

Abstract
Alzheimer's disease (AD) is a neurodegenerative brain disorder associated with the loss of synapses between neurons in the brain. Synaptic cell adhesion molecules are cell surface glycoproteins which are expressed at the synaptic plasma membranes of neurons. These proteins play key roles in formation and maintenance of synapses and regulation of synaptic plasticity. Genetic studies and biochemical analysis of the human brain tissue, cerebrospinal fluid, and sera from AD patients indicate that levels and function of synaptic cell adhesion molecules are affected in AD. Synaptic cell adhesion molecules interact with Aβ, a peptide accumulating in AD brains, which affects their expression and synaptic localization. Synaptic cell adhesion molecules also regulate the production of Aβ via interaction with the key enzymes involved in Aβ formation. Aβ-dependent changes in synaptic adhesion affect the function and integrity of synapses suggesting that alterations in synaptic adhesion play key roles in the disruption of neuronal networks in AD.
1. Synaptic Cell Adhesion Molecules
Cell adhesion molecules (CAMs) are cell surface glycoproteins located at the cell surface plasma membrane of neurons and other cells. CAMs have a large extracellular domain and are either transmembrane proteins or attached to the plasma membrane via a glycosylphosphatidylinositol (GPI) anchor. The extracellular domains of CAMs mediate cell adhesion by either forming homophilic adhesion bonds via binding to the same molecules on cell surface membranes of adjacent cells or interacting heterophilically with other proteins on the cell surface membranes of adjacent cells or in the extracellular matrix [1].
CAMs accumulating at synapses between neurons are often called synaptic CAMs and represent members of the major families of cell adhesion molecules, including immunoglobulin superfamily (IgSF) CAMs, cadherins, integrins, neuroligins, and neurexins, and also other cell surface proteins, which mediate cell adhesion, such as cellular prion protein (PrPc) and amyloid precursor protein (APP) (Figure 1).
Figure 1.

Schematic diagram illustrating examples of synaptic CAMs in glutamatergic synapses. Synaptic CAMs accumulate in synaptic membranes where they form homophilic (e.g., NCAM-NCAM, SynCAM-SynCAM, and cadherin-cadherin) or heterophilic (e.g., L1-NCAM, neuroligin-neurexin, and LRRTM3-neurexin) adhesion bonds, which are important for stabilization of the interactions between synaptic membranes. Presynaptically, CAMs are involved in regulation of synaptic vesicle recycling (blue arrows), which is mediated by coat proteins (black dashed lines) assembled on synaptic membranes to reform synaptic vesicles after exocytosis and neurotransmitter release. Postsynaptically, intracellular domains of synaptic CAMs interact with scaffolding and adaptor proteins (examples of interactions are shown with black arrows), such as spectrin, postsynaptic density protein 95 (PSD95), proteins 4.1B and 4.1N, or catenin, which link synaptic CAMs to postsynaptic glutamate receptors, the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) and N-methyl-D-aspartic acid receptors (NMDAR). Interactions with synaptic CAMs promote the recruitment of scaffolding proteins and neurotransmitter receptors to synapses and are involved in the activity-dependent remodeling of synapses. NCAM: neural cell adhesion molecule, SynCAM: synaptic cell adhesion molecule, PrP: cellular prion protein, and LRRTM3: Leucine-rich-repeat- (LRR-) containing transmembrane protein 3.
Synaptic CAMs perform numerous functions at synapses (Figure 1). In developing neurons, CAMs promote mechanical stabilization of the contacts between axons and dendrites of neurons [2] and formation of synapses [3]. Synaptic CAMs also play key roles in the establishment of neurotransmission by recruiting other synaptic components, such as synaptic scaffolding proteins, which interact with the intracellular domains of synaptic CAMs, and associated neurotransmitter receptors (Figure 1), and by inducing the maturation of the neurotransmitter release machinery [4]. In mature neurons, CAMs play a role in the stabilization of the synapse ultrastructure [5–7], regulation of the neurotransmitter release [8, 9], and synaptic remodeling and plasticity [10–13]. The multiple roles of synaptic CAMs in regulation of synapse formation and function have been described in a number of recent reviews [14, 15] and are discussed here mostly in the context of Alzheimer's disease.
Alzheimer's disease (AD) is a neurodegenerative brain disorder, which predominantly affects the aging population. One of the earliest signs of AD is the loss of synapses [16]. Synapse loss in AD has been linked at least partly to the toxicity induced by Aβ, a peptide that accumulates in the brains of AD patients [17–19]. Synaptic cell adhesion is directly involved in AD pathogenesis, since APP is a precursor protein of the Aβ peptide and also a synaptic cell adhesion molecule playing a role in regulation of synaptic morphology, synaptic plasticity, and hippocampus-dependent behavior [20]. Functions of APP in synapses and molecular mechanisms of Aβ formation are the subject of a number of recent reviews [21–23] and are not discussed here. In this review, we summarize current data on the changes in the levels and function of other synaptic CAMs in AD brains and their complex interactions with Aβ suggesting that abnormal function in different synaptic CAMs can be an important factor contributing to synapse dysfunction in AD.
2. Genetic Association between CAMs and AD
The involvement of CAMs in AD is suggested by genome-wide association studies (GWAS). Significantly altered expression of CAM pathway genes in AD was found in the samples from the cerebellum and temporal cortex of AD-affected individuals and AD-nonaffected controls [24]. Besides APP, among synaptic CAMs found to be associated with the risk of AD, PRNP gene coding for PrPc has been identified as an AD susceptibility gene by systematic meta-analysis of AD genetic association studies [25]. The methionine/valine (M/V) polymorphism at codon 129 within the PRNP gene, which represents a known risk factor for Creutzfeldt-Jakob disease (CJD), has also been reported to be a risk factor for early onset AD [26–28].
