

Abstract
Background & Aims
There is evidence from clinical studies that compromised intestinal epithelial permeability contributes to the development of non-alcoholic steatohepatitis (NASH), but the exact mechanisms are not clear. Mice with disruption of the gene (F11r) encoding junctional adhesion molecule A (JAM-A) have defects in intestinal epithelial permeability. We used these mice to study how disruption of the intestinal epithelial barrier contributes to NASH.
Methods
Male C57BL/6 (control) or F11r−/− mice were fed a normal diet or a diet high in saturated fat, fructose and cholesterol (HFCD) for 8 weeks. Liver and intestinal tissues were collected and analyzed by histology, quantitative reverse transcription PCR and flow cytometry. Intestinal epithelial permeability was assessed in mice by measuring permeability to fluorescently labeled dextran. The intestinal microbiota were analyzed using 16S rRNA sequencing. We also analyzed biopsies from proximal colon of 30 patients with non-alcoholic fatty liver disease (NAFLD) and 19 subjects without NAFLD (controls) undergoing surveillance colonoscopy.
Results
F11r−/− mice fed a HFCD, but not a normal diet, developed histologic and pathologic features of severe NASH including steatosis, lobular inflammation, hepatocellular ballooning, and fibrosis, whereas control mice fed a HFCD developed only modest steatosis. Interestingly, there were no differences in body weight, ratio of liver weight:body weight, or glucose homeostasis between control and F11r−/− mice fed a HFCD. In these mice, liver injury was associated with significant increases in mucosal inflammation, tight junction disruption and intestinal epithelial permeability to bacterial endotoxins, compared with control mice or F11r−/− mice fed a normal diet. The HFCD led to a significant increase in inflammatory microbial taxa in F11r−/− mice, compared with control mice. Administration of oral antibiotics or sequestration of bacterial endotoxins with sevelamer hydrochloride reduced mucosal inflammation and restored normal liver histology in F11r−/− mice fed a HFCD. Protein and transcript levels of JAM-A were significantly lower in the intestinal mucosa of patients with NAFLD than without NAFLD; decreased expression of JAM-A correlated with increased mucosal inflammation.
Conclusions
Mice with defects in intestinal epithelial permeability develop more severe steatohepatitis following a HFCD than control mice; colon tissues from patients with NAFLD have lower levels of JAM-A and higher levels of inflammation than subjects without NAFLD. These findings indicate that intestinal epithelial barrier function and microbial dysbiosis contribute to development of NASH. Restoration of intestinal barrier integrity and manipulation of gut microbiota might be developed as therapeutic strategies for patients with NASH.
Keywords: occuldin, claudin-4, bacterial translocation
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is a leading cause of chronic liver failure in the United States, and it’s incidence is expected to increase in the near future1. One third of the US population, and a majority of obese individuals develop non-alcoholic fatty liver (NAFL) or bland steatosis; a benign condition characterized by triglyceride deposition in hepatocytes2. Although asymptomatic, nearly twenty percent of individuals with NAFL progress to nonalcoholic steatohepatitis (NASH), and fifteen percent of NASH patients develop cirrhosis2. In addition, NASH patients are at a higher risk for developing hepatocellular carcinoma.3 Despite the growing incidence of NAFLD-related chronic liver disease, the lack of clarity in the mechanisms of NASH pathogenesis has hindered our ability to develop effective biomarkers as well as therapies for more severe forms of NAFLD. One major barrier to achieving this goal is the lack of a suitable murine model, which faithfully recapitulates human NASH.
Several recent clinical studies have highlighted the potential role for compromised intestinal epithelial permeability in NASH pathogenesis3–5. In both human and animal NASH models, increased intestinal epithelial permeability correlates with increased serum endotoxin6, 7; a potent inducer of hepatic inflammation8. Furthermore, increased intestinal epithelial permeability and small intestinal bacterial overgrowth (SIBO) are two frequently observed features of human NASH and NASH-related cirrhosis3. However, it is not clear whether intestinal epithelial barrier dysfunction is a cause or consequence of chronic systemic inflammation observed in NASH patients.
Here, we interrogated the role of intestinal epithelial permeability in NAFLD progression using junctional adhesion molecule A (JAM-A) knockout mice (F11r−/−) that have a basal defect in the intestinal epithelial barrier9. JAM-A is a component of the tight junction (TJ) complex that regulates intestinal epithelial paracellular permeability9–11. Previous studies have demonstrated that F11r−/− mice have leaky TJs that allow for translocation of gut bacteria to peripheral organs9, 12, 13. Interestingly, these mice do not develop spontaneous colitis when fed a normal diet, rendering them useful for studying the effect of a Western diet in NAFLD progression. Here we demonstrate a critical role for intestinal epithelial permeability in NASH pathogenesis and our findings underscore the complex interplay between diet, intestinal epithelial barrier and gut microbial composition in driving NAFLD progression.
METHODS
Mice
C57BL/6 (control) mice were purchased from The Jackson Laboratory and F11r−/− mice were originally a gift from T. Sato, Cornell University, New York, NY. F11r−/− mice were generated as previously described14 and were backcrossed to C57BL/6 mice for seven generations. The intestine specific JAM-A KO mice, VillinCreF11rFL/FL were obtained by crossing VillinCre mice with F11rFL/FL mice. C57BL/6, F11r−/−, F11rFL/FL and VillinCreF11rFL/FL mice were bred and maintained at Emory University Division of Animal Resources. All animal studies were approved by Institutional Animal Care and Use Committee.
NAFLD diet
To induce NAFLD, 5–6 week-old male control and F11r−/− mice were fed a high fat, high fructose and high cholesterol diet (HFCD) ad libitum for 8 weeks. The diet consisted of 0.2% cholesterol, 20% protein, 43% CHO and 23% fat (6.6% trans-fat) (TD.120330; Harlan Laboratories)15, 16. Additionally, 2.31% fructose was provided in drinking water. The normal diet is the standard mouse chow that contains 16% protein, 61% carbohydrate and 7.2% fat. The F11rFL/FL and VillinCreF11rFL/FL mice were fed HFCD containing 2.31% fructose (TD.130885; Harlan Laboratories) to eliminate the need of supplementing drinking water with fructose.
Microbiota depletion
For antibiotic treatment, mice were given a combination of ampicillin (1gm/L), vancomycin (500mg/L), neomycin sulfate (1g/L) and metronidazole (1g/L) for 8 weeks in drinking water.
In vivo permeability assay
Intestinal permeability was assessed by in vivo FITC-dextran (FD4; Sigma-Aldrich) permeability assay as described previously17. Mice fasted for 4 h were gavaged with 0.6 mg/g body weight FITC-dextran (4 KDa) solution and blood was collected by submandibular bleeding after 3 h. Fluorescence intensity in the serum was measured using Fluorescence Spectrophotometer (Synergy 2, Biotek, Winooski, VT). FITC-dextran concentrations were determined from a standard curve generated by serial dilutions of FITC-dextran.
