

Abstract
Characterization of human monoclonal antibodies is providing considerable insight into mechanisms of broad HIV-1 neutralization. Here we report an HIV-1 gp41 membrane-proximal external region (MPER)-specific antibody, named 10E8, which neutralizes ~98% of tested viruses. An analysis of sera from 78 healthy HIV-1-infected donors demonstrated that 27% contained MPER-specific antibodies and 8% contained 10E8-like specificities. In contrast to other neutralizing MPER antibodies, 10E8 did not bind phospholipids, was not autoreactive, and bound cell-surface envelope. The structure of 10E8 in complex with the complete MPER revealed a site-of-vulnerability comprising a narrow stretch of highly conserved gp41-hydrophobic residues and a critical Arg/Lys just prior to the transmembrane region. Analysis of resistant HIV-1 variants confirmed the importance of these residues for neutralization. The highly conserved MPER is a target of potent, non-self-reactive neutralizing antibodies, suggesting that HIV-1 vaccines should aim to induce antibodies to this region of HIV-1 Env.
Introduction
Induction of an antibody response capable of neutralizing diverse HIV-1 isolates is a critical goal for vaccines that protect against HIV-1 infection. Potentially the greatest obstacle to achieving this goal is the extraordinary diversity that develops in the target of neutralizing antibodies, the envelope glycoprotein (Env). Although vaccines have thus far failed to induce broadly neutralizing antibody responses, there are examples of chronically infected patients with sera that neutralize highly diverse HIV-1 isolates1–8. These individuals provide evidence that it is possible for the human antibody response to neutralize highly diverse strains of HIV-1, though the mechanisms by which such responses are induced or mediated remain incompletely understood9,10.
Recently, isolation and characterization of human monoclonal antibodies from cells of chronically infected patients have provided considerable advances in understanding the specificities and mechanisms underlying broadly neutralizing antibody responses to HIV-1. Env exists on the virion and infected-cell surface as a trimer of heterodimers made up of gp120 and gp41 subunits. For some time, only a small number of broadly neutralizing monoclonal antibodies (mAbs) had been isolated consisting of one antibody that binds the CD4-binding site on gp120 (b12), one that binds a glycan configuration on the outer domain of gp120 (2G12) and three that bind the membrane-proximal external region (MPER) on gp41 (2F5, Z13e1, and 4E10) 11–13. More recently, considerably more broad and potent antibodies have been discovered that target the CD4-binding site of the envelope protein14–17 (for which VRC01 is a prototype) and glycan containing regions of the V1/V2 and V3 regions of gp1204,18–20 (for which PG9 and PGT128 are prototypes). The specificities of these new antibodies are providing important information regarding antigen targets on Env to which the humoral immune response might be directed to mediate broad and potent neutralization. However, evidence for these specificities in many chronically infected patients within our cohort is lacking, suggesting that broad and potent neutralization may be mediated by other specificities.
Here we report isolation of a broad and potent gp41 MPER-specific human mAb, 10E8, from an HIV-1-infected individual with high neutralization titers. 10E8 is among the most broad and potent antibodies thus far described, and lacks many of the characteristics previously thought to limit the usefulness of MPER-specific antibodies in vaccines or passive therapies, including lipid binding and autoreactivity. In addition, the crystal structure of 10E8, along with biochemical binding studies, demonstrate that the breadth of 10E8 is mediated by its unique mode of recognition of a structurally conserved site-of-vulnerability within the gp41 MPER.
10E8 isolation and neutralizing properties
To understand the specificities and binding characteristics that underlie a broadly neutralizing antibody response we developed techniques that permitted isolation of human monoclonal antibodies without prior knowledge of specificity20. Serum from one donor, N152, exhibited neutralizing breadth and potency in the top 1% of our cohort against a 20 cross-clade pseudovirus panel (Supplementary Table 1) 1. Peripheral blood CD19+IgM-IgD-IgA- memory B cells from this patient were sorted and expanded for 13 days with IL-2, IL-21, and CD40-ligand expressing feeder cells. The supernatants of ~16,500 B cell cultures were screened and IgG genes from wells with neutralization activity were cloned and re-expressed21 and two novel antibodies (10E8 and 7H6) were isolated.
Nucleotide sequence analysis of DNA encoding 10E8 and 7H6 revealed that both were IgG3 antibodies and were somatic variants of the same IgG clone. These antibodies were derived from IGHV3-15*05 and IGLV3-19*01 germline genes, and were highly somatically mutated in variable genes of both heavy chain (21%) and lambda light chain (14%) compared to germline. These antibodies also possessed a long heavy-chain complementarity-determining region (CDR H3) loop composed of 22 amino acids (Fig. 1a). The heavy chains of 10E8 and 7H6 were identical and there were only two residue differences in the light chain (Supplemental Fig. 1)22.
Figure 1. Analyses of 10E8 sequence and neutralization.

a, Inferred germline genes encoding the variable regions of 10E8 and 7H6. b, Neutralizing activity of antibodies against a 181-isolate Env-pseudovirus panel. Dendrograms indicate the gp160 protein distance of HIV-1 primary isolate Envs. Data below the dendrogram show the number of tested viruses, the percentage of viruses neutralized, and the geometric mean IC50 for viruses neutralized with an IC50<50 μg/ml. Median titers are based on all tested viruses, including those with IC50 >50ug/ml, which were assigned a value of 100.
To assess neutralization activity of the clonal variants, they were initially tested against 5 Env-pseudoviruses (Supplementary Table 1a), and mAb 10E8 was selected for further study. To determine if the neutralization activity of 10E8 was representative of the overall neutralization specificity present in patient N152 donor serum, the neutralization panel was expanded to 20 Env-pseudoviruses, and 10E8 was tested in parallel with N152 donor serum. Although there were some similarities in the pattern of neutralization of highly resistant variants, a correlation of the neutralization IC50 of mAb 10E8 and ID50 of N152 serum did not achieve statistical significance (p=0.11; Supplementary Fig. 2 and Supplementary Table 1b). This finding suggests that although 10E8 may play a major role, the full breadth of neutralization of by N152 serum is likely mediated by an amalgam of 10E8-like or other antibodies.
To compare the neutralization potency and breadth of 10E8 with other broadly neutralizing anti-HIV-1 antibodies, 10E8 was then tested in a 181-isolate Env-pseudovirus panel in parallel with 4E10, 2F5, VRC01, NIH45-46, 3BNC117, PG9, and PG16 (Fig. 1b and Supplementary Table 1c). At an IC50 below 50 μg/ml, 10E8 neutralized 98% of the tested pseudoviruses compared to 98% for 4E10 and 89% for VRC01. However, at an IC50 below 1 μg/ml, 10E8 neutralized 72% of the tested viruses compared to 37% for 4E10. The median and geometric mean IC50 values for 10E8 were below 1 ug/ml. Thus, 10E8 mediates broad and potent neutralization against a large range of viruses and the potency is comparable to some of the best available monoclonal antibodies.
