

Abstract
Ubiquitin and polyubiquitin chains target proteins for a wide variety of cellular processes. Ubiquitin-mediated targeting is regulated by the lysine through which the ubiquitins are linked as well as the broader ubiquitin landscape on the protein. The mechanisms of this regulation are not fully understood. For example, the canonical proteasome-targeting signal is a lysine 48-linked polyubiquitin chain, and the canonical endocytosis signal is a lysine 63-linked polyubiquitin chain. However, lysine 63-linked polyubiquitin chains can also target substrates for degradation. Biochemical studies of ubiquitinated proteins have been limited by the difficulty of building proteins with well-defined polyubiquitin chains. Here we describe an efficient and versatile method to synthesize ubiquitin chains of defined linkage and length. The synthesized ubiquitin chains are then attached to any protein containing a ubiquitin moiety. These proteins can be used to study ubiquitin targeting in in vitro assays in the tightly controlled manner required for biochemical studies.
Graphical Abstract
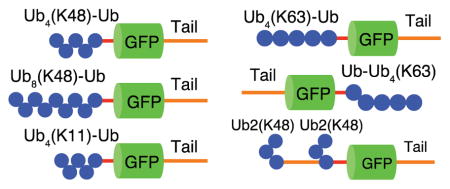
Proteins are typically ubiquitinated through the formation of an isopeptide bond between the carboxy-terminus of a ubiquitin moiety and the epsilon amino group of a lysine residue in the target protein1. Ubiquitin modification is catalyzed by a cascade of three or more enzymes, a ubiquitin activating enzyme (E1), a ubiquitin conjugating enzyme (E2), and a ubiquitin ligase (E3). The human genome is thought to encode two E1 enzymes, ~40 E2 enzymes, and more than 600 E3 enzymes2. Ubiquitin itself contains seven lysine residues and polyubiquitin chains can be built through any of these lysines as well as through the N-terminus of ubiquitin1, 3–5. The lysine through which the ubiquitin moieties are linked targets the protein to different fates. The three most abundant ubiquitin linkages in Saccharomyces cerevisiae are through lysine 48 (K48), followed by lysine 11 (K11), and lysine 63 (K63)5. The distribution of linkage patterns is similar in Arabidopsis thaliana6 and Homo sapiens7. K48-linked polyubiquitin chains are the prototypical proteasome-targeting signal8 whereas K63-linked chains are associated with cellular functions such as endocytosis, translation, and lysosomal targeting1, 9. However, many questions remain. For example, proteins modified with K63-linked polyubiquitin chains can be degraded by the proteasome in vitro and in vivo10, 11. Similarly, K11-linked chains have been shown to target for proteasomal degradation through the endoplasmic reticulum-associated protein degradation (ERAD) pathway and to play a role in progression of mitosis by inducing the degradation of cell cycle regulators5, 12, 13. K11 linkages are involved in mixed and branched ubiquitin chains14, and it is unclear if homotypic K11-linked chains can target proteins to the proteasome14, 15.
The quantitative biochemical characterization of the recognition of different ubiquitin chains has been hampered by the difficulty in making sufficient amounts of proteins modified by polyubiquitin chains with defined ubiquitin linkages and length. Methods for polyubiquitin chain synthesis have primarily focused on generating free polyubiquitin chains either enzymatically16, 17 or chemically18. Nevertheless, three main classes of technology exist to create ubiquitinated proteins. The first approach, which provided the basis for the method described here, creates ubiquitin chains by repeated rounds of enzymatic ligation followed by chemical or enzymatic de-blocking19, 20. The starting ubiquitin molecules have either the reactive lysine residue or the C-terminus blocked to limit chain growth to a single attachment step, which then generates a ubiquitin chain that is blocked on both the C-terminus and the target lysine. After each round of synthesis, the ubiquitin chains are deblocked on either their C-terminus or the lysine to form singly blocked chains, which are then reacted to create another doubly blocked ubiquitin chain of longer length19, 20. This method helped establish the minimal proteasome recognition signal3. However, this method requires multiple rounds of synthesis and de-blocking and has not been widely adopted. A second approach relies on the target protein being ubiquitinated by its cognate ubiquitination machinery. For example, to study IκBα ubiquitination a method was developed to in vitro phosphorylate IκBα with the kinase IKK3 followed by ubiquitination with E1, the E2 Ubch3, and the E3 complex SCFβ-TRCP1 (ref21). This method can create ubiquitin chains on one or more lysine residues in the target protein and the ubiquitin chains themselves are heterogeneous in length and often linkage. Therefore, this approach is most appropriate to investigate the physiological ubiquitination process but is not well suited for systematically making well-defined ubiquitin modifications. In a variation of this method, the specific recognition motif for the E3 Rsp5, the consensus sequence Pro-Pro-X-Tyr (PY motif), is inserted into a protein of interest. Rsp5 recognizes the PY motif and ubiquitinates lysine residues in the target protein22. A third set of methods uses non-enzymatic or chemoenzymatic synthesis to generate specific polyubiquitin chains (e.g., refs18, 23–27). The synthesized ubiquitin chains can then be attached to the target protein through a disulfide bridge18, 25. This method gives great control over ubiquitin chain linkage and length, but so far has been used to make only chains of up to four ubiquitin moieties. This method also requires application of chemical techniques that may not be easily amenable to biological laboratories18. Not all non-enzymatic synthesis methods provide a means to attach the ubiquitin chains to a protein target. Where they do, the attachment point in the target protein is a Cys residue that can in principle be inserted anywhere in the target as long as the target contains only a single reactive cysteine and reducing conditions will not be used downstream.
Here we present a simple method that avoids reactive chemicals and is easily implemented in biological laboratories. This method allows the synthesis of proteins with ubiquitin chains of different defined lengths and specific linkages in four steps: one, enzymatic synthesis of ubiquitin chains of defined linkage but of heterozygous length by specific E2 enzymes; two, isolation of chains of the desired length; three, enzymatic attachment of the ubiquitin chains to a ubiquitin moiety fused in-frame to a target protein; and four, isolating the ubiquitin chain attached substrate (Figure 1). Once all starting material is prepared, a substrate can be made from start to finish in one week creating milligram amounts of the desired protein. We show that this approach can be used to attach polyubiquitin chains of different lengths, two, four, or eight ubiquitin moieties; different linkages, K48, K63, and K11; and attached to three locations in a model protein, to N-terminal, C-terminal, and internal ubiquitin domains. This method makes it possible to investigate aspects of recognition and regulation of proteins modified by polyubiquitin chains, while retaining control over substrate architecture.
Figure 1. Schematic representation of generation of a ubiquitinated proteasome substrate Ub4(K48)-Ub-GFP-Tail.

(a) Synthesis of blocked polyubiquitin chains consisting of four ubiquitin chains linked through K48 (K48R-Ub4(K48)). Blocked His-tagged ubiquitin (His-HRV3C-Ub(K48R)) and wild-type ubiquitin react to form mixed polyubiquitin chains (Step 1). Polyubiquitin chains containing a blocked ubiquitin moiety at the distal end are bound to a Ni++ affinity column (Step 2) and eluted by cleavage with HRV3C protease (Step 3). The blocked polyubiquitin chains of the desired length are then purified, first through cation exchange chromatography (Step 4) and then through size exclusion chromatography (Step 5).
(b) Attachment of polyubiquitin chains (K48R-Ub4(K48)) to protein (Ub-GFP-Tail). Polyubiquitin chains blocked at the distal end (K48R-Ub4(K48)) are attached to the target protein (Ub-GFP-Tail) enzymatically with E1 and E2-25K (Step 1). Unreacted polyubiquitin chains are removed by His affinity purification of the target protein (Step 2) with and without attached ubiquitin chains (Ub4(K48)-Ub-GFP-Tail and Ub-GFP-Tail). Ubiquitinated (Ub4(K48)-Ub-GFP-Tail) and unubiquitinated target protein (Ub-GFP-Tail) are then separated by size exclusion chromatography (SEC) (Step 3).
Experimental Procedures
Molecular Biology
Ubiquitin mutants His-HRV3C-Ub(K48R), His-HRV3C-Ub(K63R), and His-HRV3C-Ub(K11R) were constructed by inserting the coding sequence for wild-type ubiquitin preceded by a HRV3C protease cleavage site into pETDuet (Novagen #71146-3) at the first multiple cloning site after the 6 × His tag. K48, K63, or K11 were mutated to arginine by site-directed mutagenesis.
