 4 μg of ChIP-grade antibodies or an equal amount of isotype control IgG overnight at 4°C. Chromatin/antibody complexes were collected using Magna ChIP protein A+G magnetic beads (16-663; Millipore) for 2 h at 4°C. Beads were then washed, and cross-links were reversed. DNA was isolated with a QIAquick PCR cleanup kit (28106; Qiagen) and quantified by quantitative PCR (qPCR) using iTaq Universal SYBR green Supermix (172-5124; Bio-Rad) with primers specific to the major immediate early promoter (MIEP) (62), IE1 (63), LUNA promoter (64), LUNA (65), B2.7 RNA (65), or cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (66). Precipitating DNA was normalized to input levels from the same lysates using the ΔCT (where CT is threshold cycle) method (67). Statistical analysis was performed using Mstat, version 6.1, software.
DNA and mRNA analysis.
Total genomic DNA was isolated using a genomic DNA minikit (IB47202; IBI). Equal amounts of total DNA were analyzed by quantitative PCR using iTaq Universal SYBR green Supermix (172-5124; Bio-Rad) with primers specific to viral (IE1) (63) or cellular (GAPDH) (66) DNA. RNA isolation and quantitative reverse-transcription PCR (qRT-PCR) were performed as previously described (54). Viral gene expression was normalized to cellular gene expression using the ΔCT method (67).
siRNA transfections.
For transient knockdown with small interfering RNAs (siRNAs), THP-1 cells were transfected with 80 pmol of siRNA per 1 million cells using TransIT-X2 (MIR6000; Mirus) according to the manufacturer's instructions. Scrambled control siRNA (siScr; D-001810-10) or siRNAs targeting Daxx (siDx1 and siDx2; J-004420-05 and J-004420-06) were purchased from Dharmacon. Viability of transfected cells was assayed at 48 h posttransfection using a CellTiter-Glo assay (G7571; Promega) according to the manufacturer's instructions.
RESULTS
Ectopically expressed H3.1 and H3.3 are deposited onto lytic and latent HCMV genomes.
Histones of the H3 class are found associated with HCMV genomes during lytic infection and latency, but specifically which form(s) of histone H3 is present in HCMV chromatin has not been investigated. We examined H3.1 and H3.3 incorporation into nucleosomal HCMV by chromatin immunoprecipitation (ChIP) assays. H3.1 and H3.3 differ by only 5 amino acids. Therefore, to ensure specificity, we generated stable cell lines in both human fibroblasts (a model for HCMV lytic infection) and THP-1 monocytes (a model for HCMV latent infection) that ectopically express H3.1 or H3.3 with C-terminal Flag and HA epitope tags under the control of the cellular EF1α promoter (eH3.1 or eH3.3, respectively). We selected populations of transduced cells that express equivalent levels of eH3.1 and eH3.3 representing approximately 15% and 7% of total histone H3 in fibroblasts and THP-1 cells, respectively (Fig. 1A). Ectopically expressed histones fractionated with chromatin in a manner similar to that of the endogenous histone H2B in both HFs (Fig. 1B) and THP-1 cells (Fig. 1C), consistent with previous reports indicating that the epitope tags do not interfere with chromatin incorporation (58).
FIG 1.
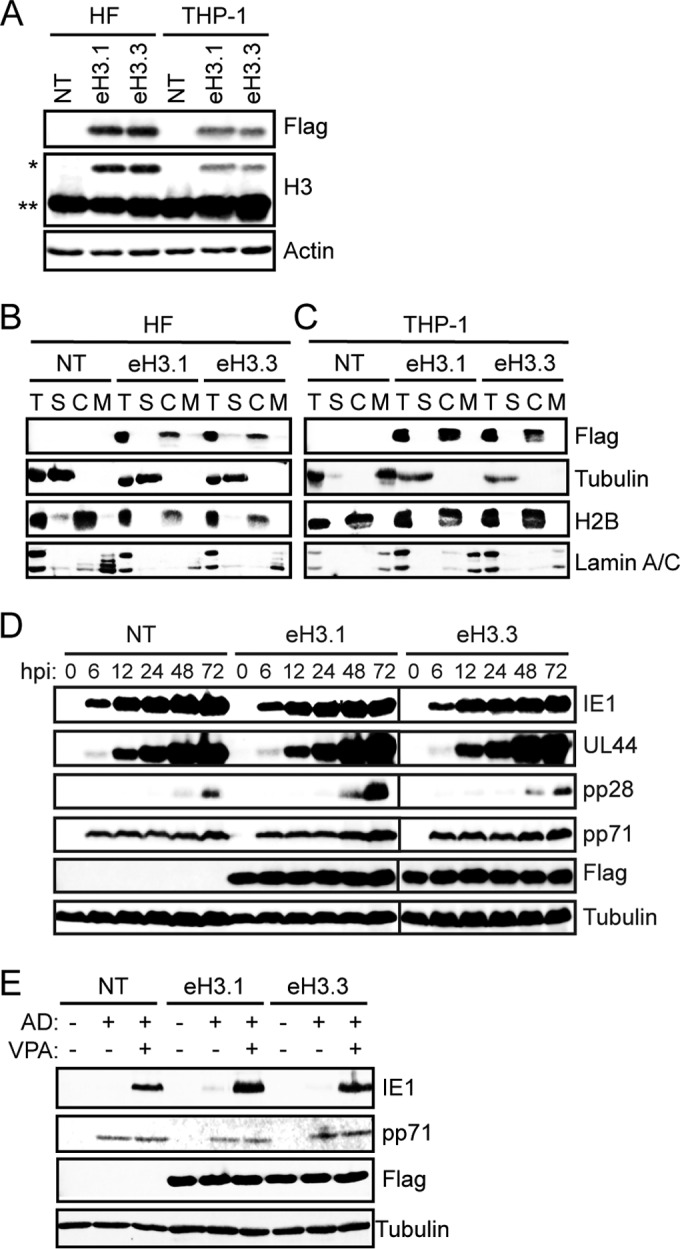
eH3-expressing cell lines support HCMV lytic infection and latency. (A) Lysates from TERT-HF (HF) and THP-1 cells nontransduced (NT) or transduced with recombinant lentivirus expressing epitope-tagged histone H3.1 (eH3.1) or H3.3 (eH3.3) were analyzed by Western blotting with the indicated antibodies. *, tagged H3; **, endogenous H3. (B and C) Nontransduced (NT) and eH3.1 or eH3.3 transduced TERT-HFs or THP-1 cells were fractionated and analyzed by Western blotting with the indicated antibodies. T, total lysate; S, soluble; C, chromatin; M, nuclear matrix. (D) Lysates from nontransduced (NT) and eH3.1 or eH3.3 transduced TERT-HFs infected with AD169 at an MOI of 1 and harvested at the indicated hour postinfection (hpi) were analyzed by Western blotting with the indicated antibodies. (E) Nontransduced (NT) and eH3.1 or eH3.3 transduced THP-1 cells were infected with AD169 (AD) at an MOI of 1 in the absence (−) or presence (+) of 1 mM VPA. At 18 h postinfection, lysates were collected and analyzed by Western blotting with the indicated antibodies. All data are representative of results from at least three experiments.