16S rRNA pyrosequencing of luminal microbiota
For high-throughput 16S rRNA gene sequencing, control and F11r−/− mice were initially co-housed for 5 weeks to ensure uniformity of intestinal microbiota18 prior to dietary intervention. Subsequently, control and F11r−/− mice were housed separately during the 8 weeks feeding period. At necropsy, cecum content was collected from all mouse cohorts and total DNA was extracted using a PowerSoil DNA isolation kit (MoBio Laboratories, Carlsbad, CA). The 16S rRNA genes were PCR amplified using Hot Master PCR mix (5Prime, Gaithersburg, MD) and 515F/806R primer pairs targeting V4 region of the 16S rRNA gene19. The sequence of the forward and the reverse primers consisted of, 515f: 5′-AATGATACGGCGACCACCGAGATCTACACTATGGTAATTGTGTGCCAGCMGCCGCGGTAA-3′ and 806r: 5′-CAAGCAGAAGACGGCATACGAGATXXXXXXXXXXXXAGTCAGTCAGCCGGACTACHVGGGTWTCTAAT-3′ respectively. Each reverse primer has a unique multiplex identifier barcode consisting of 12 bp Golay error correcting barcode, designated as XXXXXXXXXXXX, used to identify each sample in the multiplex sequencing run. The reaction conditions were 95°C for 2 min followed by 35 cycles of 95°C for 1 min, 55°C for 1 min, and 72°C for 90 s. Three independent PCRs per sample were purified with Ampure magnetic purification beads (Agencourt, Beverly, MA), pooled, and were sequenced using an Illumina Miseq Sequencing platform at the Emory Integrated Genomics Core (EIGC). Detailed protocols for bacterial 16S rRNA gene amplification and sequencing can be found at the website of Earth Microbiome Project (http://www.earthmicrobiome.org/emp-standard-protocols/)20, 21.
16S rRNA sequence analysis
Paired-end 16S rRNA-V4 region sequencing data were subjected to Quantitative Insights Into Microbial Ecology (QIIME) analysis pipeline22. Default QIIME parameters and workflow scripts were used to process raw data, normalization, clustering, taxonomic classification and visualization as described previously22. The unweighted UniFrac distances between samples were calculated using 27,000–30,000 sequences per sample23. The variation between experimental group (beta diversity) was assessed by principal coordinates analysis (PCoA) plots, and jack-knifed beta diversity was used to estimate the uncertainty in PCoA plots. Alpha diversity curves were calculated based on the number of observed species, and the Shannon diversity index was used to characterize species diversity in a community.
Statistical analysis
Statistical differences were analyzed by Student’s t-test or ANOVA and post hoc analysis for multiple group comparison. A p value < 0.05 was considered statistically significant. Except for the human data, all animal experiments were repeated at least two times on two separate occasions.
RESULTS
F11r−/− mice fed a HFCD develop severe histologic features of NASH including markers of hepatic fibrosis
To induce NAFLD, F11r−/− and control littermates were fed a HFCD for 8 weeks; F11r−/− and control mice fed a normal diet served as controls. While consumption of HFCD resulted in modest NAFL-related histologic findings in control mice, F11r−/− mice developed severe histologic features of NASH including ballooning degeneration of hepatocytes, inflammatory cell infiltration, and extensive pericentral, periportal, and sinusoidal fibrosis (Fig. 1A, B, F; Supp. Fig 1A, B). As shown in Fig. 1B, Supp. Fig 1B, hepatic fibrosis, as assessed by Sirius Red stain, in HFCD-fed F11r−/− mice was not only enhanced in the perivascular region, but was also evident in the pericellular fibers. To corroborate specificity of Sirius Red staining, we also performed immunohistochemistry for alpha smooth muscle actin (αSMA), a key hepatic stellate cell (HSC) activation marker24. Activated HSCs, or myofibroblasts, are well-established markers of both pericellular and perivascular fibrogenesis following liver injury24, 25. As observed in Fig 1C, intense immunohistochemical stain for αSMA was indicative of increased HSC activation. Additionally, the transcript levels of αSMA, as well as transcripts of key molecules associated with hepatic fibrogenesis were also significantly increased in the livers of HCFD-fed F11r−/− mice; transforming growth factor β1 (TGFβ1), tissue inhibitor of metalloproteinase 1 (TIMP-1), and collagen 1, [α1(I) and α2(I)] (Fig. 1E; Supp Fig. 1D, E). NASH-CRN scores were also significantly higher in HFCD-fed F11r−/− mice than in HFCD-fed controls (Fig. 1D). Furthermore, serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) concentrations were also significantly higher in HFCD-fed F11r−/− mice, compared with controls (Fig. 1D). In contrast, liver histopathology, serum biochemical parameters of NASH, and molecular markers of fibrosis did not significantly differ between normal diet-fed control and F11r−/− mice (Fig. 1; Supp. Figs. 1). Interestingly, despite the rapid development of NASH in F11r−/− mice and only bland steatosis in the control mice fed a HCFD, no significant differences were observed with respect to metabolic parameters including body weight, liver weight/body weight ratio and glucose homeostasis (Fig. 2A–D). Additionally, expression of genes involved in de novo lipogenesis and β-oxidation, and hepatic triglyceride levels were also not significantly different between HFCD-fed control and F11r−/− mice (Supp. Fig. 2A–E).
Figure 1. F11r−/− mice fed a high fat, high fructose and high cholesterol diet develop severe histologic features of NASH.
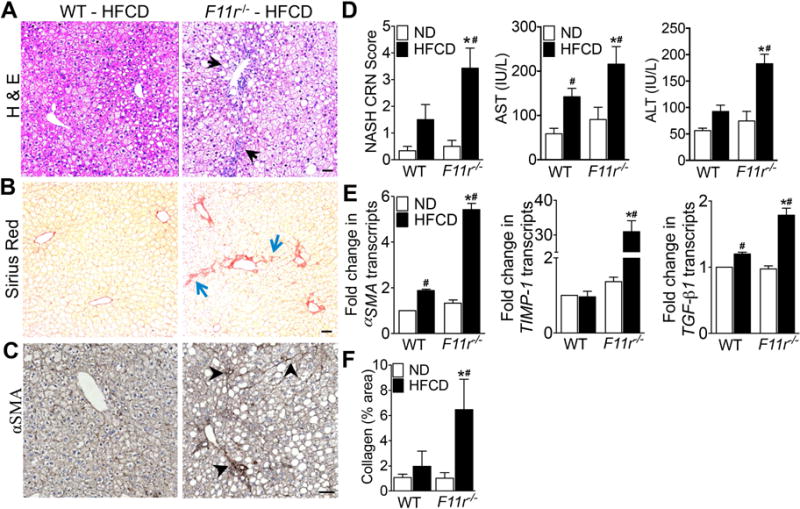
Photomicrographs of (A) Hematoxylin and Eosin (H&E) and (B) Sirius Red-stained liver tissue sections of control (WT) and F11r−/− mice fed a high fat, high fructose and high cholesterol diet (HFCD) for 8 weeks (n = 15). (C) Immunohistochemical staining of α-smooth muscle actin after 8 weeks of feeding (αSMA; n = 10). Black arrows, ballooning degeneration and macrosteatosis; blue arrows, collagen deposition; black arrowheads, αSMA expression. (D) NASH-CRN score, and serum AST and ALT levels (n = 10). (E) Expression of key hepatic stellate cell activation markers and markers of fibrosis in the liver (n = 5). (F) Quantitative analysis of Sirius Red stained liver tissue sections (n = 10). Data shown are representative of three independent experiments. Data are presented as mean ± SEM. Asterisks indicate significant differences (p < 0.05) between control and F11r−/− mice fed an identical diet. Hashtags indicate significant differences (p < 0.05) between normal diet or HFCD-fed control and F11r−/− mice. Scale bar 20 μm.
