

Abstract
Linkage disequilibrium poses a major challenge to the functional characterization of specific disease-associated susceptibility variants. Precision genome editing technologies have provided new opportunities to address this challenge. As proof-of-concept, we employed TALEN-mediated genome editing to specifically disrupt the TT>A enhancer region to mimic candidate causal variants identified in the systemic lupus erythematosus-associated susceptibility gene, TNFAIP3, in an isogenic HEK293T cell line devoid of other linkage disequilibrium-associated variants. Targeted disruption of the TT>A enhancer impaired its interaction with the TNFAIP3 promoter by long-range DNA looping, thereby reducing TNFAIP3 gene expression. Loss of TNFAIP3 mRNA and its encoded protein, A20, impaired TNFα-induced receptor-mediated downregulation of NF-κB signaling; a hallmark of autoimmunity. Results demonstrate that the TT>A enhancer variants contribute to causality and function independently of other variants to disrupt TNFAIP3 expression. Further, we believe this approach can be implemented to independently examine other candidate casual variants in the future.
Introduction
Systemic lupus erythematosus (SLE) is a chronic inheritable autoimmune disease characterized by autoantibody production and immune complex deposition that cause dramatic clinical heterogeneity including systemic inflammation and multi-organ failure (1, 2). Genome-wide association studies (GWAS) have identified more than 50 susceptibility or risk genes/loci linked to the high hereditability of SLE (1–6). Even though each susceptibility locus may contain many strongly associated single nucleotide polymorphisms (SNPs), it is generally assumed that only a few SNPs are actually responsible for disease association (causal) while the rest are associated due to linkage disequilibrium. Isolating and biologically characterizing causal variants from statistically indistinguishable non-causal variants are an arduous task requiring genetic, bioinformatic, and molecular approaches. For many susceptibility loci, characterization of the causal variants remain limited, presenting a major challenge toward understanding the molecular mechanisms that underlie the statistical associations identified using GWAS.
The tumor necrosis factor alpha-induced protein 3 (TNFAIP3) gene encodes the ubiquitin-modifying enzyme A20 that plays an important role in downregulating NF-κB signaling to limit immune responses (7). Selective loss of TNFAIP3 gene transcription and/or A20 translation in mice results in prolonged NF-κB signaling and subsequent heightened immune responses characteristic of numerous autoimmune diseases, including elevated pro-inflammatory cytokine release (8–11). GWAS studies of human patient samples have identified several genetic variants in the TNFAIP3 gene region associated with autoimmune diseases such as SLE (12–20).
We previously identified a pair of SLE-associated functional variants (rs148314165, rs200820567, collectively referred to as TT>A) in a conserved enhancer element (referred to as TT>A enhancer) that is ~42 kilobases downstream of the TNFAIP3 gene promoter (21, 22). Bioinformatic analyses identified this region to have significant regulatory functions, including several transcription factor-binding sites (22). Importantly, both rs148314165 (-T) and rs200820567 (T>A) variants are localized in an NF-κB binding site within this enhancer (21). Patient-derived cell lines carrying the SLE-associated TT>A enhancer risk allele (-A/-A) exhibited significant reductions in NF-κB binding at the TT>A enhancer site, resulting in subsequent loss of TNFAIP3 gene transcription and A20 protein translation (21). We further determined that the TT>A enhancer binds NF-κB and delivers it to the TNFAIP3 gene promoter to induce transcription via special AT-rich binding protein 1 (SATB1)-mediated long-range DNA looping (21). This study provided strong evidence suggesting that these two risk variants are likely causal variants involved in the loss of NF-κB signaling attenuation that contributes to heightened inflammatory responses associated with SLE. It is important to note that these studies were conducted in patient-derived cell lines that carry the full TNFAIP3 risk haplotype, which contains other variants that may influence TNFAIP3 expression. Whether the TT>A variants function as independent causal variants, or work in concert or in opposition with other SNPs on the risk haplotype to influence TNFAIP3 expression remains unclear.
Transcription Activation-Like Effector Nucleases (TALENs) are a precision genome editing technology that has provided new opportunities for the functional characterization of causal genetic variants and genomic elements on isogenic backgrounds, thereby eliminating the confounding effects of variants in linkage disequilibrium with the variant of interest (23). TALENs are artificial restriction enzymes generated through the fusion of a series of nucleotide-specific DNA-binding domains to a DNA-cleavage domain (FokI) that can then introduce double-strand DNA breaks (DSBs) at a specific site of interest (24, 25). DSBs trigger the non-homologous end joining (NHEJ) DNA repair response, which, during the process of repair, is prone to inducing small insertions or deletions into the targeted site. These alterations can then be used to dissect the functional importance of regulatory or coding elements in the genome. Improvements in TALEN design that enhance their performance and reduce off target effects, including reduced nuclease-associated toxicity, use of longer DNA target sites, and requirement for thymine at the 5’ end of the target site have made this technology a valuable tool to discern causal versus non-causal variants associated with inheritable diseases (23, 26).