HCMV delivered equivalent amounts of tegument and expressed representative immediate early (IE1), early (UL44), and late (pp28) lytic phase proteins with similar kinetics and to similar steady-state levels in eH3.1- and eH3.3-expressing fibroblasts compared to expression in parental cells (Fig. 1D). Similarly, HCMV entered eH3.1- and eH3.3-expressing THP-1 cells as efficiently as parental cells, as judged by tegument delivery of pp71 (Fig. 1E). IE1 gene expression was silenced in these cells, consistent with the establishment of latency (Fig. 1E). As previously reported (53, 54, 68), treatment with the histone deacetylase (HDAC) inhibitor VPA prevented silencing of IE1 gene expression during infection with AD169 in parental THP-1 cells as well as in eH3.1- and eH3.3-expressing THP-1 cells (Fig. 1E). Taken together, these results demonstrate that HCMV infections are not altered in either HF or THP-1 cells expressing ectopic eH3.1 or eH3.3, indicating that these cell lines are viable tools with which to interrogate histone variant deposition during HCMV infection.
ChIP assays were first performed in HCMV AD169-infected eH3.1- and eH3.3-expressing fibroblasts with an antibody recognizing an endogenous epitope conserved in both H3.1/2 and H3.3. As expected, in HCMV-infected parental fibroblasts, an antibody against histone H3, but not an antibody against the HA epitope tag, precipitated both viral and cellular loci (Fig. 2A). In eH3.1- and eH3.3-expressing fibroblasts, we found histone H3 associated with all three viral loci examined at both 2 (Fig. 2B) and 72 (Fig. 2C) h postinfection (hpi). The same lysates were used for immunoprecipitation with the HA antibody to detect loci associated with the ectopically expressed histone. Both eH3.1 and eH3.3 were associated with the HCMV and cellular genome at 2 (Fig. 2D) and 72 (Fig. 2E) hpi. Similarly, ChIP experiments in parental THP-1 cells infected with HCMV AD169 detected total H3, but not eH3 (HA), associated with both viral and cellular loci (Fig. 3A). Using ChIP experiments in HCMV AD169-infected eH3.1- and eH3.3-expressing THP-1 cells, we found histone H3 class proteins associated with all three viral loci examined during the establishment phase of latency (Fig. 3B). Consistent with our observations during lytic infection, both eH3.1 and eH3.3 were deposited onto the viral genome following latent infection of THP-1 cells (Fig. 3C). We conclude that both eH3.1 and eH3.3 are incorporated into chromatin associated with the HCMV genome during lytic infection and latency.
FIG 2.

eH3.1 and eH3.3 associate with the HCMV genome during lytic infection of fibroblasts. (A) ChIP assays for the indicated loci using antibodies for total H3, HA, or IgG in parental TERT-HFs infected with AD169 at an MOI of 1 for 2 h. (B and C) ChIP assays for the indicated loci using an antibody for total H3 in eH3.1- and eH3.3-expressing TERT-HFs infected with AD169 at an MOI of 1 for 2 or 72 h, as indicated. (D and E) ChIP assays for the indicated loci using an antibody for the HA epitope tag in eH3.1- and eH3.3-expressing TERT-HFs infected with AD169 at an MOI of 1 for 2 or 72 h, as indicated. Gray bars represent signal from IgG controls. Data represent the means ± standard errors of the means from at least 4 experiments. *, P < 0.05; ns, not significant (P > 0.1), by a Wilcoxon rank sum test. LUNAp, LUNA promoter.
FIG 3.

eH3.1 and eH3.3 associate with the HCMV genome during latent infection of THP-1 cells. (A) ChIP assays for the indicated loci using antibodies for total H3, HA, or IgG in parental THP-1 cells infected with AD169 at an MOI of 3 for 18 h. (B and C) ChIP assays for the indicated loci using antibodies for total H3 (B) and eH3 (HA) (C) in eH3.1- and eH3.3-expressing THP-1 cells infected with AD169 at an MOI of 3 for 18 h. Gray bars represent signal from IgG controls. Data represent the means ± standard errors of the means from three experiments. *, P < 0.05; ns, not significant (P > 0.1), by a Wilcoxon rank sum test.
Endogenous H3.1 and H3.3 are deposited onto lytic and latent HCMV genomes.
Ectopically expressed, epitope-tagged histone variants allow for easy detection and facile discrimination of histone variants and eliminate concerns emanating from the different binding affinities inherent to different antibodies. However, such ectopically expressing cells use constitutively active promoters and poly(A) addition sequences such that the de novo synthesis of canonical histones such as H3.1 can occur during cell cycle stages (e.g., G1) when they would not normally be generated efficiently. Such de novo synthesis perturbs normal free histone pools, and therefore results with ectopically expressed canonical histones (like H3.1) must be interpreted with caution. Our data indicate that H3.1 can be deposited on viral genomes when it is ectopically expressed. However, evidence that it is truly deposited (e.g., during a natural infection) requires an examination of the endogenous H3.1/2 protein.
