

ABSTRACT
Dengue virus (DENV) infects millions of people worldwide and is a major public health problem. DENV nonstructural protein 1 (NS1) is a conserved glycoprotein that associates with membranes and is also secreted into the plasma in DENV-infected patients. The present study describes a novel mechanism by which NS1 inhibits the terminal complement pathway. We first identified the terminal complement regulator vitronectin (VN) as a novel DENV2 NS1 binding partner by using a yeast two-hybrid system. This interaction was further assessed by enzyme-linked immunosorbent assay (ELISA) and surface plasmon resonance (SPR) assay. The NS1-VN complex was also detected in plasmas from DENV-infected patients, suggesting that this interaction occurs during DENV infection. We also demonstrated that the DENV2 NS1 protein, either by itself or by interacting with VN, hinders the formation of the membrane attack complex (MAC) and C9 polymerization. Finally, we showed that DENV2, West Nile virus (WNV), and Zika virus (ZIKV) NS1 proteins produced in mammalian cells inhibited C9 polymerization. Taken together, our results points to a role for NS1 as a terminal pathway inhibitor of the complement system.
IMPORTANCE Dengue is the most important arthropod-borne viral disease nowadays and is caused by dengue virus (DENV). The flavivirus NS1 glycoprotein has been characterized functionally as a complement evasion protein that can attenuate the activation of the classical, lectin, and alternative pathways. The present study describes a novel mechanism by which DENV NS1 inhibits the terminal complement pathway. We identified the terminal complement regulator vitronectin (VN) as a novel DENV NS1 binding partner, and the NS1-VN complex was detected in plasmas from DENV-infected patients, suggesting that this interaction occurs during DENV infection. We also demonstrated that the NS1-VN complex inhibited membrane attack complex (MAC) formation, thus interfering with the complement terminal pathway. Interestingly, NS1 itself also inhibited MAC activity, suggesting a direct role of this protein in the inhibition process. Our findings imply a role for NS1 as a terminal pathway inhibitor of the complement system.
INTRODUCTION
Dengue constitutes a major public health problem in tropical and subtropical countries. According to current estimates, at least 390 million cases of dengue occur annually, of which approximately 100 million are symptomatic (1). The infection is caused by dengue virus (DENV), a member of the Flaviviridae family that cocirculates in nature as four distinct antigenic serotypes (DENV1 to -4). DENV infection in humans is generally asymptomatic, but symptomatic cases can vary from a mild and self-limited fever to a potentially fatal hemorrhagic syndrome (2). The DENV genome is composed of a single positive-sense RNA that encodes a single viral polyprotein that is further processed by viral and host proteases into three structural proteins (C, prM/M, and E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (3).
NS1 is the first nonstructural protein to be translated and is essential to virus replication (4, 5). It is a conserved N-linked glycoprotein with a variable molecular mass of 46 to 55 kDa, which depends on its glycosylation status, and it is composed of three distinct structural domains: the β-roll, wing, and β-ladder (6). The NS1 protein can be found as a dimer associated with vesicular compartments within the cell, where it plays an important role as an essential cofactor in the virus replication process (4). Alternatively, NS1 can be secreted into the extracellular space as a hexameric lipoprotein particle (7) that interacts with several plasma proteins (8, 9) and activates the Toll-like receptor 4 (TLR4) response (10). Furthermore, secreted flavivirus NS1 has been characterized functionally as a complement immune evasion protein that can attenuate the activation of the classical, lectin, and alternative pathways by interacting with complement proteins and their regulators (11–13).
Vitronectin (VN) is a multifunctional glycoprotein present in the extracellular matrix and in the plasma. It consists of an N-terminal somatomedin-B domain (SMB), an RGD cell receptor binding site, four hemopexin (HPX)-like domains, and three heparin-binding domains (HBDs) (14). VN is a complement regulator that inhibits the formation of the membrane attack complex (MAC) by occupying the metastable membrane-binding site of the C5b7 complex and hindering its insertion into the cell membrane, thus preventing the completion of the C5b9 lytic pore (15). In addition, binding of C8 and C9 to the C5b7-VN complex leads to the formation of soluble C5b9 (SC5b9), which is hemolytically inactive. Furthermore, VN limits ongoing membrane-associated pore formation by inhibiting C9 polymerization (15–17). Acquisition of soluble human VN on the surfaces of microbial pathogens is a common complement evasion strategy that has been described for many bacterial pathogens (14, 18); however, this particular evasion mechanism has never been evaluated in viral pathogens.
In this work, we identified human VN as a novel DENV2 NS1 binding partner by using a yeast two-hybrid (Y2H) system. This interaction was further assessed by biochemical, biophysical, and immunoenzymatic approaches that confirmed the direct association between NS1 and VN. Moreover, we demonstrated that the NS1 protein from DENV2, West Nile virus (WNV), or Zika virus (ZIKV), by itself or in association with VN, is capable of inhibiting MAC formation and C9 polymerization. These results suggest a role for the NS1 protein as a terminal pathway inhibitor of the complement system.
MATERIALS AND METHODS
Proteins and antibodies.
Human vitronectin (VN), human hemopexin (HPX), bovine serum albumin (BSA), and heparin were purchased from Sigma-Aldrich. Human plasminogen (PLG) was purchased from Millipore. Recombinant active plasminogen activator inhibitor 1 (PAI-1) was kindly provided by Cynthia Peterson (University of Tennessee, TN), and the purified complement proteins C2, C5, C6, C7, C8, C9, and C5b6, factor H, and normal human serum (NHS) were purchased from Complement Technology.
Hexameric complex-type glycosylated NS1 proteins (NS1mam) from DENV2 (strain Thailand/16681/84), WNV (strain New York NY99), and ZIKV (strain Uganda MR 766), expressed in HEK 293 cells, were purchased from The Native Antigen (Oxfordshire, United Kingdom).
Polyclonal antiserum against purified NS1 was raised in mice and rabbits and purified using protein G HP Spin Trap columns (GE Healthcare) (19, 20). Mouse anti-human VN and PLG monoclonal antibodies were purchased from Abcam, and goat anti-human C2, C5, C6, C7, C8, and C9 and rabbit anti-VN were purchased from Complement Technology. Anti-mouse and anti-goat IgGs conjugated to horseradish peroxidase were purchased from Promega.
Production of NS1 protein.
Recombinant dimeric NS1 was expressed in Escherichia coli (NS1e). Briefly, the DENV2 ns1 gene from the New Guinea strain was inserted into the pET23b plasmid by GenScript (CA). A nucleotide sequence encoding a 6×His tag was added at the 3′ end of the ns1 gene. The recombinant plasmids were inserted into E. coli BL21(λDE3)pLysS cells, and the expression and purification steps were performed as described previously (19). Hexameric polymannose-glycosylated NS1 (NS1dros) was produced and isolated from the supernatant of a DENV2 NS1-expressing Drosophila Schneider 2 (S2) stable cell line. S2 cells were cultured in Schneider's Drosophila medium (Gibco) for 5 days at 28°C, and NS1dros expression was induced with 500 mM ZnCl2 in serum-free Sf-900 II SFM medium (Gibco). The supernatant was harvested, and NS1dros was purified using size exclusion chromatography on a Superdex 200 column (GE Healthcare).
Y2H screening.
Yeast two-hybrid (Y2H) screening was performed against a human liver cDNA library of sequences fused to the GAL4 activation domain by use of the pACT2 vector (Clontech, CA). Saccharomyces cerevisiae strain AH109 was transformed with the pGBKT7-NS1 plasmid, which encoded full-length DENV2 NS1 as bait, by using the lithium acetate method and then was grown in synthetic defined medium lacking tryptophan (SD–Trp). Autoactivation of the HIS3 reporter gene was verified by the growth of colonies in SD medium lacking histidine, leucine, and tryptophan (SD–His–Leu–Trp). The transformed cultures were then plated onto SD–His–Leu–Trp and SD–Ade–His–Leu–Trp media to select putative positive clones. The activity of the lacZ reporter gene was evaluated by a β-galactosidase assay (colony lift filter assay), using the substrate X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) on nitrocellulose membranes. To eliminate false-positive results, a plasmid linkage assay was also performed. The positive plasmids were sequenced, and their gene sequences were analyzed using the BLASTN and BLASTX software available at the NCBI website (www.ncbi.nlm.nih.gov).
