

Abstract
The contribution of epithelial-to-mesenchymal transitions (EMT) in both developmental and pathological conditions has been widely recognized and studied. In a parallel process, governed by a similar set of signaling and transcription factors, endothelial-to-mesenchymal transitions (EndoMT) contribute to heart valve formation and the generation of cancer-associated-fibroblasts. During angiogenic sprouting endothelial cells express many of the same genes and break down basement membrane, however they retain intercellular junctions and migrate as a connected “train” of cells rather than as individual cells. This has been termed a partial EndoMT. A key regulatory check-point determines whether cells undergo a full or a partial EMT/EndoMT, however, very little is known about how this switch is controlled. Here we discuss these developmental/pathologic pathways, with a particular focus on their role in vascular biology.
Keywords: EMT, EndoMT, Endothelial, angiogenesis, transcription factor
Morphological changes in tissues are invariably associated with phenotypical changes in the cells that comprise them. Often these are limited to temporary changes in protein expression patterns, but more dramatic changes can also occur, during which cells undergo changes in transcriptional programs that lead to significant changes in morphology and function. One class of such changes is called the epithelial-to mesenchymal transition (EMT), and variants of traditional EMT include endothelial-to-mesenchymal transition (EndoMT) as well as partial EMT/EndoMT. Our focus will be to highlight the distinctions among the subsets, with an emphasis on angiogenesis as a unique example of a partial EndoMT.
Endothelial-to-mesenchymal transitions
Endothelial cells (EC) have many epithelial characteristics, including strong apical-basal polarity, the ability to form tubes, and the potential to undergo a transition to a mesenchymal-like cell (EndoMT). There are many reasons, therefore, to believe that this process is related to epithelial-mesenchymal transitions and may thus share some of the same pathways and effectors, including the key transcription factors Snail, Slug, Twist, Zeb1 and Zeb2, which we describe in detail below. There have been several excellent reviews published on EMT1–4 and so we will focus on EndoMT, with reference to EMT where clear overlaps exist. During embryogenesis subsets of EC in the developing heart undergo EndoMT, acquire mesenchymal markers, invade the surrounding tissue and form the valves and septa of the adult heart5, a process that involves transforming growth factor-β (TGFβ), bone morphogenetic protein (BMP) and Notch signaling pathways6, 7. These pathways converge on a complex network of transcription factors that includes HES, HEY1/2, Twist and SOX98, 9. Pathologically, EndoMT can be reactivated in the adult heart, and has been shown to contribute to cardiac fibrosis, a characteristic common to most forms of heart failure. Using lineage-tracing techniques, Kalluri’s group demonstrated that 27 to 35% of fibroblasts present in fibrotic heart tissue were of EC origin, strongly suggesting a role for EndoMT in this process. Importantly, EndoMT was TGFβ1-dependent, whereas BMP-7 preserved the EC phenotype and consequently reduced fibrosis10. Interestingly, however, a more recent study suggests that the accumulation of cardiac fibroblasts is not due to an EndoMT, but rather, the cells derive from a previously unrecognized fibroblast population, itself derived from endothelial cells during development11. EndoMT has also been implicated as a source of fibroblasts in hypertrophic cardiomyopathy12, diabetes-induced cardiac fibrosis13, and chronic pulmonary hypertension14, 15, although these studies lacked definitive lineage-tracing analyses.
There is also evidence supporting a role for EndoMT during both acute and chronic kidney injury16. In three distinct mouse models of chronic kidney disease approximately 30 to 50% of fibroblasts co-expressed the EC marker CD31 along with markers of myofibroblasts and fibroblasts, including fibroblast specific protein-1 (FSP-1) and alpha-smooth muscle actin (α-SMA). Lineage tracing experiments confirmed the EC origin of these cells16. More recent work has suggested that only about 10% of the myofibroblasts present in kidney fibrosis derive from an EndoMT, while the remainder come from proliferation of local fibroblasts and differentiation from bone marrow cells17. Other studies, however, have suggested that these fibroblasts may be derived from pericytes18. It should be borne in mind however, that these are all mouse studies and strain-specific differences are always a possibility. Other fibrotic diseases where EndoMT has been implicated as a source of fibroblasts/stromal cells include intestinal fibrosis19 and Scleroderma20, 21.
In aggregate, these studies provide evidence that EndoMT likely provides a source of fibroblasts in both damaged heart and kidney (although the extent is unclear) and may function to facilitate tissue remodeling and fibrosis.