Single nucleotide polymorphisms (SNPs) in the neural cell adhesion molecule 2 (NCAM2), a synaptic IgSF CAM highly expressed in hippocampal synapses, have been reported as a risk factor related to the progression of AD in the Japanese population [29]. SNPs in the NCAM2 gene also show association with levels of Aβ in the cerebrospinal fluid in humans, suggesting that NCAM2 is involved in the pathogenic pathway to the senile plaques that concentrate in AD brains [30]. In another large GWAS involving over 16,000 individuals, SNPs in contactin-5, another member of the synaptic IgSF CAMs localizing to the presynaptic membranes [31], were shown to be significantly associated with AD [32]. The junctional adhesion molecule 2 (JAM2) is another member of IgSF potentially linked to AD. SNPs in JAM2 were found to be significantly associated with AD [33]. JAM2 is localized to tight junctions in epithelial and endothelial cells but is also expressed in retinal ganglion cells [34]. The link between JAM2 and AD is also suggested by a study reporting chromosomal 21 region duplication spanning 0.59 Mb and comprising JAM2, APP, and some other genes in a patient with AD [35]. Whether JAM2 functions in the regulation of synapses in neurons is, however, not known. The association with AD was also observed for SNPs in the gene coding for the leucine-rich repeat transmembrane neuronal 3 (LRRTM3) synaptic CAM, which is highly expressed in the hippocampus [36]. Meta-analysis of five GWAS also identified the gene coding for neurexin-3 as a gene playing a role in susceptibility to AD in males [37].
3. Changes in the Levels of Synaptic CAMs in AD
Changes in the levels of synaptic CAMs in AD brains have been reported in a number of studies performed over the last 25 years. Reduced levels of the largest NCAM isoform with the longest intracellular domain, NCAM180, but not total NCAM levels have been reported in one of the early studies comparing samples from control and AD frontal cortex by quantitative crossed immunoelectrophoresis [38] suggesting changes in the expression of NCAM in AD. In later studies, analysis of control and AD brain sections by immunohistochemistry with antibodies against NCAM found significantly fewer NCAM positive neurons in the frontal cortex of AD-affected individuals when compared to normal aging individuals [39]. In agreement, the levels of NCAM were shown to be reduced in frontal and temporal cortex from AD patients by ELISA [40]. Interestingly, there was little difference in the levels of NCAM in the occipital cortex and hippocampus of control and AD patients [39, 41]. However, immunohistochemical analysis of the AD hippocampus using antibodies against polysialic acid (PSA), a unique carbohydrate attached predominantly to NCAM, revealed an increase in the immunoreactivity and numbers of PSA-NCAM positive neurons in AD hippocampus and especially in the dentate gyrus indicating changes in the posttranslational processing of NCAM [42]. PSA-NCAM is highly expressed in the developing nervous system, but its expression in the mature nervous system is restricted to brain areas undergoing plastic changes [43], suggesting that an increase in PSA-NCAM in AD is related to extensive neuronal remodeling in AD brains.
Levels of contactin-2, a GPI anchored IgSF CAM also called transient axonal glycoprotein 1 (TAG-1), were shown by Western blot to be reduced in the temporal lobe of AD patients [44]. Contactin-2 is cleaved by β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) and its levels in AD brains inversely correlate with BACE1 levels and amyloid plaque density [44] suggesting that an increase in BACE1 activity observed in late-onset AD [45–49] results in increased contactin-2 cleavage. BACE-1 also cleaves other synaptic CAMs, such as IgSF CAM L1 and the close homologue of L1 (CHL1) [50, 51]. The intracellular domain of L1 is also cleaved by γ-secretase in human carcinoma cells [52], and γ-secretase induced proteolytic cleavage of L1 is increased in a mouse model of AD, which carries human APP with the pathogenic Swedish mutation and the L166P mutated human presenilin-1 [53]. Changes in the activity of BACE-1 and γ-secretase may therefore affect the expression of a number of other synaptic CAMs in AD brains.
In addition to IgSF CAMs, levels of PrPc analyzed by Western blot were also found to be decreased in the hippocampus of patients with sporadic AD but not with familial AD [54]. Levels of PrPc are also lower in the temporal cortex samples of AD patients [54, 55]. Levels of N-cadherin are also reduced in the temporal cortex of AD patients [56]. In contrast, Western blot analyses have not revealed significant changes in the levels of contactin-5 in the temporal cortex of AD patients [55] and levels of full length N-cadherin in the superior frontal gyrus of AD patients [57]. Levels of platelet endothelial cell adhesion molecule 1 (PECAM1), an IgSF CAM, were also similar in frontal and temporal cortex of control subjects and moderate to severe AD patients [58]. Therefore, expression of only a subset of synaptic CAMs appears to be affected in AD and only in some brain regions.
Interestingly, in a recent study, levels of NCAM2 were shown by Western blot to be increased in the hippocampus of AD patients but strongly reduced in synaptosomes isolated from this brain region [59] (Figure 2). Levels of NCAM2 were not significantly affected in the temporal cortex and cerebellum of AD patients. These observations indicate that changes in the total protein levels or the lack of such changes does not necessarily correlate with the changes in the subcellular localization and function of synaptic CAMs. Changes in the levels of other synaptic CAMs at synapses in AD brains and whether alterations in the overall levels of other synaptic CAMs reflect changes in their synaptic localization remain to be investigated in the future studies.
Figure 2.

Changes in NCAM2-mediated synaptic adhesion in AD-affected hippocampus. In AD-nonaffected hippocampus (a), NCAM2 accumulates in synapses and plays a role in the synapse maintenance. In AD-affected hippocampal synapses (b), levels of full length NCAM2 are decreased. This decrease is accompanied by an increase in the levels of the proteolytic cleavage products of NCAM2. The overall expression of NCAM2 is also increased probably due to the increase in the levels of extrasynaptic NCAM2.
4. Changes in the Levels of the Proteolytic Products of CAMs in AD
In addition to changes in the levels of the full length synaptic CAMs, changes in the levels of the proteolytic products of synaptic CAMs have also been found in AD brains. Interestingly, changes in the proteolytic products of synaptic CAMs do not necessarily correlate with the changes in the total protein levels. While the total levels of N-cadherin appear to be unaffected in the superior frontal gyrus of AD patients, the levels of ectodomain-shed C-terminal fragment of N-cadherin are increased [57]. The levels of the extracellular domains of NCAM2 proteolytically released from the neuronal cell surface are increased in AD hippocampus [59] (Figure 2). This increase in the levels of proteolytic products of NCAM2 inversely correlates with the levels of full length NCAM2 at synapses, while the total levels of NCAM2 are also increased in AD hippocampus [59] (Figure 2). It is therefore possible that changes in the proteolytic products of synaptic CAMs in AD brains reflect changes in their proteolysis at specific subcellular locations, such as synapses, rather than changes in the overall turnover of these proteins.