Coimmunoprecipitation.
Human hepatocellular carcinoma (HuH-7.5.1) cell lines were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (Invitrogen, NY), 2 mM l-glutamine (Gibco), penicillin and streptomycin (Gibco), 0.22% sodium bicarbonate, and 0.2% HEPES, pH 7.4, in a humidified CO2 incubation chamber at 37°C. After 2 days, HuH-7.5.1 cells were mock infected or infected with DENV2 strain 16681 at a multiplicity of infection (MOI) of 1. The HuH-7.5.1 culture supernatants were collected at 48 h postinfection. The coimmunoprecipitation assays were performed using a Pierce coimmunoprecipitation kit (Pierce). Purified anti-NS1 polyclonal antibody was attached to an N-terminus-binding resin, and the supernatant (S) of mock-infected or DENV2-infected HuH-7.5.1 cells was added to the resin. The coimmunoprecipitation flowthrough (F) and eluted (E) samples were separated by 12% SDS-PAGE and transferred to a Hybond ECL nitrocellulose membrane (GE Healthcare). The membrane was then blocked with 5% nonfat milk in TBST (0.1% Tween 20 in Tris-buffered saline [25 mM Tris-HCl, pH 7.6, 3 mM KCl, and 140 mM NaCl]) for 2 h, followed by overnight incubation with an anti-NS1 or anti-VN antibody in blocking solution. The membrane was then washed three times with TBST and incubated with horseradish peroxidase-conjugated anti-mouse IgG in blocking solution for 2 h. The membrane was washed again and developed with a Supersignal West Pico kit (Pierce, IL), and the image was acquired using ImageQuant LAS-4000 (GE Healthcare).
Direct binding assays.
Microtiter plates were coated with 100 nM VN, PAI-1, HPX, or BSA in phosphate-buffered saline (PBS; 8.06 mM Na2HPO4, 1.94 mM KH2PO4, 2.7 mM KCl, and 137 mM NaCl; pH 7.4) overnight at 4°C. After five washes with PBS, the wells were blocked with 200 μl of 2% BSA in PBS containing 0.05% Tween 20 (PBST) for 1 h at 37°C, followed by five washes with PBST. NS1e at specific concentrations was added to each well and incubated for 2 h at 37°C, followed by a 1-h incubation with anti-NS1. Peroxidase-conjugated anti-mouse IgG was then added for 1 h at 37°C. The plates were washed five times with PBST between each step.
To investigate the effect of heparin or PAI-1 on the NS1-VN interaction, the binding buffer was supplemented with 0 to 500 μg/ml heparin or with 0 to 5 μg/ml PAI-1, and bound NS1e (10 or 1 μg/ml, respectively) was detected using anti-NS1 followed by a peroxidase-conjugated antibody. To investigate the effect of PLG on NS1e binding to immobilized VN, a mixture with a constant amount of NS1e (100 nM) and PLG at different molar ratios was added to VN-coated wells (100 nM). Similarly, NS1e was used at different molar concentrations combined with a constant amount of PLG (100 nM) and added to immobilized VN. Bound NS1 and PLG were detected using anti-NS1 and anti-PLG, respectively, followed by the addition of peroxidase-conjugated antibodies.
To assess binding of complement components to NS1e-coated wells (10 μg/ml), the proteins C2, C5, C6, C7, C8, and C9 (0 to 1,000 nM [each]) were incubated for 1 h at 37°C. Bound components were detected using specific polyclonal antibodies followed by a peroxidase-conjugated anti-goat IgG antibody. To investigate whether C9 would bind to DENV2, WNV, or ZIKV NS1mam-coated wells (10 μg/ml), C9 (0 to 500 nM) was added to each well and incubated for 1 h at 37°C. Wells were then washed with PBS and incubated with a polyclonal anti-C9 antibody followed by incubation with a peroxidase-conjugated anti-goat IgG antibody for 1 h at 37°C. The reactions from all binding experiments were developed by adding 100 μl of 0.4 mg/ml o-phenylenediamine and 50 μl of 9 N H2SO4. The optical density at 490 nm (OD490) was determined by use of a 96-well plate reader.
SPR measurements.
Surface plasmon resonance (SPR) measurements were performed using a Biacore X instrument (GE Healthcare). VN was covalently immobilized to the dextran matrix of a CM5 sensor chip via the primary amine groups (amine coupling kit; GE Healthcare). The carboxymethylated dextran surface was activated by the injection of a mixture of 0.2 M N-ethyl-N′-(diethylamino-propyl)carbodiimide and 0.05 M N-hydroxysuccinimide. The ligand was injected in 10 mM sodium acetate buffer (pH 4.0). The remaining N-hydroxysuccinimide esters were blocked by injection of 1 M ethanolamine hydrochloride (pH 8.5). All immobilization steps were performed at a flow rate of 10 μl/min in 10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.005% P20 (HBS-EP; GE Healthcare). The immobilization level for VN was 8,500 response units (RU). No protein was immobilized on the control flow cell that underwent the activation and blocking steps. Binding experiments were performed at 25°C at a flow rate of 15 μl/min in HBS-EP. The NS1e protein was injected at different concentrations ranging from 0.12 μM to 3.0 μM. The data were double referenced by subtraction of the control flow cell signal and a blank run (buffer only) signal. In all experiments, association phases ran for 120 s and dissociation phases for 280 s. The surface was regenerated with pulses of 0.01 M glycine (pH 2.0). The data were analyzed by global fitting to a 1:1 Langmuir binding model of both the association and dissociation phases for several concentrations simultaneously, using BIAevaluation 4.1 software (BIAcore). In each case, the data were obtained with a χ2 value of <2. The apparent equilibrium dissociation constants (KD) were calculated from the ratio of the dissociation and association rate constants (kd/ka).
Sandwich enzyme-linked immunosorbent assay (ELISA) with plasmas from DENV-infected patients.
Wells were coated at 4°C overnight with purified anti-NS1 rabbit polyclonal antibodies (20 μg/ml) in PBS. The plates were blocked for 1 h at 37°C with 2% BSA in PBST and then washed five times with PBST. This step was performed after each period of incubation. The wells were incubated with plasmas from DENV-infected patients or healthy blood donors (HD), diluted 1:2 in PBS, for 4 h at 37°C. Subsequently, the wells were incubated with an anti-VN monoclonal antibody (1:10,000) for 1 h at 37°C, followed by incubation for 1 h at 37°C with a peroxidase-conjugated anti-mouse IgG antibody (1:10,000). The reactions were developed as described above. Each sample was assayed twice.
The use of human samples was approved by the Research Ethics Committee for Clinical Studies of the University Hospital Clementino Fraga Filho, Federal University of Rio de Janeiro (UFRJ), and registered with the Brazilian Human Ethics Committee (Plataforma Brasil, CAAE; protocol 00534012.6.0000.5257; expiration date, 3 June 2017). Informed consent was not obtained because the patient records and information were anonymized and deidentified prior to analysis.
Hemolytic assay.
The MAC-inhibitory activity of NS1 and VN was analyzed in a hemolytic assay using sheep erythrocytes and purified complement proteins (18). Erythrocytes were resuspended to 1 × 108 cells/ml in Veronal-buffered saline (VBS) and preincubated with 1 μg/ml C5b6 for 1 h at room temperature. In a separate preparation, a mixture of purified NS1e (12.5 μg/ml) and VN (20 μg/ml), NS1e (12.5 μg/ml) only, or VN (20 μg/ml) only was preincubated for 1 h at 37°C, and the complement proteins C7 (1 μg/ml), C8 (0.1 μg/ml), and C9 (1 μg/ml) (C7–9) were added to the mixture for 15 min at 37°C. Thereafter, the C5b6-coated erythrocytes were added to the NS1e-VN-C7–9 mixture and incubated for 30 min at 37°C. Alternatively, increased amounts of NS1e or NS1dros (6.25, 12.5, and 25 μg/ml) were preincubated with the C7–9 proteins for 15 min at 37°C before incubation with C5b6-coated erythrocytes. Erythrocytes were centrifuged, and the amount of hemoglobin released from the lysed cells was measured by determining the absorbance at 540 nm. The relative MAC-inhibitory activity was presented as the percentage of total hemolysis. NS1 buffer and factor H (50 μg/ml) were used as negative controls.
