

Abstract
Endothelial cells of the blood–brain barrier form a structural and functional barrier maintaining brain homeostasis via paracellular tight junctions and specific transporters such as P-glycoprotein. The blood–brain barrier is responsible for negligible bioavailability of many neuroprotective drugs. In Alzheimer’s disease, current treatment approaches include inhibitors of BACE-1 (β-site of amyloid precursor protein cleaving enzyme), a proteinase generating neurotoxic β-amyloid. It is known that BACE-1 is highly expressed in endosomes and membranes of neurons and glia. We now provide evidence that BACE-1 is expressed in blood–brain barrier endothelial cells of human, mouse, and bovine origin. We further show its predominant membrane localization by 3D-dSTORM super-resolution microscopy, and by biochemical fractionation that further shows an abluminal distribution of BACE-1 in brain microvessels. We confirm its functionality in processing APP in primary mouse brain endothelial cells. In an Alzheimer’s disease mouse model we show that BACE-1 is upregulated at the blood–brain barrier compared to healthy controls. We therefore suggest a critical role for BACE-1 at the blood–brain barrier in β-amyloid generation and in vascular aspects of Alzheimer’s disease, particularly in the development of cerebral amyloid angiopathy.
Keywords: BACE-1, blood–brain barrier, endothelium, β-amyloid, Alzheimer’s disease
Introduction
BACE-1 (β-site of amyloid precursor protein (APP) cleaving enzyme) also known as β-secretase is the rate-limiting APP processing enzyme involved in the release of neurotoxic β-amyloid (Aβ), contributing to Alzheimer’s disease (AD) in the context of the amyloid hypothesis.1
The motivation to assess BACE-1 expression at the blood–brain barrier (BBB) came from reports showing that a large percentage (∼80%) of AD cases result in cerebral amyloid angiopathy (CAA) with the deposition of Aβ at the blood vessels.2 The source of the vessel-associated amyloid peptides is however not entirely clear. Neuronal Aβ in combination with its poor clearance across the BBB and deposition at the vessels have been classically attributed to CAA plaque formation. A significant amount of APP is present in the peripheral circulation, that is expressed by platelets and more prone to toxic Aβ formation than the neuronal form of APP in the brain parenchyma.3,4 Platelet-derived APP contains the Kunitz Protease Inhibitor (KPI) domain that favors aggregation of amyloid peptides, resulting from the cleavage of APP.5 Moreover, there are reports correlating an increase in KPI domain containing APP in the circulation with incidence and severity of AD in humans.6
We hypothesized that BACE-1 present at the BBB might be involved in the cleavage of circulating APP and or neuronal APP, thus generating Aβ locally at brain vessels.
Moreover, BACE-1 at the BBB raises the novel aspect, that mutually exacerbating lesions of the BBB by acute Aβ production and oxidative stress at the endothelial level may precede neurodegeneration, and reemphasizes the vascular and peripheral aspect of AD.7 BACE-1 is considered a leading target for the development of neuroprotective drugs against AD and herein its expression and processing has so far been associated with neuronal or glial localization.8 Deletion of one copy of the BACE-1 gene is reducing, whereas deletion of both alleles is virtually abolishing Aβ production in APP overexpressing mice.9 The relatively large active site has so far hampered the development of small molecule inhibitors able to pass the BBB.10 Nevertheless, compounds for oral administration have recently entered clinical trials and showed convincing central activity.11 Although considered a relatively safe drug target, there is some concern for possible side-effects of BACE-1 inhibition. This is because BACE-1 also cleaves and thereby contributes to the release of the neurotrophic factor neuregulin-112 and processes voltage-dependent sodium channels.13 Recent studies showed presynaptic deficits and a predisposition to seizures in BACE-1 deficient animals.8 There are posttranslational modifications and several membrane-bound and soluble isoforms of BACE-1, of which the respective enzymatic activities and dynamics remain unclear.14 Moreover, there have been reports that muscarinic receptors have an impact on the degradation of BACE-1.15
In the current work, we focused on whether functional BACE-1 is present at the BBB and whether its endogenous expression is altered in a transgenic mouse model for AD expressing human APP with Swedish and London mutation (hAPPSL16). We provide evidence that BACE-1 is indeed expressed in BBB endothelial cells of human, mouse, and bovine origin. We further show a predominant membrane localization of BACE-1 by super-resolution microscopy and by fractionation that also shows a mainly abluminal distribution of BACE-1. Moreover, we confirm its functionality in processing APP in primary mouse brain endothelial cells. In an AD mouse model we show that BACE-1 is upregulated at the BBB compared to healthy controls, suggesting a critical role for BACE-1 at the BBB in amyloid-β generation.
Materials and methods
Reagents
All chemicals and reagents were obtained from Sigma (Germany) unless otherwise specified. Nylon mesh cell strainers for microvessel preparation were obtained from BD Biosciences (Germany). Glass-Teflon homogenizer was obtained from Wheaton sciences (USA). Electric overhead stirrer (VOS 14) was from VWR (Germany). Tricarboxy Ethyl Phosphine (TCEP) for Western blotting was obtained from Thermo Fisher Scientific (Germany). All the secondary antibodies for Western blotting were obtained from LI-COR Biosciences (Germany) and for immunofluorescence staining from Invitrogen (Germany). Cell culture media and its components were obtained from GIBCO® (Germany). The primary antibodies used are listed in the Table 1.
Table 1.
List of primary antibodies.
| Protein ID | Clone | Dilution | Vendor | Catalog No |
|---|---|---|---|---|
| VE-Cadherin (CDH5) | C-19 | 1:100 | Santa Cruz | SC-6458 |
| α-tubulin (TUB1A1) | DM1A | 1:1000 | Sigma | T6199 |
| APP | 22C11 | 1:500 | Millipore | MAB348 |
| Amyloid-β | 1:200 | Millipore | AB5076 | |
| GLUT-1 | 1:1000 | REF 21 | ||
| EAAT-2 | 1:500 | Abcam | AB41621 | |
| P-glycoprotein (PgP) | C219 | 1:500 | Calbiochem | 517310 |
| BACE-1 | 1:100 | Abcam | AB2077 | |
| BACE-1 | 1:200 | Sigma | B0681 | |
| BACE-1 | EPR3956 | 1:500 | Epitomics | 2882-1 |
| Claudin-5 (CLDN5) | 1:200 | Invitrogen | 35-2500 | |
| Laminin α5 | 405 | 1:300 | Dr L Sorokin | |
| GAPDH | 1:1000 | Abcam | AB9484 | |
| Lamin B1 | 1:2000 | Abcam | AB16048 |
Animal care and handling
This study was performed in strict compliance with animal handling protocols approved by the Regierungspräsidium Darmstadt, Germany (No. V54-19c20/15 – F94/18) and following the guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. All the experiments complied with the ARRIVE guidelines for conducting animal experiments. Animals were sacrificed under appropriate anesthesia and every effort was made to minimize their suffering. The number of mice was kept to the minimum required for valid results.