10E8 epitope specificity and binding
To map the epitope of the 10E8 antibody, we tested binding to different subregions of Env by enzyme-linked immunosorbent assay (ELISA). 10E8 bound strongly to gp140, gp41, and the 4E10-specific MPER peptide, but not to gp120 (Fig. 2a). To further map the 10E8 epitope within the MPER, we examined binding of 10E8 to overlapping peptides corresponding to the 2F5 (656–671), Z13e1 (666–677), and 4E10 (671–683) specificities. 10E8 bound to the full MPER and the 4E10-specific peptides, but not 2F5- or Z13e1-specific peptides. Within the 4E10 epitope, when a peptide with a truncated C-terminus was tested, 4E10.19 (671–680), 10E8 binding was weakened considerably, suggesting that the three terminal amino acids of the MPER, Tyr681, Ile682, and Arg683, were crucial for 10E8 binding (Supplementary Fig.3a). Consistent with these results, only the full MPER and 4E10-specific peptides blocked 10E8-mediated neutralization of the chimeric C1 virus, which contains the HIV-2 Env with the HIV-1 MPER (Supplementary Fig. 3b). Taken together, these data suggest that the minimal 10E8 epitope is located within residues 671–683 of the MPER although additional contacts toward the amino terminus of the MPER could not be excluded.
Figure 2. Binding specificity of 10E8.
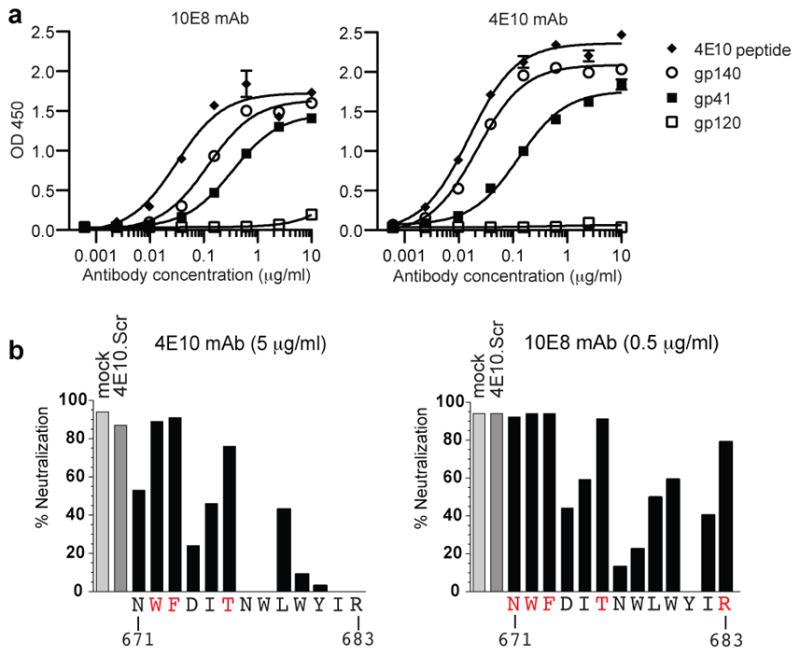
a, ELISA binding of mAb 10E8 or 4E10 to gp140, gp120, gp41, or 4E10 peptide. Error bars denote one standard error of the mean (SEM). b, Inhibition of mAb 10E8 or 4E10 neutralization of C1 HIV-2/HIV-1 MPER virus by 4E10 alanine scanning peptides. Residues shown in red indicate positions for which the alanine mutant peptide did not block neutralization.
To more precisely map the epitope of 10E8, a panel of alanine mutant peptides scanning MPER residues 671–683 was used to block 10E8 neutralization of the chimeric C1 virus in a TZM-bl assay (Fig. 2b)23. MPER peptides with alanine substitutions at Trp672, Phe673 or Thr676 failed to block 4E10 or 10E8 neutralization, suggesting that these residues are critical for both 4E10 and 10E8 binding. Residue Asn671 and residue Arg683, both of which are not required for 4E10 binding, were found to be critical for 10E8 binding and neutralization (Supplementary Table 2 and Fig. 2b). We also tested the ability of 10E8 to neutralize HIV-1JR2 pseudoviruses with alanine substitutions in MPER residues 660–683 (Supplementary Table 3). Consistent with the effects of alanine substitutions on peptide binding, residues Asn671 and Arg683 were critical for 10E8, but not 4E10, neutralization. Individual alanine substitutions at residues 671–673, 680 and 683 resulted in reduced neutralization sensitivity to 10E8 most apparent at the IC90 level rather than the IC50 level. Although the mechanism for this phenomenon is unclear, a similar effect has been observed previously when MPER mutations cause partial resistance to 4E1024. Taken together, these results suggested that 10E8 recognized a novel epitope, which overlaps the known 4E10 and Z13e1 epitopes, but differs in a critical dependence on binding to Asn671 and Arg683, the last residue of the MPER.
We next investigated whether the greater neutralization potency of 10E8 compared to other MPER antibodies was a result of higher binding affinity to the MPER. Capture of a biotinylated peptide encompassing the full MPER (656–683) to a surface-plasmon resonance chip allowed the binding kinetics of Fabs 10E8, 2F5 and 4E10 to be examined. In contrast to its higher neutralization potency, the KD of 10E8 to this MPER peptide was weaker than that of 2F5 and 4E10; 17 nM for 10E8 versus 3.8 nM for 2F5 and 0.74 nM for 4E10 (Supplementary Fig. 4). Therefore, the affinity of 10E8 for the MPER in a soluble peptide format did not explain its greater neutralization potency compared to other MPER-specific antibodies.
Prevalence of 10E8-like antibodies
We next investigated the prevalence of MPER-specific and 10E8-like neutralizing antibodies in our cohort of HIV-1-infected donors. We selected 78 sera from our cohort with a neutralization ID50>100 against at least 1 pseudovirus in a 5-virus mini-panel1. The median time since diagnosis of these donors was 13.5 years, median CD4 count was 557 cells/μl, median plasma HIV RNA 5573 copies/ml, and they were not receiving antiretrovirals. We tested neutralization against the HIV-2/HIV-1 chimera C1 (Supplementary Table 4)25. Of 78 sera, 21 exhibited neutralization activity against the HIV-2/HIV-1 C1 virus (Supplementary Table 5). To map the region that was targeted by these sera, neutralization was measured using 7 HIV-2/HIV-1 chimeras containing subdomains of the MPER (Supplementary Table 4)25. Of the 21 sera with neutralization activity against the entire MPER, 8 exhibited a neutralization pattern similar to that observed for 10E8, which entailed neutralization of only those HIV-2/HIV-1chimeric viruses that contained the terminal residue of the MPER Arg683 (C4, C4GW and C8; Supplementary Table 5). To further confirm these results, we used peptides corresponding to different portions of the MPER to block sera neutralization of the HIV-2/HIV-1 chimera C1 (Supplementary Table 6). Of the 8 sera found to have a 10E8-like pattern based upon neutralization of the chimeras, 3 were blocked by peptides consistent with 10E8-like activity. An additional 3 of the 8 10E8-like sera were blocked by peptides in a pattern consistent with a combination of 10E8 and Z13e1-like antibodies. The 6 patients whose sera had 10E8-like activity did not differ from the remaining 72 patients with regard to clinical course or HIV neutralization (Supplementary Fig. 5, legend). Overall, 27% of the tested patient sera exhibited anti-MPER neutralizing activity. This prevalence is considerably higher than observed in prior work, possibly related to selection of donors with known neutralizing activity8,26–28. Further, 8% of the tested sera had 10E8-like antibodies (Supplementary Fig. 5), suggesting that 10E8-like antibodies are not rare.