Finally, EndoMT also has a significant role to play in cancer. For example, Zeisberg and colleagues, using two different mouse models of cancer, demonstrated that EndoMT accounts for up to 40% of cancer associated fibroblasts (CAFs)22. A distinct population of fibroblasts co-expressed the EC marker CD31 along with either FSP-1 or α-SMA. Use of transgenic mice with irreversibly tagged EC revealed strikingly similar results – unique populations of fibroblasts co-expressing endothelial and mesenchymal markers. These data suggest that EndoMT is a significant source of CAFs in tumors. Remarkably, it has also been demonstrated that Twist over-expression in head and neck cancer cells can drive them into an endothelial cell phenotype23.
Angiogenesis: a partial EndoMT
When epithelial cells commit to a mesenchymal phenotype, the event is designated as a complete EMT. Partial EMT is also possible, and this occurs when one or more of the key characteristics of complete-EMT are not exhibited, such as loss of cell-cell contact. For example, during re-epithelialization of cutaneous wounds, keratinocytes undergo a series of changes reminiscent of EMT including loss of polarity, rearrangement of the actin cytoskeleton, alterations in cell-cell contacts, and breakdown of basement membrane (BM); however, these cells retain some intercellular junctions and migrate as a cohesive cell sheet24. Similarly, during Madin-Darby canine kidney (MDCK) cell tubulogenesis chains of epithelial cells migrate while again retaining intercellular junctions – a partial EMT driven by slug activity25. Angiogenesis, the formation of new blood vessels from the pre-existing vasculature, is essential during development and many normal physiological processes, but is also important in numerous pathological processes, including tumor growth. Interestingly, comparison of angiogenesis and EMT reveals several similarities. Among these, the tip cells that lead emerging sprouts lack apical-basal polarity, degrade both BM and extracellular matrix (ECM) and, by definition, are migratory. However, angiogenic EC do not usually separate from their neighbors, suggesting that angiogenesis may involve a partial EndoMT26, 27.
Although much is known about the growth factors, receptors and signaling pathways that govern angiogenesis, there is still much to learn about transcriptional changes that regulate each phase of angiogenesis, including sprouting. Our lab has recently published preliminary evidence demonstrating that the transcription factors Snail (SNAI1 in human, Snai1 in mouse) and Slug (SNAI2/Snai2) are indeed expressed and regulated by angiogenic EC during in vitro angiogenesis26. We demonstrated that inhibition of Snail or Slug expression results in a reduced ability of angiogenic EC to invade and migrate through multiple ECM environments. Importantly, lentiviral-mediated re-expression of membrane type-1 matrix metalloproteinase (MT1-MMP) rescued the inability of EC lacking Slug to migrate. This finding therefore suggests that MT1-MMP is a critical downstream target of Slug during angiogenesis. Importantly, we and others have observed increased expression of Snail and Slug in the vasculature of colon, breast28 and ovarian carcinoma29. It is interesting to speculate that the same factors that drive epithelial cells toward a mesenchymal, pro-metastatic phenotype may also drive EC toward a pro-angiogenic phenotype, which is also associated with metastasis. Key factors here may include vascular endothelial growth factor (VEGF), TGFβ, BMPs, hepatocyte growth factor (HGF) and Wnts. The “permanently activated” phenotype of tumor vasculature may well reflect the chronic activation of the EndoMT process, driven by persistently elevated VEGF and a hypoxic environment, leading to excessive sprouting and a failure to settle back into the mature, stable phenotype associated with non-tumor EC. It is possible therefore that drugs that target EMT/EndoMT, potentially through these pathways, may be doubly effective in slowing metastatic spread of epithelial tumors. Finally, we have preliminary data suggesting that Slug deficiency in mice leads both to impaired developmental and pathological angiogenesis (KMWR, NW and CCWH, unpublished data). In aggregate, these data clearly point to a role for the Snail family of transcription factors during angiogenesis. Furthermore, the findings that cell-cell contact is retained and expression of vascular endothelial-cadherin (VE-cadherin) is not reduced, are reminiscent of the partial EMT seen during keratinocyte migration in wound closure and during mammary gland or kidney epithelial cell tubule formation30. We therefore believe that angiogenic sprouting may represent a partial EndoMT.