Isolation of mouse brain microvessels (MBMV)
Brain microvessels were isolated from 2-month-old wild-type C57BL/6 mice (6–8 mice/preparation) or hAPPSL mice at an age of 10 months along with age-matched wild-type mice as described previously.17 Briefly, animals were anesthetized with isofluorane followed by sacrificing the mice by cervical dislocation and extracting the brains into ice-cold microvessel isolation buffer (1 × MVB17). The cerebellum and olfactory lobes were removed and meninges were peeled off the cortical hemispheres by rolling on Whatmann filter paper. The brains were then subjected to homogenization 20 strokes using a glass-Teflon homogenizer (0.25 mm clearance) attached to an overhead electrical stirrer, centrifuged at 1000 × g for 10 min at 4℃. The pellet was made to 17% dextran followed by centrifugation at 4000 × g for 30 min to pellet the blood vessels, which was filtered through 70, 40 micron nylon meshes to eliminate erythrocytes and pericytes and to obtain the final microvessels on the 40 micron mesh. The final sample was re-suspended in Hepes EDTA Sucrose buffer (HES; 10:1:250 mM, pH 7.4)17 containing protease inhibitor cocktail (Roche, Germany) and phosphatase inhibitor cocktail (Sigma, Germany) for Western blotting and stored at –80℃ or directly lysed for RNA in the RLT lysis buffer (Qiagen, Germany) and stored –80℃ for qRT-PCR experiments. For immunocytochemistry, the vessels were re-suspended in 1 × PBS and stored on ice and used immediately.
Isolation of primary MBMECs and culture of HBMECs, hCMEC/D3 cells
Primary mouse brain microvascular endothelial cells (MBMECs) were isolated from 2 to 6 adult C57BL/6 mice in each preparation as described previously with only modification being the use of a dounce homogenizer instead of mincing the tissue.18 Cultures were maintained and routinely characterized for endothelial markers such as claudin-5, VE-cadherin by immunofluorescence staining, and transendothelial electrical resistance (TEER) assay for barrier tightness as described previously.18,19 Primary human brain endothelial cells (HBMECs) were obtained at passage 2 from Pelobiotech (Germany) and cultured as above for MBMECs and used within passage 4. Immortalized human brain endothelial line, hCMEC/D3 cells were obtained from Prof Pierre-Olivier Couraud (INSERM, Paris, France) and cultured as we described previously.19
Fractionation of bovine and HBMECs
Briefly, the microvessels isolated from bovine cerebral cortices were digested with collagenase type IA (1800 units/g) to remove the basement membrane, pericytes, and glial fragments. The refined microvessels were homogenized and the luminal and abluminal membrane fractions were separated by discontinuous Ficoll gradient as indicated in the supplementary flow diagram (Figure S2). Sample purity was assessed as we described previously and stored at –80℃.20,21
Fractionation of cultured primary HBMECs was performed by classic differential centrifugation. Briefly, confluent HBMECs were harvested in ice-cold isotonic HES buffer plus protease, and phosphatase inhibitor cocktails (Roche) followed by homogenization in a douncer applying 40 strokes. The resultant cell lysate was centrifuged at 1000 × g for 5 min at 4℃ to obtain the nuclear pellet (NU) that was washed once with HES buffer and re-suspended in 10-fold lower SB buffer (2.3 M Urea, 1.5% SDS, 50 mM Tris, 25 mM TCEP, and 0.01% BPB) for Western blotting compared to the starting volume of HBMEC lysate. The supernatant was centrifuged at 15000 × g, 15 min, 4℃ to obtain the Golgi/mitochondrial pellet (trans-Golgi network; TGN) that was washed once as above and also re-suspended in 10-fold lower SB buffer. The supernatant was subjected to ultracentrifugation at 80,000 × g (30,000 r/min, F45L rotor (Piramoon), Sorvall WX Ultra 80 (Thermo Scientific)), 1 h, 4℃ to obtain the plasma membrane (PM) pellet which was again washed once in HES and re-suspended in SB buffer as above. The supernatant was subjected to a final round of ultracentrifugation at 220,000 × g (45,000 r/min) for 3 h, 4℃ to obtain the endosomal vesicles (EN) pellet that was re-suspended in SB buffer. The supernatant represented the cytoplasmic (CYTO) fraction.
RNA isolation and qRT-PCR
For RNA isolation, RNeasy mini kit (Qiagen, Germany) was used according to manufacturer’s protocol for low sample yields, followed by cDNA preparation using First Strand cDNA Synthesis Kit (Fermentas) also according to the manufacturer’s protocol. qRT-PCR using ABsolute QPCR SYBR Green Fluorescein Mix (Thermofisher Scientific, USA) was according to the manufacturer’s protocol applying the following conditions: 15 min at 95℃, 45 cycles of 30 s at 95℃, 30 s at 61℃, and 35 s at 72℃ in MyiQ Real-Time PCR Detection System (Bio-Rad). Analysis of the qRT-PCR data was performed with iQ5 2.1 software (Bio-Rad). Primer sequences (5′-3′, sense_antisense) for qRT-PCR: BACE-1_01(tgccatcactgaatcggacaa_tctgcttcaccagggagtcaa)/BACE-1_02(tggagatggtggacaacctga_cggaggtctcgatatgtgctg)/LRP1(atcaggcgcattgaccttcac_ggccaagccatactgaatcacc)/FcRN(gactgctaggccacctggaga_gaaagcagcacaggtcagcac)/RAGE(aagccctcctgtcagcatcag_ctctcctcacgcctgggttgt)/PgP(gctatcacggccaacatctcc_tgtccaacactgaatgctccaa)/GLUT1(tcgtcgttggcatccttattg_gtagcagggctgggatgaaga)/Cldn5(tgtcgtgcgtggtgcagagt_tgctacccgtgccttaactgg)/ZO1(tgcttctcttgctggccctaa_gggtggcttcacttgaggtttc)/VECadherin(gcccagccctacgaacctaaa_gggtgaagttgctgtcctcgt)/RNApol(atgagctggaacgggaatttga_accactttgatgggatgcaggt)/CD31(attcctcaggctcgggtcttc_ccgccttctgtcacctccttt)/G6PDX(gggtccaccactgccacttt_tttgcgttcattcagggcttt)/RPLPO(gtgtttgacaacggcagcatt_tctccacagacaatgccagga)/AQP4(agtgacagagctgcggcaagg_ttccagaaagcctgagtcca)/NG2(cctggccttggctcttacctt_gctgggatgtggagaactgga)/DCX(ggaaggggaaagctatgtctg_ ttgctgctagccaaggactg).