Analysis of 10E8 autoreactivity
A property common to the previously characterized MPER mAbs 2F5 and 4E10 is that they cross-react with self-antigens29. In addition, binding to both the cell membrane and the Env trimer is thought to be important for optimal neutralization by these antibodies and this autoreactivity may be an obstacle to the elicitation of similar antibodies by a vaccine29,30. Surface plasmon resonance analysis showed that 10E8 did not bind to anionic phospholipids, such as phosphatidyl choline-cardiolipin (PC-CLP) and phosphatidyl choline-phosphatidyl serine (PC-PS) liposomes (Fig. 3a). 10E8 also did not bind HEp-2 epithelial cells, in contrast to 2F5 and 4E10 that bound in a cytoplasmic and nuclear pattern (Fig. 3b). Additionally, 10E8 did not bind autoantigens, such as Sjogren's syndrome antigens A and B, Smith antigen, ribonucleoprotein, scleroderma 70 antigen, Jo1 antigen, centromere B and histone (Supplementary Table 7). Taken together, these results suggest that 10E8, in contrast to other MPER antibodies, is not autoreactive.
Figure 3. Analysis of 10E8 autoreactivity.

a, SPR analysis of 10E8 binding to anionic phospholipids. 10E8 was injected over PC-CLP liposomes or PC-PS liposome immobilized on the BIAcore L1 sensor chip. 4E10 and 2F5 were used as positive controls and 13H1, 17b, and anti-RSV F protein as negative controls. b, Reactivity of 10E8 with HEP-2 epithelial cells. Controls are as above with VRC01 added as an additional negative control. Antibody concentration was 25 μg/ml. All pictures are shown at 400x magnification.
Virion accessibility of 10E8
The 2F5 and 4E10 antibodies have been shown to bind relatively poorly to the HIV-1 envelope spike on the surface of infected cells or to cell-free virions, and react more efficiently after Env engagement of the CD4 receptor31. We measured binding to cleaved, full-length envelope spikes on HIVJRFL transfected cells (Supplementary Fig. 6a). Although 10E8 bound less efficiently than other antibodies such as VRC01 or F105, where accessibility is not an issue, it bound more efficiently than either 2F5 or 4E10. In contrast to results of alanine substitution, a mutation in the 4E10 (F673S) region in full-length HIVJRFL envelope spikes enhanced 10E8 binding although the mechanism remains unclear. A mutation in the 2F5 (K665E) region had no influence on 10E8 binding. These data suggest that 10E8 has modestly greater access to the MPER epitope on the cell surface than either 2F5 or 4E10.
To assess binding to cell-free virus, we incubated virions with antibody, washed out unbound antibody, and testing neutralization31–33. During washing, antibodies that cannot access their Env target on free virions will be largely removed and therefore neutralization will be diminished. As a control, neutralization of the HXBc2 isolate was not diminished by washing, because the MPER region is accessible on this laboratory adapted isolate31. Washing also had little impact on neutralization of JRFL by VRC01. Consistent with prior work, 2F5 and 4E10 neutralization of most virus isolates tested was substantially diminished after washing (Supplementary Fig. 6b)31,34. In contrast to 2F5 and 4E10, washing had a smaller effect on 10E8 neutralization of most viruses tested, as measured by the area under the curve or analysis of the fold-change in neutralization at a fixed inhibitory concentration (Supplementary Fig. 6c). Although 10E8 is not fully able to access its epitope on the native viral spike similarly to VRC01, under most experimental conditions tested it was better able to access its epitope than either 2F5 or 4E10.
Structure of 10E8-gp41 complex
To provide an atomic-level understanding of the interaction of 10E8 with HIV-1, we crystallized the antigen-binding fragment (Fab) of 10E8 in complex with a peptide encompassing the entire 28-residue gp41 MPER (residues 656–683). Monoclinic crystals diffracted to 2.1 Å resolution, and structure solution and refinement to Rcryst = 18.01% (Rfree = 21.76%) revealed two complexes in the asymmetric unit (heretofore referred to as complexes 1 and 2) (Supplementary Table 8). Overall, 10E8 bound to one face of the MPER peptide, which formed two helices, each 15–20 Å in length and oriented 100° relative to each other (Fig. 4a). Electron density was observed for the entire MPER, ranging from Asn656-Arg683 (Leu660-Arg683 for complex 2), with the highest degree of ordered density observed from residue Trp666 within the N-terminal helix through to Arg683 of the C-terminal helix (Supplementary Fig. 7). Analysis of main-chain dihedral angles (Supplementary Table 9) indicated that the N-terminal α-helix extends from residue Asn657 to Ala667, tightens into a 310-helix between residues Ser668 and Leu669, before turning at residues Trp670 and Asn671. The C-terminal α-helix, capped by Asn671, starts at residue Trp672 and extends to residue Arg683, the final residue of the MPER (Fig. 4a,b).
Figure 4. Crystal structure of 10E8 Fab in complex with its gp41 MPER epitope.

a. 10E8 recognizes a highly conserved gp41 helix to neutralize HIV-1. Fab 10E8 is shown in ribbon representation (shades of violet for heavy chain and of gray for light chain) in complex with a gp41 peptide (red) that encompasses the MPER (Asn656-Arg683). b, Interface between 10E8 and gp41 with select 10E8-side chains (green, heavy chain; cyan, light chain) and gp41-side chains (gray) in stick representation. In analogy to a hand, the hinge can be viewed as being gripped by a thumb (represented by the CDR H2), the C-terminal helix as being suspended along a corresponding extended forefinger (represented by the CDR H3), and residues that commence the C-terminal helix as being caught in the cleft between the thumb and forefinger (represented by the juncture of the CDR loops). c-d, Buried contact surfaces and epitope conservation. An examination of the buried contact surface on gp41 (gray; c) reveals that epitope residues (labeled, d) that are directly contacted by 10E8 are highly conserved across 2870 examined strains (Supplementary Tables 10–12; conservation percentages provided in parentheses). e-h, Alanine mutagenesis of paratope and epitope. Residues at the tip of the 10E8 CDR H3 loop and within the hydrophobic cleft are crucial for recognition of gp41 and for virus neutralization (Supplementary Tables 15–16), as mapped onto the buried 10E8-contact surface (e,g). These results mirror the effects of alanine scan mutations of the 10E8 epitope (Supplementary Tables 2–3), as mapped onto the buried gp41-contact surface (f,h). A comparison of the effects of the alanine mutagenesis of the paratope and epitope reveal that residues of the epitope that are most crucial for 10E8 recognition and neutralization, are also the most highly conserved (d).
The 10E8 antibody contacts the gp41 MPER primarily through its heavy chain, although crucial contacts are also mediated by the light chain CDR L3 (Fig. 4c and Supplementary Tables 10–12). Three predominant loci of interaction are observed between the antibody and gp41 (Supplementary Tables 13–14): One between residues of the tip of the CDR H3 loop and the tip of the C-terminal helix of the peptide, a second between residues of the CDR H2 loop and residues of the hinge region of the peptide, and a third at the juncture of the three heavy chain CDR loops and the light chain CDR L3, which form a hydrophobic cleft that holds residues of the beginning of the MPER C-terminal helix (Fig. 4b).