Figure 2. F11r−/− and control mice fed a high fat, high fructose and high cholesterol diet develop metabolic parameters associated with NAFLD.
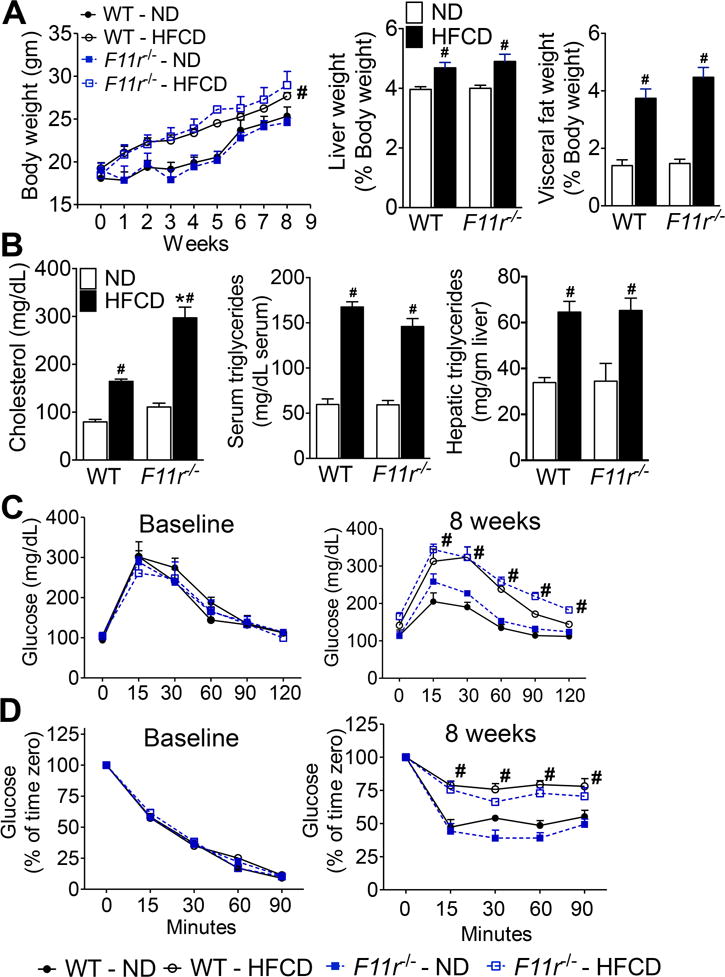
(A) Changes in body, liver and visceral fat weight in control (WT) and F11r−/− mice fed a normal diet (ND) or a high fat, high fructose and high cholesterol diet (HFCD) for 8 weeks (n = 15). Changes in the liver and visceral fat weights are reported as percent of body weight. (B) Changes in serum cholesterol and triglycerides, and hepatic triglyceride levels (n = 10). (C) Glucose and (D) insulin tolerance at baseline and after 8 weeks of feeding (n = 10). Data shown are representative of three independent experiments. Data are presented as mean ± SEM. Asterisks indicate significant differences (p < 0.05) between control and F11r−/− mice fed an identical diet. Hashtags indicate significant differences (p < 0.05) between normal diet- or HFCD-fed control and F11r−/− mice.
To verify that the severity of NASH in HFCD-fed F11r−/− mice was not due to global deletion of JAM-A protein in the liver or in the neutrophils, intestine-specific JAM-A deficient mice, VillinCreF11rFL/FL were fed a HFCD for 8 weeks. As seen in Supp. Fig. 3A–G, VillinCreF11rFL/FL mice fed a HFCD developed similar histological features of NASH as observed in HFCD-fed F11r−/− mice. No differences in liver histopathology and serum biochemical parameters of NASH were observed between VillinCreF11rFL/FL and F11rFL/FL mice fed a normal diet (Supp. Figs. 3). Taken together, these data underscore a critical role for intestinal epithelial permeability in aggravating HFCD-induced progression of NAFLD to NASH.
HFCD induces severe hepatic inflammation in F11r−/− mice
To determine the cellular and molecular players involved in HFCD-induced NASH in F11r−/− mice, we used confocal microscopy and flow cytometry to assess additional indices of hepatic inflammation. Confocal images of liver tissue sections stained with macrophage marker F4/80 revealed increased infiltration of F4/80+ hepatic macrophages in HFCD-fed F11r−/− mice, compared with controls (Fig. 3A; Supp. Fig 4B). These results strongly correlated with increased concentrations of monocyte chemoattractant protein 1 (MCP-1) (Fig. 3B) in the livers of HFCD-fed F11r−/− mice. Flow cytometry confirmed increased infiltration of CD11b+F4/80+ intrahepatic macrophages along with increased recruitment of CD11b+Ly6C+ inflammatory monocytes to the livers of HFCD-fed F11r−/− mice (Figs. 3C,D; Supp. Fig 4D,E). Additional investigation revealed significant up regulation of toll-like receptor (TLR) -2, -4, -5, and -9 transcripts, the major TLRs involved in the detection of bacterial pathogen associated molecular patterns (PAMPs), in the livers of HFCD-fed F11r−/− mice (Fig. 3E). In agreement with increased TLR expression, hepatic transcript and protein levels of key pro-inflammatory cytokines associated with NASH, IL-1β, and IL-6 were significantly higher in HFCD-fed F11r−/− mice (Fig. 3F; Supp. Fig. 5A–C). In addition, circulating levels of IL-1β, TNF-α, and IL-6 were also significantly higher in HFCD-fed F11r−/− mice suggesting that only in the context of defective intestinal epithelial barrier function did the HFCD induce profound systemic inflammation in F11r−/− mice (Supp. Fig. 5D–F). No differences in hepatic inflammatory parameters and immune cell infiltration were observed between control and F11r−/− mice fed a normal diet (Fig. 3B–F; Supp. Figs. 4D–E and 5A–F). Together, these data imply that enhanced hepatic inflammation in HFCD-fed F11r−/− mice results from TLR-mediated activation and recruitment of cellular components of the innate immune system.
Figure 3. High fat, high fructose and high cholesterol diet induces severe hepatic inflammation in F11r−/− mice.