Western blotting
Samples for Western blotting were solubilized in SB buffer for 1 h at 30℃ with shaking at 600 r/min. Electrophoresis was performed at a constant 80 V for 3 h at room temperature using 7–15% Tris-HCL Bis-acrylamide gels. The separated proteins were transferred to nitrocellulose membrane at 4℃, 36 V constant for 16 h. Reversible Ponceau S staining was performed to confirm equal protein loading and was followed by Western blotting as described previously.21 Imaging was performed in Odyssey (LI-COR, Germany) multichannel imaging device and raw images were exported in tiff format using Image Studio 2.1 software (LI-COR, Germany) and quantitation performed as described previously.21
Immunocytochemistry and image processing
MBMECs cultured on 24-well transwell inserts or freshly isolated MBMVs were subjected to the immunocytochemistry protocol exactly as described previously19 with MBMVs first subjected to brief drying on glass chamber slides followed by –20℃ methanol fixation for 4 min or 4% PFA for 10 min at room temperature. At the end of the staining protocol the slides were mounted with coverslips using Aqua-Poly/Mount mounting medium (Polysciences Inc., Germany) and visualized with an 80i microscope (Nikon) and documented with a digital DS-5Mc camera or with a C1si confocal microscope (Nikon). Images were processed in NIS Elements AR Imaging Software (Nikon) and Photoshop CS6 for Macintosh (Adobe). Deconvolution was performed on confocal Z stacks with Huygens Essential software, applying a theoretical point spread function (SVI, The Netherlands).
Immunohistochemistry from human samples
Formalin-fixed and paraffin-embedded normal and CAA brain specimens from patients (ethical review No. 0409) were cut into 4-µm thick sections and automatically stained with antibodies against human BACE-1 (Abcam) and human amyloid-β (Millipore) using standard protocols on the Ventana Discovery XT automated immunohistochemistry system (Roche, Switzerland).
3D-dSTORM super-resolution imaging and analysis
HBMECs were stained exactly as described for MBMECs followed by 3D-dSTORM imaging on a custom-built setup for single-molecule imaging as described previously.22 An astigmatic lens was placed in the emission light path in order to facilitate 3D imaging. Samples were illuminated using ∼2–3 kW/cm2 647 nm excitation (Innova 70C, Coherent) in highly-inclined laminar optical (HILO) sheet imaging mode to reduce background fluorescence; 10.000–15.000 frames were recorded at a frame rate of 33 Hz using the µManager software. Fluorophore density was adjusted by 404 nm reactivation (Coherent Cube 404). Samples were imaged in a buffer containing 50 mM Tris pH 8.0, 75 mM MEA (cysteamine hydrochloride, Sigma Aldrich), 23 mM glucose, and an oxygen scavenger system (20 units glucose oxidase and 400 units catalase, Sigma Aldrich). The final buffer included 70% D2O to increase brightness of the fluorophores.22 Acquired image series were analyzed using the rapidSTORM software (V. 2.21) and a cubic B-spline calibration followed by creating z-slices with rapidSTORM. All images were post-processed using the Fiji software package (http://fiji.sc/Fiji). Histograms of z-slices were created in Origin Pro 9.1G (Origin Labs) by counting z-positions of all localizations within the depicted region of interest (bin size 50 nm).
Statistics
Most experiments were repeated at least twice with the number of animals included in the corresponding figure legend. Quantitation was performed in Microsoft Excel (MS Office 2008) followed by exporting the data to Prism 5.0 (GraphPad). Graphs and statistics were performed in Prism with the significance level set to p < 0.05. Paired analysis was performed to account for batch-to-batch variations in primary endothelial cell isolation/culture. The specific statistical test and the p values are included in the figure legends.
Results
Expression of BACE-1 in mouse brain microvessels and cultured endothelial cells
Total RNA from mouse brain microvessels was isolated as previously published.19 We performed quantitative real time RT-PCR to obtain the expression levels of BACE-1 transcripts in vessels and compared it to that of whole brain RNA as a positive control (Figure 1). The purity of microvessel preparations was assessed by the expression of the BBB endothelial marker claudin-5 (Cldn-5) when compared to the whole brain sample (Figure S1). To eliminate the possibility of contamination from other brain cells in the vessel preparation (MBMV) such as neurons that are known to express BACE-1, we isolated and cultured murine brain endothelial cells (MBMECs) as previously described and tested BACE-1 expression in these pure endothelial cells.18 Purity analysis of MBMV and MBMEC by qRT-PCR is included in the supplementary Figure S1 using markers for astrocytes (aquaporin-4, AQP4), pericytes (neural/glial antigen 2, NG-2), and neurons (doublecortin, DCX). These data show minimal contamination from other cell types in the MBMV and the complete absence of contamination in MBMECs. Using two different primer pairs we observed a significant expression of BACE-1 in microvessels and in pure cultured endothelial cells (∼30%; Figure 1) compared to whole brain lysates. Consequently, the detected expression of BACE-1 is primarily endothelial-specific in the vessels fragments (MBMVs) and in the pure, primary cultured cells (MBMECs) it is entirely of endothelial origin. Protein analysis was first performed by Western blotting for BACE-1 after testing six commercially available anti-BACE-1 antibodies with corresponding competing peptides to show specificity and employing recombinant BACE-1 as a positive control. Three of these antibodies showed strong reactions with BACE-1 in the total protein extracts from mouse brain microvessels with an electrophoretic mobility corresponding to ∼75 kDa as predicted from the literature for the full-length protein (Figure 2b, c). Figure 2 shows that the full-length isoform (∼75 kDa), known to be the active isoform,23 is indeed expressed in brain endothelial cells. The antibody against the N-terminus that is able to detect most isoforms also showed a strong band at ∼50 kDa, likely representing the soluble isoform that lacks the intra-cellular domain recognized by the two antibodies against the C-terminus.14 The function of the soluble isoform is not entirely clear.
Figure 1.