10E8-gp41 interface
To complement the results observed for the mutagenesis of the highly conserved 10E8 epitope (Fig. 4d and Supplementary Table 2), each residue of the 10E8 paratope, as determined from the crystal structure, was individually mutated to alanine and the resulting 25 10E8 variants assessed for affinity to a soluble MPER peptide. Overall, the most pronounced effects of the alanine mutations on the binding affinity of 10E8 to a soluble MPER peptide occurred within residues of the CDR H3 loop, though mutations within the hydrophobic cleft also showed substantial effect (Fig. 4e, Supplementary Table 15, and Supplementary Fig. 8). 10E8 residues identified by alanine scan as critical for the interaction with gp41 stretched from the cleft all the way to the tip of the CDR H3 (Fig. 4e) and were mirrored by a corresponding stretch of gp41 residues which substantially affected 10E8 binding when mutated to alanine (Fig. 4f).
The same panel of 10E8 alanine mutations was tested for neutralization potency against a panel of five Env-pseudoviruses that included both Tier 1 and Tier 2 viruses (Supplementary Table 16). Similar to the binding data, residues of the 10E8 CDR H3 had dramatic effects on neutralization, as did residues of the hydropohobic cleft (Fig. 4g). Generally, KDs of paratope mutants correlated with neutralization (Supplementary Fig. 9). Backbone interactions (on both 10E8 and gp41) also contribute to the interface, especially between the CDR H2 of 10E8 and the hinge region of the MPER, though these are silent in alanine scan analyses. Overall, 10E8 utilizes a narrow band of residues (~20 x 5 Å) that stretches from the CDR H1 and H2 and extends along most of the CDR H3 to recognize a string of highly conserved hydrophobic gp41 residues, and a critical charged residue, Arg/Lys683, that occurs just prior to the transmembrane region (Fig. 4f,h).
A conserved gp41-neutralization determinant
Several structures of neutralizing antibodies in complex with the MPER of gp41 have been reported previously, including those for antibodies 2F5, Z13e1 and 4E1035–39(Supplementary Fig. 10a). The MPER adopts divergent loop conformations when bound by 2F5 and Z13e1 and an α-helix when bound by 4E10. Comparison of 2F5, Z13e1, and 4E10 epitopes with 10E8-bound gp41 revealed that only the 4E10 epitope has similar secondary structure, with superposition yielding an RMSD of 2.49 Å for all atoms of residues 671–683 and 0.98 Å for main-chain atoms (Supplementary Fig. 10b, Supplementary Table 17).
To compare further the recognition of 10E8 and 4E10, we examined their angles of epitope approach. As shown in Supplementary Fig. 10c-f, alignment of the recognized MPER helix places 10E8 and 4E10 into similar overall spatial positions. The relative orientations of the recognized helix and the heavy and light chains of the two antibodies, however, differ dramatically. With 10E8, the C-terminal helix is perpendicular to the plane bisecting heavy and light chains (Supplementary Fig. 10c,e); with 4E10, the recognized helix is at the interface between heavy and light chains (Supplementary Fig. 10d,f). Perhaps relevant to this, 10E8 utilizes CDR loops almost exclusively in its recognition of gp41, while 4E10 incorporates substantial β-strand interactions with gp41 at the interface between the heavy and light chains.
The differing modes of 10E8 and 4E10 recognition of the conserved C-terminal MPER helix result in a substantial difference in the proportion of the recognized helical face: 10E8 contacts roughly a third of the helical face, while 4E10 contacts over half (Supplementary Fig. 10g,h and Supplementary Tables 18–19). The smaller contact surface of 10E8 may provide an explanation for the reduced recognition of lipid surfaces by 10E8 versus 4E10 – providing a potential structure-based explanation for reduced autoreactivity of 10E8.
Sequence variation and 10E8 neutralization
To place the specificity and structural data into the context of known variation of the MPER, we analyzed viral sequences with resistance to neutralization by 10E8 (Fig. 5a). Of the 183 viruses tested, only 3 were highly resistant to 10E8 with IC50 >50 μg/ml. Each of these viruses had substitutions at positions found to affect neutralization by alanine scanning (Asn671, Trp672, Phe673, and Trp680). Plasma virus of the patient N152, from whom 10E8 was cloned, is also likely resistant to 10E8 mediated neutralization40. Sequence analysis of plasma viral RNA revealed rare substitutions at positions Trp680 and Lys/Arg683 (Fig. 5a). These residues are typically highly conserved with variation only occurring in 1.17% of 3,730 HIV Env sequences in the Los Alamos Database (www.hiv.lanl.gov). When the substitutions for the 3 resistant viruses and the patient viruses were placed on the background of the sensitive JR2 virus, substitutions at Asn671Thr, Trp672Leu, and Phe673Leu had a modest effect on the IC50 but raised the IC80 above 20 μg/ml. In the structural analysis, direct contacts with 10E8 were not observed at position 671 suggesting the effects on neutralization of Thr or Ala substitutions at this position are mediated by conformational or other effects within gp41. The combination of Trp672Leu and Phe673Leu conferred high-level resistance at the IC50 and IC80 level. Changes corresponding to the patient’s dominant circulating virus had a similar effect. Although Lys/Arg683Gln alone conferred resistance at the IC80 level, together Trp680Arg and Lys/Arg683Gln resulted in greater resistance to 10E8 (Fig. 5a). When taken together with the analysis of the 10E8 paratope, these data suggest that in addition to Trp672, Phe673, and Trp680 found in the 4E10 epitope, the additional 10E8-bound residue Lys/Arg683 is critical to neutralization. In addition to other differences in binding based upon structural analyses noted above, it is possible that the additional potency of 10E8 compared to 4E10 against naturally occurring viral variants may be mediated through binding of highly conserved residues Trp680 and Lys/Arg683 that directly interact with the 10E8 CDRH3.
Figure 5. A site of gp41 vulnerability.

a, Impact of sequence variation on 10E8 neutralization. Predicted amino acid sequences within the binding epitope of 10E8 for 3 10E8 resistant viruses and the patient virus are shown. The 10E8 epitope and differences in sequence compared to the JR2 virus are labeled in red. IC50 and IC80 values that are >20-fold than JR2 wild-type pseudovirus are highlighted in yellow. Error bars denote one SEM. b, Structural definition of a highly conserved region of gp41 recognized by neutralizing antibodies. Atoms of highly conserved residues that make direct contacts with 10E8 are colored red and shown in stick representation, atoms buried by 10E8 are colored purple, and main chain-contacting atoms are colored cyan. Semi-transparent surfaces of the gp41 MPER are colored according to the underlying atoms. 90° views are shown, with bound antibody 10E8 in the right panel. The 10E8 CDR H3 interacts with highly conserved hydrophobic residues, whereas the CDR H2 contacts main chain atoms at the juncture between the N- and C-terminal helices. Many of the unbound residues of the MPER (gray) are hydrophobic, especially those within the C-terminal helix. In the structure of a late fusion intermediate (Supplementary Fig. 8) these residues face towards the outside of a helical coiled-coil; in the pre-fusion conformation of the viral spike, these may interact with the viral membrane or with other hydrophobic regions of Env.