Signaling pathways governing EndoMT
EndoMT and EMT share many of the same regulators, with members of the TGFβ superfamily being arguably the most prominent players. TGFβ signaling through Smad-dependent and independent pathways leads to direct transcriptional regulation of multiple genes, including several EMT/EndoMT-inducing transcription factors31. Expression of these transcription factors subsequently drives loss of cell-cell adhesion by repression of epithelial/endothelial genes encoding junction proteins, regulation of cytoskeletal rearrangement, and increased expression and activity of both MT-MMPs and secreted MMPs32. Moreover, during EndoMT, upregulation of EC Slug by TGFβ and other growth factors results in increased migration and invasion into extracellular matrices of diverse composition, and this is due in part to the indirect activation of MT1-MMP, MMP-2 and MMP-926. Interestingly, nuclear Smads form multi-protein complexes with EMT/EndoMT-transcription factors, including Snail, Zeb1 and Zeb2, resulting in suppression or activation of promoters of epithelial (E-cadherin, Occludin, ZO-1) or mesenchymal (Vimentin, N-cadherin) genes, respectively4. TGFβ can also activate Smad-independent pathways such as MAPK/ERK/JNK, all of which are implicated in EndoMT31, 33, 34 Finally, a recent study has shown a requirement for PKCδ and c-Abl in mediating TGFβ-induced EndoMT in mouse pulmonary EC35.
Aside from TGFβ, several other signaling pathways associated with EMT have also been reported to regulate EndoMT. The relationship between canonical Wnt signaling and the onset of EMT and metastasis is well established in many cancer models. In human prostate cancer, the expression and nuclear activity of β-catenin correlates with the level of hypoxia-induced factor 1 alpha (HIF-1α), and HIF-1α-induced EMT36. The degree of hypoxia-induced EMT can also be enhanced by Wnt3a-induced activation of β-catenin in hepatic carcinoma37. Furthermore, it has been demonstrated that canonical Wnt signaling stabilizes Slug expression through regulating glycogen synthase kinase 3-β (GSK3-β) phosphorylation and βTrcp-1-mediated ubiquitination, thereby inducing EMT in triple-negative breast cancer38. In contrast, the understanding of canonical Wnt signaling in EndoMT was mostly limited to developmental processes until recently. In an experimentally induced myocardial infarction model, Aisagbonhi et. al., using lineage-tracing experiments, demonstrated that the canonical Wnt pathway is transiently activated in endothelial cells, and this in turn leads to EndoMT39. More recently, the effect of Wnt7a and its antagonist Dkk-1 on EndoMT during arteriosclerosis was explored. In contrast to its effect on myofibroblasts, the activation of Wnt signaling through Wnt7b expression preserves the phenotype of endothelial cells, while the expression of Dkk-1 promotes EndoMT40.
Fibroblast growth factor (FGF) has been proposed as a gatekeeper of partial EndoMTs through its regulation of the let-7 miRNA, which normally acts to suppress TGFβ-induced EndoMT41. When FGF signaling is reduced in a murine model of transplant arteriopathy, in this case by inflammatory signals that down-regulate the FGF receptor (FGFR), let-7-mediated suppression of TGFβ signaling is relieved and EC undergo an EndoMT leading to intimal fibrosis. Given that the sprouty genes, which regulate FGFR signaling, have previously been implicated in the regulation of EC sprouting42, it is tempting to speculate that they may be acting as a rheostat to fine-tune FGF signaling43 and thereby control whether EC undergo a partial or full EndoMT in response to TGFβ signaling.
Notch activation is a well-known regulator of angiogenesis44–47; and is linked to both EMT and EndoMT events. The cleavage and nuclear translocation of the Notch intracellular domain (NICD) can induce transcriptional alterations and hence a series of morphological and functional changes related to a mesenchymal transition48. Notch can suppress (or activate) gene expression directly or through upregulation of Snail and Slug in both epithelial cells and EC, and thus initiate EMT and EndoMT in both developmental and pathological conditions49–51. Notch ligands can also be induced by TGFβ signaling to activate Notch receptors52 and enhance EMT synergistically53. Blockage of either Jagged-1, or its downstream signaling target Hey-1, can attenuate TGFβ-induced EMT in mammary gland, kidney tubule and epidermal epithelial cells50, 54. Notch and VEGF are both induced in the hypoxic tumor environment and they work together to drive metastasis. On the one hand, interaction of Notch and HIF pathways leads to increased “stemness” of cancer cells, self-renewal ability and a complete EMT50, 55. On the other hand, hypoxia-dependent induction of VEGF expression augments tumor angiogenesis, which provides increased opportunities for tumor cell intravasation. Finally, the crosstalk between Notch and VEGF pathways in the context of hypoxic tumors also promotes partial EndoMT in angiogenic tumor EC leading to the formation of unstable, leaky vessels52. Altered vessel integrity and permeability correlates with enhanced tumor cell dissemination to distant sites56.