We therefore sought to confirm our results from eH3.1- and eH3.3-expressing cell lines indicating that both variants could be deposited on the viral genome during lytic and latent infection by examining the deposition of endogenous histone H3 variants. During the course of our work, ChIP-grade antibodies that recognize H3.1/2 or H3.3 became commercially available. The H3.1/2 antibody recognizes a linear epitope found in both H3.1 and H3.2 while the H3.3 antibody recognizes an epitope unique to H3.3. We independently verified the specificity of these antibodies with immunoprecipitation experiments in eH3.1- and eH3.3-expressing fibroblasts (Fig. 4A) and then used them for ChIP experiments during lytic and latent HCMV infections.
FIG 4.

Endogenous H3.1/2 and H3.3 associate with the HCMV genome during lytic infection of fibroblasts. (A) Nuclear extracts from eH3.1 and eH3.3 transduced TERT-HFs were subjected to immunoprecipitations (IPs) with the indicated antibody and analyzed for tagged histones by Western blotting (WB). (B to D) ChIP assays for the indicated loci using antibodies against total H3 (panH3), H3.1/2, H3.3, or an IgG control in NHDFs infected with AD169 at an MOI of 1 for 2, 6, or 72 h, as indicated. Data represent the means ± standard errors of the means from at least three experiments. *, P < 0.05; **, P < 0.01; ns, not significant (P > 0.1), by a Wilcoxon rank sum test.
As expected, ChIP assays with a pan-histone H3 antibody detected histone H3 at all viral and cellular loci tested at all time points in HCMV AD169-infected fibroblasts (Fig. 4B, C, and D). Likewise, ChIP assays with antibodies to the endogenous proteins detected both H3.1/2 and H3.3 association with the HCMV genome during lytic infection (Fig. 4B, C, and D). At first glance the data seem to imply that H3.1/2 shows a stronger association than H3.3 with the HCMV genome at early times during lytic infection (Fig. 4B and C), a difference largely neutralized at a late (72 hpi) time point (Fig. 4D). However, this interpretation is confounded by the possibility that differences in antibody affinities (not genome association) could explain the higher recovery of viral sequences with the H3.1/2 antibody than with the H3.3 antibody. Thus, the conservative conclusion is that during lytic infection, H3.1/2 and H3.3 are deposited onto the HCMV genome. However, our data raise the intriguing possibility of an earlier and more robust association of H3.1/2 histones than of H3.3 with HCMV genomes.
The incompletely differentiated myeloid lineage cells where HCMV establishes latency also chromatinize infecting viral genomes (64, 69, 70). We found that both H3.1/2 and H3.3 associated with HCMV AD169 latent genomes, similarly to lytic infection, at all three loci examined in THP-1 cells (Fig. 5A). Clinical strains such as TB40/E encode the viral UL138 gene that enforces latency maintenance by maintaining a repressive epigenetic signature at the viral MIEP (54). HCMV-TB40/E-infected THP-1 cells also showed H3.1/2 and H3.3 association with latent viral genomes (Fig. 5B), indicating that viral genes unique to clinical strains do not appear to affect the identity of histones deposited during the establishment of latency. While THP-1 cells are an invaluable, trusted, and widely used working model for HCMV latency, primary CD34+ hematopoietic progenitor cells represent a more physiologic cell type for latency studies. Our ChIP experiments indicate that both H3.1/2 and H3.3 are associated with latent AD169 (Fig. 5C) and TB40/E (Fig. 5D) genomes in primary CD34+ cells. We conclude that both H3.1/2 and H3.3 are deposited onto latent HCMV genomes.
FIG 5.

Endogenous H3.1/2 and H3.3 associate with the HCMV genome during latent infection. (A and B) ChIP assays for the indicated loci using antibodies against total H3 (panH3), H3.1/2, H3.3, or an IgG control in THP-1 cells infected with AD169 or TB40/E at an MOI of 1 for 18 h. (C and D) ChIP assays for the indicated loci using antibodies against total H3 (panH3), H3.1/2, H3.3, or an IgG control in primary CD34+ cells infected with AD169 or TB40/E at an MOI of 1 for 18 h. Data represent the means ± standard errors of the means from at least four experiments. *, P < 0.05; **, P < 0.01, by a Wilcoxon rank sum test.
Daxx promotes histone deposition onto latent HCMV genomes.
Daxx is an H3.3 chaperone (34) and participates in the transcriptional repression of HCMV lytic phase genes during latency (53, 68). We therefore asked if Daxx was responsible for depositing H3 histones on latent HCMV genomes. Transient transfection of two independent siRNAs targeting Daxx into THP-1 cells reduced Daxx steady-state protein levels (Fig. 6A) and, as previously shown (53, 68), increased viral IE1 transcription (Fig. 6B) while not altering cell viability (Fig. 6C). ChIP assays revealed decreased levels of total H3 and variant H3.3 at the MIEP (Fig. 6D) and within the transcribed region of IE1 (Fig. 6E) in Daxx-depleted cells compared to levels in cells transfected with a scrambled siRNA control. These loci are transcriptionally repressed during latency. However, Daxx depletion did not impact total H3 or H3.3 incorporation at the viral LUNA promoter (Fig. 6F) or the cellular GAPDH gene (Fig. 2G), loci that are transcriptionally active during latency. Our observations indicate that Daxx promotes H3.3 incorporation at transcriptionally repressed but not transcriptionally active viral loci during HCMV latency.
FIG 6.

