

Abstract
Colorectal cancer (CRC) is a disease in which pathogenesis is influenced by genetic and epigenetic events that occur with tumor initiation and progression. Precision oncology is becoming increasingly important in the management and therapy of CRC since large variation exists in individual patient prognosis and response to chemotherapy that is due to molecular heterogeneity. Certain biomarkers have been identified that can be utilized to predict clinical outcome beyond staging, and to inform treatment selection. Molecular testing is routinely performed in clinical practice for the selection of patients for targeted biologic agents or immunotherapy, and is advocated for prognostic stratification. Estimating prognosis can inform treatment decisions with avoidance of under or over treatment and also guide the intensity of patient follow-up. Classifiers of CRC have been developed that integrate genetic and/or epigenetic features which can provide prognostic and predictive information. The mutational status of KRAS and BRAFV600E combined with analysis of the DNA mismatch repair system with/without CIMP has been shown to identify colon cancer subtypes with distinct clinical features and prognoses. Gene expression profiling has also been used to subtype CRCs and can overcome the limitations of single/limited gene testing. A recent effort identified four consensus molecular subtypes of biological relevance that were associated with different patient outcomes. Efforts to validate and refine these subtypes to include additional genomic features are ongoing. In addition to the potential for molecular subtypes to predict therapeutic efficacy, they can inform the development of new agents in a subtype–specific manner to accelerate drug-discovery efforts. The focus of this article is to highlight molecular markers that can inform clinical decision-making in patients with CRC. Molecular profiling–based stratification
Keywords: Predictive markers, Prognostic markers, Colorectal cancer, Molecular subtypes, RAS, BRAF, MSI, DNA Mismatch Repair, Immunotherapy, Targeted Therapy, Biologics, anti-EGFR, anti-VEGF
Technological primer
Molecular testing has become routine in patients with metastatic CRC to select patients for targeted therapy. While analysis of individual genes has been the norm, the use of next-generation sequencing (NGS) platforms allows for high throughput genomic analysis that has become commonplace and is commercially available. NGS can detect most genomic alterations with analysis of the entire coding sequence of multiple genes and can probe introns from genes that are often altered in solid tumors. This technology has the ability to identify somatic alterations including-base substitutions, insertions/deletions, copy number alterations and rearrangements in a single assay from limited archival tissue. NGS of sporadic CRCs has confirmed prior identified genetic alterations and has classified new alterations. A newer technology for mutation detection is the use of circulating tumor (ct) DNA which is an alternative to tissue-based mutational analysis 1. In a recent study in metastatic CRC patients, ctDNA and patient-specific candidate tissue mutations was detectable in the cell-free DNA from the plasma in a high proportion of treatment naive patients 2. Furthermore, changes in ctDNA during first-line chemotherapy appeared to predict the later radiologic response. The applications of these ‘liquid biopsies’ has the potential to replace tumor tissue analysis in clinical practice, and can enable the monitoring of tumor burden and detect molecular resistance to therapy.
Several groups have reported molecular classifications of CRCs using gene expression data from NGS or expression arrays 3–7, and in some cases, these subtypes provided predictive 4, 7 or prognostic information 3–6. High throughput gene expression profiling reveals tumor subtypes that show overlap with previous classification systems utilizing microsatellite instability (MSI), CpG island methylator phenotype (CIMP), chromosomal instability (CIN) or KRAS/BRAF mutation status 4, 6, which cannot be identified solely based on single mutations or epigenetic alterations. While subtyping efforts have been largely limited to gene expression profiling, advances in genomic technology enable tumor characterization using whole exome (entire human coding DNA) and whole genome (all coding and noncoding regions) sequencing (WGS) that are being increasing utilized. WGS can provide additional data on chromosome or gene copy number and translocations. Advantages include the ability to achieve broad coverage of the genome and there is evolving ease of performance and declining costs. These technologies generate vast amounts of data for which bioinformatics approaches are being applied, yet important challenges remain regarding data interpretation and its clinical application. From a clinical practice standpoint, these technologies will need to produce relevant data in a relatively short time period for it have an impact on the management of individual CRC patients.