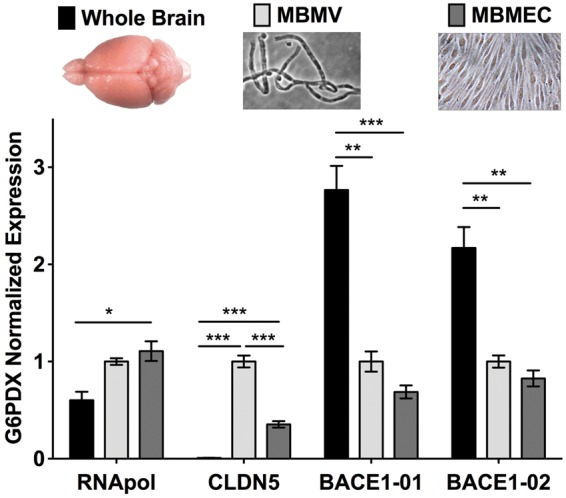
BACE-1 mRNA expression analysis in mouse brain microvessels and cultured endothelial cells. In freshly isolated mouse brain microvessels (MBMVs) and primary cultured mouse brain microvascular endothelial cells (MBMECs) mRNA expression analysis by qRT-PCR showed significant expression of BACE-1 using two different primer pairs. Whole brain mRNA served as a positive control for BACE-1 expression as neurons are known to express high levels of BACE-1. BACE-1 expression was present even in pure cultured endothelial cells that have no contamination from neurons thus indicating a specific expression of BACE-1. Cldn-5 served as a marker for endothelium, which was at much higher levels in cultured and freshly isolated brain microvascular endothelial cells. The Ct range for BACE-1 qRT-PCR was in the range 23–26 cycles with non-template control about 35 cycles indicating specificity of expression. Statistical significance was by One-way ANOVA followed by TUKEY-HSD test for multiple groups (n = 4 experiments, *p < 0.05, **p < 0.01, and ***p < 0.001 with MBMV set as 1 with 2–4 mice in each experiment).
Figure 2.

BACE-1 protein expression analysis in mouse brain microvessels. (a) Western Blots for BACE-1 using a highly specific antibody (B0681, Sigma) made against the N-terminus of BACE-1 (AA 46-62 of BACE, N-terminal) show the staining of the 75 and 50 kDa isoforms of BACE-1 (lane 1) in purified microvessels from wild-type mouse brains; the staining is specific as a blocking peptide (lane 2) completely abolished the staining of mouse brain microvessels. Lanes 3 and 4 show silver-stained 1D gels of recombinant BACE-1 (Invitrogen P2947 and Sigma S4195) as positive controls; lanes 5 and 6 show Western blots of the recombinant BACE-1 proteins with B0681. The blot is representative of three independent preparations of brain microvessels using 8–10 mice each time. The recombinant proteins were made to the extracellular domain of BACE-1 fused to Fc region of human IgG1 (Invitrogen P2947) or with a C-terminal FLAG-tag (Sigma S4195), hence they migrate at a lower molecular weight than the endogenous full-length BACE-1, which runs at 75 kDa. (b) In addition to B0681 (N-terminal epitope, Sigma) ab2077 (C-terminal epitope, Abcam) and 2882-1 (C-terminal, Epitomics) were also used to confirm the expression of BACE-1 in mouse brain microvessels. Competing peptides (for the antibody epitopes) from the vendor were used to obtain the specificity of the bands. For the Epitomics antibody for which the vendor competing peptides were not available only the C-terminal peptide for ab2077 but not the N-terminal one corresponding to B0681 abolished the specific band. The N-terminal antibody from Sigma recognizes two isoforms (full length 75 kDa, soluble 50 kDa) but the C-terminal antibodies (Abcam, Epitomics) recognize only the higher molecular weight full-length isoform. The red arrowheads indicate the specific bands and the red cross marks point the loss of these specific bands.
Localization and activity of BACE-1 at the BBB
We next performed immunofluorescence staining for BACE-1 in purified wild-type brain microvessels and in cultured MBMECs employing the N-terminal antibody (B 0681) that showed strongest immunoreactivity in Western blots for both isoforms (Figure 3a, b). We detected BACE-1 in the vessels as well as in MBMECs with an expression profile indicating localization at the PM and in intracellular compartments as described previously for other cell types.14 The staining for Cldn-5 was primarily junctional and present in all the cells, confirming the purity of MBMEC cultures and microvessel preparations (Figure 3b).18 To investigate the distribution of BACE-1 at the BBB endothelium, we utilized fractionated bovine brain microvessels as described previously.21 Using markers for luminal (blood-facing) and abluminal (brain-facing) membranes such as P-glycoprotein (PgP) and glutamate transporter (EAAT2), respectively, we obtained the localization of BACE-1 at these membranes. GLUT-1 served as a marker expressed equally on both membranes. Our data indicate a predominant localization of BACE-1 at the abluminal membrane, as the quantification showed ∼2.5-fold higher concentration on this site when compared to the luminal membrane (Figure 3c). The intracellular, endosomal vesicles showed minimal BACE-1 content (Figure S3b).
Figure 3.

Localization and activity of BACE-1 in brain microvessels and cultured endothelial cells. Immunofluorescence for BACE-1 showing specific staining for BACE-1 at the PM and intracellular vesicles in (a) freshly isolated MBMVs (representative pictures from 3 preparations using 3–4 mice) and in (b) primary cultured MBMECs (representative of three preparations using 4–6 mice each time). Claudin-5 (Cldn5) served as an endothelial marker. (c) Secondary antibody control for BACE-1, showing no staining for BACE-1 when the primary antibody was omitted. (d) Localization analysis in fractionated freshly isolated bovine brain capillary endothelial cells shows the predominant expression of BACE-1 in the abluminal membranes (*p < 0.05, 2-tailed paired t-test). PgP and EAAT-2 served as markers of luminal and abluminal membranes, respectively. GLUT-1 served as a marker expressed equally on both membranes. Quantitation was performed using blots for BACE-1 from three preparations utilizing 10 bovine brains each time. (e) Activity of BACE-1 is shown by inhibition of BACE-1 (Merck IV, 24 h) in MBMECs in medium with 20% bovine serum that is reported to contain full-length APP. The blots show an increase in the mature form of APP (mAPP) in the medium indicating reduced cleavage by BACE-1 and a concomitant increase in α-secretase activity resulting in increased levels of sAPPα. Loading differences accounted for by taking equal volume fraction from cells seeded at the same density along with a Ponceau S protein stain (not shown). Quantitation of the blots was performed from three experiments utilizing two animals in each preparation. Significance was by 2-tailed paired t-tests (*p < 0.05, **p < 0.01).