Notch-mediated EMT/EndoMT is unusual, and somewhat paradoxical, as it is contact-dependent. Importantly, the ability of cells to retain intercellular adhesion complexes while migrating as a group is crucial to tubulogenesis. As described above, processes involving tubulogenesis, such as angiogenesis and kidney tubule formation, both require a partial EMT/EndoMT, during which the participating cells temporarily lose apical/basal polarity and gain migratory capacity, but never fully acquire all mesenchymal phenotypes, nor completely lose cellular adhesion. While other signaling pathways such as TGFβ, HGF and FGF are capable of promoting this process, it is intriguing to speculate that Notch activation, perhaps in conjunction with sprouty, is a crucial determinant of a partial versus full EMT/EndoMT.
Aside from the major signaling pathways discussed above, miRNA, epigenetic regulation and histone modification have also recently emerged as regulators of EMT30, 32 and may also have a role in EndoMT41. These alterations control the expression level of the Snail/Slug, ZEB, and Twist families of transcription factors, and these in turn feed back to affect the expression and/or activity of the miRNA, or histone modifying enzymes30, 32. Clearly, the relationship(s) between the master regulators governing EMT/EndoMT are extremely complex4, 31.
Transcription factor interactions governing EndoMT
Snail (SNAI1 in human, Snai1 in mouse), Slug (SNAI2/Snai2), Zeb1 (ZEB1/Zeb1), Zeb2 (ZEB2/Zeb2) and Twist (TWIST1/Twist1) have been identified as the key transcriptional regulators of EMTs and EndoMTs. A shared function of these proteins is their ability to repress the transcription of epithelial-cadherin (E-cadherin), however, numerous studies have demonstrated that they have overlapping but non-redundant roles in EMT and tumor progression. In human carcinomas it is generally accepted that Snail plays a major role in inducing EMT, while Zeb1/2 and Twist are mainly involved in maintaining the invasive mesenchymal phenotype32. However, our recent study on EndoMT suggests that at least in the case of sprouting angiogenesis, Slug is the primary initiator of this process while the induction of Snail occurs at a much later time26. It is therefore unclear if each of these transcription factors has a distinct and specific role during EMT/EndoMT or if they rather act in symphony to promote a mesenchymal phenotype. Accumulating evidence from studies observing their expression patterns and their ability to regulate each other has begun to reveal a non-linear map that suggests these transcription factors mostly act in concert. For example, Snail can upregulate Zeb1 and Zeb2 in oral squamous carcinoma and, at the same time, negatively regulate its own expression through direct promoter binding57, 58. Moreover, Slug indirectly upregulates Snail through epithelial growth factor (EGF) and/or HGF signaling, thereby promoting mammary gland branching morphogenesis59. Slug can also activate Zeb1 and its own expression through direct transcriptional regulation60, 61. In addition, many have shown that Twist1 can regulate the expression level of Snail and Slug by either directly influencing transcription62, 63 or through post-translational regulation via the NF-κB/GSK-3β axis64.
Dynamic functions of EndoMT transcriptional regulators
The master regulators of EMT mediate repression of E-cadherin expression and this is often described as the hallmark of EMT. However, several recent studies show that in both in vitro and in vivo models, EMT master regulators can induce EMT/EndoMT-like phenotypes in cells without complete loss of membrane E-cadherin – a partial EMT. Similarly, we observed that overexpression of Slug in EC promotes EC sprouting, a process suggestive of a partial EndoMT, without altering the mRNA levels or surface expression of VE-cadherin, the EC equivalent of E-cadherin26. Interestingly, Leroy et al. and others have shown that Slug upregulation prevents apoptosis and promotes cell proliferation through p5365, two processes associated with angiogenic sprouting. The induction of Twist alone, perhaps surprisingly, is sufficient to induce single cell dissemination/local invasion without the loss of epithelial identity, and moreover, E-cadherin expression is required for this process66. Conversely, the deletion of E-cadherin alone is not sufficient to induce EMT. Indeed, in the absence of E-cadherin, and despite a reduction in multiple classes of junction proteins, epithelial cells are still able to invade an extracellular matrix as a chain rather than single cells66. Collectively these data suggest that master regulators of EMT and EndoMT serve more functions than simply acting as repressors of the epithelial/endothelial phenotype.