E2-25K (Homo sapiens), Ubc13 (Saccharomyces cerevisiae), and Mms2 (S. cerevisiae) were derived from Addgene plasmids #18892, #18894, and #18893, respectively, which were originally constructed by the Pickart lab20, and cloned into pGEX-6p-1 (GE #28-9546-48) to create GST-E2-25K, GST-Ubc13, and GST-Mms2.
Ub-GFP-Tail consisted of a wild-type yeast ubiquitin domain where the last glycine was mutated to a valine at the N terminus, followed by a BamHI site, a GSGGSG linker, a circular permutant of superfolder GFP (gift from Robert T. Sauer28, 29), a SacII site, an 35 amino acid long unstructured region, and finally a 6 × His Tag. The 35 amino acid unstructured region was derived from cytochrome b2 (ref.30). Tail-GFP-Ub consisted of a 6 × His Tag at the N terminus, followed by a BamHI site, a 35 amino acid unstructured region, a EcoRI site, a circular permutant of GFP, a GSGGSG linker, a SacII site, and finally by a ubiquitin domain where the last glycine was mutated to a valine. Ub-35-Ub-GFP-Tail was the same as Ub-GFP-Tail except that a second ubiquitin domain with the last glycine mutated to a valine followed by the same 35 amino acid unstructured region was inserted at the N terminus (see Table S1 for amino acid sequences).
Protein Expression and Purification
Wild-type ubiquitin and Ub(K48R) were purified as previously described19, 31 except that cells were lysed by two passages at 15,000 psi through an EmulsiFlex-C3 (Avestin) homogenizer. Wild-type ubiquitin and Ub(K48R) can also be purchased (Boston Biochem #U-100Sc and #UM-K48R).
Mutant ubiquitins His-HRV3C-Ub(K48R), His-HRV3C-Ub(K63R), His-HRV3C-Ub(K11R), and target proteins Ub-GFP-Tail and Tail-GFP-Ub were expressed in E. coli Rosetta(DE3)pLysS (Novagen) cells. A starter culture was diluted 1:100 and grown in 2xYT medium containing 100 μg mL−1 ampicillin and 34 μg mL−1 chloramphenicol at 37 °C to an OD600 of 0.6, then induced with 0.4 mM IPTG for 4 hours. Cells were collected by centrifugation at 6,000 × g for 10 min, resuspended in NPI-10 (50 mM sodium phosphate, 300 mM sodium chloride, and 10 mM imidazole pH 8.0), and stored at −80 °C. For purification, Protease Inhibitor Cocktail Set V, EDTA-Free (Calbiochem #539137) was added to the frozen cells; the cells were then thawed in a water bath at room temperature and lysed by two passages at 15,000 psi through an EmulsiFlex-C3 (Avestin) homogenizer. The lysate was cleared by two centrifugation steps at 30,000 × g for 20 min at 4 °C and filtered through a 0.45 μm syringe filter (Pall #4614). The clarified lysate was applied to a 5 mL HisTrap FF Crude column on an FPLC (ÄKTApurifier 10 #28-4062-64) at 2 mL min−1, washed with two column volumes (CV) NPI-10, washed with ten CV 4.2% NPI-250 (50 mM sodium phosphate, 300 mM sodium chloride, and 250 mM imidazole pH 8.0) in NPI-10, and eluted with ten CV NPI-250. The mutant ubiquitins were then concentrated and buffer exchanged in a 3 kDa molecular weight cutoff Amicon Ultra centrifugation filter (Millipore #UFC900324) into 50 mM Tris-HCl pH 7.6 to a final concentration of 20–50 mg mL−1. The target proteins were then concentrated and buffer exchanged in a 10 kDa molecular weight cutoff Amicon Ultra centrifugation filter (Millipore #UFC901024) into 50 mM Tris-HCl pH 7.6 and 5% glycerol.
The His6-Ube1 (Mus musculus) expression plasmid was a gift from Jorge Eduardo Azavedo (Addgene plasmid # 32534) and the protein was purified as previously described32. E1 can also be purchased (Boston Biochem #E-304). UBE2S-UBD and AMSH* expression plasmids were a gift from David Komander and proteins were purified as previously described33–35. E2-25K (Homo Sapiens), Ubc13 (S. cerevisiae), and Mms2 (S. cerevisiae) were purified as GST-fusion proteins and the GST tag was removed by HRV3C Protease as previously described31. E2-25K and the Ubc13/Mms2 complex can also be purchased (Boston Biochem #E2-603, #E2-664).
His-HRV3C was expressed from E. coli Rosetta(DE3)pLysS (Novagen) cells. A starter culture was diluted 1:100 and grown in 2xYT medium containing 100 μg mL−1 ampicillin and 34 μg mL−1 chloramphenicol at 37 °C to an OD600 of 0.5, cooled to 28 °C, then induced with 0.5 mM IPTG for 5 hours. Cells were collected by centrifugation at 6,000 × g for 10 min, resuspended in NPI-10, and stored at −80 °C. For purification, 1 mM DTT, 1 mM PMSF, 10 mM MgCl2, and DNAse were added to the frozen cells; the cells were then thawed in a water bath at room temperature and lysed by two passages at 15,000 psi through a EmulsiFlex-C3 (Avestin) homogenizer. The lysate was cleared by two centrifugation steps at 30,000 × g for 20 min at 4 °C and filtered through a 0.45 μm syringe filter (Pall #4614). The clarified lysate was applied to a 5 mL HisTrap FF Crude column on an FPLC (ÄKTApurifier 10 #28-4062-64) at 2 mL min−1, washed with two CV His Binding Buffer (NPI-10, 1 mM DTT, 1 mM PMSF), washed with ten CV 4.2% His Elution Buffer (NPI-250 with 1 mM DTT) in His Binding Buffer, and eluted with ten CV His Elution Buffer. The eluate was dialyzed three times for at least 4 hours in 500 mL Dialysis Buffer (50 mM Tris pH 8.0, 150 mM NaCl, 10 mM EDTA, 20% glycerol, and 1 mM DTT). His-HRV3C can also be purchased (Clonetech #7360).
Generating Blocked Ubiquitin Chains
To generate K48R-Ub4(K48), 7.5 mg mL−1 wild-type ubiquitin, 7.5 mg mL−1 His-HRV3C-Ub(K48R), one-fifth volume PBDM8 buffer (250 mM Tris-HCl pH8, 25 mM MgCl2, 50 mM creatine phosphate, 3 U mL−1 inorganic pyrophosphatase, 3 U mL−1 creatine phosphokinase), 2.5 mM ATP, 0.5 mM DTT, 20 μM E2-25K, and 0.2 μM E1 were combined and incubated at 37 °C overnight. To quench the reaction, 5 mM DTT was added and the reaction incubated at room temperature for 20 minutes. To remove precipitated protein, the reaction was centrifuged at 15,000 × g for 5 minutes. For affinity purification, the supernatant was diluted with an equal volume of NPI-10. For each 50 mg total ubiquitin, 1 mL of washed Ni-NTA beads (Quiagen #30210) was added to the reaction and allowed to bind under nutation at 4 °C for 1 hour. The mixture was then poured into an empty PD-10 column (GE #17-0435-01) and allowed to settle by gravity. The column was washed twice with ten CV NPI-10 followed by ten CV HRV3C cleavage buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, and 1 mM DTT). His-HRV3C Protease in HRV3C cleavage buffer was then added to the column, the column capped, and nutated overnight at 4 °C. The blocked but no longer 6 × His-tagged ubiquitin chains were then eluted by washing three times with one CV HRV3C cleavage buffer. To assess cleavage efficiency, the uncleaved protein that remained bound to the column was eluted with five CV NPI-250 and analyzed by SDS-PAGE.
To charge the polyubiquitin chains for binding to the cation exchange column, the mixture was acidified with 0.03 volumes 2N acetic acid to a pH of 4. The acidified chains were then applied to a 6 mL Resource S (GE #17-1180-01) or a 8 mL SOURCE 15S (GE #17-0944-10) Tricorn column equilibrated with 50 mM ammonium acetate pH 4.5, washed with two CV of 50 mM ammonium acetate, and eluted with a gradient of one CV 0–200 mM, 25 CVs 200–450 mM, and one CV 450–1000 mM NaCl in 50 mM ammonium acetate pH 4.5, taking 2 mL fractions. A fraction from each of the peaks was analyzed by SDS-PAGE, each peak was concentrated and buffer exchanged into 50 mM Tris-HCl pH 7.6 and the peak containing K48R-Ub4(K48) was concentrated to 500 μL and purified further on a Superdex Hi-Load 75-pg (GE #28-9893-33) size exclusion column at 0.25 mL min−1, collecting 2 mL fractions. The peak fractions were analyzed by SDS-PAGE and Instant Blue (expedeon #ISB1L) staining and fractions containing the purest K48R-Ub4(K48) were pooled, concentrated and buffer exchanged into 50 mM Tris-HCl pH 7.6.