In this report, we mutate the endogenous TT>A enhancer in human embryonic kidney (HEK293T) cells using TALEN-mediated genome editing to allow us to evaluate the causal effect of TT>A enhancer mutation on TNFAIP3 gene expression on a uniform genetic background. We demonstrate that the mutated TT>A enhancer reduces both the mRNA level of TNFAIP3 and the protein level of A20 through impaired long-range DNA looping, resulting in prolonged stimuli-induced NF-κB signal activation consistent with the naturally occurring SLE-associated TT>A variants (21). Moreover, our study demonstrates the utility of TALEN-mediated genome editing in the development of model systems to isolate and characterize putative causal genetic variants from other associated variants.
Results
TALEN-mediated modification of the TT>A enhancer in the HEK293T cell line
We designed a unique pair of individually epitope-tagged TALENs using TALEN-Targeter 2.0 that specifically recognize the 5’ and 3’ TALEN target sites flanking the TT>A enhancer region of the TNFAIP3 gene (Figure 1A) (27). To enhance specificity and minimize off-target effects, we designed our TALENs using a previously described mutant derivative of the nuclease domain that ensures the 5’ and 3’ TALEN monomers function as an obligate heterodimer (28). Transient expression of the hemagglutinin (HA)-tagged 5’ TALEN and FLAG-tagged 3’ TALEN in HEK293T cells 48 hours post transfection was confirmed by Western blotting (Figure 1B). Of note, HEK293T cells were transiently transfected with equal amounts of plasmid encoding HA-tagged 5’ or FLAG-tagged 3’ TALENs, thus reduced detection of the FLAG-tagged 3’ TALEN arm is likely due to differences in antibody avidity. We subsequently isolated and analyzed twelve single cell clones from the TALEN-transfected HEK293T cells using PCR amplification and high-resolution melting curve analyses (28). We identified four clones as positive for TALEN-mediated NHEJ events; indicative of an approximate 30% success rate. Consistent with the variability of TALEN-mediated genome editing (23, 24), the identified mutants varied significantly in the genomic disruptions presented at the TT>A enhancer. Two heterozygous mutant clones containing one wild type allele and one mutant allele were selected for further functional characterization of the TT>A enhancer. Mutant 1 clone contained a mutant allele with a 26 base pair deletion predicted to completely disrupt SATB1 and NF-κB binding to the TT>A enhancer (29), while Mutant 2 clone contained a mutant allele with both a 9 base pair deletion and 4 base pair insertion (indel) predicted to disrupt NF-κB binding, but not SATB1 binding (Figure 1C). In our previous work, we demonstrated that the TT>A variants did not alter the interaction of SATB1 with the TT>A enhancer, but that siRNA-mediated suppression of SATB1 did impair long-range DNA looping of the TT>A enhancer with the TNFAIP3 promoter (21). Therefore, we anticipated the effect of TALEN-editing to be more severe in Mutant 1 (both SATB1 and NF-κB sites deleted) compared with Mutant 2 (NF-κB site deleted).
Figure 1. Design of TALENs for targeting the TT>A enhancer.
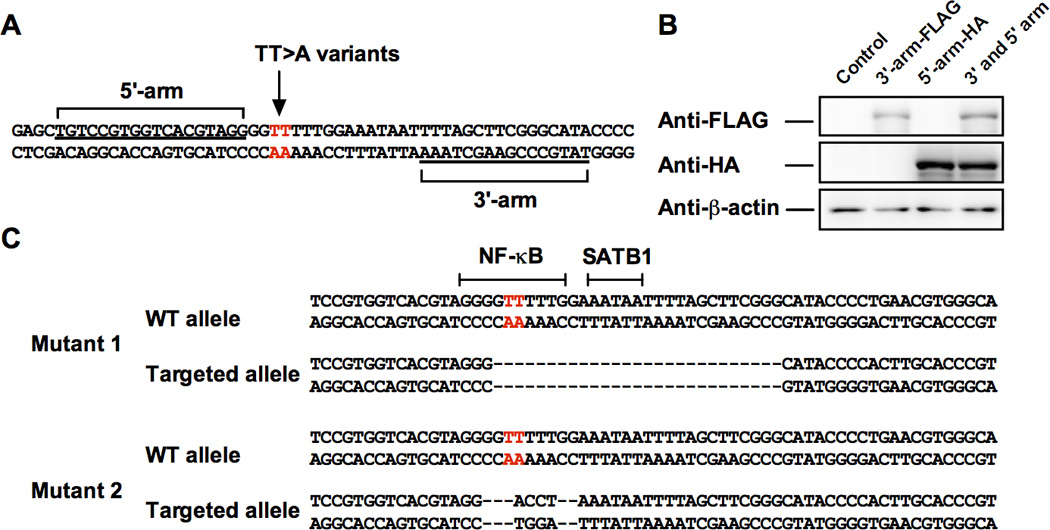
A. Location of 5’ and 3’ TALEN nuclease binding sites. B. Western blotting of transfected HA-tagged 5’ TALEN arm and FLAG-tagged 3’ TALEN arm in HEK293T cells. C. Location of TALEN-mediated mutations of the TT>A enhancer in two HEK293T cell lines. Mutant 1 has a 26 base pair deletion of the TT>A enhancer including the NF-κB and SATB1 binding sites. Mutant 2 has a 9 base pair deletion and 4 base pair insertion (indel) of the TT>A enhancer that disrupts the NF-κB binding site. Red letters in A and C indicate the SLE risk TT>A polymorphisms.