Daxx promotes histone deposition onto latent HCMV genomes. (A) Lysates from THP-1 cells transfected with a scrambled (siCtrl) or Daxx (siDx1 and siDx2)-specific siRNA for 48 h were analyzed for Daxx knockdown by Western blotting. Tubulin served as a loading control. (B) RNA from siScr- and siDx-treated THP-1s infected with HCMV AD169 at an MOI of 1 for 18 h was analyzed for IE1 transcripts by qRT-PCR. Viral gene expression was normalized to cellular GAPDH and is shown relative to that in the siScr-treated control. (C) Viability of THP-1 cells transfected with a scrambled or Daxx-specific siRNA for 48 h was measured using a CellTiter-Glo assay. Data are shown as relative viability compared to that of the siScr control. (D to G) THP-1 cells treated with a scrambled or Daxx-specific siRNA for 48 h were infected with AD169 at an MOI of 1 for 18 h. ChIP assays were then performed with the indicated antibodies, and the precipitating DNA was analyzed by qPCR for the MIEP, transcribed regions of IE1, LUNA promoter (LUNAp), or the cellular GAPDH gene. Data represent IgG-subtracted percent input relative to the level of the siScr control. All data in panels B to G are means ± standard errors of the means from four experiments. *, P < 0.05; **, P < 0.01; ns, not significant (P > 0.2), by a Student's t test.
Interestingly, Daxx depletion impaired H3.1/2 deposition at all loci tested (Fig. 6D to G) even though Daxx is not known to directly deposit or to bind H3.1/2 (34, 61). We consider it unlikely for H3.1/2 deposition to require prior H3.3 incorporation because H3.1/2 deposition in the absence of Daxx is impaired at loci where H3.3 deposition is not (Fig. 6F and G) and because other data actually suggest that H3.1/2 deposition occurs prior to H3.3 (Fig. 4). Thus, the impaired incorporation of H3.1/2 into HCMV chromatin in Daxx-depleted THP-1 cells likely indicates either an alteration of Daxx histone-binding specificity or an effect on other histone chaperone complexes. We have not examined the requirement of Daxx for histone deposition during lytic infection of fibroblasts because under these conditions Daxx is rapidly degraded by tegument-delivered pp71 (71).
Incorporation of H3.1/2 and H3.3 requires neither viral transcription nor viral DNA synthesis.
The naked DNA introduced into nuclei by infection with viruses such as HCMV does not appear to mimic a normal cellular DNA substrate for histone deposition. Regions of naked cellular DNA are generated by the processes of transcription and replication. During transcription, nucleosomes are removed from templates to permit the passage of RNA polymerases and then added back to recently transcribed DNA to reestablish chromatin (19). To determine whether viral transcription was required for the incorporation of either H3.1/2 or H3.3, we infected THP-1 cells with UV-inactivated HCMV AD169 that failed to accumulate the B2.7 RNA (Fig. 7A) normally transcribed during latency (65). Similar levels of endogenous H3.1/2 and H3.3 were found associated with three different loci of live and UV-inactivated virus (Fig. 7B), indicating that viral transcription is not required for the initial chromatinization of HCMV genomes.
FIG 7.
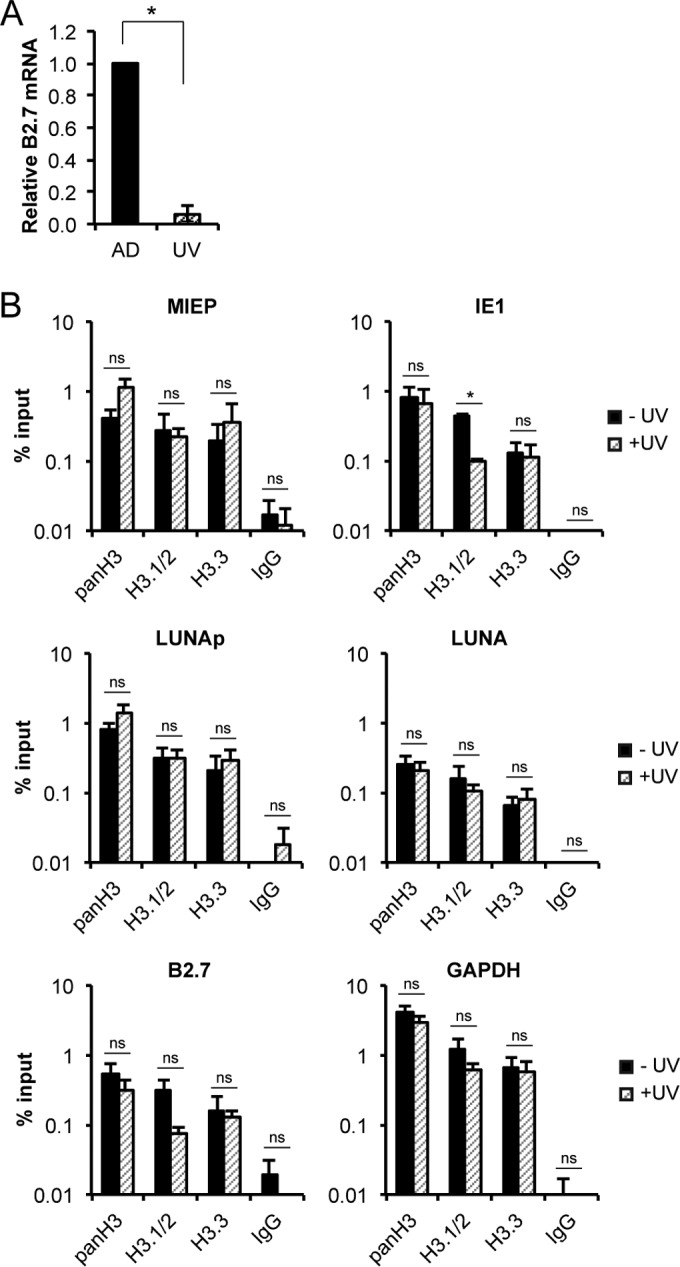
Incorporation of H3.1/2 or H3.3 into HCMV chromatin does not require viral transcription. (A) THP-1 cells infected with live (AD) or UV-inactivated (UV) AD169 at an MOI of 1 for 18 h were harvested and analyzed for viral and cellular transcripts by qRT-PCR. Viral transcripts were normalized to the level of cellular GAPDH and are shown relative to live AD169-infected controls. Data are the means ± standard errors of the means from three experiments. *, P < 0.05, by a Wilcoxon rank sum test. (B) ChIP assays for the indicated loci using antibodies against total H3 (panH3), H3.1/2, H3.3, or an IgG control in THP-1 cells infected with live (black bars) or UV-inactivated (striped bars) AD169 as described for panel A. Data represent the means ± standard errors of the means from three experiments. *, P < 0.05; ns, not significant (P > 0.1), by a Wilcoxon rank sum test.