The shorter polyubiquitin chains (K48R-Ub2(K48)) were synthesized as above except that the peak containing K48R-Ub2(K48) was selected for further purification on a Superdex Hi-Load 75-pg (GE #28-9893-33) size exclusion column at 0.25 mL min−1. The longer polyubiquitin chains (K48R-Ub8(K48)) were purified as described above for K48R-Ub4(K48) except that 11.25 mg mL−1 wild-type ubiquitin, 3.75 mg mL−1 His-HRV3C-Ub(K48R) ubiquitin were used. The longer ubiquitin chains separated less well than the smaller chains and individual elution peaks overlapped significantly. Therefore, all of the protein-containing fractions were analyzed by SDS-PAGE to identify fractions containing K48R-Ub8(K48). The K48R-Ub8(K48) chains were further purified on a Superdex Hi-Load 75-pg (GE #28-9893-33) size exclusion column at 0.25 mL min−1.
Polyubiquitin chains linked through K63 (K63R-Ub4(K63)) were synthesized by combining 10 mg mL−1 wild-type ubiquitin, and 5 mg mL−1 His-HRV3C-Ub(K63R) ubiquitin, one-fifth volume PBDM7.6 buffer (250 mM Tris-HCl pH 7.6, 25 mM MgCl2, 50 mM creatine phosphate, 3 U mL−1 inorganic pyrophosphatase, 3 U mL−1 creatine phosphokinase, and 10 mM ATP), 0.5 mM DTT, 8 μM Ubc13, 8 μM Mms2, and 0.2 μM E1 and incubating at 37 °C overnight. The rest of the process for K63R-Ub4(K63) preparation was the same as described for K48R-Ub4(K48). Polyubiquitin chains linked through K63 with each ubiquitin harboring a K48R mutation (Ub4(K63)(K48R)) were synthesized as described for K63R-Ub4(K63) except that 20 mg mL−1 Ub(K48R) was used and the Ni purification and HRV3C cleavage steps were omitted.
Polyubiquitin chains linked through K11 (K11R-Ub4(K11)) were synthesized as K63R-Ub4(K63) except that His-HRV3C-Ub(K11R) ubiquitin as used instead of His-HRV3C-Ub(K63R) and 80 μM Ube2S-UBD, 0.4 μM AMSH* was used in place of Ubc13 and Mms2.
Attaching Blocked Ubiquitin Chains to a Target Protein
To attach K48-linked polyubiquitin chains (K48R-Ub4(K48) and K48R-Ub8(K48)) to Ub-GFP-Tail, 45 μM Ub-GFP-Tail, 15 μM of the ubiquitin chain, one-fifth volume PBDM8 buffer, 2.5 mM ATP, 0.5 mM DTT, 30 μM E2-25K, and 0.2 μM E1 were incubated at 37 °C overnight. To attach two ubiquitin chains (K48R-Ub2(K48)) to Ub-35-Ub-GFP-Tail, 15 μM Ub-35-Ub-GFP-Tail, 45 μM K48R-Ub2(K48), one-fifth column PBDM8 buffer, 2.5 mM ATP, 0.5 mM DTT, 30 μM E2-25K, and 0.2 μM E1 were incubated at 37 °C overnight. To attach K63-linked chains to target proteins, specifically, (K63R-Ub4(K63)) to Ub-GFP-Tail or Ub4(K63)(K48R) to Tail-GFP-Ub), 45 μM target protein, 15 μM of the ubiquitin chain, one-fifth volume PBDM7.6 buffer, 0.5 mM DTT, 8 μM Ubc13, 8 μM Mms2, and 0.2 μM E1 were incubated at 37 °C for five hours. To attach K11-linked chains to target substrates (K11R-Ub4(K11)) to Ub-GFP-Tail) 45 μM Ub-GFP-Tail, 15 μM K11R-Ub4(K11), one-fifth volume PBDM7.6 buffer, 0.5 mM DTT, 60 μM UBE2S-UBD, 0.4 μM AMSH*, and 0.2 μM E1 were incubated at 37 °C overnight. Reactions were quenched by 5 mM DTT and incubated at room temperature for 20 minutes. Precipitated protein was removed by centrifugation at 15,000 × g for 5 minutes. Unattached ubiquitin chains were removed from the protein mixture by affinity chromatography. The reaction was diluted with an equal volume of NPI-10 and the protein was His-purified on a 5 mL HisTrap FF Crude column on an FPLC. The protein was loaded onto the column, washed with two CV NPI-10, washed with ten CV of 4.2% NPI-250 in NPI-10, and eluted with ten CV NPI-250. The eluate should contain only substrate with and without ubiquitin chain attached. The eluate was concentrated to 0.5 mL and purified on a Superdex Hi-Load 200-pg (GE #28-9893-35) size exclusion column at 0.25 mL min−1, taking 2 mL fractions. The fractions containing protein were examined by SDS-PAGE and Instant Blue (expedeon #ISB1L) staining and those containing the purest ubiquitinated substrate were pooled, concentrated and buffer exchanged into 50 mM Tris-HCl pH 7.6 containing 5% Glycerol.
Proteasome Purification
Proteasome was purified from S. cerevisiae as in ref.36 with minor modifications. The core particle (CP) and regulatory particle (RP) of the proteasome were purified separately from yeast strains YYS37 and YYS40 respectively36.
Substrate Degradation
The substrates (Ub4(K48)-Ub-CP8-Tail, Ub8(K48)-Ub-CP8-Tail, Ub4(K63)-Ub-CP8-Tail, Ub4(K11)-Ub-CP8-Tail, and Tail-CP8-Ub-Ub4(K48), Ub2(K48)-Ub-35-Ub2(K48)-Ub-GFP-Tail) were degraded in vitro by purified wild-type yeast proteasome. Purified CP and RP were mixed together at a 1:2 ratio and incubated at 30 °C for 30 minutes. After incubation the proteasome was mixed to a concentration of 50 nM CP and 100 nM RP with 8 mM DTT, two times degradation buffer (ten times degradation buffer (100 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10% glycerol)), and two times ATP Regenerating System (ARS) (25 × ARS (25 mM ATP, 250 mM CP, and 2.5 mg mL−1 CPK)). Substrate was diluted to 10 nM in protein buffer (4 mg mL−1 BSA and one-tenth volume ten times degradation buffer). Both mixtures were incubated at 30 °C for 5 minutes and then 20 μL of substrate (final substrate concentration 5 nM) was mixed with 20 μL of proteasome mix (final proteasome concentration of 25 nM) in a 384-well no bind black plate (Corning #3575). The substrates’ fluorescence (488 nm excitation 520 nm emission) was measured every minute for 90 minutes in a plate reader (infinite M1000 PRO, Tecan). Background (average fluorescence of a well containing everything except substrate) was subtracted and fluorescence plotted over time. Each assay represents the average of three replicates. The decay curves were fitted to the equation describing a single exponential decay to a constant offset using the software package KaleidaGraph (version 4.1, Synergy Software).
Results
Generating blocked lysine 48 ubiquitin chains
To create proteins modified with ubiquitin chains of different linkages and lengths we first synthesized free polyubiquitin chains. Our initial efforts focused on lysine 48 (K48)-linked ubiquitin chains, which form the canonical proteasome targeting signal and represent the most abundant linkage in Saccharomyces cerevisiae, Arabidopsis thaliana, and Homo sapiens5–7. For the synthesis we used enzymes that are part of the normal synthesis machinery and are able to act on free ubiquitin and create free polyubiquitin chains11, 37. To define linkages, we used enzymes that are specific to the linkage of interest: for K48-linked ubiquitin chains, we used the H. sapiens ubiquitin-conjugating enzyme E2-25K, which specifically generates K48-linked chains37. E2-25K only works with mammalian E1 so we used the Mus musculus E1 Ube1 (E1)38 and free ubiquitin in the presence of ATP to synthesize free polyubiquitin chains.
E2-25K can only extend polyubiquitin chains at either the proximal or distal ubiquitin; thus only the ends of the resulting chains are reactive. The ubiquitin moiety at one end of the chain has a reactive free C-terminus that can be attached to other lysine residues and is referred to as the proximal ubiquitin because it will be closest to the substrate when the chain is attached. The ubiquitin at the opposite end of the chain, referred to as the distal ubiquitin, contains a K48 that can accept the ligation of the C-terminus of additional ubiquitin molecules. The K48 residues and C-termini of all other ubiquitin moieties are part of isopeptide bonds and thus are not accessible to a K48-specific enzyme.