Discussion
10E8 is a broad and potent neutralizing antibody with important implications for efforts to stimulate such antibodies with vaccines. Previous MPER antibodies were somewhat limited in potency, and had a more limited ability to access MPER on Env of primary isolates. In addition, lipid binding and autoreactivity were thought to be characteristics of MPER antibodies and important obstacles to their elicitation by vaccines9,29,30. However, 10E8 lacks each of these characteristics. In addition, antibodies with a similar specificity were not rare in our chronically infected cohort. This suggests that 10E8-like antibodies were not deleted from the repertoire because of autoreactivity. These results further suggest that 10E8-like antibodies might be raised in a larger fraction of HIV-uninfected persons receiving a vaccine designed to elicit these antibodies without the B cell defects of chronic HIV infection. Design of such a vaccine will likely require not only presentation of an intact 10E8 epitope but also use of a platform sufficiently immunogenic to drive the evolution of 10E8-like antibodies.
The extraordinary breadth and potency of 10E8 appears to be mediated by its ability to bind highly conserved residues within MPER. Although the epitope of 10E8 overlaps those of known mAbs such as 4E10, it differs in recognition surface, angle of approach, lipid binding, and self-reactivity. Alanine scanning, structural analysis, and paratope analysis each indicate that 10E8 makes crucial contacts with highly conserved residues Trp672, Phe673, Trp676 and Lys/Arg683. The extraordinary breadth of some potent mAbs, for example that bind the CD4 binding site, is thought to be conferred by blocking a functionally important site that is critical for viral entry. Whether 10E8 impairs Env function or simply acts by binding highly conserved residues remains to be determined. Nonetheless, the breadth and potency of 10E8 demonstrates a conserved site of gp41 vulnerability (Fig. 5b) that is an important target antigen for HIV neutralization and that will likely reinvigorate interest in MPER-based HIV vaccine design.
Methods
Study patients
We selected the plasma and peripheral blood mononuclear cells (PBMC) from the HIV-1-infected patients enrolled in the National Institute of Health under a clinical protocol approved by the Investigational Review Board in the National Institute of Allergy and Infectious Diseases (NIAID-IRB). All participants signed informed consent approved by the NIAID-IRB. The criteria for enrollment were as follows: having a detectable viral load, a stable CD4 T-cell count above 400 cells/μl, being diagnosed with HIV infection for at least 4 years, and off ARV treatment for at least 5 years. Based on the locations of current and former residences, all patients were presumed to be infected with clade B virus. Donor N152 was selected for B cell sorting and antibody generation because his serum neutralizing activity is among the most potent and broad in our cohort. He is a slow progressor based on criteria described previously41. At the time of leukapheresis, he had been infected with HIV-1 for 20 years, with CD4 T-cell counts of 325 cells/μl, plasma HIV-1 RNA values of 3,811 copies/ml and was not on antiretroviral treatment.
Viruses and plasmids
HIV-1 JR2 MPER alanine mutant pseudovirus plasmids were obtained from Michael Zwick (The Scripps Research Institute, La Jolla, CA). HIV-2/HIV-1 chimeras were kindly provided by Lynn Morris (National Institute for Communicable Diseases, Johannesburg, South Africa).
Memory B-cell staining, sorting and antibody cloning
Staining and single-cell sorting of memory B cells were performed as follows. PBMCs from HIV-1 infected donor N152 were stained with antibody cocktail consisting of anti-CD19-PE-Cy7 (BD Bioscience), IgA-APC (Jackson ImmunoResearch Laboratories Inc.), IgD-FITC (BD Pharmingen), and IgM-PE (Jackson ImmunoResearch Laboratories Inc.) at 4ºC in dark for 30 min. The cells were then washed with 10 ml PBS-BSA buffer and resuspended in 500 μl PBS-BSA. 66,000 CD19+IgA-IgD-IgM- memory B cells were sorted using a FACSAria III cell sorter (Becton Dickinson) and resuspended in IMDM medium with 10% FBS containing 100 U/ml IL-2, 50 ng/ml IL-21 and 1x105/ml irradiated 3T3-msCD40L feeder cells42. B cells were seeded into 384-well microtiter plates at a density of 4 cells/well in a final volume of 50 μl. After 13 days of incubation, 40 μl of culture supernatants from each well were collected and screened for neutralization activity using a high throughput micro-neutralization assay against HIV-1MN.03 and HIV-1Bal.26. B cells in each well were lysed with 20 μl lysis buffer containing 0.25 μl of RNase inhibitor (New England Biolabs Inc.), 0.3 μl of 1M Tris pH8 (Quality Biological Inc.) and 19.45 μl DEPC-treated H2O. The plates with B cells were stored at -80°C.
The variable region of the heavy chain and the light chain of the immunoglobulin gene were amplified by RT-PCR from the wells that scored positive in both the HIV-1MN.03 and HIV-1Bal.26 neutralization assay. The cDNA product was used as template in the PCR reaction. In order to amplify the highly somatically mutated immunoglobulin gene, two sets of primers as described previously21 were used in two independent PCRs. One set of primers consisted of the forward primers and the reverse primers specific for the leader region and constant region of IgH, Igκ or Igλ, respectively. The other set of primers consisted of the forward primer mixes specific for FWR1 and respective reverse primers specific for the IgH, Igκ and Igλ J genes. All PCRs were performed in 96-well PCR plates in a total volume of 50 μl containing 20 nM each primer or primer mix, 10 nM each dNTP (Invitrogen), 10 μl 5x Q-solution (Qiagen) and 1.2 U HotStar Taq DNA polymerase (Qiagen). From the positive PCR reactions, pools of the VH or VL-region DNA were ligated to a pCR2.1-Topo-TA vector (Invitrogen) for sequencing before cloning into the corresponding Igγ1, Igκ and Igλ expression vector. 10 μg of heavy and light chain plasmids, cloned from the same well and combined in all possible heavy and light chain pairs, were mixed with 40 μl FuGENE 6 (Roche) in 1500 μl DMEM (Gibco) and co-transfected into 293T cells. The full-length IgG was purified using a recombinant protein-A column (GE Healthcare)
Neutralization assays
Neutralization of the monoclonal antibodies was measured using single-round HIV-1 Env-pseudoviruses infection of TZM-bl cells43. HIV-1 Env-pseudoviruses were generated by co-transfection of 293T cells with pSG3 delta Env backbone and a second plasmid that expressed HIV-1 Env. At 72 h post-transfection, supernatants containing pseudovirus were harvested and frozen at -80°C until further use. In the neutralization assay, 10 μl of 5-fold serially diluted patient serum or mAb was incubated with 40 μl pseudovirus in a 96-well plate at 37°C for 30 min before addition of TZM-bl cells. After 2 days of incubation, cells were lysed and the viral infectivity was quantified by measuring luciferase activity with a Victor Light luminometer (Perkin Elmer). The 50% inhibitory concentration (IC50) was calculated as the antibody concentration that reduced infection by 50%. Antibody epitopes were mapped using HIV-1 JR2 MPER alanine mutant pseudoviruses in a TZM-bl assay.