During DNA replication, nucleosomes are removed from templates to permit the passage of DNA polymerases and then added back to recently replicated DNA to reestablish chromatin (72). We infected HFs with HCMV AD169 in the presence or absence of the viral DNA polymerase inhibitor PAA and demonstrated that the drug efficiently inhibited viral DNA replication (Fig. 8A). Similar levels of endogenous H3.1/2 and H3.3 were found associated with all three viral loci in the presence or absence of PAA (Fig. 8B). In all cases, recovery from the histone antibodies was greater than that of the control IgG antibody although the increase did not always reach statistical significance. We conclude that viral DNA replication is not required for the chromatinization of HCMV genomes. In summary, our evidence demonstrates that both H3.1/2 and H3.3 are deposited onto lytic and latent HCMV genomes through processes that require neither viral transcription nor DNA replication.
FIG 8.
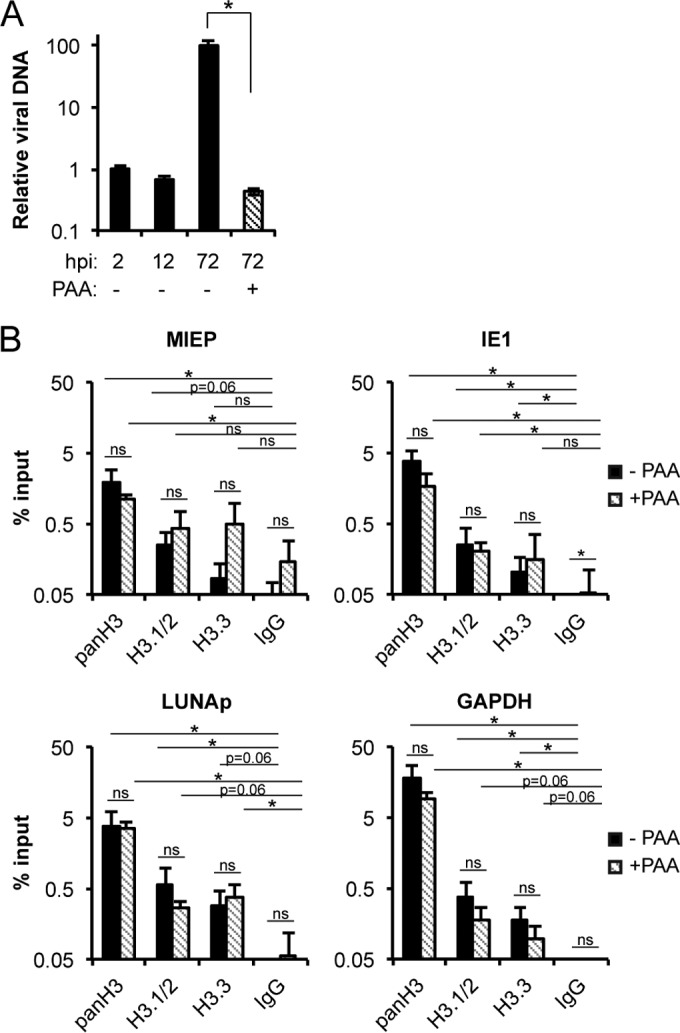
Incorporation of H3.1/2 or H3.3 into HCMV chromatin does not require viral DNA synthesis. (A) NHDFs infected with AD169 at an MOI of 1 in the absence (−) or presence (+) of 100 μg/ml PAA were harvested at the indicated hour postinfection (hpi), and viral (IE1) and cellular (GAPDH) DNA were quantitated by qPCR. Viral DNA was normalized to cellular DNA and is shown relative to the 2-hpi time point. Data represent the means ± standard errors of the means from four experiments. *, P < 0.05, by a Wilcoxon rank sum test. (B) ChIP assays for the indicated loci using antibodies against total H3 (panH3), H3.1/2, H3.3, or an IgG control from NHDFs infected with AD169 at an MOI of 1 for 72 h in the absence (black bars) or presence (striped bars) of 100 μg/ml PAA. Data represent the means ± standard errors of the means from four experiments. *, P < 0.05; ns, not significant (P > 0.1), by a Wilcoxon rank sum test.
DISCUSSION
Most cells infected with HCMV will be in the G1 phase of the cell cycle. Canonical histone proteins (such as H3.1/2) are synthesized most efficiently during the S phase and are most often deposited concomitantly with DNA replication, making them seemingly poor candidates for the initial chromatinization of infecting viral genomes. On the other hand, variant histones (such as H3.3) are efficiently expressed during G1 and efficiently deposited in the absence of DNA replication, making them prime candidates for this process.
Not surprisingly, we found H3.3 associated with both lytic and latent viral genomes. This histone variant is expressed constitutively throughout the cell cycle and deposited in a replication-independent manner. Depending on the chaperone that deposits H3.3, this histone can mark transcriptionally active (deposition by HIRA) or silent (deposition by Daxx or HIRA) chromatin (73, 74). During latency, the viral immediate early (lytic phase) genes are transcriptionally repressed, and Daxx plays a part both in this repression (53, 68) and in H3.3 deposition (Fig. 6). Likewise, histones associated with viral genomes at the very start of lytic infection show transcriptionally repressive epigenetic modifications (38), again pointing to H3.3 deposition by Daxx. Interestingly, endogenous H3.3 association with lytic viral genomes at 2 hpi is low, only to rise by 6 and then 72 hpi. This may reflect tegument-delivered pp71-mediated Daxx degradation and the subsequent reaccumulation of Daxx at later time points (68). In both adenovirus (75) and EBV (51) infections, decreases in H3.3 association with viral genomes upon Daxx depletion correlated with increased viral transcription, again pointing to Daxx-mediated H3.3 deposition as a means to silence infecting viral genomes.