C9 polymerization assay.
The effects of NS1e and NS1dros on C9 polymerization were assessed according to a previously published protocol (21). Briefly, NS1e (10 to 100 μg/ml), NS1dros (10 to 100 μg/ml), or NS1 buffer was preincubated with 3 μg of C9 at 37°C in 20 mM Tris-HCl (pH 7.2). After 40 min of incubation, 50 μM ZnCl2 in 20 mM Tris-HCl (pH 7.2) was added for 2 h at 37°C. In a separate experiment, NS1e (10 or 25 μg/ml) was preincubated with VN (10 or 25 μg/ml) for 30 min at 37°C in 20 mM Tris-HCl (pH 7.2), and 3 μg of C9 was added for 40 min at 37°C. Polymerization was induced by addition of 50 μM ZnCl2 in 20 mM Tris-HCl (pH 7.2) for 2 h at 37°C. The samples were separated in precast 4 to 20% gradient polyacrylamide gels (Bio-Rad) under nonreducing conditions, and C9 polymerization was visualized by silver staining. The C9 monomer band intensity was evaluated by densitometry using ImageJ software, version 1.49 (National Institutes of Health).
The effect of NS1mam on C9 polymerization was assessed by using proteins from DENV2, WNV, and ZIKV. Initially, BHK-21 cells were cultivated in a 24-well microplate in alpha minimal essential medium (α-MEM; Gibco) supplemented with 10% fetal bovine serum, 0.22% sodium bicarbonate, and 0.2% HEPES, pH 7.4, in a humidified CO2 incubation chamber at 37°C. After 2 days, cells were washed three times with VBS and preincubated with 5 μg/ml purified C5b6 for 2 h at 37°C in VBS. The NS1mam protein from DENV2, WNV, or ZIKV was preincubated with 5% NHS for 30 min at 37°C. Rabbit polyclonal anti-NS1 (50 μg/ml), which cross-reacts with the WNV and ZIKV NS1 proteins (data not shown), was mixed with 50 μg/ml NS1mam to serve as a control. Cells were washed with VBS three times and then incubated with NS1mam mixed with NHS for 50 min at 37°C. Thereafter, cells were washed three times with VBS and lysed with cell lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40). The samples were separated in a 10% polyacrylamide gel under nonreducing conditions and then transferred to a 0.45-μm Immobilon-P polyvinylidene difluoride (PVDF) membrane (GE Healthcare) at 15 V for 16 h. The membrane was blocked with 5% nonfat milk in TBST for 2 h, followed by overnight incubation with an anti-C9 antibody in blocking solution. The membrane was then washed three times with TBST and incubated with anti-goat IgG conjugated to horseradish peroxidase in blocking solution for 2 h. The membrane was washed again and developed with a Supersignal West Pico kit (Pierce, IL), and the image was acquired using ImageQuant LAS-4000 (GE Healthcare).
To evaluate the effect of VN preincubation with NS1mam on C9 polymerization, VN (0 to 25 μg/ml) was preincubated with the NS1mam protein from DENV2, WNV, or ZIKV (25 μg/ml) for 30 min at 37°C, incubated with 5% NHS for 30 min at 37°C, and added to C5b6-treated BHK-21 cells for 50 min at 37°C. Thereafter, cells were washed three times with VBS and lysed with cell lysis buffer, and the C9 polymer was analyzed by Western blotting using anti-C9.
Statistical analysis.
The data sets were compared using two-tailed, unpaired Student's t test, and statistical significance was achieved when P values were <0.05. Multiple comparisons were performed using one-way or two-way analysis of variance (ANOVA) (with the Bonferroni posttest), and asterisks in figures indicate significant differences from the control, as indicated in the legends.
RESULTS
Identification of VN as an NS1-interacting partner.
Using full-length NS1 as bait, we performed a Y2H screen of a human liver cDNA library as previously described by our group (8). Putative DENV NS1 protein-interacting partners were identified by activation of the HIS3 and ADE2 reporter genes, and the authenticity of the interaction was confirmed by colony lift filter assays of positive clones. True positive transformants activated the β-galactosidase (lacZ) reporter gene and appeared blue, whereas false-positive clones remained colorless (Fig. 1A). One positive clone identified by the colony lift assay was subjected to nucleotide sequencing and identified as carrying the human VN gene.
FIG 1.

DENV2 NS1 interacts with vitronectin (VN) in yeast and mammalian cells. (A) Transformants carrying the bait (pGBKT7-NS1) and prey (pACT2-VN) plasmids were visualized by their growth on double (SD–Leu–Trp; column 1), triple (SD–His–Leu–Trp; column 2) and quadruple (SD–Ade–His–Leu–Trp; column 3) dropout media and by blue staining in the colony lift filter assay (column 4), indicating yeast growth and HIS3, ADE2, and lacZ reporter gene activation, respectively. AH109 yeast cells cotransformed with the plasmids pGBKT7-53 (encoding murine p53 fused to the GAL4 DNA-binding domain) and pGADT7-T (encoding the simian virus 40 [SV40] large T antigen fused to the GAL4 activation domain) served as the positive control (C+). AH109 cotransformed with the plasmids pGBKT7-NS1 and pGADT7-AD served as the negative control (NC). (B) Supernatants from mock- or DENV2-infected HuH7.5.1 cells were immunoprecipitated with purified anti-NS1 polyclonal antibody. The supernatant (S), flowthrough (F), and eluted (E) fractions were analyzed by Western blotting with anti-VN and anti-NS1 polyclonal antibodies. Purified VN protein and DENVsup were used as band mass controls (C). The presence of two bands, of 75 and 65 kDa, corresponding to human VN was observed in the elution fraction of the DENV2 samples.
To assess whether this interaction occurs in DENV-infected cells, we first infected HuH7.5.1 cells, which have been shown to express VN (22), with DENV2 strain 16681 at an MOI of 1 for 48 h. The mock- or DENV2-infected HuH7.5.1 cell supernatants (S) were then loaded onto a column containing anti-NS1 polyclonal antibodies immobilized on a resin to collect all coimmunoprecipitated proteins. The elution fractions (E) were then analyzed using anti-NS1 and anti-VN polyclonal antibodies. We observed two bands, of 75 and 65 kDa, corresponding to VN (Fig. 1B, upper panel) and one band, of approximately 50 kDa, corresponding to DENV2 NS1 (Fig. 1B, lower panel). This result indicates that VN was coimmunoprecipitated with the NS1 protein.
DENV NS1 directly binds VN.
To assess whether NS1 directly interacts with VN, purified human VN was immobilized onto microtiter plates, and serial dilutions of recombinant dimeric NS1e (19) were added. NS1e interacted with VN in a dose-dependent manner, whereas no binding was observed with BSA, used as a negative control (Fig. 2A). This interaction was further confirmed by SPR analysis, in which VN was immobilized on a CM5 sensor chip dextran surface and the NS1e protein was injected at different concentrations, ranging from 0.12 μM to 3.0 μM. A concentration-dependent binding of NS1e to immobilized VN was observed (Fig. 2B). An evaluation of the global rates of association and dissociation (ka and kd, respectively) revealed a ka of 4.23 × 104 ± 0.23 × 104 M−1 s−1 and a kd of 6.45 × 10−2 ± 0.92 × 10−2 s−1. The global dissociation equilibrium constant (KD) was 1.52 ± 0.17 μM, assuming a simple 1:1 Langmuir binding mode. Taken together, the ELISA and SPR data indicate that DENV2 NS1 directly binds VN.
FIG 2.