HIV-2/HIV-1 chimera neutralization
HIV-2/HIV-1 C1 chimera (HIV-2 virus 7312A with HIV-1 gp41 MPER)25 was used in the competition assay. A fixed concentration of MPER peptide was incubated with serially diluted 2F5, 4E10, Z13e1 or 10E8 antibody at 37°C for 30 min before incubation with HIV-2/HIV-1 C1 chimera. Wild-type HIV-2 virus 7312A was used as a control. Antibody epitope mapping was completed by adding 10 μl 10E8 mAb to 5 μl serial dilutions of 4E10 peptide or its alanine mutants at 37°C for 30 min prior to the addition of HIV-2/HIV-1 C1 chimera. The degree to which peptides blocked antibody-mediated neutralization was calculated as the fold change in the IC50 value of the antibody in the presence of 4E10 alanine mutants compared to the wild-type peptide. The precise binding region within the MPER targeted by patient serum or antibodies was determined using the HIV-2/HIV-1 chimeras containing different portions of HIV-1 MPER, such as C1 (HIV-2 Env with HIV-1 MPER), C1C (HIV-2 Env with clade C MPER), C3 (HIV-2 Env with 2F5 epitope), C4 (HIV-2 Env with 4E10 epitope), C6 (HIV-2 Env with short 4E10 epitope NWFDIT), C7 (HIV-2 Env with short 2F5 epitope ALDKWA) and C8 (HIV-2 Env with both Z13 and 4E10 epitope). 5-fold diluted patient serum or mAb was incubated with chimera in a 96-well plate at 37°C for 30 min before addition of TZM-bl cells. The specificities within patient sera were confirmed by blocking neutralization of the C1 chimera with 25 μg/ml of 2F5, 4E10, MPER, Bal.V3, control peptide, or 50 μg/ml of Z13 peptide.
ELISA assays
Each antigen at 2 μg/ml was coated on 96-well plates overnight at 4°C. Plates were blocked with BLOTTO buffer (PBS, 1% FBS, 5% non-fat milk) for 1 h at room temperature (RT), followed by incubation with antibody serially diluted in disruption buffer (PBS, 5% FBS, 2% BSA, 1% Tween-20) for 1 h at RT. 1:10,000 dilution of horseradish peroxidase (HRP)-conjugated goat anti-human IgG antibody was added for 1 h at RT. Plates were washed between each step with 0.2% Tween 20 in PBS. Plates were developed using 3,3′,5,5′-tetramethylbenzidine (TMB) (Sigma) and read at 450 nm.
Autoreactivity assays
Binding of 10E8 to phospholipid was measured by SPR conducted on a BIAcore 3000 instrument and data analyses were performed using the BIAevaluation 4.1 software (BIAcore) as described previously30. Phospholipid-containing liposomes were captured on a BIAcore L1 sensor chip, which uses an alkyl linker for anchoring lipids. Before capturing lipids, the surface of the L1 chip was cleaned with a 60-s injection of 40 mM octyl-β-D-glucopyranoside, at 100 μl/minute, and the chip and fluidics were washed with excess buffer to remove any traces of detergent. mAbs were then injected at 100 μg/ml at a flow rate of 30 μl/min. After each Ab injection, the surface was again cleaned with octyl -D-glucopyranoside, and 5-s injections of each 5 mM HCl, then 5 mM NaOH, to clean any adherent protein from the chip.
Reactivity to HIV-1 negative human epithelial (HEp-2) cells was determined by indirect immunofluorescence on slides using Evans Blue as a counterstain and FITC-conjugated goat anti-human IgG (Zeus Scientific, Raritan N.J.) 29. Slides were photographed on a Nikon Optiphot fluorescence microscope. Regarding Figure 3b, kodachrome slides were taken of each MAb binding to HEp-2 cells at a 32 second exposure, and the slides scanned into digital format. The Luminex AtheNA Multi-Lyte ANA test (Wampole Laboratories, Princeton, NJ) was used to test for MAb reactivity to SSA/Ro, SS-B/La, Sm, ribonucleoprotein (RNP), Jo-1, double-stranded DNA (dsDNA), centromere B, and histone and was performed per the manufacturer's specifications and as previously described29. MAb concentrations assayed were 50, 25, 12.5 and 6.25 μg/ml. 10 μl of each concentration were incubated with the luminex fluorescent beads and the test performed per manufacturer’s specifications.
Fluorescence-activated cell sorting (FACS) staining of cell-surface HIV-1 Env
FACS staining was performed as previously described31,44. 48 h following transfection, cells were harvested and washed in FACS buffer (PBS, 5% HIFBS, 0.02% azide) and stained with monoclonal antibodies. The transfected cells were suspended in FACS buffer and were incubated with the antibodies for 1 h at RT. The monoclonal antibody-cell mixture was washed extensively in FACS buffer and phycoerythrin (PE)-conjugated goat anti-human secondary antibody (Sigma) was added for 1 h at a 1:200 dilution, followed by extensive washing to remove unbound secondary antibody. The antibody-PE-stained cells were acquired on a BD LSRII instrument and analyzed by FlowJo.
Antibody-virus washout experiments
From a starting concentration of 2 mg/ml, 12.5 μl of 5-fold serially diluted antibodies in PBS were added to 487.5 μl of DMEM containing 10% HIFCS and 15 μl of pseudovirus such that the final concentrations of antibodies were 50 μg/ml to 0.08 μg/ml in a total volume of 500 μl. In the “no inhibitor” control, the same volume of PBS was added instead of antibody. The reaction mixture was incubated for 30 min at 37°C. The 250 μl reaction mixture was diluted to 10 ml with complete DMEM, centrifuged at 25,000 rpm in a SW41 rotor, for 2 h at 4°C. The virus pellet was then washed two additional times with 10 ml of PBS. During the washing steps, the virus-antibody complex was centrifuged at 40,000 rpm for 20 min at 4°C. After the final wash, 250 μl of DMEM was added to the washed virus pellet and it was resuspended by gentle shaking at 4°C for 30 min. 100 μl of the suspended virus was used to infect 100 μl of TZM-bl cells (0.2 million/ml), in duplicate. From the remaining 250 μl of reaction mixture, an equal volume of the antibody virus mixture was used as a “no washout” control. Plates were incubated at 37°C in a CO2 incubator for 2 days. After 2 days, the luciferase assay was done as described previously45. The data was then plotted to determine the neutralization mediated by the antibodies in “wash” or “no wash” condition.s
Structure determination and analysis
The antigen binding fragment of 10E8 (Fab) was prepared using LysC digestion, as previously described46. The IgG was first reduced with 100 mM DTT for 1 h at 37 °C, followed by 1 h of dialysis in Hepes, pH 7.6, to reduce the DTT concentration to 1 mM. Antibodies were then dialyzed against 2 mM iodoacetamide for 48 h at 4 °C, and subjected to a final dialysis against Hepes, pH 7.6, for 2 h. After reduction and alkylation, antibodies were cleaved with Lys-C (Roche), run over a Protein A column to segregate away the Fc fragment, and then subjected to ion exchange (Mono S) and size-exclusion chromatography (S200). Purified 10E8 Fab was incubated with 10-fold excess peptide RRR-NEQELLELDKWASLWNWFDITNWLWYIR-RRR (American Peptide, CA) and the complex then set up set for 20°C vapor diffusion sitting drop crystallizations on the Honeybee 963 robot. 576 initial conditions adapted from the commercially available Hampton (Hampton Research), Precipitant Synergy (Emerald Biosystems), and Wizard (Emerald Biosystems) crystallization screens were set up and imaged using the Rockimager (Formulatrix), followed by hand optimization of crystal hits. Crystals were grown in 40% PEG 400, 0.1 M NaCitrate, 0.1 M Tris pH 7.5 diffracted to 2.1 Å resolution in a cryoprotectant composed of mother liquor supplemented with15% 2R-3R-butanediol and excess peptide. After mounting the crystals on a loop, they were flash cooled and data was collected at 1.00 Å wavelength at SER CAT ID-22 or BM-22 beamlines (APS) and processed using HKL-200047. Structures were solved through molecular replacement with Phaser48,49, using a previously obtained free structure of 10E8 as a search model. Refinement of the structure was undertaken with Phenix50, with iterative model building using Coot51. The structure was validated with MolProbity52, yielding 97% and 99.8% of residues falling within most favored Ramachandran regions and allowed Ramachandran regions, respectively. The structure was analyzed with APBS53 for electrostatics, Ligplot54 for direct contacts, PISA55 for buried surface areas, and lsqkab (ccp4 Package56) for RMSD alignments. Helical wheels were generated using the program Pepwheel (http://150.185.138.86/cgi-bin/emboss/pepwheel). All graphics were prepared with Pymol (PyMOL Molecular Graphics System).