In order to elucidate if BBB endothelial BACE-1 is enzymatically active, we treated MBMECs from adult wild-type mice (C57BL/6) with a commercial BACE-1 inhibitor (10 µM inhibitor IV, Merck) in medium containing 20% bovine serum that is known to contain full length APP.3,4 The endothelium as a source of APP, however, cannot be ruled out. The Western blot data indicate an increase in the expression of both the mature APP and the soluble APP-α (sAPP-α) isoforms in the medium upon BACE-1 inhibition (Figure 3d). The higher levels of mature APP in BACE-1-inhibited cells indicates that the majority of the full-length APP is not cleaved, which indicates that active BACE-1 has been inhibited in brain endothelial cells. Soluble sAPP-α is a cleavage product of α-secretase, whose activity has been shown to be increased upon BACE-1 inhibition by a competition mechanism in vivo, again suggesting that brain endothelial cells do possess active BACE-1.24
In order to understand if BACE-1 is also expressed in a comparable fashion in human brain ECs, primary cultured human brain endothelial cells (HBMECs) were stained for BACE-1 (Figure 4a). The punctate expression pattern was similar to those in MBMECs, and the endothelial adherens junction marker VE-cadherin displayed the expected junctional localization. Western blot analyses for BACE-1 in HBMECs and hCMEC/D3 (transformed human brain endothelial cells) also showed the specific BACE-1 band at ∼75 kDa, Cldn-5 served as an endothelial-specific marker (Figure 4b). The expression of BACE-1 in D3 cells was higher than in HBMECs that could be explained by potential differences between the transformed cell line and primary cells. Scientifically more attractive but difficult to recapitulate in these cells might be age-related differences in BACE-1 levels between the donors of these cell types.
Figure 4.

Subcellular localization of BACE-1 in human brain endothelial cells (a) Primary human brain endothelial cells (HBMECs, P4) were stained for BACE-1 (B0681, Sigma), the expression being similar to murine brain endothelial cells showing a punctate pattern. VE-cadherin served as an endothelial marker staining the adherens-junctions. (b) Western blots for BACE-1 in HBMECs (P3) and hCMEC/D3 (P28), a human brain endothelial line confirmed the staining data. Cldn-5 was used as an endothelial marker. (a, b) are representative of two experiments using P3-P4 HBMECs and P28-P30 D3 cells. (c) 3D-dSTORM-imaging of Alexa-Fluor 647 labeled BACE-1. Diffraction-limited widefield fluorescence image shows the spindle shape of differentiated HBMECs (left image). BACE-1 is preferentially located close to the PM as shown by the cross-sections of small insets (XZ). The yellow and white dotted lines mark the apical (top) and basal (bottom) PMs, respectively. Insets are color-coded according to the relative z-position in nm, as indicated by the numbers. Scale bar 10 and 1 µm (overview and insets, respectively). The histograms (on the right) show quantification for the localization. (d) Western blotting on fractionated HBMECs shows enrichment of BACE-1 in PM and TGN fractions with significantly lower levels in cytoplasmic (CYTO) and endosomal vesicle fraction (EN). TUB1A1, lamin B1, and CDH5 served as markers for CYTO, NU, and PM fractions, respectively. (N = 3 sets of fractionation using HBMECs from three different donors, significance was by One-way ANOVA followed by TUKEY-HSD test for multiple groups. **p < 0.01, ***p < 0.001).
To microscopically elaborate the subcellular localization of BACE-1 in HBMECs, we subjected these cells to super-resolution imaging. 3D-dSTORM imaging of BACE-1 in HBMECs showed an axial distribution of BACE-1 corresponding primarily to the PM (Figure 4c, Figure S5). This distribution was similar to that of laminin α5, a PM marker (Figure S4). The apico-basal distribution of BACE-1 in cultured HBMECs is however not comparable to freshly isolated/fractionated bovine endothelial cells (Figure 3c) due to a loss of endothelial polarity in culture resulting from a downregulation of several BBB-specific markers.25 To more quantitatively assess the subcellular localization, HBMECs were fractionated by differential centrifugation followed by Western blotting. We observed a significant enrichment of BACE-1 in the PM and the TGN fractions (Figures 4d and S3a). It was previously reported that BACE-1 could cycle between the TGN, endosomes, and the PM. BACE-1 and APP from the PM location are known to be segregated into endocytic vesicles that are targeted to endosomal compartments for optimal activity of BACE-1, ensured by the acidic environment.8 BACE-1 was also detectable in endosomal (EN) and cytoplasmic (CYTO) fractions albeit to a much lesser extent. BACE-1 in the nuclear fraction (NU) is most likely a result of unbroken cell membranes resulting from incomplete homogenization that contaminate the nuclear fraction.
Expression and regulation of endothelial BACE-1 in vivo in healthy and CAA/AD brain
In order to investigate whether human brain vessels also express BACE-1, we performed immunohistochemistry on human brains sections. The data show expression of BACE-1 in the microvessels of both the white and grey matter (Figure 5a, b). We also observed BACE-1 staining in larger vessels of the leptomeninges (Figure 5c), which are more frequently associated with CAA.2 Neurons served as a positive control since they are known to express functional BACE-1 (Figure 5d). We further analyzed a single human CAA case for the expression of BACE-1 at the BBB in a diseased state. We observed a robust BACE-1 staining in the vessels of the CAA brain that showed intense amyloid-β deposits, implicating BBB-localized BACE-1 in the amyloid angiopathy (Figure 5e).
Figure 5.

BACE-1 expression in normal and CAA human brain vessels. (a–d) Immunohistochemistry for BACE-1 (C-terminal epitopes, Abcam antibody) shows expression of BACE-1 in the human brain vessels from both the white and grey matter. Leptomeningeal vessels also were stained indicating that even the non-BBB forming vessels express BACE-1. Neurons served as a positive control. Figures (a–d) are representative of five cases. (e) Arteriole and capillary of a single human CAA case. Double staining for amyloid-β (red) and BACE-1 (brown) shows intense staining of amyloid-β around vessels and endothelial as well as neuronal staining for BACE-1.