To attach polyubiquitin chains to a target protein, it is convenient to block the remaining acceptor lysine residue in the distal ubiquitin moiety so that the free ubiquitin chains can only be attached to the target protein and not to each other, creating substrates with homogenous polyubiquitin chains. To this end, we generated a mutant ubiquitin in which lysine 48 was replaced by an arginine (K48R). This mutant ubiquitin, when reacted with wild-type ubiquitin, was always incorporated into the polyubiquitin chain at the distal end because the K48R mutation blocks further growth at the relevant residue. The reaction also created some polyubiquitin chains that did not incorporate the mutant ubiquitin and it was important to separate these two populations of chains. For this purpose we also attached an N-terminal 6 × His-tag to the mutant ubiquitin through a linker containing a specific cleavage site for the Human rhinovirus 3C (HRV3C) protease (His-HRV3C-Ub(K48R)). This tag permits purification of only the blocked ubiquitin chains and subsequent removal of the 6 × His tag by proteolytic cleavage with HRV3C protease (Figure 1A).
Generating and separating blocked K48 ubiquitin chains of different lengths
To synthesize ubiquitin chains of different lengths, we incubated a mixture of blocked ubiquitin (His-HRV3C-Ub(K48R)) and wild-type ubiquitin with E1 and E2-25K in the presence of ATP. The reaction created polyubiquitin chains of mixed lengths, some of which had incorporated the blocked ubiquitin (His-HRV3C-Ub(K48R)) at their distal end (Figure 1A). Chains containing the distal blocked ubiquitin were then bound to a nickel column and eluted by on-column cleavage with a His-tagged HRV3C protease on the column. The eluate was a mixed population of ubiquitin chains of different lengths but the same linkage, each chain with a distal ubiquitin harboring the K48R mutation as well as three additional amino acids remaining on the N-terminus created the HRV3C cleavage site (Figure 1A).
Polyubiquitin chains of specific length were then purified by ion exchange chromatography followed by size exclusion chromatography. At low pH ubiquitin is positively charged and will bind to a cation exchange column. Longer ubiquitin chains carry a larger total charge and therefore require a higher salt concentration to elute. Separation on a cation exchange column at a pH 4.5, eluted with a linear salt gradient, separated chains of different lengths progressively (Figure 2A). K48R-Ub4(K48) (peak #4 in Figure 2A) was then purified further by size exclusion chromatography (SEC) (Figure 2B). Ubiquitin chain linkage was confirmed by mass spectrometry (Table S2). The synthesis of K48R-Ub4(K48) shown in Figure 2A and 2B used 56 mg of each ubiquitin and produced ~5.8 mg K48R-Ub4(K48) (Table S3).
Figure 2. Synthesis and purification of blocked K48R-Ub4(K48) and K48R-Ub8(K48) ubiquitin chains.

(a) Cation exchange (Resource S) separation of blocked K48-linked polyubiquitin chains to isolate tetraubiquitin chains (K48R-Ub4(K48)). Blocked (His-HRV3C-Ub(K48R)) and wild-type ubiquitin at a molar ratio of 1:1 were reacted to form mixed polyubiquitin chains. The Ni++ and HRV3C purified chains were applied to a Resource S cation exchange column and eluted with linear salt gradients (1 CV 0 – 200 mM NaCl, 25 CV 200 – 550 mM NaCl, 3 CV 550 – 1000 mM NaCl). Peak fractions were analyzed by SDS-PAGE to determine chain length of each peak (inset).
(b) Size exclusion (Superdex 75) separation of tetraubiquitin chains. Peak 4 in (a) was concentrated to 500 μL and then separated on a Superdex 75 16/600 PG column at 0.25 mL min−1. The protein containing fractions were analyzed by SDS-PAGE (inset) and fractions 30 and 31 were pooled, concentrated, and buffer exchanged.
(c) Cation exchange (Resource S) separation of longer Lys48-linked polyubiquitin chains to isolate octaubiquitin chains (K48R-Ub8(K48)). Blocked (His-HRV3C-Ub(K48R)) and wild-type ubiquitin at a molar ratio of 1:3 were reacted to form mixed polyubiquitin chains. The His and HRV3C purified chains were applied to a Resource S cation exchange column and eluted with linear gradients of salt (3 CV 0 – 200 mM NaCl, 25 CV 200 – 450 mM NaCl, 3CV 450 – 1000 mM NaCl) (top). Fractions containing protein were analyzed by SDS-PAGE to determine which fractions contained the desired length ubiquitin chain (bottom).
(d) Superdex 75 separation of octaubiquitin chains (K48R-Ub8(K48)). K48R-Ub8(K48) containing fractions 62–65 in (c) were concentrated to 500 μL and then separated on a Superdex 75 16/600 PG column at 0.25 mL min−1. The protein-containing fractions were analyzed by SDS-PAGE (inset) and fractions 24 and 25 were pooled, concentrated, and buffer exchanged.
(e) (Left) Purified K48R-Ub4(K48). Purified product from Figure 2A and B. (Right) Purified K48R-Ub8(K48). Purified product from Figure 2C and D
The distribution of the polyubiquitin chain length depends on the ratio of blocked to wild-type ubiquitin in the reaction mixture with lower proportions of blocked ubiquitin leading to longer chain lengths. To create chains of different lengths we adjusted the ratios of the ubiquitins used: equal amounts of blocked and free ubiquitin proved suitable for the synthesis of chains consisting of two or four ubiquitin moieties (Figure 2A), whereas a ratio of one part blocked ubiquitin to three parts wild-type ubiquitin worked well to synthesize ubiquitin chains consisting of eight ubiquitin moieties (Figure 2C). The quality of the separation by cation exchange chromatography and thus the purity of individual fractions deteriorated for longer ubiquitin chains, requiring SDS-PAGE analysis of all fractions to assess which fractions contained the desired chain length (compare Figures 2A and 2C). K48R-Ub8(K48) was then further purified by SEC (Figure 2D). The longer length of these polyubiquitin chains led the separation and purification to yield a slightly heterogeneous mixture of lengths (Figure 2E). The synthesis of K48R-Ub8(K48) (Figure 2C and 2D) used 22.5 mg blocked ubiquitin and 67.5 mg wild-type ubiquitin and yielded ~4.3 mg K48R-Ub8(K48) (Table S3). The eluted blocked ubiquitin chains were pure as judged by SDS-PAGE analysis (Figure 2E).
Attaching blocked K48 ubiquitin chains to proteins
Polyubiquitin chains were attached to a target protein by ligating the chain to a ubiquitin domain that was encoded in frame in the target protein. The same enzymes used to create the polyubiquitin chains were used to ligate the chains to the target to create polyubiquitin chains with homogeneous K48 linkages. We chose a fluorescent protein as the target protein so that the substrate could be easily monitored. Here we used a circular permutant of superfolder GFP28, 29 (GFP) in the target protein and fused a ubiquitin moiety to its N-terminus. We also attached an unstructured region (Tail) to the C-terminus of the GFP domain, consisting of a 35 amino acid-long sequence derived from cytochrome b2, to serve as an initiation region for proteasomal degradation followed by a C-terminal 6 × His tag for purification30. The final substrate was a fusion protein consisting of an N-terminal ubiquitin domain, followed by a GFP domain variant, and a C-terminal unstructured region (Ub-GFP-Tail; Figure 1B).
For attachment to a protein, ubiquitin chains with a blocked distal ubiquitin moiety (K48R-Ub4(K48) or K48R-Ub8(K48)) were incubated with a three-fold molar excess of the target protein as well as E1, E2-25K, and ATP at 37 °C overnight. The reaction created a mixed population of ubiquitinated substrate (Ub4(K48)-Ub-GFP-Tail or Ub8(K48)-Ub-GFP-Tail), unattached ubiquitin chains (K48R-Ub4(K48) or K48R-Ub8(K48)), and unreacted target protein (Ub-GFP-Tail). We removed the unattached ubiquitin chains by purifying the target protein through its C-terminal 6 × His tag by affinity chromatography. The attached and unattached substrates now differed in size by 34 or 68 kDa for Ub4 or Ub8 substrates respectively, and could be separated from each other by SEC (Figure 3A and 3B). The final ubiquitinated substrate proteins were pure as judged by SDS-PAGE analysis (Figure 3C and 3D). Substrate modified with K48R-Ub8(K48) chains appears as a blurry band by SDS-PAGE (Figure 3D). There are two likely reasons for this behavior. 1) Ub8 chains are slightly heterogeneous in length (Figure 2E); and 2) GFP denatures only partially in SDS sample buffer without boiling and runs at two distinct sizes (Figure S1). Boiling polyubiquitin chains in SDS sample buffer leads to their multimerization and therefore the samples were not boiled. The synthesis of Ub4(K48)-Ub-GFP-Tail (Figure 3A and 3C) used 1 mg K48R-Ub4(K48) and 3.7 mg Ub-GFP-Tail to yield 1.2 mg Ub4(K48)-Ub-GFP-Tail. The synthesis of Ub8(K48)-Ub-GFP-Tail (Figure 3B and 3D) used 4 mg K48R-Ub8(K48) and 7.4 mg Ub-GFP-Tail to yield 1.2 mg Ub8(K48)-Ub-GFP-Tail (Table S3).