Remaining questions and perspectives
Our knowledge of the mechanisms underlying EMT and EndoMT is rapidly advancing, however, there are still a number of critical questions that have to be answered, including:
Are Snail, Slug, Twist, Zeb1 and Zeb2 all required for an EndoMT?
Do these genes work sequentially, in parallel and/or in feedback loops?
What regulates expression of EndoMT-promoting transcription factors?
What are the target genes for EndoMT transcription factors?
What controls whether cells undergo a full or partial EndoMT?
How is VE-cadherin “protected” from down-regulation in partial EndoMT?
Are there fundamental differences between EMT and EndoMT or are the basic mechanisms identical?
Do the same factors that promote EMT in cancer promote EndoMT in angiogenic EC?
How does EndoMT contribute to progression of diseases such as cancer, arteriosclerosis and fibrosis?
Are there fundamental differences between pathological EndoMT and developmental EndoMT?
Can EndoMT be targeted therapeutically in cancer and other diseases involving pathologic angiogenesis?
Answers to these questions have the potential to fundamentally affect how we target pathologic angiogenesis.
Figure 1.
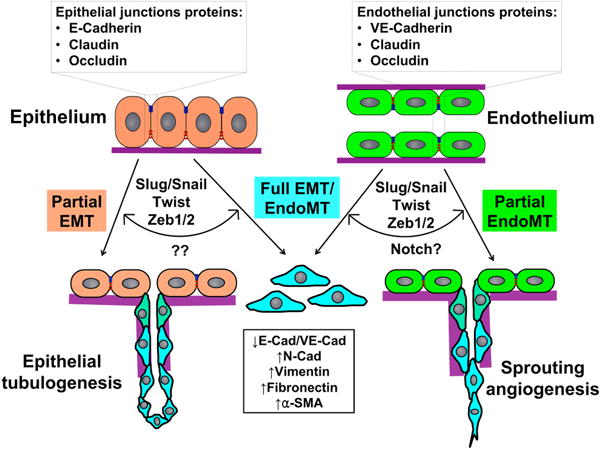
Complete vs. partial EMT/EndoMT. Epithelial and endothelial cells comprise the quiescent epithelium and endothelium respectively and utilize junctional proteins to maintain connections. Once transcriptional reprogramming is initiated, an event led by the EMT/EndoMT-transcription factors Slug, Snail, Twist and Zeb1/2, the epithelial/endothelial cells lose apical-basal polarity, sever intercellular junctions and become motile cells. However, the regulatory signal(s) that determine whether these cells undergo a complete EMT/EndoMT or partial EMT/EndoMT remains unclear. In the case of sprouting angiogenesis, the contact-dependent Notch signaling pathway may have a major role to play in this process.
Significance.
As a single cell multiplies and differentiates to generate a fully-developed multi-cellular organism daughter cells often undergo phenotypic changes that can be either permanent or temporary. One such change is termed an epithelial-to-mesenchymal transition (EMT) and this has been widely studied in both developmental and pathological conditions. It contributes to gastrulation and neural crest formation during development, and metastasis of epithelial tumors is also thought to involve an EMT. In a somewhat similar process, governed by an overlapping set of signaling and transcription factors, endothelial-to-mesenchymal transitions (EndoMT) contribute to heart valve formation, the generation of cancer-associated-fibroblasts, and the activated endothelial cells that drive angiogenic sprouting. A key regulatory check-point determines whether cells undergo a full EndoMT (heart valve development) or a partial EndoMT (angiogenesis), however very little is known about how this switch is controlled. Here we discuss these developmental/pathologic pathways, with a particular focus on their role in vascular biology.
Acknowledgments
We thank members of the Hughes lab for helpful discussions
Sources of Funding
This work was supported by US National Institutes of Health grant RO1HL60067. K.M.W-R. was supported by a pre-doctoral award from the American Heart Association. C.C.W.H. receives support from the Chao Family Comprehensive Cancer Center (CFCCC) through an NCI Center Grant award P30A062203.