Assessment of binding affinities of 10E8 and 10E8 variants to the gp41 MPER
Surface–Plasmon Resonance (SPR) (Biacore T200, GE Healthcare) was used to assess binding affinity of wild type 10E8 to a gp41 MPER peptide. A biotinylated peptide composed of residues 656–683 of the gp41 MPER (RRR-NEQELLELDKWASLWNWFDITNWLWYIR-RRK-biotin; American Peptide, CA) was coupled to a biacore SA chip to a surface density of 20–50 Response Units (RU). The 10E8 fragment of antigen binding (Fab) was then flowed over as analyte at concentrations ranging from 0.25 nM to 125 nM, at 2-fold serial dilutions, with association and dissociation phases of up to 5 min, at a flow rate of 30 ml/min. The binding of the 2F5 and 4E10 Fab controls to the same peptide were examined under identical conditions.
Binding affinities of the 10E8 paratope alanine mutants to the MPER were also assessed with SPR, but using an antibody capture method. A Biacore CM5 chip was amine-coupled with anti-human Fc antibody to high surface densities of ~10,000 RU. The 10E8 paratope variant IgGs were then captured to between 1500–2500 RU and a peptide composed of residues 656–683 of the gp41 MPER (RRR-NEQELLELDKWASLWNWFDITNWLWYIR-RRR) flowed over as analyte at 2-fold serial dilutions starting at 500 nM (with the exception of HC D30A, W100bA, S100cA, P100fA, which started at 250 nM). Association and dissociation phases spanned 3 min and 5 min, respectively, at a flow rate of 30 μl/min. Binding sensograms were fit with 1:1 Langmuir models using Biacore BiaEvaluation Software (GE Healthcare). In all cases, Biacore HBSEP+ buffer was used (10 mM Hepes, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.1% P-20).
PCR amplification and sequencing
Extraction of viral RNA from plasma and cDNA synthesis were performed as previously described57. Single molecules of a 588 bp fragment, encompassing the MPER region of the HIV-1 envelope gene, obtained through limiting dilution, were PCR-amplified with the Expand High Fidelity PCR System (Roche Applied Science, Indianapolis, IN) using the following primer sets: +7789 (sense) 5’-TCTTAGGAGCAGCAGGAAGCACTATGGG-3’ and -8524 (antisense) 5’-GTAAGTCTCTCAAGCGGTGGTAGC-3’ in a first round reaction; +7850 (sense) 5’-ACAATTATTGTCTGGTATAGTGCAACAGCA-3’ and -8413 (antisense) 5’-CCACCTTCTTCTTCGATTCCTTCGG-3’ in a second round reaction. Each round of PCR consisted of 25 cycles, with the initial denaturation at 94 °C for 2 min, followed by 25 cycles of denaturation at 94 °C for 15s, annealing at 50 °C for 30s, and extension at 72 °C for 1 min, with the final extension at 72 °C for 7 min. The PCR products were purified with the QIA quick PCR purification kit (QIAGEN, Valencia, CA), and then cloned into pCR2.1-TOPO vector (TOPO TA Cloning it, Invitrogen, Carlsbad, CA) for sequence analysis of individual molecular clones. The DNAs from 18 independent clones were sequenced with the ABI BigDye Terminator v3.1 Ready Reaction Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and analyzed with the ABI PRISM 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA).
Statistical analysis
The relationship between the potency of N152 patient serum and 10E8, and the relationship between 10E8 variant binding and neutralization were evaluated by the Spearman rank method.
Supplementary Material
Acknowledgments
We thank Claire W. Hallahan (Biostatistics Research Branch, National Institute of Allergy and Infectious Diseases, Bethesda, MD) for statistical analyses. We thank Krissey Lloyd. Robert Parks, Josh Eudailey, and Jukie Blinn for performing autoantibody assays. We also thank M. Zwick (Department of Immunology and Microbial Science, The Scripps Research Institute, La Jolla, CA, USA.) for providing us the HIV-1 JR2 MPER alanine mutant pseudovirus plasmids. HIV-2/HIV-1 chimeras were kindly provided by L. Moore (National Institute for Communicable Diseases, Johannesburg, South Africa). We thank J. Stuckey for assistance with figures, and members of the Structural Biology Section and Structural Bioinformatics Core, Vaccine Research Center, for discussions and comments on the manuscript. This project has been funded in part with federal funds from the Intramural Research Programs of NIAID and the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. Use of sector 22 (Southeast Region Collaborative Access team) at the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under contract number W-31-109-Eng-38. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Author Contributions M.C., J.H., L.L., G.O., J.M., P.D.K., designed the study, analyzed the data, and prepared this manuscript. J.H., L.L. performed B cell sorting, antibody cloning, epitope mapping assay, MPER-specific neutralizing sera screening and assessed the impact of sequence variation on 10E8 neutralization. M.L., J. M. tested the 10E8 breadth and potency. B.C., S.K.S, and R. W. performed the infected cell surface staining and antibody-virion washout assays. S. A., B. H. performed the autoreactivity assays. G.O., Y.Y. and P.D.K. performed 10E8 structural analysis, with T.W. and B.Z. assisting with paratope alanine scanning. R.T.B. screened the B-cell culture supernatants for neutralization activity. H.I. sequenced the patient N152 virus. S. M. led the clinical care of the patients. M.C., L.L., N.R. and N.S.L. optimized B-cell culture protocol.
Author Information The nucleotide sequence of 10E8 heavy and light chains have been submitted to GenBank under accession numbers JX645769 and JX645770. Coordinates and structure factors for 10E8 Fab in complex with the gp41 MPER have been deposited with the Protein Data Bank under accession code 4G6F.
Competing financial interests The authors declare no competing financial interests.