A number of studies indicate that the proteolytic products of CAMs are also present at varying levels in the cerebrospinal fluid (CSF) and serum of humans. Western blot analyses with antibodies specific to different portions of these molecules show that these proteolytic products are detectable with the antibodies against the epitopes within their extracellular domains while the intracellular domains are not detectable [60]. These observations indicate that the proteolytic products of CAMs in CSF and serum represent fragments of the extracellular domains of CAMs possibly released to CSF by shedding from the cell surface of neurons in the brain. Proteolytic products of several CAMs have been reported to be increased in CSF and serum of AD patients. For example, CSF levels of L1 analyzed by ELISA have been reported to be significantly increased in AD [60]. This study also reported an increase in the CSF levels of NCAM, which, however, was not statistically significant when compared to normal controls. Increased levels of several proteolytic products of NCAM were also found in the sera of AD patients [61]. In contrast, ELISA analysis has not revealed significant differences in the levels of neuronal cell adhesion molecule (NrCAM), L1 family member, in CSF samples from healthy controls and AD patients [62].
Analysis of the levels of the proteolytic products of CAMs has therefore been proposed to be useful in diagnostics of AD. It should be noted, however, that levels of the proteolytic products of such CAMs as NCAM or L1 in CSF and serum samples of healthy individuals and AD patients overlap considerably [60, 61]. Also, changes in CSF levels of these products are often not specific to AD. For example, levels of the proteolytic products of L1 are also increased in vascular dementia and dementia of mixed type [60]. Levels of the proteolytic products of NCAM are increased in CSF of people suffering from schizophrenia [63] and bipolar mood disorder type I or recurrent unipolar major depression [64], but not in bipolar mood disorder type II patients [64]. However, levels of NCAM are not changed in the serum of patients with autism, although levels of NCAM180 protein but not mRNA are reduced in the brains of these patients [65]. Also, in contrast to AD, CSF levels of L1 are decreased in schizophrenia [63]. Therefore, analysis of specific isoforms and cleavage products derived via different proteolysis pathways might be required to establish proteolytic products of CAMs as markers of specific neurologic conditions including AD.
5. Synaptic CAMs as Receptors for Aβ Oligomers
A number of observations indicate that synaptic CAMs act as receptors for Aβ oligomers at the synaptic sites. The extracellular domain of L1 but not the extracellular domain of CHL1 interacts with Aβ in a label-free binding assay [66] (Figure 3). The fibronectin type III homologous repeats 1–3 of the extracellular domain of L1 mediate this effect. Interestingly, the recombinant extracellular domain of L1, but not the recombinant extracellular domain of CHL1, inhibits aggregation of Aβ in vitro. Furthermore, overexpression of L1 by injection of adenoassociated virus encoding L1 decreases the Aβ plaque load, levels of Aβ42, Aβ42/40 ratio, and astrogliosis in a mouse model of AD, which carries human APP with the pathogenic Swedish mutation and the L166P mutated human presenilin-1 [66]. The extracellular domain of NCAM2 also binds to Aβ oligomers both in vitro and in vivo in cultured mouse hippocampal neurons and in the hippocampus of Aβ generating transgenic mice overexpressing human APP containing the pathogenic Swedish mutation [59].
Figure 3.
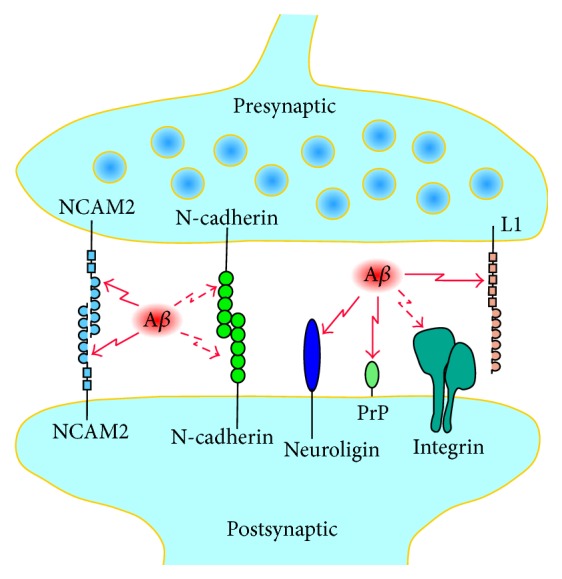
Synaptic CAMs function as receptors for Aβ. Schematic representation of a synapse showing presynaptic and postsynaptic CAMs, which bind to Aβ. Direct interaction with Aβ has been demonstrated for NCAM2, neuroligin, PrPc, and L1 (solid red arrows). Binding of Aβ to integrins and N-cadherins is suggested by indirect observations and remains to be confirmed in a direct binding assay (dashed red arrows).
Aβ oligomers also directly associate with the N-terminus of PrPc both in vitro and in the human AD brain, with the binding sites located within residues 23–27 and 95–110 of PrPc [67–71]. Interaction of PrPc with Aβ is a function of Aβ load in the brain and does not depend on PrPc levels [71]. The pathological relevance of this interaction remains, however, to be established. PrPc interacts with other synaptic proteins, including N-methyl-D-aspartic acid- (NMDA-) type glutamate receptors [72] and NCAM [73], and binding of Aβ oligomers to PrPc can interrupt the physiological interactions of PrPc at synapses, resulting in disturbed neuronal communication [74]. PrPc deficient mice are resistant to the neurotoxic effect of Aβ oligomers, and antibodies against PrPc or PrPc peptides prevent Aβ oligomer-induced neurotoxicity indicating that PrPc is involved in the molecular pathways activated by Aβ oligomers to induce neuronal cell death [75]. PrPc traps and concentrates Aβ in an oligomeric form and disassembles mature Aβ fibers [70]. The cleavage fragment of PrPc containing binding sites for Aβ strongly suppresses Aβ toxicity in cultured mouse hippocampal neurons and in vivo in mice after intracerebroventricular injections of Aβ [69, 76]. However, memory impairment induced by injection of Aβ oligomers is not reduced in PrPc knockout mice [77], ablation or overexpression of PrPc has no effect on the impairment of hippocampal synaptic plasticity in a transgenic model of AD [78], and synaptic depression, reduction in spine density, or blockade of LTP is induced by Aβ in organotypic hippocampal slice neurons from both wild type and PrPc knockout mice [79]. Therefore, the Aβ-mediated synaptic defects do not require PrPc.