DENV2 NS1 directly binds human vitronectin (VN). (A) Microtiter plates were coated with purified VN (100 nM), and increasing amounts of recombinant DENV2 NS1e were added. Bound NS1e was detected using an anti-NS1 polyclonal antibody. BSA was used as the negative control. Error bars indicate standard deviations for three independent experiments, and asterisks indicate significant differences from the control based on two-way ANOVA and the Bonferroni posttest. ***, P < 0.001. (B) Replicates (black solid lines) and 1:1 fitting (gray dashed lines) of the binding of recombinant DENV2 NS1e to immobilized VN as analyzed by surface plasmon resonance analysis. Increasing concentrations of NS1e (0.12 to 3.0 μM) were injected onto a VN-coated CM5 sensor chip. The amount of NS1 associated with VN was measured in response units (RU). (Inset) The equilibrium response units (Req) obtained for binding of NS1 to immobilized VN were plotted against the concentration of NS1e.
DENV NS1-VN interaction is increased by PAI-1 and inhibited by plasminogen.
VN can bind several molecules, such as heparin, PAI-1, HPX, and PLG, and these interactions occur at distinct sites in the VN molecule (23–27). Therefore, to identify the putative binding sites of NS1 on the VN molecule, competitive ELISAs were performed in which VN ligands were used as competitors (28). VN-coated wells were incubated with fixed concentrations of NS1e and various concentrations of heparin (15.6 to 500.0 μg/ml) or PAI-1 (0.16 to 5.00 μg/ml). The addition of increasing amounts of heparin did not affect binding (Fig. 3A), suggesting that the NS1-VN interaction does not occur through the HBDs. However, when PAI-1 was coincubated with NS1e, an unexpected dose-dependent increase in the binding of NS1 to VN was observed (Fig. 3B), thus indicating that PAI-1 facilitates VN-NS1 interactions. To evaluate whether NS1 directly interacts with PAI-1, purified human PAI-1 was immobilized and incubated with increasing concentrations of NS1e. A dose-dependent interaction was observed, thus confirming direct binding between these molecules (Fig. 3C).
FIG 3.

The NS1-VN interaction is affected by PAI-1 and PLG but not by heparin. Microtiter plates were coated with VN (100 nM), and the effects of increasing concentrations of heparin (A) and PAI-1 (B) on the binding of NS1e to VN were analyzed. Bound NS1e was detected using an anti-NS1 polyclonal antibody. One-way ANOVA and the Bonferroni posttest were performed to calculate the significance of differences compared to the binding without heparin or PAI-1. (C) DENV NS1 directly interacts with PAI-1 but does not interact with HPX. Microtiter plates were coated with purified PAI-1 (100 nM) or HPX (100 nM), and increasing amounts of NS1e were added. Bound NS1e was detected using an anti-NS1 polyclonal antibody. BSA was used as the negative control. Two-way ANOVA and the Bonferroni posttest were used to calculate the significance of differences. (D and E) NS1 and PLG compete for VN binding. VN was immobilized on microtiter plates, and a constant amount of NS1e (100 nM) together with increasing amounts of PLG (D) or a constant amount of PLG (100 nM) with increasing amounts of NS1e (E) was added. Bound NS1e and PLG were detected using specific antibodies. One-way ANOVA and the Bonferroni posttest were used to calculate the significance of differences compared to the binding without NS1 or PLG. Error bars indicate standard deviations for three independent experiments, and the asterisks indicate significant differences from the control. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
A canonical feature of the VN structure is the homology with hemopexin-type repeats found in human HPX. Computationally predicted models indicate that the VN central domain adopts a full four-bladed β-propeller fold similar to that described for HPX (29). Moreover, both HPX and VN hemopexin domains are recognized by surface proteins of Streptococcus pyogenes, and these interactions are important for bacterial attachment (30). Based on these findings, we assessed whether NS1 interacts directly with HPX or, alternatively, if NS1 competes with HPX for binding to VN. First, HPX-coated wells were incubated with increasing concentrations of NS1e. No significant binding was observed (Fig. 3C). In a competitive binding assay in which VN-coated wells were incubated with fixed concentrations of NS1e and increasing concentrations of HPX (1.8 to 30.0 μg/ml), the addition of HPX did not alter the NS1-VN interaction (data not shown). These two assays indicate that NS1 does not bind HPX and does not interact with VN via the hemopexin domains.
Because PLG binds VN, we hypothesized that PLG-binding sites on VN may overlap the NS1-binding sites. A constant amount of NS1e (100 nM) with increasing amounts of PLG or a constant amount of PLG (100 nM) with increasing amounts of NS1e was added to immobilized VN. NS1 binding to VN was affected in the presence of PLG and was reduced ∼80% at a 10-fold molar excess (Fig. 3D). Similarly, the presence of increasing concentrations of NS1 affected the binding of PLG to immobilized VN (Fig. 3E). Taken together, these results indicate that NS1 and PLG have overlapping binding sites on VN.
Identification of the NS1-VN complex in plasmas from DENV-infected patients.
During DENV infection, the NS1 protein is found at high concentrations in the sera of infected patients (31, 32). Because alterations in vascular homeostasis are the basis of dengue pathophysiology and severity, we evaluated whether NS1 interacts with circulating VN in the plasma in DENV-infected patients. A capture ELISA probed with an anti-VN monoclonal antibody was performed using a plasma panel composed of 14 samples from DENV-infected patients (Table 1) and 40 samples from healthy blood donors (HD). All DENV-positive samples were from DENV1 infections and were NS1 positive according to the Platelia Dengue NS1 Ag assay. The captured NS1-VN complex was significantly increased in DENV-positive samples compared to HD samples (P < 0.001) (Fig. 4A), clearly suggesting that this interaction occurs during infection in the plasma. We then evaluated whether this complex formation correlates with disease severity. DENV-positive samples were divided according to the new dengue classification guidelines (2), into samples from cases of dengue without warning signs (DWO; n = 8) and those from cases of dengue with warning signs (DW; n = 6). Although both groups showed higher mean OD values than those for HD (P < 0.001 [for each group]) (Fig. 4B), we did not observe a significant difference in these values between DWO and DW samples (P = 0.439) (Fig. 4B), indicating that the presence of the NS1-VN complex during infection does not correlate with disease progression. Nonetheless, we cannot exclude the possibility that the lack of a significant difference between dengue patients with different severities of disease may have been due to the small sample size.
TABLE 1.
Characteristics of DENV-infected patientsa
| Patient no. | DENV serotype | WHO classification | Day of illness | Presence of IgM | Presence of IgG | Platelet count (1,000/mm3) | TGO/AST concn (IU/liter) | TGP/ALT concn (IU/liter) |
|---|---|---|---|---|---|---|---|---|
| 1 | DENV1 | DW | 4 | + | − | 137 | 48 | 47 |
| 2 | DENV1 | DW | 3 | − | − | 222 | 98 | 162 |
| 3 | DENV1 | DW | 5 | + | − | 132 | 142 | 142 |
| 4 | DENV1 | DW | 3 | − | − | 168 | 41 | 59 |
| 5 | DENV1 | DWO | 6 | + | − | 88 | 109 | 106 |
| 6 | DENV1 | DWO | 12 | − | − | 185 | 28 | 31 |
| 7 | DENV1 | DW | 3 | + | + | 143 | 35 | 32 |
| 8 | DENV2 | DWO | 2 | − | − | 202 | ||
| 9 | DENV1 | DW | 3 | − | + | 284 | 33 | 44 |
| 10 | DENV1 | DWO | 1 | − | + | 250 | 13 | 29 |
| 11 | DENV1 | DWO | 1 | − | + | 153 | 15 | 31 |
| 12 | DENV1 | DWO | 4 | + | − | 109 | ||
| 13 | DENV1 | DWO | 2 | − | − | 116 | 43 | 56 |
| 14 | DENV1 | DWO | + | + | 153 |
All patients were positive for DENV NS1. DW, dengue with warning signs; DWO, dengue without warning signs; ALT, alanine aminotransferase; AST, aspartate aminotransferase; TGO, glutamicoxalacetic transaminase; TGP, glutamic-pyruvic transaminase.
FIG 4.