Figure 3. Attachment of K48-linked ubiquitin chains to a target protein (Ub-GFP-Tail) and purification of the ubiquitinated proteins (Ub4(K48)-Ub-GFP-Tail and Ub8(K48)-Ub-GFP-Tail).

(a) Purification of K48-linked tetraubiquitin substrate (Ub4(K48)-Ub-GFP-Tail). K48R-Ub4(K48) was attached to Ub-GFP-Tail enzymatically and affinity purified on a Ni column. The eluate was concentrated to 500 μL and then separated on a Superdex 200 16/600 PG column at 0.25 mL min−1. The protein-containing fractions were analyzed by SDS-PAGE (bottom) and fractions containing Ub4(K48)-Ub-GFP-Tail (36–39) were pooled, concentrated, and buffer exchanged.
(b) Purification of K48-linked octaubiquitin substrate (Ub8(K48)-Ub-GFP-Tail). Same as (a) except that the size difference between attached and unattached substrates is greater, so the shift on SEC is greater: Ub8(K48)-Ub-GFP-Tail elutes at ~68 mL and Ub-GFP-Tail at ~82 mL. Fractions 33–35 were pooled, concentrated, and buffer exchanged.
(c) K48R-Ub4(K48) attachment to Ub-GFP-Tail and purification of Ub4(K48)-Ub-GFP-Tail. K48R-Ub4(K48) was attached to Ub-GFP-Tail with E1 and E2-25K creating Ub4(K48)-Ub-GFP-Tail (compare lanes 1 and 2). The resulting product was His-purified by Ni affinity chromatography to remove unreacted polyubiquitin chains leaving only Ub4(K48)-Ub-GFP-Tail and Ub-GFP-Tail (compare lanes 2 and 3). The eluate (lane 3) was further purified on an S200 SEC (compare lane 3 to 4).
(d) K48R-Ub8(K48) attachment to Ub-GFP-Tail and purification of Ub8(K48)-Ub-GFP-Tail. K48R-Ub8(K48) was attached to Ub-GFP-Tail with E1 and E2-25K, creating Ub8(K48)-Ub-GFP-Tail (compare lanes 1 and 2). The resulting product was affinity purified on a Ni column to remove unreacted chains leaving only Ub4(K48)-Ub-GFP-Tail and Ub-GFP-Tail. The eluate from the Ni column was further purified on an S200 SEC (lane 3 and 4).
K63 ubiquitin proteins
Lysine 63 (K63)-linked polyubiquitin chains were synthesized with the S. cerevisiae E2/E2 variant complex Ubc13/Mms2, which generates K63-linked polyubiquitin chains39. We used a similar mutant ubiquitin as we employed for K48-linked chains, except that K63 instead of K48 was mutated from a lysine to an arginine (His-HRV3C-Ub(K63R)). Distally-blocked Ub4 chains linked through K63 (K63R-Ub4(K63)), were synthesized using a ratio of one part blocked (His-HRV3C-Ub(K63R)) to two parts wild-type ubiquitin with E1, Ubc13, and Mms2 overnight at 37 °C. The remainder of the purification of the polyubiquitin chains was the same as for K48R-Ub4(K48) (Figure 4A and 4B). The eluted blocked ubiquitin chains were pure as judged by to SDS-PAGE analysis (Figure 4C). Ubiquitin chain linkage was confirmed by mass spectrometry (Table S2). The synthesis of K63R-Ub4(K63) (Figure 4A, 4B and 4C) used 50 mg blocked ubiquitin and 100 mg wild-type ubiquitin and yielded ~5.4 mg K63R-Ub4(K63) (Table S3).
Figure 4. Synthesis and purification of K63-linked polyubiquitin chains and attachment to a target protein.

(a) Cation exchange (Resource S) separation of blocked K63 ubiquitin chains for K63R-Ub4(K63). A 1:2 ratio of blocked (His-HRV3C-Ub(K63R)) to wild-type ubiquitin were reacted to form mixed chains. The His- and HRV3C-purified chains were applied to a 8 mL SOURCE 15S cation exchange column and eluted with linear gradients of salt (1 CV 0 – 200 mM NaCl, 20 CV 200 – 450 mM NaCl, 1 CV 450 – 1000 mM NaCl). Peaks were analyzed by SDS-PAGE to determine chain length of each peak (inset).
(b) Size exclusion (Superdex 75) separation of K63R-Ub4(K63). Peak 4 in (a) was concentrated to 500 μL and then separated on a Superdex 75 16/600 PG column at 0.25 mL min−1. Fractions 29–30 were pooled, concentrated, and buffer exchanged. The protein containing fractions were analyzed by SDS-PAGE (inset) and fractions 28–30 were pooled, concentrated, and buffer exchanged.
(c) Purified K48R-Ub4(K63). Purified product from Figure 4A and B
(d) Purification of K63 linked tetraubiquitin substrate (Ub4(K63)-Ub-GFP-Tail). K63R-Ub4(K63) was attached to Ub-GFP-Tail with E1, Ubc13, and Mms2 and purified by Ni-affinity chromatography. The eluate from the Ni column was concentrated to 500 μL and then separated on a Superdex 200 16/600 PG column at 0.25 mL min−1. The protein-containing fractions were analyzed by SDS-PAGE (inset) and fractions containing pure Ub4(K63)-Ub-GFP-Tail (35–36) were pooled, concentrated, and buffer exchanged.
(e) K63R-Ub4(K63) attachment to Ub-GFP-Tail and purification of Ub4(K63)-Ub-GFP-Tail. K63R-Ub4(K63) was attached to Ub-GFP-Tail with E1, Ubc13, and Mms2 creating Ub4(K63)-Ub-GFP-Tail (compare lanes 1 and 2). The resulting product was purified by Ni affinity chromatography to remove unreacted polyubiquitin chains leaving only Ub4(K63)-Ub-GFP-Tail and Ub-GFP-Tail (compare lanes 2 and 3). The eluate from the Ni column (lane 3) was further purified on an S200 SEC (compare lane 3 to 4).
For protein attachment, the blocked K63-linked polyubiquitin chains (K63R-Ub4(K63)) were incubated with a three-fold molar excess of the target protein as well as E1, Ubc13, Mms2, and ATP for five hours at 37 °C (in contrast to the overnight incubation used for K48-linked chains). The reaction created a mixed population of ubiquitinated target protein (Ub4(K63)-Ub-GFP-Tail), unattached ubiquitin chains (K63R-Ub4(K63)), and unreacted target protein (Ub-GFP-Tail). The remainder of the purification was the same as for Ub4(K48)-Ub-GFP-Tail (Figure 4D and 4E). The synthesis of Ub4(K63)-Ub-GFP-Tail (Figure 4D and 4E) used 0.5 mg K63R-Ub4(K63) and 1.9 mg Ub-GFP-Tail to yield ~0.4 mg Ub4(K63)-Ub-GFP-Tail (Table S3).