TT>A enhancer knockout reduces gene expression of TNFAIP3
We previously demonstrated in patient-derived cells carrying the TT>A risk allele that SATB1-dependent long-range DNA looping functioned to deliver NF-κB bound by the TT>A enhancer to the TNFAIP3 promoter (21). To determine whether our TALEN-mediated disruptions in HEK293T cells were sufficient to mimic the disease risk allele, we examined whether TNFAIP3 gene transcription and A20 protein expression was significantly altered in the Mutant 1 and 2 cells lines using real time quantitative PCR (RT-PCR) and Western blotting. Mutant 1 and 2 cell lines exhibited significant reduction in TNFAIP3 mRNA (Figure 2A) and A20 protein (Figure 2B) relative to the parental HEK293T cell line. Additionally, Mutant 1 cells with the heterozygous 26 base pair deletion exhibited a trend for greater reduction of TNFAIP3 expression when compared to Mutant 2 cells with the preserved SATB1 site. Albeit not statistically significant, this supports our previous finding that SATB1-mediated long-range DNA looping and NF-κB binding are both important for efficient TT>A enhancer-mediated TNFAIP3 gene expression (21), and further suggests that our TALEN-mediated genome editing approach is capable of producing relevant allelic series to examine specific molecular and cellular effects of predicted causal variants.
Figure 2. Mutating the TT>A enhancer region by TALEN engineering impairs TNFAIP3 expression.

A. Quantitative RT-PCR analysis of TNFAIP3 mRNA expression normalized to control HMBS mRNA in control and clonal mutant HEK293T cell lines. B. Western blotting analysis of A20 protein normalized to β-actin in parental (control) and clonal mutant HEK293T cell lines. Statistical significance was calculated using paired student t-test. For all experiments: n = 3, *p < 0.05.
Mutated TT>A enhancer leads to the chromatin conformation changes at the TNFAIP3 locus
To test whether TALEN-mediated disruptions in the TT>A enhancer disrupted long-range DNA looping, we employed an allele-specific chromatin conformation capture (3C) assay (Figure 3A) (21). Our 3C assay utilizes a 3’ PCR primer located in the TT>A enhancer near the TT>A variants in combination with a 5’ PCR primer located in the TNFAIP3 promoter to measure relative crosslinking frequencies (RCF) between TT>A enhancer and the promoter. Since each mutated cell line is heterozygous for the mutated TT>A enhancer, we used a clonal assay in which the PCR products from 3C were cloned using the pBluescript KS II vector and amplified by E. coli. We randomly selected 72 clones transformed with PCR constructs from each mutant cell line to amplify and isolate PCR inserts from DNA looping events. A melting curve analysis was used to quantify the number of looping events attributed to each wild type or mutant allele for each TALEN-edited cell line. As a control for the clonal assay, genomic DNA not subjected to 3C, was PCR amplified across the TALEN-edited mutations and processed in parallel with the 3C amplicons. As expected with a heterozygous cell line, we found approximately equal clonal allele frequencies of the wild type allele and the mutant allele with genomic DNA from each cell line not subjected to 3C (Figure 3B). In contrast, Mutant 1 and 2 cells subjected to 3C from both revealed significant reductions in the clonal frequency of DNA looping events involving the mutant allele, relative to the wild type allele (Figure 3C). Moreover, 3C analysis of the Mutant 1 allele with disruptions in both the TT>A enhancer and the SATB1 binding site failed to detect any DNA looping events. These findings demonstrate the importance of the TT>A enhancer binding sequence in induction of TNFAIP3 expression, and support the combined role of SATB1 and NF-κB binding to the TT>A enhancer for efficient induction of TNFAIP3 expression.