Glossary
NONSTANDARD ABBREVIATIONS AND ACRONYMS
- BM
basement membrane
- BMP
bone morphogenetic protein
- CAF
cancer associated fibroblasts
- E-cadherin
epithelial cadherin
- EC
endothelial cells
- EGF
epidermal growth factor
- EMT
epithelial-to-mesenchymal transition
- EndoMT
endothelial-to-mesenchymal transition
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- FSP-1
fibroblast specific protein-1
- GSK3-β
glycogen synthase kinase 3-β
- HGF
hepatocyte growth factor
- HIF-1α
hypoxia-induced factor 1 alpha
- MMP
matrix metalloproteinase
- MT1-MMP
membrane type-1 matrix metalloproteinase
- N-cadherin
neural cadherin
- NICD
notch intracellular domain
- α-SMA
α-smooth muscle actin
- TGFβ
transforming growth factor β
- VE-cadherin
vascular endothelial - cadherin
- VEGF
vascular endothelial growth factor
Footnotes
Disclosure
The authors declare no conflicts of interest
References
- 1.Kalluri R, Weinberg RAs. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiery JP, Acloque H, Huang RY, Nieto MAs. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Tsai JH, Yang Js. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes & development. 2013;27:2192–2206. doi: 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamouille S, Xu J, Derynck Rs. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews. Molecular cell biology. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armstrong EJ, Bischoff Js. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakajima Y, Yamagishi T, Hokari S, Nakamura Hs. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP) Anat Rec. 2000;258:119–127. doi: 10.1002/(SICI)1097-0185(20000201)258:2<119::AID-AR1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 7.Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JLs. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes & development. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer A, Gessler Ms. Delta-Notch–and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic acids research. 2007;35:4583–4596. doi: 10.1093/nar/gkm477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wirrig EE, Yutzey KEs. Conserved transcriptional regulatory mechanisms in aortic valve development and disease. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:737–741. doi: 10.1161/ATVBAHA.113.302071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri Rs. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 11.Moore-Morris T, Guimaraes-Camboa N, Banerjee Is, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. The Journal of clinical investigation. 2014;124:2921–2934. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teekakirikul P, Eminaga S, Toka Os, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. The Journal of clinical investigation. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, Yagi K, Miyagawa K, Rikitake Y, Suzuki T, Kisanuki YY, Yanagisawa M, Hirata Ks. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121:2407–2418. doi: 10.1161/CIRCULATIONAHA.110.938217. [DOI] [PubMed] [Google Scholar]
- 14.Arciniegas E, Frid MG, Douglas IS, Stenmark KRs. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1–8. doi: 10.1152/ajplung.00378.2006. [DOI] [PubMed] [Google Scholar]
- 15.Zhu P, Huang L, Ge X, Yan F, Wu R, Ao Qs. Transdifferentiation of pulmonary arteriolar endothelial cells into smooth muscle-like cells regulated by myocardin involved in hypoxia-induced pulmonary vascular remodelling. Int J Exp Pathol. 2006;87:463–474. doi: 10.1111/j.1365-2613.2006.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri Rs. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri Rs. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schrimpf C, Xin C, Campanholle G, Gill SE, Stallcup W, Lin SL, Davis GE, Gharib SA, Humphreys BD, Duffield JSs. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J Am Soc Nephrol. 2012;23:868–883. doi: 10.1681/ASN.2011080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM, Gopalan B, Stylianou E, Fiocchi Cs. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. The American journal of pathology. 2011;179:2660–2673. doi: 10.1016/j.ajpath.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jimenez SAs. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN rheumatology. 2013;2013:835948. doi: 10.1155/2013/835948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leach HG, Chrobak I, Han R, Trojanowska Ms. Endothelial cells recruit macrophages and contribute to a fibrotic milieu in bleomycin lung injury. American journal of respiratory cell and molecular biology. 2013;49:1093–1101. doi: 10.1165/rcmb.2013-0152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri Rs. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 23.Chen HF, Huang CH, Liu CJ, Hung JJ, Hsu CC, Teng SC, Wu KJs. Twist1 induces endothelial differentiation of tumour cells through the Jagged1-KLF4 axis. Nature communications. 2014;5:4697. doi: 10.1038/ncomms5697. [DOI] [PubMed] [Google Scholar]