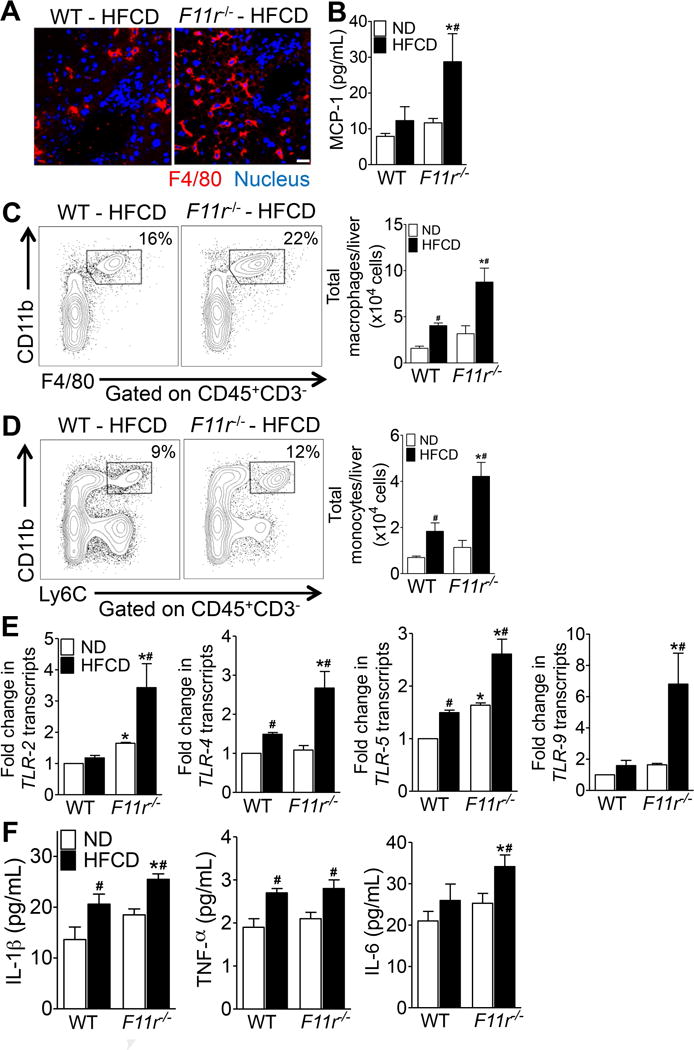
(A) Confocal microscopic images of F4/80+ Kupffer cells (red) in the liver of control (WT) and F11r−/− mice fed a high fat, high fructose and high cholesterol diet (HFCD) for 8 weeks (n = 5). Nuclei are stained blue. Scale 20 μm. (B) Quantification of hepatic MCP-1 expression in control and F11r−/− mice fed a normal diet (ND) or a HFCD for 8 weeks (n = 5). (C, D) Flow cytometric analysis of percent and absolute number of (C) CD11b+F4/80+ macrophages and (D) CD11b+Ly6C+ monocytes in the liver of control and F11r−/− mice fed a normal diet (n = 5) or a HFCD (n = 8) for 8 weeks. (E) Expression of key bacterial toll-like receptors (TLRs) in the liver and (F) quantification of hepatic pro-inflammatory cytokines after 8 weeks of feeding (n = 5). Data shown are representative of two independent experiments. Data are presented as mean ± SEM. Asterisks indicate significant differences (p < 0.05) between control and F11r−/− mice fed an identical diet. Hashtags indicate significant differences (p < 0.05) between normal diet- or HFCD-fed control and F11r−/− mice.
HFCD exacerbates intestinal epithelial permeability in F11r−/− mice allowing increased bacterial translocation
The excessive hepatic inflammation in F11r−/− mice in the setting of an impaired intestinal epithelial barrier prompted us to determine whether HFCD consumption resulted in increased translocation of gut-associated microbial products. We measured intestinal epithelial permeability with an in vivo FITC-dextran permeability assay, and used serum lipopolysaccharide (LPS) concentrations as an indicator of increased microbial product translocation. Consistent with previous reports12, the dextran flux was 3-fold higher in normal diet-fed F11r−/− mice, compared with control mice, confirming a basal defect in intestinal epithelial barrier function in the absence of JAM-A (Fig. 4A)9, 12. Interestingly, dextran flux significantly increased in HFCD-fed F11r−/− mice, compared with normal diet-fed F11r−/− mice, indicating that consumption of HFCD was responsible for further deterioration of intestinal epithelial barrier function (Fig. 4A) and was essential for NASH development in F11r−/− mice. We also observed a two-fold increase in serum LPS in HFCD-fed F11r−/− mice, compared with HFCD-fed control mice, suggesting heightened translocation of gut microbial products (Fig. 4B). Importantly, we failed to observe significant differences in intestinal epithelial permeability measurements or serum LPS concentrations in the control mice fed a normal diet or control mice fed a HFCD (Fig. 4A–B). Thus, in addition to demonstrating a role for gut microbial products in NASH pathogenesis, these results provide an explanation as to why short-term dietary manipulation is insufficient to induce NASH development in murine models.
Figure 4. High fat, high fructose and high cholesterol diet induces intestinal epithelial barrier disruption, mucosal inflammation and translocation of gut microbial products in F11r−/− mice.
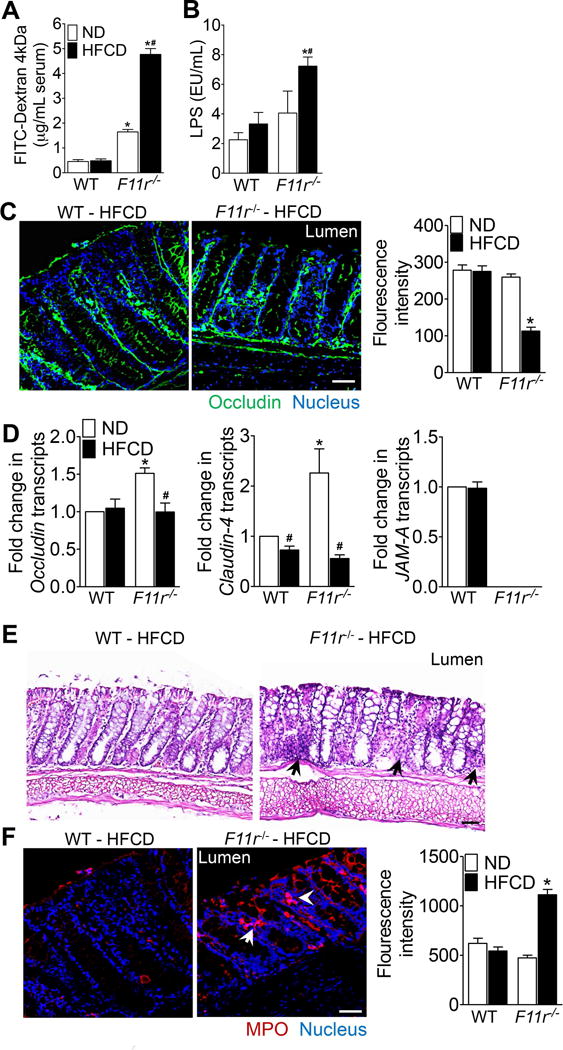
(A) Intestinal permeability to FITC-dextran in control (WT) and F11r−/− mice fed a normal diet (ND) or a high fat, high fructose and high cholesterol diet (HFCD) for 8 weeks (n = 8). (B) Serum LPS levels after 8 weeks of normal diet (n = 5) and HFCD (n = 7) feeding. (C) Confocal images and quantification of occludin (green) immunofluorescence in the colonic mucosa (n = 5). Nuclei are stained blue. Scale bar 100 μm. (D) Photomicrographs of H&E-stained colonic tissue (n = 5). Black arrows, immune cells. Scale 20 μm. (E) Confocal images with quantification of myeloperoxidase (MPO, red) immunofluorescence in the colonic mucosa (n = 5). Nuclei are stained blue. White arrows, cells expressing MPO. Scale bar 100 μm. Data shown are representative of two independent experiments. Data are presented as mean ± SEM. Asterisks indicate significant differences (p < 0.05) between control and F11r−/− mice fed an identical diet. Hashtags indicate significant differences (p < 0.05) between the normal diet- or HFCD-fed control and F11r−/− mice.