Figure 3. TALEN mutagenesis of the TT>A enhancer reduces interaction with the TNFAIP3 promoter.
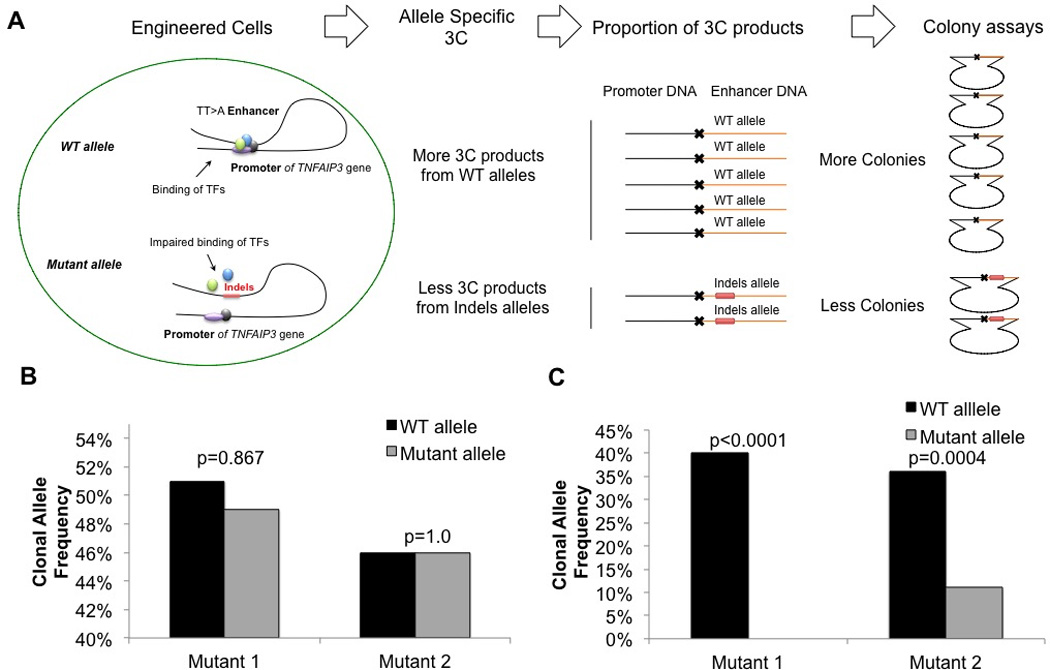
A. Schematic representation of the allele-specific 3C and clonal assay analyses of the engineered heterozygous clonal mutant HEK293T cell lines. Wild type (WT) allele experiences higher frequencies of long-range DNA looping, thus higher proportions of 3C events are observed relative to Mutant 1 or Mutant 2 alleles. B. Genomic DNA isolated from 72 clonal expansions from Mutant 1 or Mutant 2 HEK293T cells without 3C was analyzed for wild type and mutant allele frequencies. The expected clonal frequency of approximately 50% wild type and 50% mutant allele was observed. C. Analysis of 72 mutant 1 or 2 clonal expansions from the 3C capture were analyzed for relative crosslinking frequencies between wild-type or mutated TT>A enhancer and TNFAIP3 promoter. P values were calculated using the Fisher’s exact test and are as indicated.
Mutations in the TT>A enhancer reduces the ability of TNFAIP3 to attenuate the activation of NF-κB signaling
TNFAIP3-encoded A20 limits immune responses by negatively regulating NF-κB activity (7). Given that disruptions in the TT>A enhancer disrupts TNFAIP3 expression, we next evaluated whether the disruption in TNFAIP3 alters receptor mediated NF-κB signaling and, more importantly, A20-mediated NF-κB signaling attenuation. Western blotting analysis of the mutant HEK293T cells harvested at various time points following TNFα stimulation revealed prolonged IκBα phosphorylation (p-IκBα), relative to parental HEK293T cells (Figure 4). Phosphorylation of IκBα dissociates it from NF-κB, and promotes its degradation (30). Therefore, sustained p-IκBα expression in the mutant cell lines suggests that disruptions in the TT>A enhancer is sufficient to prevent efficient receptor-mediated NF-κB signaling attenuation (Figure 4). Also, consistent with the RT-PCR and 3C analyses, Mutant 1 demonstrated more sustained NF-κB signaling relative to Mutant 2 cells (Figure 4B). Collectively, our findings confirm, using TALEN-mediated TT>A enhancer mutation in HEK293T cells, that the TT>A enhancer is an important regulator of TNFAIP3 expression, thereby promoting a negative feedback mechanism to suppress NF-κB-mediated immune responses.