We were surprised to find significant amounts of H3.1/2 associated with both lytic and latent HCMV genomes. These canonical histones are normally deposited during the S phase in a replication-dependent manner. However, there is some expression of H3.1/2 outside S phase (76, 77), and histone eviction and exchange occurring during cellular transcription (19) or induced by viral infection (78) could contribute to the pool of H3.1/2 available for viral genome chromatinization. Furthermore, the small amount of DNA introduced by HCMV infection (compared to that of the cellular genome) combined with the low nucleosome density of viral genomes (42) means that relatively little H3.1/2 is needed for chromatinization of the virus.
Our results diverge somewhat from a prior study of HSV-1 chromatinization with ectopically expressed histone variants in HeLa cells (50). That study reported that H3.1 incorporation required viral DNA replication and correlated H3.3 deposition by HIRA with transcriptional activation. The recent realization that the human papillomavirus oncoprotein E7 expressed in HeLa cells modulates DNA sensing pathways (79) and the potential link between infecting viral genome sensing and chromatinization (80) may explain why our results differ.
Single ChIP experiments (the type we performed) cannot determine if H3.1/2 and H3.3 decorate the same viral genomes or if some genomes incorporate both H3.1/2 and H3.3. The chromatinization of different viral genomes with different histone proteins could have profound effects on the transcriptional capabilities of the different genomes. Indeed, during infection at high multiplicities of infection (MOIs), only a subset of infecting viral genomes becomes transcriptionally active (81). While it would be interesting to try and correlate individual histone protein occupancy with transcriptional competence during such infections, such experiments are technically challenging due to the population-based nature of ChIP assays.
Here, we demonstrate that the identity of the H3 class of histones that associate with HCMV genomes is identical between lytic and latent infections. Previous work has shown that the patterns of histone PTMs at the MIEP at the very onset of lytic infection and during latency are also indistinguishable (38, 69, 70). Taken together, these results support a model wherein the initial assembly of chromatin on the viral genome occurs similarly or identically during both lytic and latent infection. This suggests that cellular proteins and/or components of the virion drive the initial assembly of viral chromatin. Our finding here that neither de novo viral transcription nor viral DNA replication is required for the association of either H3.1/2 or H3.3 with the viral genome is consistent with such a model.
We do not fully understand what triggers viral chromatin assembly during lytic or latent infections. The observation that transfected DNA is also assembled into nucleosomes (82, 83) suggests that chromatinization of infecting viral genomes may simply be a broad cellular response to naked DNA entering the nucleus that is common to all cells. Alternatively, it may be a generalized response to damaged DNA or the presence of repetitive sequences, both of which are found within infecting viral genomes and both of which prompt histone deposition (35, 84). The events that initiate the process of histone deposition on infecting viral genomes and the processes that mediate histone deposition remain to be explored.
Another question that needs to be addressed is whether viral genome chromatinization is beneficial or deleterious to viral infections. Beneficial consequences of viral genome chromatinization include protection from nucleases, avoiding detection as nonself, imparting structure needed for transcription or replication, and permitting epigenetic control of viral transcription by the virus. Deleterious consequences of viral genome chromatinization might include an inhibition of DNA replication or packaging into capsids and permitting epigenetic control of viral transcription by the cell. Understanding how the process of viral genome chromatinization is initiated, maintained, and modified should begin to answer this intriguing question.
In summary, we have shown that assembly of viral chromatin on HCMV lytic and latent genomes involves both variant and canonical histones. This finding raises the possibility that different histone proteins, as well as their PTMs, may play distinct roles during viral infection.
ACKNOWLEDGMENTS
We thank Phil Balandyk and Diccon Fiore for technical assistance, Yoshihiro Nakatani for tagged H3.1/H3.3 constructs, and members of the Kalejta lab for helpful discussions.
This study was supported by a National Institutes of Health grant to R.F.K. (AI074984). E.R.A. was supported by an NSF Graduate Research Fellowship.
We declare that we have no competing interests.