HFCD enhances intestinal epithelial permeability by disrupting TJ assembly in F11r−/− mice
To ascertain whether diet-was responsible for an increase in the intestinal epithelial permeability in F11r−/− mice, we performed studies to examine whether dietary manipulation resulted in further disruption of TJs. Colonic expression and distribution of the TJ protein occludin was determined by immunofluorescence microscopy. As shown in Fig. 4C, occludin expression was significantly reduced in HFCD-fed F11r−/− mice, compared with HFCD-fed control mice. However, no significant difference in occludin expression was observed between control and F11r−/− mice fed a normal diet (Supp. Fig. 6B). Further analyses revealed significantly higher expression of occludin and claudin-4 transcripts in the colonic mucosa of normal diet-fed F11r−/− mice than the control mice suggesting a possible compensatory mechanism to maintain intestinal epithelial barrier function in the absence of JAM-A (Fig. 4D). HFCD significantly reduced occludin and claudin-4 expression in F11r−/− mice, compared with normal diet-fed mice, however transcript levels were not significantly different from control mice. In the control mice, except for claudin-4 expression, which is lower in HFCD-fed mice, no significant differences in occludin and JAM-A expression was observed between normal diet- and HFCD-fed mice (Fig. 4D). In contrast to the colonic TJ proteins, no significant differences in occludin and claudin-4 expression were observed in the ileum of control and F11r−/− mice fed either a HFCD or a normal diet (Supp. Fig. 7A–C). In addition to TJ disruption, severe mucosal inflammation was noted in F11r−/− mice fed a HFCD evidenced by increased number of immune cell infiltrates and myeloperoxidase (MPO) activity in the colonic mucosa (Fig. 4E–F; Supp. Fig. 6A, C). Together, these data provide evidence that diet-induced exacerbation of mucosal inflammation and intestinal epithelial barrier disruption increases the likelihood of translocation of microbial products in HFCD-fed F11r−/− mice.
NAFLD patients have decreased JAM-A expression in the colonic mucosa
To investigate whether our findings have potential significance in human NASH, biopsies obtained from proximal colon of 30 patients with NAFLD and 19 subjects without NAFLD (controls) undergoing surveillance colonoscopy were examined for JAM-A protein expression by quantitative reverse transcription PCR (RT-qPCR) and by Western Blot. Patients were considered to have NAFLD if they had a body mass index (BMI) ≥25 kg/m2, elevated serum AST and ALT levels, and had a right upper quadrant ultrasound (RUQ US) that was consistent with hepatic steatosis (Supp. Fig. 8B–C). Since liver biopsies were not obtained, we did not classify patients as specifically having NASH, and therefore collectively termed the study cohort as NAFLD. We defined controls as those subjects having BMI <25 kg/m2, normal serum ALT levels (≤40 U/L), and a negative liver US. As seen in Supp. Fig. 8D–F, both protein and transcript levels of JAM-A were significantly lower in the intestinal mucosa of NAFLD patients, compared with controls. Interestingly, decreased JAM-A expression in the intestinal mucosa correlated with increased mucosal inflammation in NAFLD patients as revealed by increased infiltration of immune cells in the mucosa (Supp. Fig. 8G). Together these data suggest that JAM-A may play a role in the pathogenesis of human NASH.
Rapid NAFLD progression in F11r−/− mice correlates with diet-mediated alteration in gut microbiota
Alterations in gut microbiota play a prominent role in obesity, NAFLD and colitis26–31. Therefore, to determine the effect of HFCD on gut microbiota in F11r−/− mice, the luminal microbiota was analyzed using 16s rRNA sequencing followed by phylogenetic analysis, and a comparison of the microbial community structure using the unweighted UniFrac algorithm. As seen in Fig 5A–E, the luminal microbial composition of normal diet-fed control and F11r−/− mice matched that of lean mice; a microbial composition consisting of an increased abundance of bacteria belonging to the phylum Bacteroidetes and reduced abundance of Firmicutes and Proteobacteria26–28. As expected, HFCD consumption resulted in decreased gut microbial diversity in both control and F11r−/− mice (Fig. 5A, B). HFCD consumption increased abundance of Proteobacteria and Firmicutes and reduced abundance of Bacteroidetes in the luminal content of control and F11r−/− mice (Fig. 5C–E); a gut microbial composition associated with obesity and colitis26, 29–31. However, compared with control mice, the abundance of Firmicutes and Proteobacteria were significantly higher in the luminal content of HFCD-fed F11r−/− mice (Fig. 5C). Additional microbial analysis of HFCD-fed F11r−/− flora revealed a significant increase in Desulfovibrionaceae, a bacterial taxa associated with colitis32, and a significant decrease in Akkermansia, an anti-obesogenic, anti-inflammatory bacterial taxa32–34, in HFCD-fed F11r−/− mice, compared with controls (Supp. Table 1). Taken together, these results suggest that in the context of a defective intestinal epithelial barrier, HFCD-induced an abundance of pro-inflammatory pathobionts, which appear to have contributed to rapid NASH development in F11r−/− mice.
Figure 5. Diet mediated alteration in gut microbiota promotes NAFLD progression in F11r−/− mice.
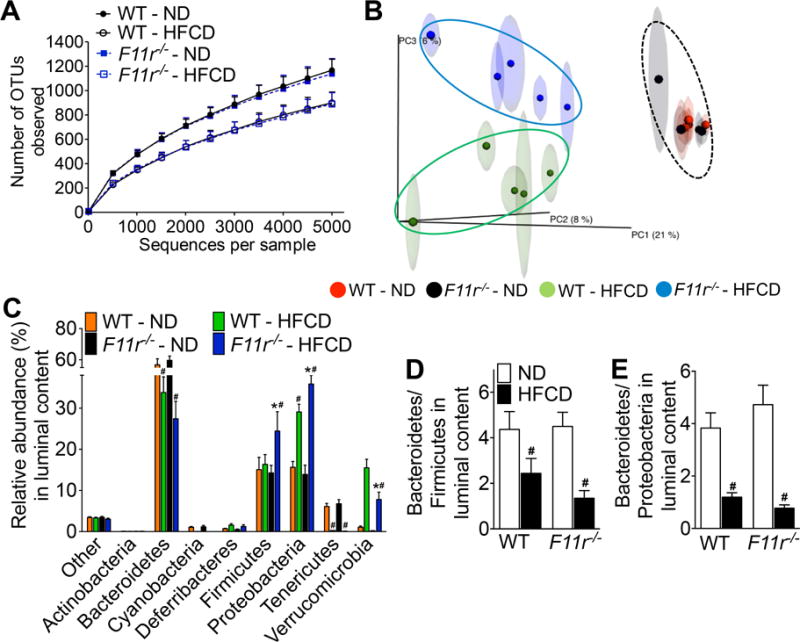
Luminal microbiota from control (WT) and F11r−/− mice fed a normal diet (ND) or a high fat, high fructose and high cholesterol diet (HFCD) for 8 weeks were analyzed using 16S rRNA sequencing followed by phylogenetic analysis and a comparison of the microbial community structure using the unweighted UniFrac algorithm (n = 5). (A) Microbiota richness and diversity in the luminal content (n = 5). (B) Jackknifed principal coordinate analysis (PCoA) of the un-weighted UniFrac distance matrix of the luminal microbiota. The ovals represent clustering by treatment groups (n = 5). (C) Relative abundance of luminal bacterial phyla in control and F11r−/− mice after 8 weeks of feeding (n = 5). The ratios of (D) Bacteroidetes to Firmicutes and (E) Bacteroidetes to Proteobacteria in the luminal content (n = 5). Data are presented as mean ± SEM. Asterisks indicate significant differences (p < 0.05) between control and F11r−/− mice fed an identical diet. Hashtags indicate significant differences (p < 0.05) between the normal diet- or HFCD-fed control and F11r−/− mice.