Figure 4. TALEN mutagenesis of the TT>A enhancer region delayed TNFα-inducible inactivation of NF-κB signaling.

A. Western blotting of phospho-IκBα at designated time points following TNFα stimulation of parental (wild type) and clonal mutant HEK293T cells. B. Densitometry analyses of Western blotting data represented in (A) normalized to β-actin levels; n = 3, P value calculated by two-way ANOVA and are as indicated.
Discussion
Our previous work identified the mechanistic basis for how the TT>A enhancer regulates TNFAIP3 expression by engaging NF-κB and delivering it to the TNFAIP3 promoter through SATB1-mediated long-range DNA looping (21). Naturally occurring disease-associated TT>A variants (rs148314165 and rs200820567) in this enhancer fail to bind NF-κB, leading to impaired DNA looping and reduced TNFAIP3 expression. This results in defective A20-mediated NF-κB signal attenuation and hyperactive NF-κB pathway responses. Given the importance of NF-κB regulation in maintaining immune homeostasis, these results suggest that the TT>A variants may be driving the association with SLE (21). However, as a result of strong linkage disequilibrium, the impact of other SLE-associated gene variants carried on the risk haplotype may influence the molecular phenotype defined for the TT>A variants. Therefore, the potential for these additional gene variants to modulate TNFAIP3 expression independent of the TT>A variants could not previously be ruled out using patient-derived cell lines.
In this study, we employed precision genome-editing technology to induce targeted disruptions to the TT>A enhancer of HEK293T cells in order to validate the effect of the TT>A variants in isolation. We demonstrated that TALEN-mediated disruption of the TT>A enhancer impaired TNFα-induced TNFAIP3 expression by interfering with long-range DNA looping of the TT>A enhancer with the TNFAIP3 promoter, resulting in elevated and sustained NF-κB signaling (Figure 5). Interestingly, our results from the two clonal mutants varied such that Mutant 1 yielded more severe inhibition of long-range DNA looping, TNFAIP3 expression, and NF-κB signal termination. Given that Mutant 1 exhibited disruptions in both the predicted TT>A-containing NF-κB binding site and adjacent SATB1 binding site, relative to Mutant 2 with only NF-κB binding disruption, it is not unreasonable to speculate that this is due to the combined inhibition of NF-κB binding and loss of SATB1-mediated long-range DNA looping of bound NF-κB to the TNFAIP3 promoter. Thus, the TT>A variants identified on the TNFAIP3 risk haplotype are likely causal variants that contribute to heightened NF-κB-mediated inflammation shared among autoimmune diseases.
Figure 5. SLE-associated TT>A enhancer risk allele is causal for SLE-associated A20 protein reduction and elevated NF-κB signaling.
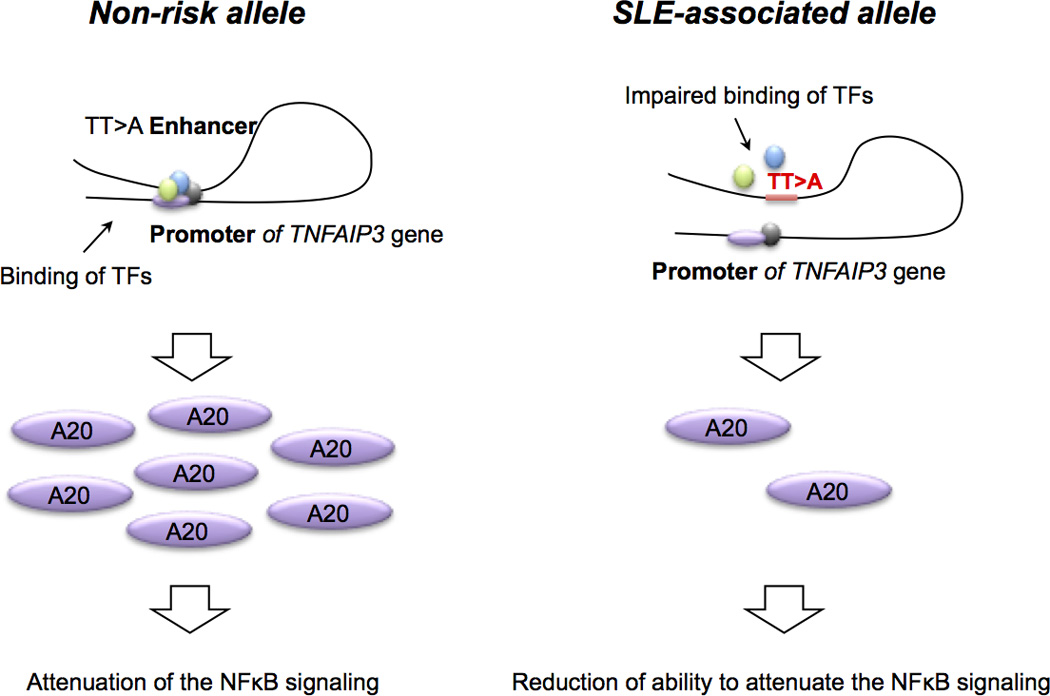
Schematic depiction of the TT>A enhancer function in non-risk and SLE-associated risk alleles. SLE-associated polymorphisms in the TT>A enhancer impairs long-range DNA looping and TT>A enhancer interaction with, and delivery of transcription factors including NF-κB, to the TNFAIP3 gene promoter. Subsequent reductions in TNFAIP3 mRNA transcription and A20 protein translation impair A20-mediated ubiquitination and degradation of NF-κB, thus preventing attenuation of pro-inflammatory NF-κB signaling.