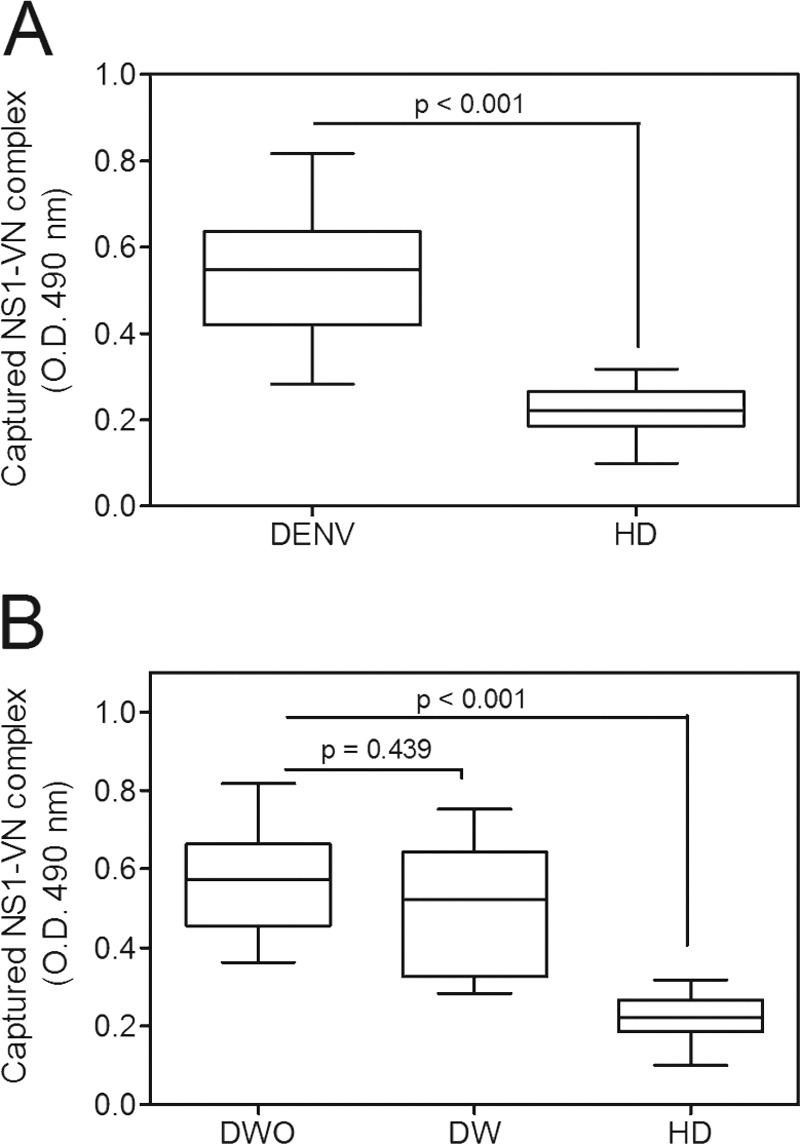
The NS1-VN interaction complex was captured in plasmas from DENV-infected patients. (A) Microtiter plates were coated with purified anti-NS1 polyclonal antibody (10 μg/ml), and NS1-positive plasma samples from DENV-infected patients and from healthy blood donors (HD) were added. Bound VN was detected using a specific monoclonal anti-VN antibody. (B) The data obtained from DENV-positive samples were separated into cases of dengue without warning signs (DWO) and dengue with warning signs (DW) and compared to the results for HD. The boxes indicate medians and interquartile ranges, and the whiskers indicate the 5th to 95th percentiles. To calculate the significance of differences between groups, Student's t test was performed, and a significant difference was achieved when the P value was <0.001.
DENV NS1 binds to MAC components and inhibits its formation.
To assess whether the NS1-VN interaction would contribute to subverting the complement attack as described for bacterial pathogens (28, 33), we evaluated whether this complex blocks MAC formation by using purified MAC components. NS1e was preincubated with VN, mixed with purified C7, C8, and C9, and then added to C5b6-treated sheep erythrocytes. In the presence of both NS1 and VN, erythrocyte lysis was significantly inhibited, whereas the NS1 buffer did not block the MAC cytolytic activity (Fig. 5A). VN inhibited MAC formation, as expected (17). Interestingly, recombinant NS1e also inhibited MAC activity, by 50%. However, erythrocyte lysis inhibition was more pronounced in the presence of both NS1e and VN (P < 0.05) (Fig. 5A), suggesting that the NS1-VN interaction is important for avoiding MAC activation.
FIG 5.

DENV2 NS1 binds MAC components and inhibits MAC formation. (A) NS1 inhibits erythrocyte lysis in the presence or absence of VN. NS1e (12.5 μg/ml) and VN (20 μg/ml) were preincubated separately or together for 1 h at 37°C. The proteins were then incubated with C7, C8, and C9 and added to C5b6-coated sheep erythrocytes. Cell lysis was measured by determining the free hemoglobin absorbance at 540 nm. Lysis in the absence of inhibitor (MAC) was set to 100%. NS1 buffer was used as a negative control. (B) Both NS1 produced in E. coli (NS1e; nonglycosylated) and NS1 produced in Drosophila S2 cells (NS1dros; polymannose glycosylated) inhibited MAC formation in a dose-dependent manner. NS1e or NS1dros (6.25, 12.5, and 25 μg/ml) was preincubated with C7, C8, and C9 and added to C5b6-coated sheep erythrocytes. After incubation, cell lysis was measured. Incubation with factor H (50 μg/ml) or NS1 buffer was included as a negative control. For panels A and B, error bars indicate standard deviations for three independent experiments, and the asterisks indicate the significance of differences from the control, determined using one-way ANOVA and the Bonferroni posttest. ns, not significant; *, P < 0.05; ***, P < 0.001. (C) NS1 binds the terminal complement components C5, C6, C7, and C9. Microtiter plates were coated with NS1e (10 μg/ml), and increasing amounts of C2, C5, C6, C7, C8, and C9 (0 to 1,000 nM) were added. Binding was detected using specific polyclonal antibodies. C2 was used as an NS1-complement binding negative control. The error bars indicate standard deviations for three independent experiments, and the asterisks indicate the significance of differences from the control, determined using two-way ANOVA and the Bonferroni posttest. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To investigate whether NS1 glycosylation and oligomerization have similar inhibitory effects on MAC formation, purified NS1e expressed in E. coli, which is in its dimeric conformation and is not glycosylated, and purified NS1 expressed in Drosophila S2 cells (NS1dros), which is in its hexameric conformation and is polymannose glycosylated, were incubated with purified C7, C8, and C9 and then added to C5b6-treated sheep erythrocytes. Both proteins significantly inhibited erythrocyte lysis in a dose-dependent manner, whereas factor H and NS1 buffer, included as negative controls, did not inhibit MAC-dependent lysis (Fig. 5B). Therefore, it seems that NS1 glycosylation and oligomerization are not required for the protein to inhibit MAC activity.
We also evaluated whether NS1 interacts with purified MAC components. Microtiter plates coated with NS1e were incubated separately with purified C5, C6, C7, C8, and C9. NS1 strongly bound to C5, C6, and C9, whereas a weaker interaction was observed between NS1 and C7. Furthermore, no interaction was detected with C8. C2 was included as a negative control because it does not interact with NS1 (Fig. 5C). From these data, it is plausible to conclude that NS1 may inhibit MAC formation through direct interaction with MAC components.
The DENV2 NS1-VN complex inhibits C9 polymerization in vitro.
Because DENV NS1 interacts with C9, we next investigated the effects of NS1e and DENV2 NS1dros on C9 polymerization. Both NS1e (Fig. 6A) and NS1dros (Fig. 6B) inhibited C9 polymerization in a concentration-dependent manner, indicating that the NS1-C9 complex affects MAC formation. We also evaluated whether the NS1-VN complex affects C9 polymerization. A more pronounced inhibition was observed in the presence of both NS1e and VN than that with NS1e or VN alone, suggesting that this interaction enhances the inhibition of C9 polymerization (Fig. 6C and D). We also performed a competitive ELISA using a constant amount of NS1e in the presence of increasing amounts of VN. NS1 binding to C9 was not affected by VN even at a 10-fold molar excess (Fig. 6E), thus indicating that NS1 and VN do not compete for the same binding sites on C9. Taken together, our data strongly suggest that NS1 and VN bind C9 simultaneously and interfere with its polymerization.