REFERENCES
- 1.Campos EI, Reinberg D. 2009. Histones: annotating chromatin. Annu Rev Genet 43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 2.Cutter AR, Hayes JJ. 2015. A brief review of nucleosome structure. FEBS Lett 589:2914–2922. doi: 10.1016/j.febslet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 4.Goldberg AD, Allis CD, Bernstein E. 2007. Epigenetics: a landscape takes shape. Cell 128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Li G, Reinberg D. 2011. Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev 21:175–186. doi: 10.1016/j.gde.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luger K, Dechassa ML, Tremethick DJ. 2012. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 13:436–447. doi: 10.1038/nrm3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knezetic JA, Luse DS. 1986. The presence of nucleosomes on a DNA template prevents initiation by RNA polymerase II in vitro. Cell 45:95–104. doi: 10.1016/0092-8674(86)90541-6. [DOI] [PubMed] [Google Scholar]
- 8.Teves SS, Weber CM, Henikoff S. 2014. Transcribing through the nucleosome. Trends Biochem Sci 39:577–586. doi: 10.1016/j.tibs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Li B, Carey M, Workman JL. 2007. The role of chromatin during transcription. Cell 128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Polo SE. 2015. Reshaping chromatin after DNA damage: the choreography of histone proteins. J Mol Biol 427:626–636. doi: 10.1016/j.jmb.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luger K. 2003. Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev 13:127–135. doi: 10.1016/S0959-437X(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 12.Gurard-Levin ZA, Almouzni G. 2014. Histone modifications and a choice of variant: a language that helps the genome express itself. F1000Prime Rep 6:76. doi: 10.12703/P6-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melters DP, Nye J, Zhao H, Dalal Y. 2015. Chromatin dynamics in vivo: a game of musical chairs. Genes 6:751–776. doi: 10.3390/genes6030751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang GG, Allis CD, Chi P. 2007. Chromatin remodeling and cancer, part II: ATP-dependent chromatin remodeling. Trends Mol Med 13:373–380. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y-M, Du J-Y, Lau ATY. 2014. Posttranslational modifications of human histone H3: an update. Proteomics 14:2047–2060. doi: 10.1002/pmic.201300435. [DOI] [PubMed] [Google Scholar]
- 17.Gardner KE, Allis CD, Strahl BD. 2011. Operating on chromatin, a colorful language where context matters. J Mol Biol 409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 19.Venkatesh S, Workman JL. 2015. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16:178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- 20.Skene PJ, Henikoff S. 2013. Histone variants in pluripotency and disease. Development 140:2513–2524. doi: 10.1242/dev.091439. [DOI] [PubMed] [Google Scholar]
- 21.Osley MA. 1991. The regulation of histone synthesis in the cell cycle. Annu Rev Biochem 60:827–861. doi: 10.1146/annurev.bi.60.070191.004143. [DOI] [PubMed] [Google Scholar]
- 22.Stillman B. 1986. Chromatin assembly during SV40 DNA replication in vitro. Cell 45:555–565. doi: 10.1016/0092-8674(86)90287-4. [DOI] [PubMed] [Google Scholar]
- 23.Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ. 2002. The human and mouse replication-dependent histone genes. Genomics 80:487–498. doi: 10.1006/geno.2002.6850. [DOI] [PubMed] [Google Scholar]
- 24.Wu RS, Perry LJ, Bonner WM. 1983. Fate of newly synthesized histones in G1 and G0 cells. FEBS Lett 162:161–166. doi: 10.1016/0014-5793(83)81070-9. [DOI] [PubMed] [Google Scholar]
- 25.Wu RS, Tsai S, Bonner WM. 1982. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell 31:367–374. doi: 10.1016/0092-8674(82)90130-1. [DOI] [PubMed] [Google Scholar]
- 26.Franklin SG, Zweidler A. 1977. Non-allelic variants of histones 2a, 2b and 3 in mammals. Nature 266:273–275. doi: 10.1038/266273a0. [DOI] [PubMed] [Google Scholar]
- 27.Talbert PB, Henikoff S. 2010. Histone variants—ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 28.Ederveen TH, Mandemaker IK, Logie C. 2011. The human histone H3 complement anno 2011. Biochim Biophys Acta 1809:577–586. doi: 10.1016/j.bbagrm.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Verdaasdonk JS, Bloom K. 2011. Centromeres: unique chromatin structures that drive chromosome segregation. Nat Rev Mol Cell Biol 12:320–332. doi: 10.1038/nrm3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szenker E, Ray-Gallet D, Almouzni G. 2011. The double face of the histone variant H3.3. Cell Res 21:421–434. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver RC, Miller JC, Lee Y-L, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D, Allis CD. 2010. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elsaesser SJ, Goldberg AD, Allis CD. 2010. New functions for an old variant: no substitute for histone H3.3. Curr Opin Genet Dev 20:110–117. doi: 10.1016/j.gde.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong LH, Ren H, Williams E, McGhie J, Ahn S, Sim M, Tam A, Earle E, Anderson MA, Mann J, Choo KHA. 2009. Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res 19:404–414. doi: 10.1101/gr.084947.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drané P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. 2010. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 24:1253–1265. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elsässer SJ, Noh K-M, Diaz N, Allis CD, Banaszynski LA. 2015. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522:240–244. doi: 10.1038/nature14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He Q, Kim H, Huang R, Lu W, Tang M, Shi F, Yang D, Zhang X, Huang J, Liu D, Songyang Z. 2015. The Daxx/Atrx complex protects tandem repetitive elements during DNA hypomethylation by promoting H3K9 trimethylation. Cell Stem Cell 17:273–286. doi: 10.1016/j.stem.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maxwell KL, Frappier L. 2007. Viral proteomics. Microbiol Mol Biol Rev 71:398–411. doi: 10.1128/MMBR.00042-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groves IJ, Reeves MB, Sinclair JH. 2009. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic “pre-immediate-early” repression of viral gene expression mediated by histone post-translational modification. J Gen Virol 90:2364–2374. doi: 10.1099/vir.0.012526-0. [DOI] [PubMed] [Google Scholar]
- 39.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol 79:4952–4964. doi: 10.1128/JVI.79.8.4952-4964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nitzsche A, Paulus C, Nevels M. 2008. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J Virol 82:11167–11180. doi: 10.1128/JVI.01218-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oh J, Fraser NW. 2008. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J Virol 82:3530–3537. doi: 10.1128/JVI.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leinbach SS, Summers WC. 1980. The structure of herpes simplex virus type 1 DNA as probed by micrococcal nuclease digestion. J Gen Virol 51:45–59. doi: 10.1099/0022-1317-51-1-45. [DOI] [PubMed] [Google Scholar]