Neuroligin-1 is enriched in excitatory synapses and its extracellular domain binds to Aβ in vitro and in cultured rat hippocampal neurons and rat cerebral cortex [80, 81]. Aβ does not interact with neuroligin-2, which is enriched in inhibitory synapses. Neuroligin-1 acts as a nucleating factor during the Aβ aggregation process, stimulating the formation of Aβ oligomers [81]. The soluble extracellular α/β-hydrolase-fold (ChE-like) domain of neuroligin-1 reduces the Aβ-induced reduction in synaptic density in cultured rat hippocampal neurons and in field excitatory postsynaptic potentials (fEPSP) in rat hippocampal slices possibly by competing with the synaptic neuroligin-1 for binding to Aβ [80].
Indirect observations also suggest that Aβ interacts with integrins since Aβ toxicity was inhibited in human neurons pretreated with adhesion-blocking antibodies against different subunits of integrins, and in particular β1, α2, and αV [82]. Inhibition of α1β1 integrin has also been shown to reduce Aβ toxicity in rat hippocampal cultures [83]. Aβ toxicity was also inhibited by disintegrin echistatin, a peptide isolated from snake venom that has been shown to inhibit RGD-dependent integrins such as αVβ1 and by integrin ligands such as vitronectin, fibronectin, and superfibronectin suggesting that integrin ligands compete with Aβ for binding to integrins [82]. Application of Aβ also reduces the overall expression of N-cadherin in cultured mouse cortical neurons suggesting that Aβ can bind to N-cadherins [56], although a reduction in N-cadherin proteolysis after application of Aβ has been reported in another study [84].
Interestingly, some CAMs have been shown to interact also with APP. The association of N-cadherin with APP in mouse brains has been shown by coimmunoprecipitation experiments [85]. In an unbiased search for the binding partners of APP using time-controlled transcardiac perfusion cross-linking followed by high stringency immunoaffinity purification and tandem mass spectrometry, several other cell adhesion molecules were identified including PrPc and IgSF CAMs Thy-1, contactin, NCAM1, and neurofascin [86]. In spite of homology to NCAM2, NCAM1 binds to a region of APP which is different to the Aβ-containing region [87] indicating that these interactions may play a role in physiological functions of both molecules. In agreement, contactin-2 has been shown to be a functional ligand of APP. Binding of contactin-2 to APP increases the release of the intracellular domain of APP through γ-secretase-dependent cleavage [88]. Contactin-2 competitively inhibits the binding of APP to transforming growth factor β2 (TGFβ2) [89]. Binding of TGFβ2 to APP induces neuronal cell death [90] and this effect is inhibited by TAG-1 [89] suggesting that TAG-1 regulates interactions of APP with extracellular ligands.
6. Synaptic CAMs in Regulation of Aβ Production
BACE1 is a potential therapeutic target for AD since BACE1 cleavage of APP is the rate limiting step in Aβ production [91]. Synaptic cell adhesion molecules have been shown to play a role in regulation of BACE1 activity. In a high-throughput siRNA screen assessing 15,200 genes for their role in Aβ secretion, LRRTM3 has been identified as a neuronal gene that promotes APP processing by BACE1 [92]. Knockdown of LRRTM3 expression using siRNA results in reduced secretion of Aβ in cultured cells and primary neurons, while overexpression of LRRTM3 increases Aβ secretion [92] suggesting that LRRTM3 promotes BACE1 activity.
In contrast, overexpression of PrPc results in inhibited BACE1-mediated cleavage of APP and reduced Aβ production, while Aβ production is increased in the brains of PrPc knockout mice and in cultured N2a cells after siRNA mediated knockdown of PrPc expression [93] suggesting that PrPc inhibits BACE1 activity. In agreement, in a follow-up study, PrPc has been shown to interact with the prodomain of BACE1 in the trans-Golgi network and regulate targeting of BACE1 to the cell surface and endosomes where it preferentially cleaves APP [94]. PrPc reduces BACE1-mediated cleavage of wild type APP, but not human APP with the Swedish and Indiana familial mutations, suggesting that PrPc may play a role in sporadic AD but not in familial AD [94]. Interestingly, the region at the extreme N-terminus of PrPc, which is critical for the interaction or PrPc with BACE-1 and PrPc-dependent inhibition of APP-cleaving activity [93], also contains the binding site for Aβ oligomers [67]. These observations suggest that PrPc can play a protective role (inhibition of BACE1) and pathogenic role (binding of toxic Aβ oligomers) in AD and also suggest that the protective function of PrPc can be affected by Aβ oligomers.
An increase in Aβ secretion is also observed in cells cotransfected with N-cadherin [85, 95]. N-cadherin promotes cell surface expression of γ-secretase and increases accessibility of γ-secretase to APP [95]. Altogether, these observations thus indicate that synaptic CAMs are involved in regulation of the key enzymes involved in Aβ production.
7. Effects of Disruptions of Synaptic Adhesion on the Synapse Integrity in AD
Inhibition of N-cadherin function by blocking INP peptides, which mimic a short sequence in the EC1 domain of N-cadherin and thus impair the homophilic transsynaptic interaction of N-cadherin molecules, accelerates the Aβ-induced synapse impairment characterized by a reduction in the frequency of the AMPA receptor-mediated miniature excitatory postsynaptic currents (AMPA mEPSCs) and reduced density of synaptic boutons along dendrites in cultured cortical neurons [57]. Similar effects are observed when N-cadherin function is inhibited by expression of the dominant-negative, truncated N-cadherin lacking the extracellular cadherin domains, or by overexpression of the ectodomain-shed C-terminal fragment of human N-cadherin, which accumulates in AD brains. It is noteworthy that the ectodomain-shed C-terminal fragment of human N-cadherin is further cleaved by γ-secretase, and inhibition of γ-secretase activity also accelerates the Aβ-induced synapse impairment [57].
Inhibition of N-cadherin function alone has no effect on the numbers of synapses and frequency of AMPA mEPSCs [57]. Interestingly, disruption of NCAM2-mediated synaptic adhesion using recombinant extracellular domains of NCAM2 (NCAM2-ED) results in a reduction in synapse density along dendrites of hippocampal neurons and dispersion of AMPA receptors from synapses [59]. NCAM2-ED accumulates in AD hippocampus and its effect on the synapse integrity is similar to and not additive with the effect of Aβ [59]. It is therefore possible that Aβ-dependent proteolysis of NCAM2 is one of the initial synapse-destabilizing effects of Aβ, which is then followed by disruption of N-cadherin containing adhesion complexes resulting in the complete synapse disassembly.