FIG 6.

NS1 by itself or in association with vitronectin (VN) inhibits C9 polymerization. C9 was incubated with different amounts of NS1e (10 to 100 μg/ml) (A) or NS1dros (10 to 100 μg/ml) (B) or with NS1 buffer (B; negative control) at 37°C for 40 min before the addition of 50 μM ZnCl2 for 2 h at 37°C. (C) C9 was incubated separately or with a mixture of NS1e and VN (10 and 25 μg/ml) at 37°C for 40 min before the addition of 50 μM ZnCl2 for 2 h at 37°C. All samples were subjected to SDS-PAGE in a gradient gel (4 to 20%) under nonreducing conditions, and C9 polymerization was visualized by silver staining. (D) The C9 monomer band intensities from panel C were measured by densitometry and reported in arbitrary units. The error bars indicate standard deviations for three independent experiments, and the asterisks indicate the significance of differences from the C9 band intensity in the absence of NS1 and VN (C9 + ZnCl2), determined using one-way ANOVA and the Bonferroni posttest. (E) NS1 and VN do not compete for C9 binding. C9 was immobilized on microtiter plates, and a constant amount of NS1e (100 nM) with increasing amounts of VN was added. Bound NS1e was detected using an anti-NS1 polyclonal antibody. One-way ANOVA and the Bonferroni posttest were used to calculate the significance of differences compared to the binding without NS1. Error bars indicate standard deviations for three independent experiments, and the asterisks indicate the significance of differences from the control. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. MW, molecular weight.
Flavivirus NS1 binds C9 and inhibits C9 polymerization in serum.
To address whether NS1 secreted by mammalian cells (NS1mam) is capable of inhibiting C9 polymerization, we used recombinant NS1mam proteins from DENV2, WNV, and ZIKV expressed in HEK 293 cells. Initially, BHK-21 cells were preincubated with C5b6, and DENV2, WNV, or ZIKV NS1mam was preincubated with 5% NHS in a separate preparation. C5b6-treated BHK-21 cells were then incubated with NS1mam plus 5% NHS, washed, and lysed. Samples were analyzed by Western blotting with anti-C9 antibody. As the NS1mam concentration increased, the band intensity corresponding to the C9 polymer decreased (Fig. 7A) for all tested flavivirus proteins. Surprisingly, the C9 polymerization showed the largest inhibitory effect when cells were incubated with the ZIKV NS1mam protein. When anti-NS1 was preincubated with NS1mam at 50 μg/ml, the inhibition of C9 polymerization was abolished, thus confirming a specific role of NS1 in inhibition of C9 polymerization.
FIG 7.

Flavivirus NS1 from mammalian cells binds and inhibits C9 polymerization in serum. (A) Flavivirus NS1 secreted by mammalian cells (NS1mam) inhibits C9 polymerization. Initially, BHK-21 cells were preincubated with 5 μg/ml purified C5b6 for 2 h at 37°C in VBS. DENV2, WNV, or ZIKV NS1mam was preincubated with 5% NHS for 30 min at 37°C. Rabbit polyclonal anti-NS1 (50 μg/ml) was mixed with 50 μg/ml NS1mam to serve as a control. Cells were washed and incubated with NS1mam mixed with NHS for 50 min at 37°C. Thereafter, cells were washed and lysed. Samples were separated in a 10% polyacrylamide gel under nonreducing conditions, and the C9 bands were analyzed by Western blotting using anti-C9. (B) Flavivirus NS1mam interacts with purified C9. C9 (0 to 500 nM) was incubated in DENV2, WNV, or ZIKV NS1mam-coated plates (10 μg/ml) for 2 h at 37°C. Wells were washed with PBS and then incubated with polyclonal anti-C9 antibody, followed by incubation with peroxidase-conjugated anti-goat IgG antibody for 1 h at 37°C. The error bars indicate standard deviations for three independent experiments, and the asterisks indicate the significance of differences from the control, determined using two-way ANOVA and the Bonferroni posttest. ***, P < 0.001. (C) Flavivirus NS1mam in complex with VN inhibited C9 polymerization. DENV2, WNV, or ZIKV NS1mam (25 μg/ml) was preincubated with increasing concentrations of VN (0 to 25 μg/ml) for 30 min at 37°C, incubated with 5% NHS for 30 min at 37°C, and added to C5b6-treated BHK-21 cells for 50 min at 37°C. Thereafter, cells were washed three times with VBS and lysed with cell lysis buffer, and the C9 polymer was analyzed by Western blotting using anti-C9 antibody. Experiments were repeated three times.
Because ZIKV NS1mam inhibited C9 polymerization more strongly than the DENV2 and WNV NS1mam proteins did, we assessed binding of purified C9 to immobilized NS1 proteins. As shown in Fig. 7B, the DENV2 and WNV NS1mam proteins bound C9 similarly, whereas ZIKV NS1mam bound C9 more avidly, suggesting that the pronounced effect of ZIKV NS1 on C9 polymerization may be explained by its binding to C9. In addition, preincubation of flavivirus NS1mam (25 μg/ml) with increasing concentrations of VN was more effective at preventing C9 polymerization in serum (Fig. 7C).
DISCUSSION
During the course of evolution, pathogens have developed strategies to evade complement system activation in order to coexist with the human host. Flavivirus NS1 has been described as an immune evasion protein that can attenuate the activation of the classical, lectin, and alternative pathways by interacting with complement proteins and their regulators. It has been shown that WNV NS1 recruits the complement regulator factor H as a cofactor for C3b inactivation by factor I (11). Additionally, flavivirus NS1 binds C1s/pro-C1s and C4 in a complex to cleave C4 to C4b in solution, thus reducing the deposition of C4b (12). NS1 also recruits C4BP as a cofactor for factor I-mediated cleavage of C4b in solution (13). In this study, we defined a novel mechanism by which DENV NS1 inhibits MAC formation by interacting with VN and terminal complement proteins.
VN is a multifunctional glycoprotein that plays an important role in many biological processes, including pericellular proteolysis, fibrinolysis, and regulation of the terminal pathway of the complement cascade. Several pathogens, including Gram-negative and -positive bacteria, possess proteins that recruit VN to help them become serum resistant and adhere to host cells. By recruiting VN to their membranes, they avoid lysis through inhibition of C5b9 complex formation (14, 18). Whereas the majority of the described interactions are for bacterial proteins, there are a few viral proteins that have been shown to interact with human VN, including the HIV gp120/160 protein (34, 35) and the hepatitis C virus (HCV) F protein (36). This is the first study to report the interaction of DENV NS1 with VN as a complement evasion strategy.
Our findings indicate that both recombinant and native NS1 proteins from DENV-infected patients form complexes with VN. Most pathogenic bacteria bind VN at either its N-terminal region (37), its central domain (containing HPX-like domains) (30), or the basic carboxy-terminal HBD-3 (38). Usually, bacterium-VN binding does not interfere with the VN domain involved in the inhibition of the C5b9 complex, thus allowing this complement regulator to remain active in inhibiting MAC formation. Our data suggest that the NS1-VN interaction does not involve the SMB domain, HBDs, or HPX-like domains of the VN molecule. However, DENV NS1 competes with PLG for binding to VN, suggesting that these molecules have overlapping binding sites on VN.
The multifunctional nature of VN can be attributed to its recognition of various ligands, including PAI-1, which regulates PLG activation by inhibiting tissue-plasminogen activator (tPA) and urokinase-plasminogen activator (uPA) (39, 40). VN regulates the half-life of PAI-1, keeping its inhibitory form stable for a longer period (41, 42). Several studies have demonstrated increased PAI-1 levels in severe cases of dengue. Interestingly, none of them found a correlation between elevated levels of PAI-1 and disease severity. However, both elevated PAI-1 levels and disease severity did correlate with poor clinical outcome and platelet count (43–45). Additionally, it has been demonstrated that the EIII domain of the DENV2 E protein induces PAI-1 gene expression (46). Here we demonstrate that NS1 directly binds PAI-1 and that this association seems to facilitate the NS1-VN interaction. Competition assays suggest that NS1 and PAI-1 do not have overlapping binding sites on VN. Therefore, we speculate that these molecules may form a ternary complex. However, the physiological significance of NS1 binding simultaneously to PAI-1 and VN and the consequences of such an interaction to the success of viral infectivity remain to be elucidated.