K11 ubiquitin proteins
Lysine 11 (K11)-linked polyubiquitin chains were synthesized with the E2 UBE2S-UBD and the deubiquitinating (DUB) enzyme AMSH* as described previously33–35. UBE2S-UBD is a fusion protein of the E2 UBE2S where the lysine-rich tail of UBE2S is replaced with the ubiquitin-binding zinc finger ubiquitin binding domain (UBD) of the ubiquitin specific protease USP5 to increase the affinity of UBE2S for ubiquitin33, 40. The UBE2S-UBD fusion protein generates K11-linked ubiquitin chains with some contaminating K63 linkages. Therefore, the K63 specific deubiquitinating enzyme AMSH*, a fusion of the VHS and ubiquitin interacting domains of mouse STAM2 and the catalytic domain of human AMSH, was used to remove those contaminating linkages as previously described33–35. We used a similar mutant ubiquitin to block chain growth at the distal end as we employed for the synthesis of K48- and K63-linked chains, except that K11 instead of K48 or K63 was mutated from a lysine to an arginine (His-HRV3C-Ub(K11R)). Distally-blocked Ub4 chains linked through K11 (K11R-Ub4(K11)), were synthesized using a ratio of one part blocked (His-HRV3C-Ub(K11R)) to two parts wild-type ubiquitin with E1, UBE2S-UBD, and AMSH* overnight at 37 °C. The remainder of the purification for the ubiquitin chains was the same as for K48R-Ub4(K48) and K63R-Ub4(K63) (Figure 5A). The purified blocked ubiquitin chains were pure according to SDS-PAGE analysis (Figure 5A, last lane). Ubiquitin chain linkage was confirmed by mass spectrometry (Table S2).
Figure 5. Synthesis and purification of K11-linked polyubiquitin chains and attachment to a target protein.
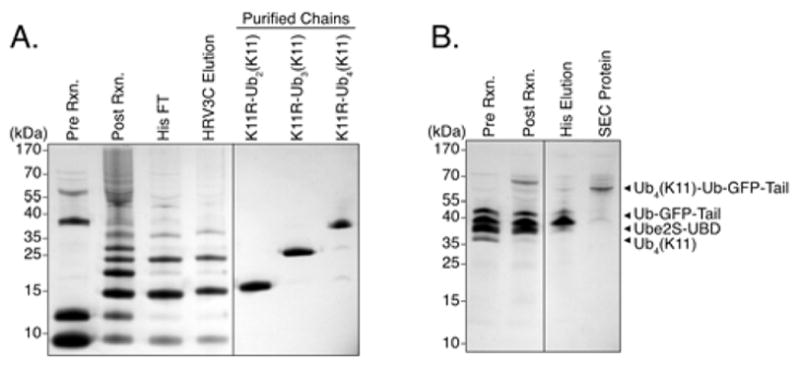
(a) Synthesis of K11R-Ub4(K11). A 1:2 ratio of blocked (His-HRV3C-Ub(K11R)) to wild-type ubiquitin were reacted to form mixed chains (compare lanes 1 and 2). The non-blocked chains are removed by His-purification (lane 3) and the blocked chains are collected by HRV3C-cleavage (lane 4). The chains were then separated by length on a cation exchange column with linear salt gradients (lanes 5–7).
(b) K11R-Ub4(K11) attachment to Ub-GFP-Tail and purification of Ub4(K11)-Ub-GFP-Tail. K11R-Ub4(K11) was attached to Ub-GFP-Tail with E1, Ube2S-UBD, and AMSH* creating Ub4(K11)-Ub-GFP-Tail (compare lanes 1 and 2). The resulting product was purified by Ni affinity chromatography to remove unreacted polyubiquitin chains leaving only Ub4(K11)-Ub-GFP-Tail and Ub-GFP-Tail (compare lanes 2 and 3). The eluate from the Ni column (lane 3) was further purified on an S200 SEC (compare lane 3 to 4).
For attachment of the polyubiquitin chains to the protein, the blocked K11-linked chains (K11R-Ub4(K11)) were incubated with three-fold molar excess of the target protein as well as E1, UBE2S-UBD, AMSH*, and ATP overnight at 37 °C. This reaction created a mixed population of ubiquitinated target protein (Ub4(K11)-Ub-GFP-Tail), unattached ubiquitin chains (K11R-Ub4(K11)), and unreacted target protein (Ub-GFP-Tail). The remainder of the purification was the same as for Ub4(K48)-Ub-GFP-Tail and Ub4(K63)-Ub-GFP-Tail (Figure 5B). Attachment of K11-linked chains was significantly less efficient than achieved for the other chains with their associated E2 enzymes (Ubc13/Mms2 for K63-linked chains and E2-25K for K48-linked chains). To increase yields, it may be possible to synthesize the ubiquitin chains with UBE2S-UBD and then attach with an alternative enzyme as described below for C-terminal K63 chains.
Alternate protein configurations
Ubiquitin chains can be attached at different locations in the target protein by modifying the location of the fused ubiquitin. The ubiquitin moiety can be fused in-frame at the N-terminus as shown above (Figure 1B), at the C-terminus, or internally. The Ubc13/Mms2 complex will only attach ubiquitin efficiently to a ubiquitin with a free N-terminus and therefore cannot be used to attach chains to C-terminal or internal ubiquitin domains11. E2-25K does not have this restriction and can be used to attach K48 chains to proteins with internal or C-terminal ubiquitin domains. To attach K63-linked polyubiquitin chains at the C terminus of a protein, we generated a K63-linked ubiquitin chain with the Ubc13/Mms2 complex using ubiquitin harboring a K48R mutation. This method does not require a mixture of blocked and WT ubiquitins, therefore the mutant ubiquitin does not contain an N-terminal 6 × His tag and the affinity and HRV3C cleavage steps in the purification can be omitted (Figure 6A). The resulting chain was then attached to the C-terminal or internal ubiquitin domain within the target protein with E2-25K, creating a substrate with one proximal K48 linkage while retaining K63 linkages throughout the rest of the chain11 (Figure 6B and 6C). The synthesis of Tail-GFP-Ub-Ub4(K63) (Figure 6C) used 1 mg Ub4(K63)(K48R) and 3.8 mg Tail-GFP-Ub to yield ~0.4 mg Tail-GFP-Ub-Ub4(K63) (Table S3).
Figure 6. Synthesis and purification of C-terminally K63-linked tetraubiquitin substrate (Tail-GFP-Ub-Ub4(K63)).

(a) Synthesis of polyubiquitin chains consisting of four (K48R) ubiquitins linked through K63 (Ub4(K63)(K48R)). Ub(K48R) is synthesized into K63-linked chains by E1, Ubc13, and Mms2 (Step 1). The polyubiquitin chains of the desired length are then purified, first through cation exchange chromatography (Step 2) and then through size exclusion chromatography (Step 3).
(b) Attachment of polyubiquitin chains (Ub4(K63)(K48R)) to protein (Tail-GFP-Ub). Polyubiquitin chains are attached to the target protein (Tail-GFP-Ub) enzymatically with E1 and E2-25K (Step 1). Unreacted polyubiquitin chains are removed by His affinity purification of the target protein with and without attached ubiquitin chains (Tail-GFP-Ub-Ub4(K63) and Tail-GFP-Ub). Ubiquitinated (Tail-GFP-Ub-Ub4(K63)) and unubiquitinated target protein (Tail-GFP-Ub) are then separated by size exclusion chromatography (SEC) (Step 2).
(c) Ub4(K63)(K48R) attachment to Tail-GFP-Ub and purification of Tail-GFP-Ub-Ub4(K63). Ub4(K63)(K48R) was attached to Ub-GFP-Tail with E1, and E2-25K creating Tail-GFP-Ub-Ub4(K63) (compare lanes 1 and 2). The resulting product was purified by Ni affinity chromatography to remove unreacted polyubiquitin chains leaving only Tail-GFP-Ub-Ub4(K63) and Tail-GFP-Ub (compare lanes 2 and 3). The eluate from the Ni column (lane 3) was further purified on an S200 SEC (compare lane 3 to 4).
To generate a protein modified with multiple ubiquitin chains, we synthesized a substrate containing two ubiquitin domains in frame, one N-terminal and one internal, separated by the same 35 amino acid unstructured region used in the tail (Ub-35-Ub-GFP-Tail). We then attached a K48-linked di-ubiquitin chain (K48R-Ub2(K48)) to each ubiquitin domain by incubating the target protein (Ub-35-Ub-GFP-Tail) with a three-fold molar excess of ubiquitin chain (K48R-Ub2(K48)) as well as E1, E2-25K, and ATP at 37 °C overnight to generate Ub2(K48)-Ub-35-Ub2(K48)-Ub-GFP-Tail. The doubly ubiquitinated protein was purified in the same manner as the other substrates (Figure 7). The use of an excess of free polyubiquitin chains over the target protein Ub-35-Ub-GFP-Tail led to the complete conversion of the target protein to the modified protein Ub2(K48)-Ub-35-Ub2(K48)-Ub-GFP-Tail (Figure 7). The synthesis of Ub2(K48)-Ub-35-Ub2(K48)-Ub-GFP-Tail (Figure 7) used 1.1 mg K48R-Ub2(K48) and 1.2 mg Ub-35-Ub-GFP-Tail to yield ~0.7 mg Ub2(K48)-Ub-35-Ub2(K48)-Ub-GFP-Tail (Table S3). Other strategies to synthesize proteins with internal or multiple ubiquitin chains may be feasible. For example, one or more ubiquitin moieties could be introduced into a target protein by click chemistry to an alkyne or an azide that had been incorporated with an unnatural amino acid41. Polyubiquitin chains could then be attached by the protocols described above.