After establishing BACE-1 expression and its activity at the healthy BBB, we addressed the significance of BACE-1 at the BBB in a mouse model for AD. To this end, we isolated brain microvessels from transgenic mice (hAPPSL) over-expressing the 751 amino acid form of human amyloid precursor protein (hAPP) with London (V717I) and Swedish (KM670/671NL) mutations under the control of the murine Thy-1 promoter.16 The hAPPSL mice show an age-dependent increase of Aβ peptides and develop amyloid plaques at an early stage of 3–4 months. In these mice, severity of the brain pathology correlates with increasing age and is characterized by behavioral deficits. In addition, the mice develop CAA with amyloid plaques on SMA-positive blood vessels. In qRT-PCR experiments, we observed a ∼4-fold increase of BACE-1 expression in the BBB microvessels isolated from 10-month-old hAPPSL mice when compared to age-matched wild-type mice, suggesting an increased APP processing activity by BACE-1 at the BBB in mutant animals (Figure 6a). Several endothelial marker genes such as VE-cadherin and tight junction molecules such as ZO-1 and Cldn-5 were unchanged whereas GLUT-1, the primary glucose transporter at the BBB, was downregulated similar to what was observed in the brains of AD patients, indicating an impairment of the BBB.26 Interestingly, the transcript for luminal Aβ transporter receptor for advanced glycation end products (RAGE) was upregulated in the AD mice. This might suggest that blood-derived Aβ could potentially contribute to vascular and brain amyloidosis, which was demonstrated by Eisele and colleagues27 by peripheral intraperitoneal administration of β-amyloid extracts in mice. Furthermore, the luminally expressed PgP, known to be involved in the efflux of Aβ into the circulation, was downregulated in AD MBMVs, suggesting a decreased clearance of Aβ from the brain by these ECs. The involvement of PgP in Aβ clearance has previously been observed by Cirrito and colleagues28 in PgP-null mice upon intrathecal Aβ administration. In contrast to what was observed in AD, the abluminally located LRP-1 transporter, conferring Aβ clearance from the brain into the BBB endothelium, was upregulated in the hAPPSL transgenic mice.29 As LRP-1 is also involved in APP endocytosis,30 increased BACE-1 activity together with increased endocytosis of APP from neuronal sources31 may result in more Aβ production in brain endothelial cells. Finally, we did not observe any change in the transcript for the neonatal Fc receptor (FcRN), an abluminal transporter, which mediates brain clearance of antibody-bound Aβ.32 A schematic for amyloid processing and transport at the BBB including the potential role of BACE-1 in the endothelium is shown in Figure 6(b).
Figure 6.

Expression analysis in brain microvessels from an AD mouse model and schematic for APP/Aβ processing at the BBB. (a) MBMVs from transgenic mice (hAPPSL) over-expressing the 751 amino acid form of human amyloid precursor protein (hAPP) with London (V717I) and Swedish (KM670/671NL) mutations under the control of the murine Thy-1 promoter were compared with wild-type mice. We observed a four-fold increase of BACE-1 expression in the BBB microvessels isolated from hAPPSL mice when compared to age-matched wild-type mice suggesting an increase in the APP cleavage activity of BACE-1 at the BBB in the mutant animals. Endothelial marker genes such as VE-cadherin and tight junction molecules ZO-1, claudin-5 were unchanged whereas GLUT-1, the primary glucose transporter at the BBB was downregulated. Interestingly, the luminal Aβ transporter RAGE was upregulated in the AD mice suggesting that circulating Aβ could potentially contribute to the brain amyloidosis. Furthermore the luminally expressed p-glycoprotein (PgP) known to be involved in efflux of Aβ into the circulation was downregulated suggesting a decreased clearance of amyloid peptides from the brain. The abluminally located LRP-1 was upregulated, which is known to endocytose APP, also supporting an increase in the BBB BACE-1 activity in this AD model. Abluminal Aβ antibody transporter FcRN was unchanged. Statistical significance was obtained from three qRT-PCR experiments using six transgenic mice (10 months age) or age-matched wild-type animals (***p < 0.001 using 2-tailed paired t-tests). (b) APP internalized from circulation via LRP2 or from brain parenchyma via LRP-1 or from the ECs is cleaved by BACE-1 to form Aβ. The predominant localization of BACE-1 at the abluminal membrane supports neuronal APP processing within the BBB endothelium. This Aβ is deposited as cerebrovascular plaques and/or is cleared from the brain parenchyma into circulation via LRP-1, FcRn from the abluminal side, and PgP from the luminal side. Additionally Aβ influx via RAGE can potentially regulate the Aβ transport across the BBB.
Discussion
While the role of the BBB in the clearance of brain-derived Aβ has been studied in detail, the contribution of the BBB in amyloidogenic processing of APP, leading to local Aβ production and deposition and ultimately to vascular amyloidosis disorders, is not yet known.
We therefore investigated the presence of BACE-1 at the BBB in vitro and in vivo in mice, bovine, and human brain endothelium and its regulation in an AD mouse model. We unequivocally demonstrate the presence of BACE-1 in all the above species and show its predominant localization on the abluminal membrane that is potentially able to cleave the circulating and or neuronal APP as demonstrated in BACE-1-inhibited primary mouse brain endothelial cells. We further show that BACE-1 expression at the BBB is upregulated in an AD mouse model (hAPPSL), in which the overexpression of mutated human APP is only in neurons driven by the murine Thy1 promoter. The increase in BACE-1 expression at the BBB in this model suggests that even for a parenchymal source of APP that is potentially a cargo of exosomes released by neurons,31 the BBB is one of the compartments for Aβ generation. This could also be explained by the formation of vascular plaques in the hAPPSL model.16
One of the proposed mechanisms of Aβ-induced neurotoxicity is the mitochondrial cascade hypothesis put forth by Swerdlow and Khan.33 The reactive oxygen species generated by Aβ lead to mitochondrial dysfunction that sets in a vicious cycle of oxidative stress, leading to BBB damage that in turn reduces Aβ clearance and increases Aβ deposition.34 Given that brain endothelial cells have abundant mitochondria, Aβ generated at the BBB by the enzymatic activity of endothelial BACE-1 may serve as the initial seeding point that leads to a cascade of events starting with oxidative stress, mitochondrial dysfunction, disruption of BBB function, and subsequent neurodegeneration. In vitro studies indicate that BACE-1 expression is upregulated by agonists of muscarinic receptors (mChR)15 1, 3 both expressed at the BBB.35 These studies may explain the observed deterioration in AD patients after an initial treatment with mChR agonists and further implicate BACE-1 at the BBB in Aβ generation and ensuing AD pathogenesis.