The complex formed by Aβ and neuroligin-1 also contains GluN2B but not GluN2A subunits of NMDA receptors [80] suggesting that Aβ can directly affect the function of neuroligin-1 in anchoring NMDA receptors at synapses. Whether binding of Aβ to other synaptic CAMs directly contributes to the synapse loss has to be investigated in the future studies.
8. Future Directions
While a number of observations indicate that synaptic cell adhesion molecules are affected in AD, our understanding of the molecular and cellular mechanisms underlying these changes and their role in the disease progression is still very incomplete. Further studies assessing levels of synaptic CAMs specifically at synapses are needed to understand whether changes in the overall levels of these CAMs reflect changes in the synaptic adhesion. Whether an increase in the levels of specific proteolytic products of CAMs in CSF and sera of AD patients reflect the Aβ-dependent proteolysis of CAMs at synapses is an interesting possibility which can be analyzed in the future studies. Since synaptic CAMs play key roles in the maintenance of synapse integrity and function by interacting with synaptic scaffolding proteins and neurotransmitter receptors, further analysis of the effects of Aβ-dependent disruption of synaptic adhesion at the synaptic level may help to understand the molecular mechanisms of the initial stages of AD. Furthermore, a number of reports showing that the Aβ toxicity can be reduced by targeting synaptic CAMs indicate that synaptic CAMs deserve further consideration as molecular targets in designing new treatments of AD.
Competing Interests
The authors declare that they have no competing interests.
References
- 1.Shapiro L., Love J., Colman D. R. Adhesion molecules in the nervous system: structural insights into function and diversity. Annual Review of Neuroscience. 2007;30:451–474. doi: 10.1146/annurev.neuro.29.051605.113034. [DOI] [PubMed] [Google Scholar]
- 2.Sytnyk V., Leshchyns'ka I., Delling M., Dityateva G., Dityatev A., Schachner M. Neural cell adhesion molecule promotes accumulation of TGN organelles at sites of neuron-to-neuron contacts. Journal of Cell Biology. 2002;159(4):649–661. doi: 10.1083/jcb.200205098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dityatev A., Dityateva G., Sytnyk V., et al. Polysialylated neural cell adhesion molecule promotes remodeling and formation of hippocampal synapses. The Journal of Neuroscience. 2004;24(42):9372–9382. doi: 10.1523/jneurosci.1702-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shetty A., Sytnyk V., Leshchyns'ka I., Puchkov D., Haucke V., Schachner M. The neural cell adhesion molecule promotes maturation of the presynaptic endocytotic machinery by switching synaptic vesicle recycling from adaptor protein 3 (AP-3)- to AP-2-dependent mechanisms. The Journal of Neuroscience. 2013;33(42):16828–16845. doi: 10.1523/jneurosci.2192-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puchkov D., Leshchyns'ka I., Nikonenko A. G., Schachner M., Sytnyk V. NCAM/spectrin complex disassembly results in PSD perforation and postsynaptic endocytic zone formation. Cerebral Cortex. 2011;21(10):2217–2232. doi: 10.1093/cercor/bhq283. [DOI] [PubMed] [Google Scholar]
- 6.Mendez P., De Roo M., Poglia L., Klauser P., Muller D. N-cadherin mediates plasticity-induced long-term spine stabilization. The Journal of Cell Biology. 2010;189(3):589–600. doi: 10.1083/jcb.201003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benson D. L., Huntley G. W. Synapse adhesion: a dynamic equilibrium conferring stability and flexibility. Current Opinion in Neurobiology. 2012;22(3):397–404. doi: 10.1016/j.conb.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andreyeva A., Leshchyns'ka I., Knepper M., et al. CHL1 is a selective organizer of the presynaptic machinery chaperoning the SNARE complex. PLoS ONE. 2010;5(8) doi: 10.1371/journal.pone.0012018.e12018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leshchyns'ka I., Sytnyk V., Richter M., Andreyeva A., Puchkov D., Schachner M. The adhesion molecule CHL1 regulates uncoating of clathrin-coated synaptic vesicles. Neuron. 2006;52(6):1011–1025. doi: 10.1016/j.neuron.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 10.Sytnyk V., Leshchyns'Ka I., Nikonenko A. G., Schachner M. NCAM promotes assembly and activity-dependent remodeling of the postsynaptic signaling complex. Journal of Cell Biology. 2006;174(7):1071–1085. doi: 10.1083/jcb.200604145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schachner M. Neural recognition molecules and synaptic plasticity. Current Opinion in Cell Biology. 1997;9(5):627–634. doi: 10.1016/S0955-0674(97)80115-9. [DOI] [PubMed] [Google Scholar]
- 12.Gerrow K., El-Husseini A. Cell adhesion molecules at the synapse. Frontiers in Bioscience. 2006;11(2):2400–2419. doi: 10.2741/1978. [DOI] [PubMed] [Google Scholar]
- 13.Südhof T. C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455(7215):903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seong E., Yuan L., Arikkath J. Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adhesion & Migration. 2015;9(3):202–213. doi: 10.4161/19336918.2014.994919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang X., Hou D., Jiang W., Zhang C. Intercellular protein-protein interactions at synapses. Protein and Cell. 2014;5(6):420–444. doi: 10.1007/s13238-014-0054-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheff S. W., Price D. A. Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiology of Aging. 2003;24(8):1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Wilcox K. C., Lacor P. N., Pitt J., Klein W. L. Aβ oligomer-induced synapse degeneration in Alzheimer's disease. Cellular and molecular neurobiology. 2011;31(6):939–948. doi: 10.1007/s10571-011-9691-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lacor P. N., Buniel M. C., Furlow P. W., et al. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. The Journal of Neuroscience. 2007;27(4):796–807. doi: 10.1523/jneurosci.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pham E., Crews L., Ubhi K., et al. Progressive accumulation of amyloid-β oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS Journal. 2010;277(14):3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klevanski M., Herrmann U., Weyer S. W., et al. The APP intracellular domain is required for normal synaptic morphology, synaptic plasticity, and hippocampus-dependent behavior. Journal of Neuroscience. 2015;35(49):16018–16033. doi: 10.1523/jneurosci.2009-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lassek M., Weingarten J., Wegner M., Volknandt W. The amyloid precursor protein—a novel player within the molecular array of presynaptic nanomachines. Frontiers in Synaptic Neuroscience. 2015;7, article 21 doi: 10.3389/fnsyn.2015.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nhan H. S., Chiang K., Koo E. H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathologica. 2015;129(1):1–19. doi: 10.1007/s00401-014-1347-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicolas M., Hassan B. A. Amyloid precursor protein and neural development. Development. 2014;141(13):2543–2548. doi: 10.1242/dev.108712. [DOI] [PubMed] [Google Scholar]