According to our data, NS1 has a MAC-inhibitory activity that can be attributed to its interaction with MAC components. NS1 binds the complement proteins C5, C6, C7, and C9 and may thereby inhibit the assembly and membrane insertion of the MAC. The functional consequences of the NS1-C9 interaction were evaluated. NS1 by itself or complexed with VN inhibited C9 polymerization, thus preventing lytic pore formation. By interacting with VN, NS1 enhanced MAC inhibition. This additive effect may be attributed to simultaneous binding of NS1 and VN to C9. We also demonstrated that glycosylation of NS1 is not a determinant factor in inhibiting MAC formation because both nonglycosylated and glycosylated forms of NS1 hindered erythrocyte lysis and C9 polymerization. We also compared the C9 polymerization inhibition levels by DENV2, WNV, and ZIKV NS1 proteins produced in mammalian cells. Our data showed that these three flavivirus proteins inhibited C9 polymerization in a way similar to that for the NS1e and NS1dros proteins used in this study, confirming our results. It is worth mentioning that ZIKV NS1 bound and inhibited C9 polymerization more strongly than the DENV2 and WNV proteins did, suggesting a possible role of ZIKV NS1 complement evasion in assisting ZIKV infection in the brain (47, 48).
This study provides novel insights into the mechanisms of flavivirus manipulation of the host complement system. NS1 is a pathogenic immune evasion protein that interacts with more than one human complement molecule and controls multiple steps of complement activation and function. In summary, NS1 by itself or in association with VN is capable of inhibiting C9 polymerization and, consequently, MAC formation. These results suggest a role for NS1 in dengue pathogenesis as a terminal pathway inhibitor of the complement system.
ACKNOWLEDGMENTS
We thank Cynthia Peterson and her student Letitia Olson at the University of Tennessee for providing the PAI-1 protein.
This work was supported by Rio de Janeiro Research Foundation (FAPERJ) (grants 010.002879/2014, 010.001597/2014, 102.356/2013, 110.196/2013, and 112.671/2012) and from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grants 400213/2014-33 1 and 475104/2012-9).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. 2009. Dengue: guidelines for diagnosis, treatment, prevention and control, new ed. WHO, Geneva, Switzerland. [PubMed] [Google Scholar]
- 3.Lindenbach BD, Rice CM. 2003. Molecular biology of flaviviruses. Adv Virus Res 59:23–61. doi: 10.1016/S0065-3527(03)59002-9. [DOI] [PubMed] [Google Scholar]
- 4.Mackenzie JM, Jones MK, Young PR. 1996. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 220:232–240. doi: 10.1006/viro.1996.0307. [DOI] [PubMed] [Google Scholar]
- 5.Lindenbach BD, Rice CM. 1997. trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J Virol 71:9608–9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akey DL, Brown WC, Dutta S, Konwerski J, Jose J, Jurkiw TJ, DelProposto J, Ogata CM, Skiniotis G, Kuhn RJ, Smith JL. 2014. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 343:881–885. doi: 10.1126/science.1247749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gutsche I, Coulibaly F, Voss JE, Salmon J, d'Alayer J, Ermonval M, Larquet E, Charneau P, Krey T, Megret F, Guittet E, Rey FA, Flamand M. 2011. Secreted dengue virus nonstructural protein NS1 is an atypical barrel-shaped high-density lipoprotein. Proc Natl Acad Sci U S A 108:8003–8008. doi: 10.1073/pnas.1017338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silva EM, Conde JN, Allonso D, Nogueira ML, Mohana-Borges R. 2013. Mapping the interactions of dengue virus NS1 protein with human liver proteins using a yeast two-hybrid system: identification of C1q as an interacting partner. PLoS One 8:e57514. doi: 10.1371/journal.pone.0057514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller DA, Young PR. 2013. The flavivirus NS1 protein: molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antiviral Res 98:192–208. doi: 10.1016/j.antiviral.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, Hume DA, Stacey KJ, Young PR. 2015. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci Transl Med 7:304ra142. doi: 10.1126/scitranslmed.aaa3863. [DOI] [PubMed] [Google Scholar]
- 11.Chung KM, Liszewski MK, Nybakken G, Davis AE, Townsend RR, Fremont DH, Atkinson JP, Diamond MS. 2006. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc Natl Acad Sci U S A 103:19111–19116. doi: 10.1073/pnas.0605668103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, Atkinson JP. 2010. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J Exp Med 207:793–806. doi: 10.1084/jem.20092545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Avirutnan P, Hauhart RE, Somnuke P, Blom AM, Diamond MS, Atkinson JP. 2011. Binding of flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J Immunol 187:424–433. doi: 10.4049/jimmunol.1100750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh B, Su YC, Riesbeck K. 2010. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Mol Microbiol 78:545–560. doi: 10.1111/j.1365-2958.2010.07373.x. [DOI] [PubMed] [Google Scholar]
- 15.Milis L, Morris CA, Sheehan MC, Charlesworth JA, Pussell BA. 1993. Vitronectin-mediated inhibition of complement: evidence for different binding sites for C5b-7 and C9. Clin Exp Immunol 92:114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Podack ER, Preissner KT, Muller-Eberhard HJ. 1984. Inhibition of C9 polymerization within the SC5b-9 complex of complement by S-protein. Acta Pathol Microbiol Immunol Scand 284(Suppl):89–96. [PubMed] [Google Scholar]
- 17.Sheehan M, Morris CA, Pussell BA, Charlesworth JA. 1995. Complement inhibition by human vitronectin involves non-heparin binding domains. Clin Exp Immunol 101:136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.da Silva LB, Miragaia LDS, Breda LC, Abe CM, Schmidt MC, Moro AM, Monaris D, Conde JN, Jozsi M, Isaac L, Abreu PA, Barbosa AS. 2015. Pathogenic Leptospira species acquire factor H and vitronectin via the surface protein LcpA. Infect Immun 83:888–897. doi: 10.1128/IAI.02844-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allonso D, da Silva Rosa M, Coelho DR, da Costa SM, Nogueira RM, Bozza FA, Santos FB, de Barcelos Alves AM, Mohana-Borges R. 2011. Polyclonal antibodies against properly folded dengue virus NS1 protein expressed in E. coli enable sensitive and early dengue diagnosis. J Virol Methods 175:109–116. doi: 10.1016/j.jviromet.2011.04.029. [DOI] [PubMed] [Google Scholar]
- 20.Allonso D, Meneses MD, Fernandes CA, Ferreira DF, Mohana-Borges R. 2014. Assessing positivity and circulating levels of NS1 in samples from a 2012 dengue outbreak in Rio de Janeiro, Brazil. PLoS One 9:e113634. doi: 10.1371/journal.pone.0113634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Yang J, Wei J, Yang Y, Chen X, Zhao X, Gu Y, Cui S, Zhu X. 2011. Trichinella spiralis paramyosin binds to C8 and C9 and protects the tissue-dwelling nematode from being attacked by host complement. PLoS Negl Trop Dis 5:e1225. doi: 10.1371/journal.pntd.0001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yasumitsu H, Seo N, Misugi E, Morita H, Miyazaki K, Umeda M. 1993. Vitronectin secretion by hepatic and non-hepatic human cancer cells. In vitro cellular & developmental biology. Animal 29A:403–407. [DOI] [PubMed] [Google Scholar]