In our proof-of-concept study, we employed a modified TALEN-technology that has reduced nuclease-associated cytotoxic effects relative to other genome-editing technologies and a requirement for the 5’ and 3’ TALEN monomers to form obligate heterodimers in order to function (28). Utilizing this approach increases the targeting specificity and likely reduces the off-target effects of our targeting strategy. Although we have not specifically interrogated our engineered cell lines for off target effects, our TALEN-mediated TT>A enhancer mutations closely mimicked the effects of naturally occurring disease-associated TT>A enhancer risk variants, strongly suggesting that our system is valid for studying the effects of this enhancer on NF-κB function. In addition to confirming the TT>A enhancer variants as functional variants that are likely responsible, in part, for the association with SLE, our study provides strong support for the use of genome-editing technology to functionally characterize predicted causal variants in other disease models for which linkage disequilibrium complicates functional variant identification.
Materials and Methods
Antibodies, cell lines, restriction enzymes, and plasmid DNAs
Anti-FLAG antibody was purchased from Sigma (St. Louis, MO); anti-HA and anti-phospho-IκBα antibodies were purchased from Cell Signaling (Danvers, MA); anti-β-actin antibody was purchased from Abcam (Cambridge, MA); anti-A20 antibody was purchased from Ebioscience (San Diego, CA). HEK293T cell line was purchased and maintained per instructions from American Type Culture Collection (ATCC; Manassas, VA). Golden Gate cloning kit and expression vectors, pCS2TAL3DD and pCS2TAL3RR, were purchased from Addgene (Cambridge, MA). DH5α competent cells were purchased from Life Technologies (Grand Island, NY). QuantiTect SYBR Green PCR Master Mix was purchased from QIAGEN (Valencia, CA). Restriction enzymes NlaIII, EcoRV, and NEB Standard Buffer 4 were purchased from New England Biolabs (Ipswich, MA).
Generation of TT>A enhancer knockout in HEK293T cells
Genomic DNA sequence surrounding the TT>A enhancer was scanned for potential TALEN target sites using TALEN-Targeter 2.0 (27). A modified TALEN Golden Gate assembly system (Addgene, Cambridge, MA) was then used to generate repeat variable di-residue (RVD) arrays to target the TT>A enhancer. The RVD sequence was subcloned into the expression vectors pCS2TAL3DD (5’ arm) and pCS2TAL3RR (3’ arm) (Addgene, Cambridge MA). Sanger sequencing confirmed the correct insertion into each expression vector (31, 32). TALENs were expressed in HEK293T cells by transient transfection using Fugene HD (VWR, Radnor, PA) with 2 µg of each plasmid DNA encoding HA-tagged 5’-arm and FLAG-tagged 3’-arm. After 48 h, protein expressions of 5’ and 3’ TALEN arms were detected using Western blotting with antibodies against the respective HA (5’ arm) and FLAG (3’ arm) tags. Single cell clones of the HEK293T cells expressing TALENs were isolated and genomic DNA harvested by phenol-chloroform extraction. The genomic DNA of the TT>A enhancer in each cell clone was amplified with PCR (Supplemental Table 1) and screened using high-resolution melting curve analysis (28). PCR amplicon sequences from each clone were further validated by Sanger sequencing (31, 32).