Depletion of gut microbiota or sequestration of intestinal LPS improves NASH histopathology and metabolic parameters in HFCD-fed control and F11r−/− mice
To corroborate the role of gut microbial translocation, or microbial product transfer in NAFLD progression, gut microbiota were depleted first using a combination of broad-spectrum antibiotics. Depletion of microbiota protected F11r−/− mice from diet-induced NASH as indicated by a marked decrease in hepatic steatosis, inflammation and fibrosis (Fig. 6A–C; Supp. Figs. 9 and 10). As anticipated, antibiotic treatment significantly reduced body weight, liver/body weight ratio, and visceral fat/body weight ratio, and improved glucose homeostasis in both control and F11r−/− mice fed a HFCD (Fig. 6D–F). Antibiotic treatment also significantly lowered serum ALT and LPS levels, and reduced mucosal inflammation in control and F11r−/− mice fed a HFCD (Fig. 6C; Supp. Fig. 11) suggesting a role of gut microbiota in diet-mediated mucosal inflammation.
Figure 6. Depletion of gut microbiota improves NASH histopathology and metabolic parameters in a high fat, high fructose and high cholesterol diet-fed control and F11r−/− mice.
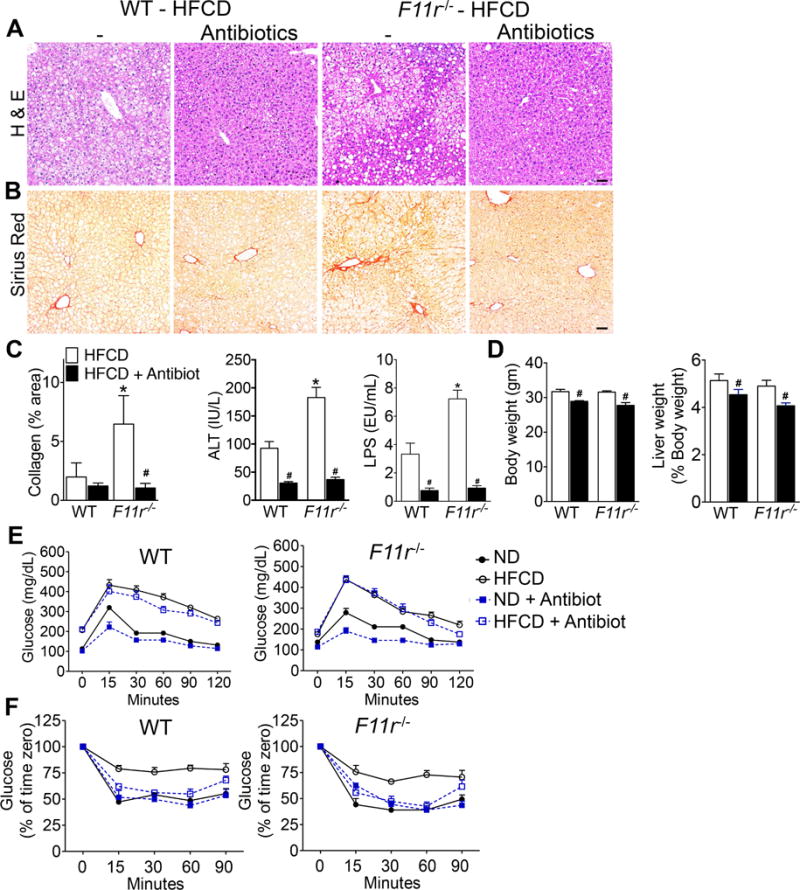
Photomicrographs of (A) H&E and (B) Sirius Red-stained liver tissue sections of control (WT) and F11r−/− mice fed a high fat, high fructose, and high cholesterol diet (HFCD) or a HFCD plus antibiotics for 8 weeks (n = 10). Scale 20 μm. (C) Quantitative analysis of Sirius Red stained liver tissue sections (n = 10), and serum ALT and LPS levels after 8 weeks of treatment (n = 5). Changes in (C) body, liver and visceral fat weights after 8 weeks of treatment (n = 10). Changes in the liver and visceral fat weights are reported as the percentages of the body weight. (D) Glucose and (E) insulin tolerance after 8 weeks of treatment (n = 10). Data shown are representative of two independent experiments. Data are presented as mean ± SEM. Asterisks indicate significant differences (p < 0.05) between control and F11r−/− mice fed an identical diet. Hashtags indicate significant differences (p < 0.05) between HFCD or HFCD plus antibiotic-treated mice.
Sequestration of intestinal LPS by sevelamer hydrochloride treatment attenuates hepatic steatosis and inflammation in HFCD-fed F11r−/− mice
Sevelamer hydrochloride is an FDA-approved phosphate binding non-absorbent polymer regularly used for the treatment of hyperphosphatemia in patients with chronic renal failure35, 36. Sevelamer has been demonstrated to bind LPS and plays a potential role in reducing inflammation37, 38. Therefore we postulated that sevelamer could lower LPS levels in our model and would likely attenuate NASH in HFCD-fed F11r−/− mice. As seen in Supp. Fig 12A, sevelamer treatment significantly reduced serum LPS levels, and more importantly, nearly eliminated NASH in HFCD-fed F11r−/− mice (Supp. Fig. 12D). These findings were further corroborated by decreased serum ALT levels in sevelamer treated mice (Supp. Fig. 12B). Of note, sevelamer-treated mice gained significantly less body weight, and were resistant to HFCD-induced glucose intolerance and insulin resistance (Supp. Fig. 12C, E–F). Collectively, these data demonstrate a central role for translocated gut microbial products in driving NAFLD progression in HFCD-fed F11r−/− mice.
DISCUSSION
In the current study, we provide experimental evidence that intestinal epithelial barrier function plays a major role in NAFLD progression. The HFCD-fed F11r−/− model overcomes key limitations that have hindered reliable induction of NASH along with typical features of Metabolic Syndrome (MetS) in murine models even after long-term feeding. To date, investigators have struggled to reliably replicate MetS and typical features of human NASH in murine liver, including substantial evidence for hepatic inflammation and fibrosis. The methionine-choline deficient (MCD) diet is the only major model that hastens robust hepatic inflammation and fibrosis, however MCD diet-fed mice do not develop MetS. Prolonged diets high in saturated fat, fructose, and cholesterol do induce MetS, but normally result in the development of bland steatosis or mild NASH15, 39 We demonstrate that the HFCD-fed F11r−/− model combats the drawbacks of currently utilized rodent NASH models by combining features of MetS, hepatic inflammation, and fibrosis. Furthermore, the data reported here were generated after 8 weeks of feeding in F11r−/− mice compared to significantly longer feeding periods currently used. Hence this model is highly compelling for additional studies to gain mechanistic insights into the natural history of NAFLD progression. Since JAM-A is a critical, but ubiquitous protein in various organs and leukocytes, the identical findings of NASH in HFCD-fed VillinCreF11rFl/Fl mice as in the global KO mice, F11r−/− mice, confirms that intestinal deficiency of JAM-A, and not hepatic JAM-A deficiency, is responsible for the phenotype which develops following HFCD feeding.