- 24.Savagner P, Kusewitt DF, Carver EA, Magnino F, Choi C, Gridley T, Hudson LGs. Developmental transcription factor slug is required for effective re-epithelialization by adult keratinocytes. J Cell Physiol. 2005;202:858–866. doi: 10.1002/jcp.20188. [DOI] [PubMed] [Google Scholar]
- 25.Leroy P, Mostov KEs. Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. Mol Biol Cell. 2007;18:1943–1952. doi: 10.1091/mbc.E06-09-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welch-Reardon KM, Ehsan SM, Wang K, Wu N, Newman AC, Romero-Lopez M, Fong AH, George SC, Edwards RA, Hughes CCs. Angiogenic sprouting is regulated by endothelial cell expression of Slug. J Cell Sci. 2014;127:2017–2028. doi: 10.1242/jcs.143420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Potenta S, Zeisberg E, Kalluri Rs. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parker BS, Argani P, Cook BP, Liangfeng H, Chartrand SD, Zhang M, Saha S, Bardelli A, Jiang Y, St Martin TB, Nacht M, Teicher BA, Klinger KW, Sukumar S, Madden SLs. Alterations in vascular gene expression in invasive breast carcinoma. Cancer Res. 2004;64:7857–7866. doi: 10.1158/0008-5472.CAN-04-1976. [DOI] [PubMed] [Google Scholar]
- 29.Lu C, Bonome T, Li Y, Kamat AA, Han LY, Schmandt R, Coleman RL, Gershenson DM, Jaffe RB, Birrer MJ, Sood AKs. Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res. 2007;67:1757–1768. doi: 10.1158/0008-5472.CAN-06-3700. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Weinberg RAs. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Heldin CH, Vanlandewijck M, Moustakas As. Regulation of EMT by TGFbeta in cancer. FEBS letters. 2012;586:1959–1970. doi: 10.1016/j.febslet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 32.Peinado H, Olmeda D, Cano As. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature reviews. Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 33.Medici D, Potenta S, Kalluri Rs. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. The Biochemical journal. 2011;437:515–520. doi: 10.1042/BJ20101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Meeteren LA, ten Dijke Ps. Regulation of endothelial cell plasticity by TGF-beta. Cell and tissue research. 2012;347:177–186. doi: 10.1007/s00441-011-1222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z, Jimenez SAs. Protein kinase Cdelta and c-Abl kinase are required for transforming growth factor beta induction of endothelial-mesenchymal transition in vitro. Arthritis and rheumatism. 2011;63:2473–2483. doi: 10.1002/art.30317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang YG, Luo Y, He DL, Li X, Zhang LL, Peng T, Li MC, Lin YHs. Role of Wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. International journal of urology: official journal of the Japanese Urological Association. 2007;14:1034–1039. doi: 10.1111/j.1442-2042.2007.01866.x. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q, Bai X, Chen W, Ma T, Hu Q, Liang C, Xie S, Chen C, Hu L, Xu S, Liang Ts. Wnt/beta-catenin signaling enhances hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1alpha signaling. Carcinogenesis. 2013;34:962–973. doi: 10.1093/carcin/bgt027. [DOI] [PubMed] [Google Scholar]
- 38.Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG, Weiss SJs. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16654–16659. doi: 10.1073/pnas.1205822109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AKs. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Disease models & mechanisms. 2011;4:469–483. doi: 10.1242/dmm.006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng SL, Shao JS, Behrmann A, Krchma K, Towler DAs. Dkk1 and MSX2-Wnt7b signaling reciprocally regulate the endothelial-mesenchymal transition in aortic endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1679–1689. doi: 10.1161/ATVBAHA.113.300647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, Ali R, Medina PP, Yu J, Slack FJ, Anderson DG, Kotelianski V, Wang F, Tellides G, Simons Ms. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell reports. 2012;2:1684–1696. doi: 10.1016/j.celrep.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee SH, Schloss DJ, Jarvis L, Krasnow MA, Swain JLs. Inhibition of angiogenesis by a mouse sprouty protein. The Journal of biological chemistry. 2001;276:4128–4133. doi: 10.1074/jbc.M006922200. [DOI] [PubMed] [Google Scholar]
- 43.Impagnatiello MA, Weitzer S, Gannon G, Compagni A, Cotten M, Christofori Gs. Mammalian sprouty-1 and -2 are membrane-anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells. The Journal of cell biology. 2001;152:1087–1098. doi: 10.1083/jcb.152.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor KL, Henderson AM, Hughes CCs. Notch activation during endothelial cell network formation in vitro targets the basic HLH transcription factor HESR-1 and downregulates VEGFR-2/KDR expression. Microvascular research. 2002;64:372–383. doi: 10.1006/mvre.2002.2443. [DOI] [PubMed] [Google Scholar]