RNA Isolation, Quantitative RT-PCR, A20 protein expression
Parental and clonal mutant HEK293T cell lines were maintained according to ATCC (Cambridge, MA) guidelines and harvested during the logarithmic growth phase. Total RNAs of parental and mutant cell lines were isolated using Trizol total RNA isolation reagent (Invitrogen Inc., Carlsbad, CA). RNA concentrations were quantified using nano-drop and diluted to a final concentration of 0.5µg/µL using MS2-RNA (Hoffmann-La Roche, Inc., Nutley NJ). RNAs were treated with DNase, and then cDNAs were synthesized using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Inc., Hercules, CA). Quantitative real time PCR (RT-PCR) was carried out using the SYBR Green method to determine the mRNA expression of TNFAIP3 (33) using the following primer pair: sense, 5'-GATAGAAATCCCCGTCCAAGG-3'; anti-sense, 5'-CGTCCATTTCTTGTACTCATGC-3' (Supplemental Table 1). The human hydroxymethylbilane synthase (HMBS) gene was used in quantitative RT-PCR as a reference. Messenger RNA expression of TNFAIP3 was normalized to mRNA expression of the reference HMBS gene. Three independent experiments were performed. Statistical significance of TNFAIP3 transcript expression was tested between parental cells and both mutant cell lines using the paired student t-test.
For Western blot analyses, the parental HEK293T, Mutant 1, and Mutant 2 cells were harvested and lysed in Whole Cell Extraction Buffer (25mM Tris-HCl, 1% Triton X-100, 150mM NaCl, 1mM EDTA and protease inhibitors). Proteins were separated by SDS-PAGE and transferred to PVDF membranes according to standard protocols. Protein expression of A20 was determined by Western blotting with anti-A20 antibody (Ebioscience, San Diego, CA) and normalized to the expression of β-actin. Three independent experiments were performed. P values were calculated using paired student t-test.
Allele-specific 3C assays
Allele-specific 3C assay was performed as described in our previous study on the above engineered HEK293T cell lines heterozygous for the Mutant 1 or Mutant 2 TT>A variants (21). In brief, two TT>A enhancer-mutated HEK293T cell lines were crosslinked with 1% buffered formaldehyde at room temperature for 10 min. Crosslinking was halted with glycine, then cells were lysed for 10 min at 4 °C in lysis buffer (10mM Tris-HCl pH 7.5; 10mM NaCl; 5mM MgCl2; 0.1mM EGTA; Protease and Phosphatase Inhibitor Cocktail tablets (Roche Applied Science)). Nuclei were suspended in 1.2 × restriction buffer (1 × NEB Standard Buffer 4 ; 1 × bovine serum albumin (BSA) (New England BioLabs, Inc, Ipswich, MA)) containing 0.3 % SDS for 1 h at 37 °C while shaking at 900 rpm. SDS was sequestered by adding Triton X-100 to 2% and incubating an additional hour at 37 °C while shaking. Chromatin was digested with restriction enzyme (NlaIII) (New England BioLabs, Inc, Ipswich, MA) for 24 hours and ligated with T4 DNA ligase overnight to generate 3C DNA as previously described (21). After ligation, the crosslinking was reversed by incubating overnight with proteinase K at a final concentration of 50µg/mL at 50 °C. Resulted 3C DNA was purified by phenol-chloroform extraction and amplified by PCR using primers listed in Supplemental Table 1. PCR amplification of the genomic DNA for the TT>A enhancer of TNFAIP3 gene was generated using the same cells with primers indicated in Supplemental Table 1. PCR products were used in the clonal assay below.
Clonal assays
PCR products derived from either genomic DNAs or the 3C assays were ligated into the pBluescript KS II vector opened with EcoRV and used to transform DH5α competent cells. 72 colonies were picked randomly from each of the following condition: genomic DNA-mutant 1; 3C DNA-mutant 1; genomic DNA-mutant 2, and 3C DNA-mutant 2. Each clone was lysed in 20µL of colony lysis buffer containing 20mM Tris-HCl pH 8.0, 1% Triton X-100, 2mM EDTA. 0.5µL of lysate from each colony was amplified with T7 and T3 primers using QuantiTect SYBR Green PCR Master Mix (QIAGEN, Valencia, CA) for 40 cycles. The resulted PCR product from each colony was analyzed by high-resolution melting curve analysis to distinguish origin of the insert (28). To ensure accuracy of the predicted insert in each clone using melting curve analysis, 12 of 72 randomly selected colonies were sequenced with T7 primer. Sanger sequencing results demonstrate 100% accuracy of prediction by melting curve analysis for both 3C and genomic DNA. Numbers of colonies carrying either wild type allele or mutant alleles were summarized and compared in each pair of matched genomic DNA and 3C DNA for each mutant cell lines. Statistical differences were calculated using the Fisher’s exact test.