- 45.Lacasse JJ, Schang LM. 2012. Herpes simplex virus 1 DNA is in unstable nucleosomes throughout the lytic infection cycle, and the instability of the nucleosomes is independent of DNA replication. J Virol 86:11287–11300. doi: 10.1128/JVI.01468-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinclair J. 2010. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim Biophys Acta 1799:286–295. doi: 10.1016/j.bbagrm.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 47.Reeves MB. 2011. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res 157:134–143. doi: 10.1016/j.virusres.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 49.Nevels M, Nitzsche A, Paulus C. 2011. How to control an infectious bead string: nucleosome-based regulation and targeting of herpesvirus chromatin. Rev Med Virol 21:154–180. doi: 10.1002/rmv.690. [DOI] [PubMed] [Google Scholar]
- 50.Placek BJ, Huang J, Kent JR, Dorsey J, Rice L, Fraser NW, Berger SL. 2009. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J Virol 83:1416–1421. doi: 10.1128/JVI.01276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai K, Chan L, Gibeault R, Conn K, Dheekollu J, Domsic J, Marmorstein R, Schang LM, Lieberman PM. 2014. Viral reprogramming of the Daxx histone H3.3 chaperone during early Epstein-Barr virus infection. J Virol 88:14350–14363. doi: 10.1128/JVI.01895-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weber CM, Henikoff S. 2014. Histone variants: dynamic punctuation in transcription. Genes Dev 28:672–682. doi: 10.1101/gad.238873.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saffert RT, Penkert RR, Kalejta RF. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol 84:5594–5604. doi: 10.1128/JVI.00348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee SH, Albright ER, Lee J-H, Jacobs D, Kalejta RF. 2015. Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci Adv 1:e1501164. doi: 10.1126/sciadv.1501164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 69:7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nowak B, Sullivan C, Sarnow P, Thomas R, Bricout F, Nicolas JC, Fleckenstein B, Levine AJ. 1984. Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 132:325–338. doi: 10.1016/0042-6822(84)90039-4. [DOI] [PubMed] [Google Scholar]
- 57.Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23:1885–1895. doi: 10.1128/MCB.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. 2004. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116:51–61. doi: 10.1016/S0092-8674(03)01064-X. [DOI] [PubMed] [Google Scholar]
- 59.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 60.Albright ER, Kalejta RF. 2013. Myeloblastic cell lines mimic some but not all aspects of human cytomegalovirus experimental latency defined in primary CD34+ cell populations. J Virol 87:9802–9812. doi: 10.1128/JVI.01436-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elsässer SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. 2012. DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition. Nature 491:560–565. doi: 10.1038/nature11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Winkler LL, Kalejta RF. 2014. The 19S proteasome activator promotes human cytomegalovirus immediate early gene expression through proteolytic and nonproteolytic mechanisms. J Virol 88:11782–11790. doi: 10.1128/JVI.01720-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hwang J, Saffert RT, Kalejta RF. 2011. Elongin B-mediated epigenetic alteration of viral chromatin correlates with efficient human cytomegalovirus gene expression and replication. mBio 2:e00023-11. doi: 10.1128/mBio.00023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reeves MB, Sinclair JH. 2010. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J Gen Virol 91:599–604. doi: 10.1099/vir.0.015602-0. [DOI] [PubMed] [Google Scholar]
- 65.Rossetto CC, Tarrant-Elorza M, Pari GS. 2013. cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14+ monocytes and CD34+ cells. PLoS Pathog 9:e1003366. doi: 10.1371/journal.ppat.1003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Juckem LK, Boehme KW, Feire AL, Compton T. 2008. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J Immunol 180:4965–4977. doi: 10.4049/jimmunol.180.7.4965. [DOI] [PubMed] [Google Scholar]
- 67.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 68.Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol 81:9109–9120. doi: 10.1128/JVI.00827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reeves MB, Lehner PJ, Sissons JGP, Sinclair JH. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J Gen Virol 86:2949–2954. doi: 10.1099/vir.0.81161-0. [DOI] [PubMed] [Google Scholar]
- 70.Reeves MB, MacAry PA, Lehner PJ, Sissons JGP, Sinclair JH. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A 102:4140–4145. doi: 10.1073/pnas.0408994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.MacAlpine DM, Almouzni G. 2013. Chromatin and DNA replication. Cold Spring Harb Perspect Biol 5:a010207. doi: 10.1101/cshperspect.a010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Campos EI, Reinberg D. 2010. New chaps in the histone chaperone arena. Genes Dev 24:1334–1338. doi: 10.1101/gad.1946810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hamiche A, Shuaib M. 2013. Chaperoning the histone H3 family. Biochim Biophys Acta 1819:230–237. [DOI] [PubMed] [Google Scholar]
- 75.Schreiner S, Bürck C, Glass M, Groitl P, Wimmer P, Kinkley S, Mund A, Everett RD, Dobner T. 2013. Control of human adenovirus type 5 gene expression by cellular Daxx/ATRX chromatin-associated complexes. Nucleic Acids Res 41:3532–3550. doi: 10.1093/nar/gkt064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hereford L, Bromley S, Osley MA. 1982. Periodic transcription of yeast histone genes. Cell 30:305–310. doi: 10.1016/0092-8674(82)90036-8. [DOI] [PubMed] [Google Scholar]
- 77.Gunjan A, Paik J, Verreault A. 2005. Regulation of histone synthesis and nucleosome assembly. Biochimie 87:625–635. doi: 10.1016/j.biochi.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 78.Conn KL, Hendzel MJ, Schang LM. 2013. The differential mobilization of histones H3.1 and H3.3 by herpes simplex virus 1 relates histone dynamics to the assembly of viral chromatin. PLoS Pathog 9:e1003695. doi: 10.1371/journal.ppat.1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lau L, Gray EE, Brunette RL, Stetson DB. 2015. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350:568–571. doi: 10.1126/science.aab3291. [DOI] [PubMed] [Google Scholar]
- 80.Knipe DM. 2015. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology 479-480:153–159. doi: 10.1016/j.virol.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kobiler O, Lipman Y, Therkelsen K, Daubechies I, Enquist LW. 2010. Herpesviruses carrying a Brainbow cassette reveal replication and expression of limited numbers of incoming genomes. Nat Commun 1:146. doi: 10.1038/ncomms1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cereghini S, Yaniv M. 1984. Assembly of transfected DNA into chromatin: structural changes in the origin-promoter-enhancer region upon replication. EMBO J 3:1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, Knipe DM. 2013. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci U S A 110:E4492–E4501. doi: 10.1073/pnas.1316194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adam S, Polo SE, Almouzni G. 2014. How to restore chromatin structure and function in response to DNA damage—let the chaperones play: delivered on 9 July 2013 at the 38th FEBS Congress in St Petersburg, Russia. FEBS J 281:2315–2323. doi: 10.1111/febs.12793. [DOI] [PubMed] [Google Scholar]