References
- 1.Doria-Rose NA, et al. Breadth of human immunodeficiency virus-specific neutralizing activity in sera: clustering analysis and association with clinical variables. J Virol. 2010;84:1631–1636. doi: 10.1128/JVI.01482-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stamatatos L, Morris L, Burton DR, Mascola JR. Neutralizing antibodies generated during natural HIV-1 infection: good news for an HIV-1 vaccine? Nature medicine. 2009 doi: 10.1038/nm.1949. [DOI] [PubMed] [Google Scholar]
- 3.Sather DN, et al. Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J Virol. 2009;83:757–769. doi: 10.1128/JVI.02036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker LM, et al. A limited number of antibody specificities mediate broad and potent serum neutralization in selected HIV-1 infected individuals. PLoS Pathog. 2010;6:e1001028. doi: 10.1371/journal.ppat.1001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simek MD, et al. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol. 2009;83:7337–7348. doi: 10.1128/JVI.00110-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Binley J. Specificities of broadly neutralizing anti-HIV-1 sera. Curr Opin HIV AIDS. 2009;4:364–372. doi: 10.1097/COH.0b013e32832e06fe. [DOI] [PubMed] [Google Scholar]
- 7.Moore PL, et al. Potent and broad neutralization of HIV-1 subtype C by plasma antibodies targeting a quaternary epitope including residues in the V2 loop. J Virol. 2011;85:3128–3141. doi: 10.1128/JVI.02658-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gray ES, et al. Antibody specificities associated with neutralization breadth in plasma from human immunodeficiency virus type 1 subtype C-infected blood donors. J Virol. 2009;83:8925–8937. doi: 10.1128/JVI.00758-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haynes BF, Kelsoe G, Harrison SC, Kepler TB. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol. 2012;30:423–433. doi: 10.1038/nbt.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker LM, Burton DR. Rational antibody-based HIV-1 vaccine design: current approaches and future directions. Curr Opin Immunol. 2010;22:358–366. doi: 10.1016/j.coi.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zwick MB, et al. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol. 2001;75:10892–10905. doi: 10.1128/JVI.75.22.10892-10905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burton DR, et al. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science. 1994;266:1024–1027. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 13.Muster T, et al. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J Virol. 1993;67:6642–6647. doi: 10.1128/jvi.67.11.6642-6647.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonsignori M, et al. Two distinct broadly neutralizing antibody specificities of different clonal lineages in a single HIV-1-infected donor: implications for vaccine design. J Virol. 2012;86:4688–4692. doi: 10.1128/JVI.07163-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333:1593–1602. doi: 10.1126/science.1207532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheid JF, et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. 2011;333:1633–1637. doi: 10.1126/science.1207227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu X, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker LM, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonsignori M, et al. Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J Virol. 2011;85:9998–10009. doi: 10.1128/JVI.05045-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker LM, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tiller T, et al. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. Journal of immunological methods. 2008;329:112–124. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabat, E. A. & National Institutes of Health (U.S.). Office of the Director. Sequences of proteins of immunological interest : tabulation and analysis of amino acid and nucleic acid sequences of precursors, V-regions, C-regions, J-chain, T-cell receptors for antigenm T-cell surface antigens, [beta] 2-microglobulins ,major histocompatibility antigens, Thy-1, complement, C-reactive protein, thymopoietin, integrins, post-gamme globulin, [alpha]2-macroglobulins, and other related proteins. 5. U.S. Dept. of Health and Human Services, Public Health Service, National Institutes of Health; 1991. [Google Scholar]
- 23.Brunel FM, et al. Structure-function analysis of the epitope for 4E10, a broadly neutralizing human immunodeficiency virus type 1 antibody. J Virol. 2006;80:1680–1687. doi: 10.1128/JVI.80.4.1680-1687.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zwick MB, et al. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. J Virol. 2005;79:1252–1261. doi: 10.1128/JVI.79.2.1252-1261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray ES, et al. Neutralizing antibody responses in acute human immunodeficiency virus type 1 subtype C infection. J Virol. 2007;81:6187–6196. doi: 10.1128/JVI.00239-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomaras GD, et al. Polyclonal B cell responses to conserved neutralization epitopes in a subset of HIV-1-infected individuals. J Virol. 2011;85:11502–11519. doi: 10.1128/JVI.05363-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris L, et al. Isolation of a human anti-HIV gp41 membrane proximal region neutralizing antibody by antigen-specific single B cell sorting. PLoS ONE. 2011;6:e23532. doi: 10.1371/journal.pone.0023532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray ES, et al. Broad neutralization of human immunodeficiency virus type 1 mediated by plasma antibodies against the gp41 membrane proximal external region. J Virol. 2009;83:11265–11274. doi: 10.1128/JVI.01359-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haynes BF, et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science. 2005;308:1906–1908. doi: 10.1126/science.1111781. [DOI] [PubMed] [Google Scholar]
- 30.Alam SM, et al. Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20234–20239. doi: 10.1073/pnas.0908713106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakrabarti BK, et al. Direct antibody access to the HIV-1 membrane-proximal external region positively correlates with neutralization sensitivity. J Virol. 2011;85:8217–8226. doi: 10.1128/JVI.00756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frey G, et al. A fusion-intermediate state of HIV-1 gp41 targeted by broadly neutralizing antibodies. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3739–3744. doi: 10.1073/pnas.0800255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rathinakumar R, Dutta M, Zhu P, Johnson WE, Roux KH. Binding of anti-membrane-proximal gp41 monoclonal antibodies to CD4-liganded and -unliganded human immunodeficiency virus type 1 and simian immunodeficiency virus virions. J Virol. 2012;86:1820–1831. doi: 10.1128/JVI.05489-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruprecht CR, et al. MPER-specific antibodies induce gp120 shedding and irreversibly neutralize HIV-1. J Exp Med. 2011;208:439–454. doi: 10.1084/jem.20101907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Julien JP, Bryson S, Nieva JL, Pai EF. Structural details of HIV-1 recognition by the broadly neutralizing monoclonal antibody 2F5: epitope conformation, antigen-recognition loop mobility, and anion-binding site. J Mol Biol. 2008;384:377–392. doi: 10.1016/j.jmb.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 36.Cardoso RM, et al. Structural basis of enhanced binding of extended and helically constrained peptide epitopes of the broadly neutralizing HIV-1 antibody 4E10. J Mol Biol. 2007;365:1533–1544. doi: 10.1016/j.jmb.2006.10.088. [DOI] [PubMed] [Google Scholar]
- 37.Cardoso RMF, et al. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005;22:163–173. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 38.Ofek G, et al. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pejchal R, et al. A conformational switch in human immunodeficiency virus gp41 revealed by the structures of overlapping epitopes recognized by neutralizing antibodies. J Virol. 2009;83:8451–8462. doi: 10.1128/JVI.00685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu X, et al. Selection pressure on HIV-1 envelope by broadly neutralizing antibodies to the conserved CD4-binding site. J Virol. 2012;86:5844–5856. doi: 10.1128/JVI.07139-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Migueles SA, et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity. 2008;29:1009–1021. doi: 10.1016/j.immuni.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kershaw MH, et al. Immunization against endogenous retroviral tumor-associated antigens. Cancer Res. 2001;61:7920–7924. [PMC free article] [PubMed] [Google Scholar]
- 43.Li M, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch M, et al. Structure-based, targeted deglycosylation of HIV-1 gp120 and effects on neutralization sensitivity and antibody recognition. Virology. 2003;313:387–400. doi: 10.1016/s0042-6822(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 45.Mascola JR, et al. Human immunodeficiency virus type 1 neutralization measured by flow cytometric quantitation of single-round infection of primary human T cells. J Virol. 2002;76:4810–4821. doi: 10.1128/JVI.76.10.4810-4821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ofek G, et al. Elicitation of structure-specific antibodies by epitope scaffolds. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17880–17887. doi: 10.1073/pnas.1004728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 48.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 52.Davis IW, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 55.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 56.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Imamichi H, et al. Human immunodeficiency virus type 1 quasi species that rebound after discontinuation of highly active antiretroviral therapy are similar to the viral quasi species present before initiation of therapy. J Infect Dis. 2001;183:36–50. doi: 10.1086/317641. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.