Detection of NF-κB activities in parental HEK293T and engineered cell lines
Parental HEK293T, mutant 1, and mutant 2 cells were cultured according to ATCC (Cambridge, MA) guidelines and stimulated with TNFα for 15 minutes to activate NF-κB signaling. Culture medium was subsequently replaced with pre-warmed fresh culture medium to removed TNFα. Cells were harvested and lysed at indicated time points, and then phosphorylation of IκBα was examined by Western blotting. Phosphorylation of IκBα was used as an indicator of NF-κB signaling. Densitometry analyses of Western blotting of phospho-IκBα were normalized to expression levels of β-actin. Three independent experiments were performed; P value was calculated by two-way ANOVA, as indicated.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health 5 R01 AR056360-05 and 5 R01 AR063124-04, and P30 GM110766.
Footnotes
Conflict of Interest: The authors have no conflicts of interest to disclose.
References
- 1.Cui Y, Sheng Y, Zhang X. Genetic susceptibility to SLE: recent progress from GWAS. J Autoimmun. 2013;41:25–33. doi: 10.1016/j.jaut.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Relle M, Weinmann-Menke J, Scorletti E, Cavagna L, Schwarting A. Genetics and novel aspects of therapies in systemic lupus erythematosus. Autoimmun Rev. 2015 doi: 10.1016/j.autrev.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Adrianto I, Wang S, Wiley GB, Lessard CJ, Kelly JA, Adler AJ, et al. Association of two independent functional risk haplotypes in TNIP1 with systemic lupus erythematosus. Arthritis Rheum. 2012;64(11):3695–3705. doi: 10.1002/art.34642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang S, Adrianto I, Wiley GB, Lessard CJ, Kelly JA, Adler AJ, et al. A functional haplotype of UBE2L3 confers risk for systemic lupus erythematosus. Genes Immun. 2012;13(5):380–387. doi: 10.1038/gene.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham RR, Cotsapas C, Davies L, Hackett R, Lessard CJ, Leon JM, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40(9):1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bentham J, Morris DL, Cunninghame Graham DS, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47(12):1457–1464. doi: 10.1038/ng.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. 2012;12(11):774–785. doi: 10.1038/nri3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33(2):181–191. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wojtowicz E, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood. 2011;117(7):2227–2236. doi: 10.1182/blood-2010-09-306019. [DOI] [PubMed] [Google Scholar]
- 11.Hovelmeyer N, Reissig S, Xuan NT, Adams-Quack P, Lukas D, Nikolaev A, et al. A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol. 2011;41(3):595–601. doi: 10.1002/eji.201041313. [DOI] [PubMed] [Google Scholar]
- 12.Yang W, Tang H, Zhang Y, Tang X, Zhang J, Sun L, et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet. 2013;92(1):41–51. doi: 10.1016/j.ajhg.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Genetic Analysis of Psoriasis C, the Wellcome Trust Case Control C. Strange A, Capon F, Spencer CC, Knight J, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42(11):985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kochi Y, Okada Y, Suzuki A, Ikari K, Terao C, Takahashi A, et al. A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat Genet. 2010;42(6):515–519. doi: 10.1038/ng.583. [DOI] [PubMed] [Google Scholar]
- 16.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42(6):508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42(4):295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41(11):1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 19.Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008;40(9):1062–1064. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39(12):1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang S, Wen F, Wiley GB, Kinter MT, Gaffney PM. An enhancer element harboring variants associated with systemic lupus erythematosus engages the TNFAIP3 promoter to influence A20 expression. PLoS Genet. 2013;9(9):e1003750. doi: 10.1371/journal.pgen.1003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet. 2011;43(3):253–258. doi: 10.1038/ng.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright DA, Li T, Yang B, Spalding MH. TALEN-mediated genome editing: prospects and perspectives. Biochem J. 2014;462(1):15–24. doi: 10.1042/BJ20140295. [DOI] [PubMed] [Google Scholar]
- 24.Kim Y, Kweon J, Kim JS. TALENs and ZFNs are associated with different mutation signatures. Nat Methods. 2013;10(3):185. doi: 10.1038/nmeth.2364. [DOI] [PubMed] [Google Scholar]
- 25.Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31(7):397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39(21):9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40(Web Server issue):W117–W122. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, et al. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012;8(8):e1002861. doi: 10.1371/journal.pgen.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, et al. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nat Immunol. 2012;13(1):95–102. doi: 10.1038/ni.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25(51):6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 31.Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94(3):441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 32.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schefe JH, Lehmann KE, Buschmann IR, Unger T, Funke-Kaiser H. Quantitative real-time RT-PCR data analysis: current concepts and the novel "gene expression's CT difference" formula. J Mol Med (Berl) 2006;84(11):901–910. doi: 10.1007/s00109-006-0097-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.