- 23.Kost C, Stuber W, Ehrlich HJ, Pannekoek H, Preissner KT. 1992. Mapping of binding sites for heparin, plasminogen activator inhibitor-1, and plasminogen to vitronectin's heparin-binding region reveals a novel vitronectin-dependent feedback mechanism for the control of plasmin formation. J Biol Chem 267:12098–12105. [PubMed] [Google Scholar]
- 24.Lane DA, Flynn AM, Pejler G, Lindahl U, Choay J, Preissner K. 1987. Structural requirements for the neutralization of heparin-like saccharides by complement S protein/vitronectin. J Biol Chem 262:16343–16348. [PubMed] [Google Scholar]
- 25.Liang OD, Rosenblatt S, Chhatwal GS, Preissner KT. 1997. Identification of novel heparin-binding domains of vitronectin. FEBS Lett 407:169–172. doi: 10.1016/S0014-5793(97)00330-X. [DOI] [PubMed] [Google Scholar]
- 26.Schar CR, Jensen JK, Christensen A, Blouse GE, Andreasen PA, Peterson CB. 2008. Characterization of a site on PAI-1 that binds to vitronectin outside of the somatomedin B domain. J Biol Chem 283:28487–28496. doi: 10.1074/jbc.M804257200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Preissner KT. 1990. Specific binding of plasminogen to vitronectin. Evidence for a modulatory role of vitronectin on fibrin(ogen)-induced plasmin formation by tissue plasminogen activator. Biochem Biophys Res Commun 168:966–971. [DOI] [PubMed] [Google Scholar]
- 28.Su YC, Jalalvand F, Morgelin M, Blom AM, Singh B, Riesbeck K. 2013. Haemophilus influenzae acquires vitronectin via the ubiquitous protein F to subvert host innate immunity. Mol Microbiol 87:1245–1266. doi: 10.1111/mmi.12164. [DOI] [PubMed] [Google Scholar]
- 29.Xu D, Baburaj K, Peterson CB, Xu Y. 2001. Model for the three-dimensional structure of vitronectin: predictions for the multi-domain protein from threading and docking. Proteins 44:312–320. doi: 10.1002/prot.1096. [DOI] [PubMed] [Google Scholar]
- 30.Liang OD, Preissner KT, Chhatwal GS. 1997. The hemopexin-type repeats of human vitronectin are recognized by Streptococcus pyogenes. Biochem Biophys Res Commun 234:445–449. doi: 10.1006/bbrc.1997.6663. [DOI] [PubMed] [Google Scholar]
- 31.Alcon S, Talarmin A, Debruyne M, Falconar A, Deubel V, Flamand M. 2002. Enzyme-linked immunosorbent assay specific to dengue virus type 1 nonstructural protein NS1 reveals circulation of the antigen in the blood during the acute phase of disease in patients experiencing primary or secondary infections. J Clin Microbiol 40:376–381. doi: 10.1128/JCM.40.02.376-381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Libraty DH, Young PR, Pickering D, Endy TP, Kalayanarooj S, Green S, Vaughn DW, Nisalak A, Ennis FA, Rothman AL. 2002. High circulating levels of the dengue virus nonstructural protein NS1 early in dengue illness correlate with the development of dengue hemorrhagic fever. J Infect Dis 186:1165–1168. doi: 10.1086/343813. [DOI] [PubMed] [Google Scholar]
- 33.Hallstrom T, Blom AM, Zipfel PF, Riesbeck K. 2009. Nontypeable Haemophilus influenzae protein E binds vitronectin and is important for serum resistance. J Immunol 183:2593–2601. doi: 10.4049/jimmunol.0803226. [DOI] [PubMed] [Google Scholar]
- 34.Su HR, Boackle RJ. 1994. Heparin mediates binding of S-protein/vitronectin to the envelope glycoprotein of the human immunodeficiency virus and CD4. Int Arch Allergy Immunol 105:238–244. doi: 10.1159/000236763. [DOI] [PubMed] [Google Scholar]
- 35.Bozzini S, Falcone V, Conaldi PG, Visai L, Biancone L, Dolei A, Toniolo A, Speziale P. 1998. Heparin-binding domain of human fibronectin binds HIV-1 gp120/160 and reduces virus infectivity. J Med Virol 54:44–53. doi:. [DOI] [PubMed] [Google Scholar]
- 36.Huang YP, Cheng J, Zhang SL, Wang L, Guo J, Liu Y, Yang Y, Zhang LY, Bai GQ, Gao XS, Ji D, Lin SM, Shao Q. 2005. Screening of hepatocyte proteins binding to F protein of hepatitis C virus by yeast two-hybrid system. World J Gastroenterol 11:5659–5665. doi: 10.3748/wjg.v11.i36.5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sa E Cunha C, Griffiths NJ, Virji M. 2010. Neisseria meningitidis Opc invasin binds to the sulphated tyrosines of activated vitronectin to attach to and invade human brain endothelial cells. PLoS Pathog 6:e1000911. doi: 10.1371/journal.ppat.1000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohler S, Hallstrom T, Singh B, Riesbeck K, Sparta G, Zipfel PF, Hammerschmidt S. 2015. Binding of vitronectin and factor H to Hic contributes to immune evasion of Streptococcus pneumoniae serotype 3. Thromb Haemost 113:125–142. doi: 10.1160/TH14-06-0561. [DOI] [PubMed] [Google Scholar]
- 39.Lawrence D, Strandberg L, Grundstrom T, Ny T. 1989. Purification of active human plasminogen activator inhibitor 1 from Escherichia coli. Comparison with natural and recombinant forms purified from eucaryotic cells. Eur J Biochem 186:523–533. [DOI] [PubMed] [Google Scholar]
- 40.Zhou A, Carrell RW, Huntington JA. 2001. The serpin inhibitory mechanism is critically dependent on the length of the reactive center loop. J Biol Chem 276:27541–27547. doi: 10.1074/jbc.M102594200. [DOI] [PubMed] [Google Scholar]
- 41.Declerck PJ, De Mol M, Alessi MC, Baudner S, Paques EP, Preissner KT, Muller-Berghaus G, Collen D. 1988. Purification and characterization of a plasminogen activator inhibitor 1 binding protein from human plasma. Identification as a multimeric form of S protein (vitronectin). J Biol Chem 263:15454–15461. [PubMed] [Google Scholar]
- 42.Hansen M, Busse MN, Andreasen PA. 2001. Importance of the amino-acid composition of the shutter region of plasminogen activator inhibitor-1 for its transitions to latent and substrate forms. Eur J Biochem 268:6274–6283. doi: 10.1046/j.0014-2956.2001.02582.x. [DOI] [PubMed] [Google Scholar]
- 43.Wills BA, Oragui EE, Stephens AC, Daramola OA, Dung NM, Loan HT, Chau NV, Chambers M, Stepniewska K, Farrar JJ, Levin M. 2002. Coagulation abnormalities in dengue hemorrhagic fever: serial investigations in 167 Vietnamese children with dengue shock syndrome. Clin Infect Dis 35:277–285. doi: 10.1086/341410. [DOI] [PubMed] [Google Scholar]
- 44.Mairuhu AT, Setiati TE, Koraka P, Hack CE, Leyte A, Faradz SM, ten Cate H, Brandjes DP, Osterhaus AD, Reitsma PH, van Gorp EC. 2005. Increased PAI-1 plasma levels and risk of death from dengue: no association with the 4G/5G promoter polymorphism. Thromb J 3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Djamiatun K, Faradz SM, Setiati TE, Netea MG, van der Ven AJ, Dolmans WM. 2011. Increase of plasminogen activator inhibitor-1 and decrease of transforming growth factor-b1 in children with dengue haemorrhagic fever in Indonesia. J Trop Pediatr 57:424–432. doi: 10.1093/tropej/fmq122. [DOI] [PubMed] [Google Scholar]
- 46.Shyu HW, Lin YY, Chen LC, Wang YF, Yeh TM, Su SJ, Cheng WC, Chen CY, Lin KH, Chou MC. 2010. The dengue virus envelope protein induced PAI-1 gene expression via MEK/ERK pathways. Thromb Haemost 104:1219–1227. doi: 10.1160/TH10-05-0302. [DOI] [PubMed] [Google Scholar]
- 47.Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, Mysorekar IU, Diamond MS. 2016. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165:1081–1091. doi: 10.1016/j.cell.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veerhuis R, Nielsen HM, Tenner AJ. 2011. Complement in the brain. Mol Immunol 48:1592–1603. doi: 10.1016/j.molimm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]