Furthermore, there is a significant amount of APP in the circulation derived from platelets, which is more prone to Aβ formation due to the presence of the KPI domain.5 A recent report shows that peripherally applied Aβ can indeed result in plaque formation in the brain.27 It was previously shown that BACE-1 is upregulated in AD in the brain parenchyma.36 The increase in BACE-1 predominantly in neurons explains the neuronal plaque formation via an increase in proteolytic cleavage of the neuronal APP to Aβ. Interestingly, it has been concluded that the endogenous BACE-1 expression levels in brain are sufficient for maximal Aβ production. In transgenic mice overexpressing BACE-1, Aβ production did not increase,37 whereas in hemi knock-out mice, 50% reduction in BACE-1 activity resulted only in a modest inhibition of Aβ production.38 Since we observed that BACE-1 expression at the BBB amounts to about 30% of those measured in brain parenchyma, upregulation of BACE-1 at this site may well have a significant impact in the pathogenesis of vascular amyloidosis in AD. Our current work strongly suggests that the BBB is an additional site for regulation of Aβ generation, transport, and subsequent plaque formation by the endothelial-specific expression of BACE-1.
Consequently, the prominent expression of BACE-1 at the BBB raises interesting novel possibilities of drug design. So far, the evolution of β-secretase inhibitors as therapeutic targets went slowly, because many did not reach the brain or were quickly re-transported to the blood stream by PgP. In transgenic animal models of AD, continuous administration of BACE-1 inhibitors has shown some benefit in terms of lowering Aβ plaque load and cognitive performance. Some of these inhibitors are in early clinical phases now.39 Pharmacological targeting of specific extracellular epitopes of BACE-1 on the blood-facing luminal side of the endothelium might facilitate drug design as the need for brain penetration and resistance to the drug efflux transport can be circumvented. Alternatively, BBB transporters such as transferrin receptors could be targeted to internalize the inhibitor drug into the BBB endothelium, where it can interact with the BACE-1 protein to inhibit its activity. Along this line the group of Ryan Watts at Genentech generated a bi-specific antibody with one arm containing the transferrin receptor specificity for transcytosis across the BBB and the other arm containing specificity for BACE-1.40 The authors show increased bioavailability of the BACE-1 antibody in the parenchyma and reduced Aβ burden in a mouse model of AD. Our recent data/modeling of iron transport at the BBB indicates that the majority of the transferrin receptors do not undergo transcytosis.41 Therefore, it would be interesting to investigate the effects of BACE-1/transferrin receptor bi-specific antibody at the BBB where it might have a significant effect on APP processing by BACE-1 expressed in the endothelium.
If an effective BACE-1 inhibitor is successfully targeted to the BBB endothelium, the drug-impermeability to this barrier might be in fact a blessing in disguise as some studies show that the complete knock out of BACE-1 results in myelination defects in the brain.8 The whole transport and processing of APP/Aβ from the brain parenchyma to endothelial cells (schematic - Figure 6b) via putative transporters such as FcRN, LRP-114 could be used to create a specific niche of bioavailability, avoiding unwanted side-effects. Based on the presented results on BACE-1 in brain endothelial cells we suggest BACE-1 at the BBB as a pivotal target against vascular aspects of AD. We further propose that drugs targeting BACE-1 within the BBB and not just crossing it might develop into novel AD therapeutics that potentially lead to better clinical outcomes.
Supplementary Material
Acknowledgments
We acknowledge Ms Ashwini Mokashi for assistance in bovine microvessel preparations. The authors thank Prof Dr Pierre-Olivier Couraud (INSERM, Paris, France) for providing the hCMEC/D3 cell line. The laminin α5 antibody (Ab #405) was a kind gift from Prof Dr Lydia Sorokin (Pathobiochemistry Univ. of Münster, Germany). The authors thank Dr. Klaus Nettesheim and Dr. Ulrike Engel (Nikon Imaging Center, Heidelberg, Germany) for assistance with N-SIM super resolution imaging.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: SFB TR23 consortium grant awarded to Stefan Liebner and Frankfurt clinic foundation grant awarded to Kavi Devraj funded this work.
Disclosure of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
KD isolated brain microvessels/endothelial cells and also designed, performed, analyzed the qRT PCR, Western blot and immunocytochemistry experiments, and wrote the manuscript. CS designed and performed dSTORM imaging and along with MH analyzed the data. SP and GS designed and performed Western blots. PNH designed, performed, and analyzed the immunohistochemistry experiments from human cases; MM analyzed the immunohistochemistry experiments from human cases. KA contributed in isolating brain microvessels from transgenic AD mice, PP, and MM partly analyzed the qRT-PCR results from AD mice. RH provided the luminal and abluminal bovine brain endothelial membranes. SL performed confocal imaging/deconvolution and along with KD and AS supervised the overall project and contributed to the manuscript preparation.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Probst G, Xu Y-Z. Small-molecule BACE1 inhibitors: a patent literature review (2006-2011). Expert Opin Ther Pat 2012; 22: 511–540. [DOI] [PubMed] [Google Scholar]
- 2.Revesz T, Holton JL, Lashley T, et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 2009; 118: 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bush AI, Martins RN, Rumble B, et al. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J Biol Chem 1990; 265: 15977–15983. [PubMed] [Google Scholar]
- 4.Bush AI, Beyreuther K, Masters CL. The beta A4 amyloid protein precursor in human circulation. Ann N Y Acad Sci 1993; 695: 175–182. [DOI] [PubMed] [Google Scholar]
- 5.Ho L, Fukuchi KI, Younkin SG. The alternatively spliced Kunitz protease inhibitor domain alters amyloid beta protein precursor processing and amyloid beta protein production in cultured cells. J Biol Chem 1996; 271: 30929–30934. [DOI] [PubMed] [Google Scholar]
- 6.Moir RD, Lynch T, Bush AI, et al. Relative increase in Alzheimer's disease of soluble forms of cerebral Aβ amyloid protein precursor containing the Kunitz protease inhibitory domain. J Biol Chem 1998; 273: 5013–5019. [DOI] [PubMed] [Google Scholar]