Figure 7. Synthesis and purification of doubly ubiquitinated substrate (Ub2(K48)-Ub-35-Ub2(K48)-GFP-Tail).
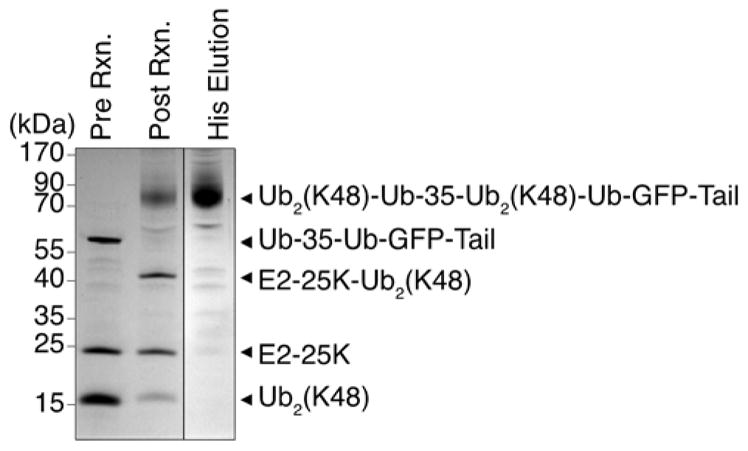
K48R-Ub2(K48) attachment to Ub-35-Ub-GFP-Tail and purification of Ub2(K48)-Ub-35-Ub2(K48)-GFP-Tail. K48R-Ub2(K48) was attached to Ub-35-Ub-GFP-Tail with E1, and E2-25K creating Ub2(K48)-Ub-35-Ub2(K48)-GFP-Tail (compare lanes 1 and 2). The resulting product was purified by Ni affinity chromatography to remove unreacted polyubiquitin chains and enzymes leaving only Ub2(K48)-Ub-35-Ub2(K48)-GFP-Tail and Ub-35-Ub-GFP-Tail (compare lanes 2 and 3).
Substrate degradation by the proteasome
The ubiquitinated target proteins created were competent for degradation by the proteasome, but the extent and rate of degradation varied for different substrates. Under single turn over conditions, Ub8(K48)-Ub-GFP-Tail and Ub4(K48)-Ub-GFP-Tail reacted similarly with proteasome with ~75% becoming degraded and with initial rates of 17 % min−1 and 15 % min−1 respectively. Ub4(K63)-Ub-GFP-Tail was degraded less well with ~50% degradation and an initial rate of 4 % min−1. Interestingly, the inverted Ub4(K63) substrate (Tail-Ub-Ub4(K63)) was degraded similarly to the K48 substrate with ~70% degradation and an initial rate of 12 % min−1. However, Ub4(K11)-Ub-GFP-Tail barely degraded with ~10% degradation and an initial rate of 0.4 % min−1. The substrate generated with two short ubiquitin chains degraded the best with 90% degradation and an initial rate of 21 % min−1 (Figure 8).
Figure 8. The ubiquitinated substrate proteins are degraded by the proteasome with different efficiencies.

The substrate proteins were presented to purified yeast proteasome and their degradation followed measuring GFP fluorescence. 25 nM proteasome was incubated with 5 nM of the indicated substrate in the presence of 2 mM ATP. The fluorescence was measured in 384-well plate (infinite M1000 PRO, Tecan) every minute for 90 minutes. Data points represent mean values determined from at least three repeat experiments.
Discussion
We present a simple, robust method that allows the synthesis of milligram amounts of a target protein modified with polyubiquitin chains of defined lengths and specific linkages. The method is versatile and we have demonstrated the synthesis of proteins modified with polyubiquitin chains with the three most common linkage patterns in S. cerevisiae5 and A. thaliana6 (K11-linked chains, K48-linked chains, and K63-linked chains), with polyubiquitin chains of three different lengths, and at three different locations within the target protein. The proteins were active in a relevant biochemical assay, here proteasomal degradation. Proteins comprised of the same components but modified with different ubiquitin chains were degraded with different rates. This result suggest that the method outlined here for creating proteins modified with defined ubiquitin chains should make it possible to study how differences in ubiquitin chains and substrate configurations affect proteasomal degradation.
In principle, it should be possible to expand the method described here to include any other homotypic ubiquitin chain once linkage specific enzymes are identified. Several protocols have been published describing synthesis methods for lysine 29- and lysine 33-linked ubiquitin chains35, 42. These protocols can, in theory, be incorporated into the method described here by creating an appropriately blocked ubiquitin in the same fashion. In this study polyubiquitin chains were attached to a target protein derived from GFP, but the same method should be applicable to other proteins. The method produces sufficiently large quantities of ubiquitinated protein such that assays requiring even high protein concentrations should be accessible. The limitation of the method described is that it requires the presence of a ubiquitin domain in the target protein for attachment of the polyubiquitin chain and it does not allow the synthesis of polyubiquitin chains with mixed linkages at specific branch points. Nevertheless, this method distinguishes itself from other synthesis methods through its simplicity and expedience. We propose the method presented here will facilitate biochemical studies of processes that involve the recognition of proteins modified by polyubiquitin chains.
Supplementary Material
Acknowledgments
Funding Source
This work was supported by US National Institutes of Health (U54GM105816 and Training Program in Oncogenesis and Developmental Biology T32 CA080621) as well as the Welch Foundation (F-1817).
We thank Drs. Robert T. Sauer and Andrew R. Nager for providing the GFP circular permutant plasmid and Drs. David Komander and Jonathan Pruneda for providing the UBE2S-UBD and AMSH* plasmids. Michelle Gadush and Maria D. Person identified the ubiquitin modifications at the Proteomics Facility in the Institute for Cellular and Molecular Biology at The University of Texas at Austin with support from CPRIT grant RP110782.