- 24.Bao X., Liu G., Jiang Y., et al. Cell adhesion molecule pathway genes are regulated by cis-regulatory SNPs and show significantly altered expression in Alzheimer's disease brains. Neurobiology of Aging. 2015;36(10):2904–2904.e7. doi: 10.1016/j.neurobiolaging.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 25.Bertram L., McQueen M. B., Mullin K., Blacker D., Tanzi R. E. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nature Genetics. 2007;39(1):17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 26.Del Bo R., Scarlato M., Ghezzi S., et al. Is M129V of PRNP gene associated with Alzheimer's disease? A case-control study and a meta-analysis. Neurobiology of Aging. 2006;27(5):770.e1–770.e5. doi: 10.1016/j.neurobiolaging.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 27.Dermaut B., Croes E. A., Rademakers R., et al. PRNP Val129 homozygosity increases risk for early-onset Alzheimer's disease. Annals of Neurology. 2003;53(3):409–412. doi: 10.1002/ana.10507. [DOI] [PubMed] [Google Scholar]
- 28.Riemenschneider M., Klopp N., Xiang W., et al. Prion protein codon 129 polymorphism and risk of Alzheimer disease. Neurology. 2004;63(2):364–366. doi: 10.1212/01.WNL.0000130198.72589.69. [DOI] [PubMed] [Google Scholar]
- 29.Kimura R., Kamino K., Yamamoto M., et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Human Molecular Genetics. 2007;16(1):15–23. doi: 10.1093/hmg/ddl437. [DOI] [PubMed] [Google Scholar]
- 30.Han M. R., Schellenberg G. D., Wang L. S. Genome-wide association reveals genetic effects on human Abeta42 and tau protein levels in cerebrospinal fluids: a case control study. BMC Neurology. 2010;10, article 90 doi: 10.1186/1471-2377-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimoda Y., Koseki F., Itoh M., Toyoshima M., Watanabe K. A cis-complex of NB-2/contactin-5 with amyloid precursor-like protein 1 is localized on the presynaptic membrane. Neuroscience Letters. 2012;510(2):148–153. doi: 10.1016/j.neulet.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 32.Harold D., Abraham R., Hollingworth P., et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nature Genetics. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khondoker M., Newhouse S., Westman E., et al. Linking genetics of brain changes to Alzheimer's disease: sparse whole genome association scan of regional MRI volumes in the ADNI and AddNeuroMed cohorts. Journal of Alzheimer's Disease. 2015;45(3):851–864. doi: 10.3233/jad-142214. [DOI] [PubMed] [Google Scholar]
- 34.Kim I.-J., Zhang Y., Yamagata M., Meister M., Sanes J. R. Molecular identification of a retinal cell type that responds to upward motion. Nature. 2008;452(7186):478–482. doi: 10.1038/nature06739. [DOI] [PubMed] [Google Scholar]
- 35.Antonell A., Gelpi E., Sánchez-Valle R., Martínez R., Molinuevo J. L., Lladó A. Breakpoint sequence analysis of an AβPP locus duplication associated with autosomal dominant Alzheimer's disease and severe cerebral amyloid angiopathy. Journal of Alzheimer's Disease. 2012;28(2):303–308. doi: 10.3233/jad-2011-110911. [DOI] [PubMed] [Google Scholar]
- 36.Martin E. R., Bronson P. G., Li Y.-J., et al. Interaction between the α-T catenin gene (VR22) and APOE in Alzheimer's disease. Journal of Medical Genetics. 2005;42(10):787–792. doi: 10.1136/jmg.2004.029553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez-Mir A., González-Pérez A., Gayán J., et al. Genetic study of neurexin and neuroligin genes in Alzheimer's disease. Journal of Alzheimer's Disease. 2013;35(2):403–412. doi: 10.3233/JAD-122257. [DOI] [PubMed] [Google Scholar]
- 38.Jørgensen O. S., Brooksbank B. W., Balázs R. Neuronal plasticity and astrocytic reaction in Down syndrome and Alzheimer disease. Journal of the Neurological Sciences. 1990;98(1):63–79. doi: 10.1016/0022-510x(90)90182-m. [DOI] [PubMed] [Google Scholar]
- 39.Yew D. T., Li W. P., Webb S. E., Lai H. W. L., Zhang L. Neurotransmitters, peptides, and neural cell adhesion molecules in the cortices of normal elderly humans and Alzheimer patients: a comparison. Experimental Gerontology. 1999;34(1):117–133. doi: 10.1016/s0531-5565(98)00017-5. [DOI] [PubMed] [Google Scholar]
- 40.Aisa B., Gil-Bea F. J., Solas M., et al. Altered NCAM expression associated with the cholinergic system in Alzheimer's disease. Journal of Alzheimer's Disease. 2010;20(2):659–668. doi: 10.3233/jad-2010-1398. [DOI] [PubMed] [Google Scholar]
- 41.Gillian A. M., Brion J. P., Breen K. C. Expression of the neural cell adhesion molecule (NCAM) in Alzheimer's disease. Neurodegeneration. 1994;3(4):283–291. [PubMed] [Google Scholar]
- 42.Mikkonen M., Soininen H., Tapiola T., Alafuzoff I., Miettinen R. Hippocampal plasticity in Alzheimer's disease: changes in highly polysialylated NCAM immunoreactivity in the hippocampal formation. European Journal of Neuroscience. 1999;11(5):1754–1764. doi: 10.1046/j.1460-9568.1999.00593.x. [DOI] [PubMed] [Google Scholar]
- 43.Schnaar R. L., Gerardy-Schahn R., Hildebrandt H. Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiological Reviews. 2014;94(2):461–518. doi: 10.1152/physrev.00033.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gautam V., D'Avanzo C., Hebisch M., Kovacs D. M., Kim D. Y. BACE1 activity regulates cell surface contactin-2 levels. Molecular Neurodegeneration. 2014;9(1, article 4) doi: 10.1186/1750-1326-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukumoto H., Cheung B. S., Hyman B. T., Irizarry M. C. β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Archives of Neurology. 2002;59(9):1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- 46.Tyler S. J., Dawbarn D., Wilcock G. K., Allen S. J. α- and β-secretase: profound changes in Alzheimer's disease. Biochemical and Biophysical Research Communications. 2002;299(3):373–376. doi: 10.1016/s0006-291x(02)02635-9. [DOI] [PubMed] [Google Scholar]