NASH in humans is a multi-factorial disease of complex etiology. Our work supports the hypothesis that ‘you are what you eat’, however, the collective data raise important questions about the sequence of events leading to NAFLD progression. While dietary components, and by extension, dysbiosis, play a critical role in setting the stage for NASH; multiple non-dietary factors resulting in hepatic inflammation, as demonstrated here, are crucial for NAFLD progression40–42. There is compelling evidence linking elevated levels of serum endotoxin to intestinal epithelial permeability and hepatic inflammation in NAFLD progression3, 6, 8, 43–46.
However, whether intestinal epithelial permeability to gut microbial endotoxin represents a cause, or a consequence of diet-induced dysbiosis has not clearly been delineated. In this regard, the present findings significantly advance our understanding of the gut-liver axis in NASH progression. Our results demonstrate that the mice fed a HCFD developed MetS; however, only the F11r−/− mice with a basal defect in intestinal epithelial barrier function developed significant NASH. Only F11r−/− mice had elevated serum endotoxin levels, which were further elevated by the HFCD. The HFCD-induced increase in the intestinal epithelial permeability in F11r−/− mice provides a direct relationship between gut permeability and serum endotoxin levels. However, apart from endotoxins, increased intestinal epithelial permeability could permit enhanced translocation of a multitude of gut microbial products (PAMPs) that we did not explore in the present study. Nonetheless, our data demonstrate robust hepatic inflammation along with significant transcriptional activation of bacterial TLRs suggesting the involvement of additional gut microbial products beside endotoxin. The depletion of gut microbiota by antibiotic treatment or sequestration of LPS by sevelamer administration reduced hepatic inflammation and abolished aggressive NASH further confirming a major role for gut microbial products in driving hepatic inflammation. These results provide significant mechanisms related to newfound interest in the gut-liver axis in NASH by demonstrating that in the context of defective intestinal epithelial barrier function, HFCD-induced increase in intestinal epithelial permeability allows for increased translocation of gut microbial products, which drive hepatic inflammation in NASH. The effectiveness of sevelamer is considerable, and future work to understand the exact mechanisms of action should be forthcoming.
The intestinal epithelium serves as a barrier to prevent intestinal microbiota and microbial products from entering the systemic circulation47. Three intercellular protein complexes namely tight junction, adherens junction, and desmosomes connect epithelial cells with each other to form this selectively permeable epithelial barrier48. Previous studies have shown that mice deficient in JAM-A protein, a component of the tight junction complex, which maintains normal intestinal epithelial paracellular permeability, suffer from increased intestinal epithelial permeability due to a defective tight junction complex9. Strikingly, as observed here, F11r−/− mice fed a standard mouse chow do not develop spontaneous hepatitis, nor have they been reported to develop spontaneous colitis9; a phenomenon supported by induction of compensatory mechanisms to curb excessive mucosal inflammation in these mice12. Our results suggest that under HFCD-fed conditions, this protective mechanism fails resulting in increased mucosal inflammation, which further aggravates TJ disruption as evidenced by redistribution of the TJ protein, occludin, and increased permeability to dextran. Taken together, our work demonstrates a critical role for intestinal epithelial permeability in NASH progression, and underscores the complex role of diet in regulating gut homeostasis, inflammation, and liver health. In the light of our data showing decreased JAM-A in colonic biopsies from NAFLD patients, we speculate that genetic or epigenetic vulnerability in proteins regulating intestinal epithelial barrier integrity may predispose to NAFLD progression in humans. Alternatively, baseline SNPs in such proteins could render normal structure-function relationships at greater risk from harmful host-bacterial interactions resulting from dysbiosis.
Accumulating evidence points to the role of HFCD in altering microbial composition, which in turn profoundly influences intestinal permeability, inflammation, and metabolic complications associated with obesity27, 49. We have identified a number of pro and anti-inflammatory bacterial taxa that were differentially represented in HFCD-fed control and F11r−/− mice. HFCD induced a proinflammatory, procolitogenic microbiota in F11r−/− mice characterized by a decrease in Bacteroidetes but an increase in Firmicutes and Proteobacteria; a microbial composition associated with obesity26, 50 mucosal inflammation29 and NASH51. Interestingly, HFCD also increased Desulfovibrionaceae, a bacterial taxa associated with colitis32, but decreased Akkermansia, an anti-obesogenic, anti-inflammatory bacterial taxa32–34. Thus, these data point to a pivotal role played by impaired intestinal epithelial permeability in promoting an abundance of inflammatory microbial taxa under HFCD-fed conditions, which was unique only to F11r−/− mice. These observations provide impetus for further research to dissect the respective contributions of inflammatory gut microbial taxa in NASH progression as well as to focus on the various components of the intestinal epithelial barrier that, if impaired, may increase host susceptibility to NAFLD progression.
In summary, we have developed a dietary model of NASH, which not only recapitulates key features of MetS, but also mimics hepatic inflammation and fibrosis observed in human NASH. Our data demonstrate that defective intestinal epithelial barrier, dysbiosis, and the associated activation of the innate immune system by increased translocation of gut microbial products are all risk factors associated with NAFLD progression. In the F11r−/− mice, HFCD provided a superimposing ‘hit’ driving a pro-inflammatory gut microbial composition, which exacerbated gut permeability. In turn, enhanced gut leakiness resulted in microbial product translocation, which induced hepatic inflammation and injury ultimately resulting in the progression of NAFLD to NASH (Fig. 7). The present findings strongly warrant consideration of therapies targeting intestinal epithelial barrier, dysbiosis, innate immune function, or a combination thereof to significantly slow or halt NAFLD progression.
Figure 7. Dietary modulation of intestinal epithelial permeability and microbiota promote NAFLD progression.
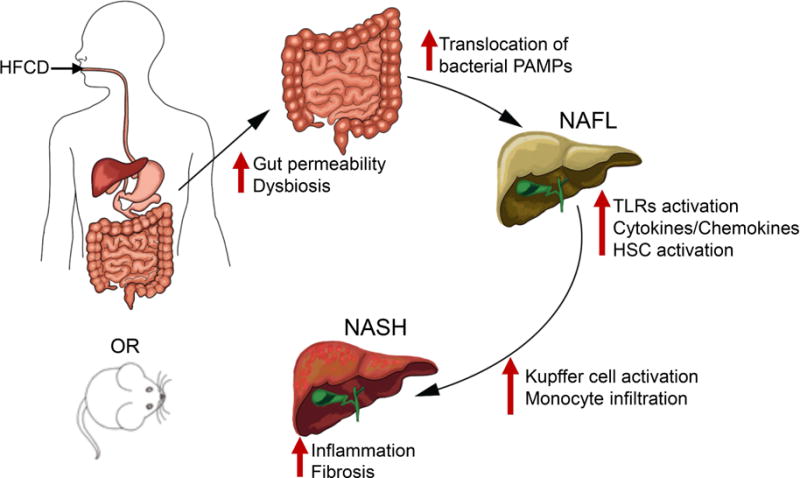
Proposed model highlights how diet mediated changes in the intestinal epithelial permeability and microbial dysbiosis result in the translocation of gut microbial products that drive hepatic inflammation and fibrosis in NASH. HFCD, high fat, high fructose and high cholesterol diet; PAMP, pathogen associated molecular patterns; NAFL, non-alcoholic fatty liver; HSC, hepatic stellate cells; NASH, non-alcoholic steatohepatitis.