- 45.Sainson RC, Aoto J, Nakatsu MN, Holderfield M, Conn E, Koller E, Hughes CCs. Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1027–1029. doi: 10.1096/fj.04-3172fje. [DOI] [PubMed] [Google Scholar]
- 46.Siekmann AF, Lawson NDs. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature. 2007;445:781–784. doi: 10.1038/nature05577. [DOI] [PubMed] [Google Scholar]
- 47.Hellstrom M, Phng LK, Hofmann JJs, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–780. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 48.Espinoza I, Miele Ls. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer letters. 2013;341:41–45. doi: 10.1016/j.canlet.2013.08.027. [DOI] [PubMed] [Google Scholar]
- 49.Becker KF, Rosivatz E, Blechschmidt K, Kremmer E, Sarbia M, Hofler Hs. Analysis of the E-cadherin repressor Snail in primary human cancers. Cells, tissues, organs. 2007;185:204–212. doi: 10.1159/000101321. [DOI] [PubMed] [Google Scholar]
- 50.Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl Us. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan As. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. The Journal of cell biology. 2008;182:315–325. doi: 10.1083/jcb.200710067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holderfield MT, Hughes CCs. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ Res. 2008;102:637–652. doi: 10.1161/CIRCRESAHA.107.167171. [DOI] [PubMed] [Google Scholar]
- 53.Niimi H, Pardali K, Vanlandewijck M, Heldin CH, Moustakas As. Notch signaling is necessary for epithelial growth arrest by TGF-beta. The Journal of cell biology. 2007;176:695–707. doi: 10.1083/jcb.200612129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EPs. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. The EMBO journal. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson Ms. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 56.Sullivan R, Graham CHs. Hypoxia-driven selection of the metastatic phenotype. Cancer metastasis reviews. 2007;26:319–331. doi: 10.1007/s10555-007-9062-2. [DOI] [PubMed] [Google Scholar]
- 57.Takkunen M, Grenman R, Hukkanen M, Korhonen M, Garcia de Herreros A, Virtanen Is. Snail-dependent and -independent epithelial-mesenchymal transition in oral squamous carcinoma cells. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2006;54:1263–1275. doi: 10.1369/jhc.6A6958.2006. [DOI] [PubMed] [Google Scholar]
- 58.Peiro S, Escriva M, Puig I, Barbera MJ, Dave N, Herranz N, Larriba MJ, Takkunen M, Franci C, Munoz A, Virtanen I, Baulida J, Garcia de Herreros As. Snail1 transcriptional repressor binds to its own promoter and controls its expression. Nucleic acids research. 2006;34:2077–2084. doi: 10.1093/nar/gkl141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee K, Gjorevski N, Boghaert E, Radisky DC, Nelson CMs. Snail1, Snail2, and E47 promote mammary epithelial branching morphogenesis. The EMBO journal. 2011;30:2662–2674. doi: 10.1038/emboj.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wels C, Joshi S, Koefinger P, Bergler H, Schaider Hs. Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. The Journal of investigative dermatology. 2011;131:1877–1885. doi: 10.1038/jid.2011.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sakai D, Suzuki T, Osumi N, Wakamatsu Ys. Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development. 2006;133:1323–1333. doi: 10.1242/dev.02297. [DOI] [PubMed] [Google Scholar]
- 62.Yu W, Zhang Y, Ruest LB, Svoboda KKs. Analysis of Snail1 function and regulation by Twist1 in palatal fusion. Frontiers in physiology. 2013;4:12. doi: 10.3389/fphys.2013.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang Js. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71:245–254. doi: 10.1158/0008-5472.CAN-10-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lander R, Nasr T, Ochoa SD, Nordin K, Prasad MS, Labonne Cs. Interactions between Twist and other core epithelial-mesenchymal transition factors are controlled by GSK3-mediated phosphorylation. Nature communications. 2013;4:1542. doi: 10.1038/ncomms2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leroy P, Mostov KEs. Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. Molecular biology of the cell. 2007;18:1943–1952. doi: 10.1091/mbc.E06-09-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shamir ER, Pappalardo E, Jorgens DM, Coutinho K, Tsai WT, Aziz K, Auer M, Tran PT, Bader JS, Ewald AJs. Twist1-induced dissemination preserves epithelial identity and requires E-cadherin. The Journal of cell biology. 2014;204:839–856. doi: 10.1083/jcb.201306088. [DOI] [PMC free article] [PubMed] [Google Scholar]