- 47.Yang L.-B., Lindholm K., Yan R., et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nature Medicine. 2003;9(1):3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- 48.Miners J. S., Van Helmond Z., Kehoe P. G., Love S. Changes with age in the activities of β-secretase and the aβ-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathology. 2010;20(4):794–802. doi: 10.1111/j.1750-3639.2010.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li R., Lindholm K., Yang L., et al. Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer's disease patients. Proceedings of the National Academy of Sciences. 2004;101(10):3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou L., Barão S., Laga M., et al. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. The Journal of Biological Chemistry. 2012;287(31):25927–25940. doi: 10.1074/jbc.M112.377465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hitt B., Riordan S. M., Kukreja L., Eimer W. A., Rajapaksha T. W., Vassar R. β-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. The Journal of Biological Chemistry. 2012;287(46):38408–38425. doi: 10.1074/jbc.m112.415505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riedle S., Kiefel H., Gast D., et al. Nuclear translocation and signalling of L1-CAM in human carcinoma cells requires ADAM10 and presenilin/γ-secretase activity. Biochemical Journal. 2009;420(3):391–402. doi: 10.1042/bj20081625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lutz D., Wolters-Eisfeld G., Joshi G., et al. Generation and nuclear translocation of sumoylated transmembrane fragment of cell adhesion molecule L1. The Journal of Biological Chemistry. 2012;287(21):17161–17175. doi: 10.1074/jbc.m112.346759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitehouse I. J., Jackson C., Turner A. J., Hooper N. M. Prion protein is reduced in aging and in sporadic but not in familial Alzheimer's disease. Journal of Alzheimer's Disease. 2010;22(3):1023–1031. doi: 10.3233/jad-2010-101071. [DOI] [PubMed] [Google Scholar]
- 55.Whitehouse I. J., Miners J. S., Glennon E. B. C., et al. Prion protein is decreased in Alzheimer's brain and inversely correlates with BACE1 activity, amyloid-β levels and Braak stage. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0059554.e59554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ando K., Uemura K., Kuzuya A., et al. N-cadherin regulates p38 MAPK signaling via association with JNK-associated leucine zipper protein: implications for neurodegeneration in Alzheimer disease. The Journal of Biological Chemistry. 2011;286(9):7619–7628. doi: 10.1074/jbc.m110.158477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andreyeva A., Nieweg K., Horstmann K., et al. C-terminal fragment of N-cadherin accelerates synapse destabilization by amyloid-β . Brain. 2012;135(7):2140–2154. doi: 10.1093/brain/aws120. [DOI] [PubMed] [Google Scholar]
- 58.Lepelletier F. X., Mann D. M., Robinson A. C., Pinteaux E., Boutin H. Early changes in extracellular matrix in Alzheimer's disease. Neuropathology and Applied Neurobiology. 2015 doi: 10.1111/nan.12295. [DOI] [PubMed] [Google Scholar]
- 59.Leshchyns'ka I., Liew H. T., Shepherd C., et al. Aβ-Dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer’s disease. Nature Communications. 2015;6, article 8836 doi: 10.1038/ncomms9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Strekalova H., Buhmann C., Kleene R., et al. Elevated levels of neural recognition molecule L1 in the cerebrospinal fluid of patients with Alzheimer disease and other dementia syndromes. Neurobiology of Aging. 2006;27(1):1–9. doi: 10.1016/j.neurobiolaging.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 61.Todaro L., Puricelli L., Gioseffi H., et al. Neural cell adhesion molecule in human serum. Increased levels in dementia of the Alzheimer type. Neurobiology of Disease. 2004;15(2):387–393. doi: 10.1016/j.nbd.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 62.Müller M., Claassen J. A., Kuiperij H. B., Verbeek M. M. Cerebrospinal fluid NrCAM is not a suitable biomarker to discriminate between dementia disorders—a pilot study. Journal of Alzheimer's Disease. 2015;46(3):605–609. doi: 10.3233/jad-142901. [DOI] [PubMed] [Google Scholar]
- 63.Poltorak M., Hemperly J. J., Williams J. R., El-Mallakh R., Freed W. J. Disturbances in cell recognition molecules (N-CAM and L1 antigen) in the CSF of patients with schizophrenia. Experimental Neurology. 1995;131(2):266–272. doi: 10.1016/0014-4886(95)90048-9. [DOI] [PubMed] [Google Scholar]
- 64.Poltorak M., Frye M. A., Wright R., et al. Increased neural cell adhesion molecule in the CSF of patients with mood disorder. Journal of Neurochemistry. 1996;66(4):1532–1538. doi: 10.1046/j.1471-4159.1996.66041532.x. [DOI] [PubMed] [Google Scholar]
- 65.Purcell A. E., Rocco M. M., Lenhart J. A., Hyder K., Zimmerman A. W., Pevsner J. Assessment of neural cell adhesion molecule (NCAM) in autistic serum and postmortem brain. Journal of Autism and Developmental Disorders. 2001;31(2):183–194. doi: 10.1023/a:1010751232295. [DOI] [PubMed] [Google Scholar]
- 66.Djogo N., Jakovcevski I., Müller C., et al. Adhesion molecule L1 binds to amyloid beta and reduces Alzheimer's disease pathology in mice. Neurobiology of Disease. 2013;56:104–115. doi: 10.1016/j.nbd.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 67.Chen S., Yadav S. P., Surewicz W. K. Interaction between human prion protein and amyloid-β (Aβ) oligomers: role of N-terminal residues. The Journal of Biological Chemistry. 2010;285(34):26377–26383. doi: 10.1074/jbc.m110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zou W.-Q., Xiao X., Yuan J., et al. Amyloid-β42 interacts mainly with insoluble prion protein in the Alzheimer brain. The Journal of Biological Chemistry. 2011;286(17):15095–15105. doi: 10.1074/jbc.m110.199356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fluharty B. R., Biasini E., Stravalaci M., et al. An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. The Journal of Biological Chemistry. 2013;288(11):7857–7866. doi: 10.1074/jbc.m112.423954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Younan N. D., Sarell C. J., Davies P., Brown D. R., Viles J. H. The cellular prion protein traps Alzheimer's Aβ in an oligomeric form and disassembles amyloid fibers. The FASEB Journal. 2013;27(5):1847–1858. doi: 10.1096/fj.12-222588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dohler F., Sepulveda-Falla D., Krasemann S., et al. High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer's disease. Brain. 2014;137, part 3:873–886. doi: 10.1093/brain/awt375. [DOI] [PubMed] [Google Scholar]