Supplementary Material
Acknowledgments
We acknowledge the contribution of graphic artist Deepali Gupta at deepali.gupta@pentadesk.com for the generation of the summary figure. This research project was supported in part by the Emory University Integrated Cellular Imaging Microscopy Core of the Emory and Children’s Pediatric Research Center.
Funding: This work was supported by NIH grant DK062092, VA grant I01BX001746, and funds from Emory University School of Medicine, to FAA.
Abbreviations
- MetS
Metabolic Syndrome
- NAFLD
non-alcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: All authors declare no conflicting interests.
Author Contributions.
F.A.A. conceived the project, designed experiments, and edited the manuscript.
K.R. designed and performed all experiments, analyzed data and wrote the manuscript.
C.D. analyzed microbial data.
S.S.I. planned and performed flow cytometric analysis and edited the manuscript.
N.T. and Y.L. maintained mouse colonies, collected metabolic data
T.S. performed histochemical staining.
P.K. provided valuable suggestions.
S. T. suggested the protocol for human study.
H.L., X. L., P. W., and Y. Y. collected human samples and analyzed data.
A.B.F NASH-CRN scoring.
A.N. provided valuable suggestions and feedback on the manuscript.
C.A.P. provided valuable suggestions, provided knockout mice.
A.S.N. provided valuable suggestions for the gut microbiota analysis.
Author names in bold designate shared co-first authorship
References
- 1.Mahady SE, George J. Management of nonalcoholic steatohepatitis: an evidence-based approach. Clin Liver Dis. 2012;16:631–45. doi: 10.1016/j.cld.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–31. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 3.Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–87. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 4.Wigg AJ, Roberts-Thomson IC, Dymock RB, et al. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–11. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lorenzo-Zuniga V, Bartoli R, Planas R, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology. 2003;37:551–7. doi: 10.1053/jhep.2003.50116. [DOI] [PubMed] [Google Scholar]
- 6.Shanab AA, Scully P, Crosbie O, et al. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci. 2011;56:1524–34. doi: 10.1007/s10620-010-1447-3. [DOI] [PubMed] [Google Scholar]
- 7.Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 8.Chitturi S, Farrell GC. Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis. 2001;21:27–41. doi: 10.1055/s-2001-12927. [DOI] [PubMed] [Google Scholar]
- 9.Laukoetter MG, Nava P, Lee WY, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–76. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monteiro AC, Sumagin R, Rankin CR, et al. JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol Biol Cell. 2013;24:2849–60. doi: 10.1091/mbc.E13-06-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menard S, Cerf-Bensussan N, Heyman M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. 2010;3:247–59. doi: 10.1038/mi.2010.5. [DOI] [PubMed] [Google Scholar]
- 12.Khounlotham M, Kim W, Peatman E, et al. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity. 2012;37:563–73. doi: 10.1016/j.immuni.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vetrano S, Rescigno M, Cera MR, et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135:173–84. doi: 10.1053/j.gastro.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Cera MR, Del Prete A, Vecchi A, et al. Increased DC trafficking to lymph nodes and contact hypersensitivity in junctional adhesion molecule-A-deficient mice. J Clin Invest. 2004;114:729–38. doi: 10.1172/JCI21231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mells JE, Fu PP, Kumar P, et al. Saturated fat and cholesterol are critical to inducing murine metabolic syndrome with robust nonalcoholic steatohepatitis. J Nutr Biochem. 2014 doi: 10.1016/j.jnutbio.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charlton M, Krishnan A, Viker K, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011;301:G825–34. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Napolitano LM, Koruda MJ, Meyer AA, et al. The impact of femur fracture with associated soft tissue injury on immune function and intestinal permeability. Shock. 1996;5:202–7. doi: 10.1097/00024382-199603000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Rogers GB, Kozlowska J, Keeble J, et al. Functional divergence in gastrointestinal microbiota in physically-separated genetically identical mice. Sci Rep. 2014;4:5437. doi: 10.1038/srep05437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4516–22. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert JA, Meyer F, Jansson J, et al. The Earth Microbiome Project: Meeting report of the “1 EMP meeting on sample selection and acquisition” at Argonne National Laboratory October 6 2010. Stand Genomic Sci. 2010;3:249–53. doi: 10.4056/aigs.1443528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lozupone C, Hamady M, Knight R. UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7:371. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 27.Backhed F, Ley RE, Sonnenburg JL, et al. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–20. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 28.Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gophna U, Sommerfeld K, Gophna S, et al. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. 2006;44:4136–41. doi: 10.1128/JCM.01004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carvalho FA, Koren O, Goodrich JK, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12:139–52. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukhopadhya I, Hansen R, El-Omar EM, et al. IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol. 2012;9:219–30. doi: 10.1038/nrgastro.2012.14. [DOI] [PubMed] [Google Scholar]
- 32.Devkota S, Wang Y, Musch MW, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–8. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gauffin Cano P, Santacruz A, Moya A, et al. Bacteroides uniformis CECT 7771 ameliorates metabolic and immunological dysfunction in mice with high-fat-diet induced obesity. PLoS One. 2012;7:e41079. doi: 10.1371/journal.pone.0041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Everard A, Belzer C, Geurts L, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110:9066–71. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cozzolino M, Staniforth ME, Liapis H, et al. Sevelamer hydrochloride attenuates kidney and cardiovascular calcifications in long-term experimental uremia. Kidney Int. 2003;64:1653–61. doi: 10.1046/j.1523-1755.2003.00284.x. [DOI] [PubMed] [Google Scholar]
- 36.Perry CM, Plosker GL. Sevelamer carbonate: a review in hyperphosphataemia in adults with chronic kidney disease. Drugs. 2014;74:771–92. doi: 10.1007/s40265-014-0215-7. [DOI] [PubMed] [Google Scholar]
- 37.Perianayagam MC, Jaber BL. Endotoxin-binding affinity of sevelamer hydrochloride. Am J Nephrol. 2008;28:802–7. doi: 10.1159/000135691. [DOI] [PubMed] [Google Scholar]
- 38.Tzanno-Martins C, Biavo BM, Ferreira-Filho O, et al. Clinical efficacy, safety and anti-inflammatory activity of two sevelamer tablet forms in patients on low-flux hemodialysis. Int J Immunopathol Pharmacol. 2014;27:25–35. doi: 10.1177/039463201402700105. [DOI] [PubMed] [Google Scholar]
- 39.Machado MV, Michelotti GA, Xie G, et al. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One. 2015;10:e0127991. doi: 10.1371/journal.pone.0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wree A, Broderick L, Canbay A, et al. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10:627–36. doi: 10.1038/nrgastro.2013.149. [DOI] [PubMed] [Google Scholar]
- 41.Tetri LH, Basaranoglu M, Brunt EM, et al. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol. 2008;295:G987–95. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–88. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 43.Imajo K, Fujita K, Yoneda M, et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012;16:44–54. doi: 10.1016/j.cmet.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 44.Harte AL, da Silva NF, Creely SJ, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (Lond) 2010;7:15. doi: 10.1186/1476-9255-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kudo H, Takahara T, Yata Y, et al. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J Hepatol. 2009;51:168–75. doi: 10.1016/j.jhep.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 46.Luther J, Garber JJ, Khalili H, et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol Gastroenterol Hepatol. 2015;1:222–232. doi: 10.1016/j.jcmgh.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–53. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 48.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 49.Hooper LV. Bacterial contributions to mammalian gut development. Trends Microbiol. 2004;12:129–34. doi: 10.1016/j.tim.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–9. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.