- 7.Tamagno E, Guglielmotto M, Monteleone D, et al. Amyloid-β production: major link between oxidative stress and BACE1. Neurotox Res 2012; 22: 208–219. [DOI] [PubMed] [Google Scholar]
- 8.Kandalepas PC, Vassar R. Identification and biology of β-secretase. J Neurochem 2012; 120(Suppl 1): 55–61. [DOI] [PubMed] [Google Scholar]
- 9.Ohno M, Sametsky EA, Younkin LH, et al. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron 2004; 41: 27–33. [DOI] [PubMed] [Google Scholar]
- 10.Klaver DW, Wilce MCJ, Cui H, et al. Is BACE1 a suitable therapeutic target for the treatment of Alzheimer's disease? Current strategies and future directions. Biol Chem 2010; 391: 849–859. [DOI] [PubMed] [Google Scholar]
- 11.May PC, Dean RA, Lowe SL, et al. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J Neurosci 2011; 31: 16507–16516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleck D, Garratt AN, Haass C, et al. BACE1 dependent neuregulin processing: review. Curr Alzheimer Res 2012; 9: 178–183. [DOI] [PubMed] [Google Scholar]
- 13.Huth T, Alzheimer C. Voltage-dependent Na+ channels as targets of BACE1 - implications for neuronal firing and beyond. Curr Alzheimer Res 2012; 9: 184–188. [DOI] [PubMed] [Google Scholar]
- 14.Venugopal C, Demos CM, Rao KSJ, et al. Beta-secretase: structure, function, and evolution. CNS Neurol Disord Drug Targets 2008; 7: 278–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Züchner T, Perez-Polo JR, Schliebs R. Beta-secretase BACE1 is differentially controlled through muscarinic acetylcholine receptor signaling. J Neurosci Res 2004; 77: 250–257. [DOI] [PubMed] [Google Scholar]
- 16.Rockenstein E, Mallory M, Mante M, et al. Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42). J Neurosci Res 2001; 66: 573–582. [DOI] [PubMed] [Google Scholar]
- 17.Fisher J, Devraj K, Ingram J, et al. Ferritin: a novel mechanism for delivery of iron to the brain and other organs. Am J Physiol, Cell Physiol 2007; 293: C641–649. [DOI] [PubMed] [Google Scholar]
- 18.Liebner S, Corada M. Wnt/β-catenin signaling controls development of the blood–brain barrier. J Cell Biol 2008; 183: 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czupalla CJ, Liebner S, Devraj K. In vitro models of the blood-brain barrier. Methods Mol Biol 2014; 1135: 415–437. [DOI] [PubMed] [Google Scholar]
- 20.Sánchez del Pino MM, Hawkins RA, Peterson DR. Biochemical discrimination between luminal and abluminal enzyme and transport activities of the blood-brain barrier. J Biol Chem 1995; 270: 14907–14912. [DOI] [PubMed] [Google Scholar]
- 21.Devraj K, Klinger ME, Myers RL, et al. GLUT-1 glucose transporters in the blood-brain barrier: differential phosphorylation. J Neurosci Res 2011; 89: 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spahn C, Cella-Zannacchi F, Endesfelder U, et al. Correlative super-resolution imaging of RNA polymerase distribution and dynamics, bacterial membrane and chromosomal structure in Escherichia coli. Meth Appl Fluoresc 2015; 3: 014005. [DOI] [PubMed] [Google Scholar]
- 23.Mowrer KR, Wolfe MS. Promotion of BACE1 mRNA alternative splicing reduces amyloid beta-peptide production. J Biol Chem 2008; 283: 18694–18701. [DOI] [PubMed] [Google Scholar]
- 24.Sankaranarayanan S, Price EA, Wu G, et al. In vivo beta-secretase 1 inhibition leads to brain Abeta lowering and increased alpha-secretase processing of amyloid precursor protein without effect on neuregulin-1. J Pharmacol Exp Ther 2008; 324: 957–969. [DOI] [PubMed] [Google Scholar]
- 25.Lyck R, Ruderisch N, Moll AG, et al. Culture-induced changes in blood-brain barrier transcriptome: implications for amino-acid transporters in vivo. J Cereb Blood Flow Metab 2009; 29: 1491–1502. [DOI] [PubMed] [Google Scholar]
- 26.Simpson IA, Davies P. Reduced glucose transporter concentrations in brains of patients with Alzheimer's disease. Ann Neurol 1994; 36: 800–801. [DOI] [PubMed] [Google Scholar]
- 27.Eisele YS, Obermüller U, Heilbronner G, et al. Peripherally applied A -containing inoculates induce cerebral – amyloidosis. Science 2010; 330: 980–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cirrito JR, Deane R, Fagan AM, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 2005; 115: 3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000; 106: 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cam JA, Zerbinatti CV, Li Y, et al. Rapid endocytosis of the low density lipoprotein receptor-related protein modulates cell surface distribution and processing of the beta-amyloid precursor protein. J Biol Chem 2005; 280: 15464–15470. [DOI] [PubMed] [Google Scholar]
- 31.Perez-Gonzalez R, Gauthier SA, Kumar A, et al. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J Biol Chem 2012; 287: 43108–43115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deane R, Sagare A, Hamm K, et al. IgG-assisted age-dependent clearance of Alzheimer's amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J Neurosci 2005; 25: 11495–11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swerdlow RH, Khan SM. A ‘mitochondrial cascade hypothesis’ for sporadic Alzheimer's disease. Med Hypotheses 2003; 63: 8–20. [DOI] [PubMed] [Google Scholar]
- 34.Boyd-Kimball D, Sultana R, Mohmmad-Abdul H, et al. Rodent Abeta(1-42) exhibits oxidative stress properties similar to those of human Abeta(1-42): implications for proposed mechanisms of toxicity. J Alzheimers Dis 2004; 6: 515–525. [DOI] [PubMed] [Google Scholar]
- 35.Elhusseiny A, Cohen Z, Olivier A, et al. Functional acetylcholine muscarinic receptor subtypes in human brain microcirculation: identification and cellular localization. J Cereb Blood Flow Metab 1999; 19: 794–802. [DOI] [PubMed] [Google Scholar]
- 36.Yang L-B, Lindholm K, Yan R, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 2003; 9: 3–4. [DOI] [PubMed] [Google Scholar]
- 37.Bodendorf U, Danner S, Fischer F, et al. Expression of human beta-secretase in the mouse brain increases the steady-state level of beta-amyloid. J Neurochem 2002; 80: 799–806. [DOI] [PubMed] [Google Scholar]
- 38.Roberds SL, Anderson J, Basi G, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet 2001; 10: 1317–1324. [DOI] [PubMed] [Google Scholar]
- 39.Ghosh AK, Brindisi M, Tang J. Developing β-secretase inhibitors for treatment of Alzheimer's disease. J Neurochem 2011; 120(Suppl 1): 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu YJ, Zhang Y, Kenrick M, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med 2011; 3: 84ra44. [DOI] [PubMed] [Google Scholar]
- 41.Simpson IA, Ponnuru P, Klinger ME, et al. A novel model for brain iron uptake: introducing the concept of regulation. J Cereb Blood Flow Metab 2015; 35: 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.