- 72.Khosravani H., Zhang Y., Tsutsui S., et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. The Journal of Cell Biology. 2008;181(3):551–565. doi: 10.1083/jcb.200711002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Santuccione A., Sytnyk V., Leshchyns'ka I., Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. Journal of Cell Biology. 2005;169(2):341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Black S. A., Stys P. K., Zamponi G. W., Tsutsui S. Cellular prion protein and NMDA receptor modulation: protecting against excitotoxicity. Frontiers in Cell and Developmental Biology. 2014;2, article 45 doi: 10.3389/fcell.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kudo W., Lee H.-P., Zou W.-Q., et al. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Human Molecular Genetics. 2012;21(5):1138–1144. doi: 10.1093/hmg/ddr542.ddr542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guillot-Sestier M.-V., Sunyach C., Ferreira S. T., et al. α-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid β (Aβ)-associated cell death. The Journal of Biological Chemistry. 2012;287(7):5021–5032. doi: 10.1074/jbc.m111.323626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Balducci C., Beeg M., Stravalaci M., et al. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(5):2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Calella A. M., Farinelli M., Nuvolone M., et al. Prion protein and Aβ-related synaptic toxicity impairment. EMBO Molecular Medicine. 2010;2(8):306–314. doi: 10.1002/emmm.201000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kessels H. W., Nguyen L. N., Nabavi S., Malinow R. The prion protein as a receptor for amyloid-β . Nature. 2010;466(7308):E3–E5. doi: 10.1038/nature09217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dinamarca M. C., Di Luca M., Godoy J. A., Inestrosa N. C. The soluble extracellular fragment of neuroligin-1 targets Aβ oligomers to the postsynaptic region of excitatory synapses. Biochemical and Biophysical Research Communications. 2015;466(1):66–71. doi: 10.1016/j.bbrc.2015.08.107. [DOI] [PubMed] [Google Scholar]
- 81.Dinamarca M. C., Weinstein D., Monasterio O., Inestrosa N. C. The synaptic protein neuroligin-1 interacts with the amyloid β-peptide. Is there a role in Alzheimer's disease? Biochemistry. 2011;50(38):8127–8137. doi: 10.1021/bi201246t. [DOI] [PubMed] [Google Scholar]
- 82.Wright S., Malinin N. L., Powell K. A., Yednock T., Rydel R. E., Griswold-Prenner I. α2β1 and αVβ1 integrin signaling pathways mediate amyloid-β-induced neurotoxicity. Neurobiology of Aging. 2007;28(2):226–237. doi: 10.1016/j.neurobiolaging.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Anderson K. L., Ferreira A. α1 Integrin activation: a link between β-amyloid deposition and neuronal death in aging hippocampal neurons. Journal of Neuroscience Research. 2004;75(5):688–697. doi: 10.1002/jnr.20018. [DOI] [PubMed] [Google Scholar]
- 84.Uemura K., Kuzuya A., Aoyagi N., et al. Amyloid β inhibits ectodomain shedding of N-cadherin via down-regulation of cell-surface NMDA receptor. Neuroscience. 2007;145(1):5–10. doi: 10.1016/j.neuroscience.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 85.Asada-Utsugi M., Uemura K., Noda Y., et al. N-cadherin enhances APP dimerization at the extracellular domain and modulates Aβ production. Journal of Neurochemistry. 2011;119(2):354–363. doi: 10.1111/j.1471-4159.2011.07364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bai Y., Markham K., Chen F., et al. The in vivo brain interactome of the amyloid precursor protein. Molecular & Cellular Proteomics. 2008;7(1):15–34. doi: 10.1074/mcp.M700077-MCP200. [DOI] [PubMed] [Google Scholar]
- 87.Chen K.-P., Dou F. Selective interaction of amyloid precursor protein with different isoforms of neural cell adhesion molecule. Journal of Molecular Neuroscience. 2012;46(1):203–209. doi: 10.1007/s12031-011-9578-3. [DOI] [PubMed] [Google Scholar]
- 88.Ma Q.-H., Futagawa T., Yang W.-L., et al. A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nature Cell Biology. 2008;10(3):283–294. doi: 10.1038/ncb1690. [DOI] [PubMed] [Google Scholar]
- 89.Tachi N., Hashimoto Y., Nawa M., Matsuoka M. TAG-1 is an inhibitor of TGFβ2-induced neuronal death via amyloid β precursor protein. Biochemical and Biophysical Research Communications. 2010;394(1):119–125. doi: 10.1016/j.bbrc.2010.02.127. [DOI] [PubMed] [Google Scholar]
- 90.Hashimoto Y., Chiba T., Yamada M., et al. Transforming growth factor beta2 is a neuronal death-inducing ligand for amyloid-beta precursor protein. Molecular and Cellular Biology. 2005;25(21):9304–9317. doi: 10.1128/mcb.25.21.9304-9317.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cole S. L., Vassar R. The role of amyloid precursor protein processing by BACE1, the β-secretase, in Alzheimer disease pathophysiology. The Journal of Biological Chemistry. 2008;283(44):29621–29625. doi: 10.1074/jbc.r800015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Majercak J., Ray W. J., Espeseth A., et al. LRRTM3 promotes processing of amyloid-precursor protein by BACE1 and is a positional candidate gene for late-onset Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(47):17967–17972. doi: 10.1073/pnas.0605461103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Parkin E. T., Watt N. T., Hussain I., et al. Cellular prion protein regulates β-secretase cleavage of the Alzheimer's amyloid precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(26):11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Griffiths H. H., Whitehouse I. J., Baybutt H., et al. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. The Journal of Biological Chemistry. 2011;286(38):33489–33500. doi: 10.1074/jbc.m111.278556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Uemura K., Lill C. M., Banks M., et al. N-cadherin-based adhesion enhances Abeta release and decreases Abeta42/40 ratio. Journal of Neurochemistry. 2009;108(2):350–360. doi: 10.1111/j.1471-4159.2008.05760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]