Abbreviations
- AMSH
associated molecule with the SH3 domain of STAM
- ARS
ATP regenerating system
- ATP
adenosine triphosphate
- CP
core particle
- CPK
creatine phosphokinase
- CV
column volume
- DTT
dithiothreitol
- DUB
deubiquitinating enzyme
- E1
Ube1 ubiquitin activating enzyme
- E2
ubiquitin-conjugating enzyme
- E2-25K
Ubiquitin-conjugating Enzyme E2K
- E3
ubiquitin ligase
- EDTA
ethylenediaminetetraacetic acid
- ERAD
endoplasmic reticulum-associated protein degradation
- GFP
circular permutant of superfolder green fluorescent protein
- GST
glutathione S-transferase
- HRV3C
human rhinovirus 3C
- IκBα
nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- K11
lysine 11
- K48
lysine 48
- K63
lysine 63
- PMSF
phenylmethylsulfonyl fluoride
- PY motif
Pro-Pro-X-Tyr
- RP
regulatory particle
- SCF
Skp1-cullin-F-box protein
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEC
size exclusion chromatography
- STAM2
signal transducing adaptor molecule
- UBD
ubiquitin binding domain
- UBE2S
ubiquitin-conjugating enzyme E2S
Footnotes
Supporting Information Available
Materials and methods and results for ubiquitin chain conformation by mass spectrometry. Gel demonstrating that GFP proteins run as two species un-boiled folded and denatured (Figure S1). Amino acid sequences of target proteins to which ubiquitin chains are attached (Table S1). Mass spectrometric conformation of ubiquitin chain linkages (Table S2). Yield of ubiquitin chain synthesis and attachment reactions (Table S3)
References
- 1.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 2.Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. 2009;37:937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 3.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDowell GS, Philpott A. Non-canonical ubiquitylation: mechanisms and consequences. Int J Biochem Cell Biol. 2013;45:1833–1842. doi: 10.1016/j.biocel.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 5.Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim D-Y, Scalf M, Smith LM, Vierstra RD. Advanced proteomic analyses yield a deep catalog of ubiquitylation targets in Arabidopsis. Plant Cell. 2013;25:1523–1540. doi: 10.1105/tpc.112.108613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dammer EB, Na CH, Xu P, Seyfried NT, Duong DM, Cheng D, Gearing M, Rees H, Lah JJ, Levey AI, Rush J, Peng J. Polyubiquitin linkage profiles in three models of proteolytic stress suggest the etiology of Alzheimer disease. J Biol Chem. 2011;286:10457–10465. doi: 10.1074/jbc.M110.149633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 9.Galan JM, Haguenauer-Tsapis R. Ubiquitin lys63 is involved in ubiquitination of a yeast plasma membrane protein. EMBO J. 1997;16:5847–5854. doi: 10.1093/emboj/16.19.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saeki Y, Kudo T, Sone T, Kikuchi Y, Yokosawa H, Toh-E A, Tanaka K. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J. 2009;28:359–371. doi: 10.1038/emboj.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofmann RM, Pickart CM. In vitro assembly and recognition of Lys-63 polyubiquitin chains. J Biol Chem. 2001;276:27936–27943. doi: 10.1074/jbc.M103378200. [DOI] [PubMed] [Google Scholar]
- 12.Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653–665. doi: 10.1016/j.cell.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, King RW, Gygi SP. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006;8:700–710. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 14.Meyer H-J, Rape M. Enhanced protein degradation by branched ubiquitin chains. Cell. 2014;157:910–921. doi: 10.1016/j.cell.2014.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grice GL, Lobb IT, Weekes MP, Gygi SP, Antrobus R, Nathan JA. The Proteasome Distinguishes between Heterotypic and Homotypic Lysine-11-Linked Polyubiquitin Chains. Cell Rep. 2015;12:545–553. doi: 10.1016/j.celrep.2015.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faggiano S, Alfano C, Pastore A. The missing links to link ubiquitin: Methods for the enzymatic production of polyubiquitin chains. Anal Biochem. 2016;492:82–90. doi: 10.1016/j.ab.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Castañeda CA, Liu J, Kashyap TR, Singh RK, Fushman D, Cropp TA. Controlled enzymatic synthesis of natural-linkage, defined-length polyubiquitin chains using lysines with removable protecting groups. Chem Commun. 2011;47:2026–2028. doi: 10.1039/c0cc04868b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemantha HP, Bavikar SN, Herman-Bachinsky Y, Haj-Yahya N, Bondalapati S, Ciechanover A, Brik A. Nonenzymatic polyubiquitination of expressed proteins. J Am Chem Soc. 2014;136:2665–2673. doi: 10.1021/ja412594d. [DOI] [PubMed] [Google Scholar]
- 19.Pickart CM, Raasi S. Controlled synthesis of polyubiquitin chains. Meth Enzymol. 2005;399:21–36. doi: 10.1016/S0076-6879(05)99002-2. [DOI] [PubMed] [Google Scholar]
- 20.Raasi S, Pickart CM. Ubiquitin chain synthesis. Methods Mol Biol. 2005;301:47–55. doi: 10.1385/1-59259-895-1:047. [DOI] [PubMed] [Google Scholar]
- 21.Xu S, Patel P, Abbasian M, Giegel D, Xie W, Mercurio F, Cox S. In vitro SCFbeta-Trcp1-mediated IkappaBalpha ubiquitination assay for high-throughput screen. Meth Enzymol. 2005;399:729–740. doi: 10.1016/S0076-6879(05)99048-4. [DOI] [PubMed] [Google Scholar]
- 22.Saeki Y, Isono E, Toh-E A. Preparation of ubiquitinated substrates by the PY motif-insertion method for monitoring 26S proteasome activity. Meth Enzymol. 2005;399:215–227. doi: 10.1016/S0076-6879(05)99014-9. [DOI] [PubMed] [Google Scholar]
- 23.Dixon EK, Castañeda CA, Kashyap TR, Wang Y, Fushman D. Nonenzymatic assembly of branched polyubiquitin chains for structural and biochemical studies. Bioorg Med Chem. 2013;21:3421–3429. doi: 10.1016/j.bmc.2013.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trang VH, Rodgers ML, Boyle KJ, Hoskins AA, Strieter ER. Chemoenzymatic Synthesis of Bifunctional Polyubiquitin Substrates for Monitoring Ubiquitin Chain Remodeling. ChemBioChem. 2014;15:1563–1568. doi: 10.1002/cbic.201402059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bavikar SN, Spasser L, Haj-Yahya M, Karthikeyan SV, Moyal T, Kumar KSA, Brik A. Chemical synthesis of ubiquitinated peptides with varying lengths and types of ubiquitin chains to explore the activity of deubiquitinases. Angew Chem Int Ed Engl. 2012;51:758–763. doi: 10.1002/anie.201106430. [DOI] [PubMed] [Google Scholar]
- 26.Mcginty RK, Chatterjee C, Muir TW. Semisynthesis of ubiquitylated proteins. Meth Enzymol. 2009;462:225–243. doi: 10.1016/S0076-6879(09)62011-5. [DOI] [PubMed] [Google Scholar]
- 27.Castañeda C, Liu J, Chaturvedi A, Nowicka U, Cropp TA, Fushman D. Nonenzymatic assembly of natural polyubiquitin chains of any linkage composition and isotopic labeling scheme. J Am Chem Soc. 2011;133:17855–17868. doi: 10.1021/ja207220g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 29.Wohlever ML, Nager AR, Baker TA, Sauer RT. Engineering fluorescent protein substrates for the AAA+ Lon protease. Protein Eng Des Sel. 2013;26:299–305. doi: 10.1093/protein/gzs105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol. 2004;11:830–837. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]
- 31.Cannon JR, Martinez-Fonts K, Robotham SA, Matouschek A, Brodbelt JS. Top-down 193-nm ultraviolet photodissociation mass spectrometry for simultaneous determination of polyubiquitin chain length and topology. Anal Chem. 2015;87:1812–1820. doi: 10.1021/ac5038363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho AF, Pinto MP, Grou CP, Vitorino R, Domingues P, Yamao F, Sá-Miranda C, Azevedo JE. High-yield expression in Escherichia coli and purification of mouse ubiquitin-activating enzyme E1. Mol Biotechnol. 2012;51:254–261. doi: 10.1007/s12033-011-9463-x. [DOI] [PubMed] [Google Scholar]
- 33.Bremm A, Freund SMV, Komander D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol. 2010;17:939–947. doi: 10.1038/nsmb.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bremm A, Komander D. Synthesis and analysis of K11-linked ubiquitin chains. Methods Mol Biol. 2012;832:219–228. doi: 10.1007/978-1-61779-474-2_15. [DOI] [PubMed] [Google Scholar]
- 35.Michel MA, Elliott PR, Swatek KN, Simicek M, Pruneda JN, Wagstaff JL, Freund SMV, Komander D. Assembly and specific recognition of k29- and k33-linked polyubiquitin. Mol Cell. 2015;58:95–109. doi: 10.1016/j.molcel.2015.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sone T, Saeki Y, Toh-E A, Yokosawa H. Sem1p is a novel subunit of the 26 S proteasome from Saccharomyces cerevisiae. J Biol Chem. 2004;279:28807–28816. doi: 10.1074/jbc.M403165200. [DOI] [PubMed] [Google Scholar]
- 37.Chen Z, Pickart CM. A 25-kilodalton ubiquitin carrier protein (E2) catalyzes multi-ubiquitin chain synthesis via lysine 48 of ubiquitin. J Biol Chem. 1990;265:21835–21842. [PubMed] [Google Scholar]
- 38.Haldeman MT, Xia G, Kasperek EM, Pickart CM. Structure and function of ubiquitin conjugating enzyme E2-25K: the tail is a core-dependent activity element. Biochemistry. 1997;36:10526–10537. doi: 10.1021/bi970750u. [DOI] [PubMed] [Google Scholar]
- 39.Hofmann RM, Pickart CM. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999;96:645–653. doi: 10.1016/s0092-8674(00)80575-9. [DOI] [PubMed] [Google Scholar]
- 40.Reyes-Turcu FE, Horton JR, Mullally JE, Heroux A, Cheng X, Wilkinson KD. The ubiquitin binding domain ZnF UBP recognizes the C-terminal diglycine motif of unanchored ubiquitin. Cell. 2006;124:1197–1208. doi: 10.1016/j.cell.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 41.Rösner D, Schneider T, Schneider D, Scheffner M, Marx A. Click chemistry for targeted protein ubiquitylation and ubiquitin chain formation. Nat Protoc. 2015;10:1594–1611. doi: 10.1038/nprot.2015.106. [DOI] [PubMed] [Google Scholar]
- 42.Kristariyanto YA, Choi SY, Rehman SAA, Ritorto MS, Campbell DG, Morrice NA, Toth R, Kulathu Y. Assembly and structure of Lys33-linked polyubiquitin reveals distinct conformations. Biochem J. 2015;467:345–352. doi: 10.1042/BJ20141502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.