Findings
CRC develops as a consequence of genetic and epigenetic alterations whose analysis in individual patients has the potential to indicate risk, inform prognosis, and predict treatment outcome. In patients with this malignancy, there is considerable stage-independent variability in patient outcomes that is due, in part, to molecular heterogeneity. This heterogeneity also makes it difficult to determine which patients will benefit most from existing therapies including targeted agents. Analysis of somatic alterations in CRCs by the The Cancer Genome Atlas (TCGA) included whole-genome sequencing that identified 24 genes that were significantly mutated and included the expected APC, TP53, SMAD4, PIK3CA and KRAS mutations 8. Commonly observed alterations enable a broad classification into 1) hypermutated tumors (∼16%) of which three-quarters show high frequency MSI (MSI-H) and one-quarter have somatic MMR gene and polymerase ε (POLE) mutations 8, 2) nonhypermutated tumors (∼84%) with multiple somatic copy number alterations and aneuploidy that contain activating mutations in KRAS and PIK3CA and loss of heterozygosity of APC and TP53 tumor suppressor genes, and 3) CIMP(∼20%) that is commonly observed in MSI-H tumors and in some nonhypermutated CRCs. CIMP is characterized by a high frequency of genome-wide DNA methylation of CpG islands 9, 10 that is believed to promote tumorigenesis by gene silencing due to methylation-mediated transcriptional repression. Sporadic MSI-H tumors are considered to be a subset of CIMP due to aberrant hypermethylation that inactivates MLH1 10. Occurring more often in women and older individuals, CIMP-positive tumors are typically proximal, higher grade, and have deficient DNA mismatch repair (dMMR) resulting in MSI-H 9, 11, 12. BRAFV600E mutations are strongly enriched in sporadic CRCs with dMMR and CIMP 8, 13, 14. CRCs with dMMR/ MSI-H include both familial and sporadic types 15. Approximately two-thirds are sporadic 9, 10 and the other third of dMMR CRCs are due to germline mutations in MMR genes (MLH1, MSH2, MSH6, PMS2) that cause Lynch Syndrome (LS). The presence of MLH1 promoter hypermethylation and/or a BRAFV600E mutation essentially excludes LS 13. Components from these classifications of CRC outlined above are now used as diagnostic, prognostic, and predictive biomarkers. Additional common biomarkers may come from expression profiling and further genomic analyses.
Predictive and Prognostic Biomarkers
A prognostic factor is a clinical or biologic characteristic that is objectively measurable and that provides information on the likely outcome of the cancer in an untreated individual. Prognostic markers are helpful for identifying patients with cancer who are at high risk of metastatic relapse and therefore, potential candidates for adjuvant systemic therapy. In contrast, a predictive biomarker is defined as a marker which can be used to identify subgroups of patients who are most likely to respond (either in terms of tumor shrinkage or survival) to a given therapy. Importantly, prognostic factors define the effects of patient or tumor characteristics on patient outcome, whereas predictive factors define the effect of treatment on the tumor.
The personalized therapy of CRC has seen major advances in the setting of metastatic disease (stage IV) whereby monoclonal antibodies (mAbs) targeting tumor angiogenesis or the epidermal growth factor receptor (EGFR) signaling pathway have significantly improved patient survival when given in combination with cytotoxic chemotherapy 16–19. Antibodies targeting vascular endothelial growth factor (VEGF) [bevacizumab] or its receptor (VEGFR; aflibercept) or EGFR (panitumumab/cetuximab) are now standard of care when combined with FOLFOX [5-fluorouracil (5-FU), leucovorin (LV) and oxaliplatin) or FOLFIRI (5-FU/LV and irinotecan) in stage IV patients 20. To date, the identification of a predictive biomarker for anti-VEGF therapy remains elusive. In contrast to their benefit in the metastatic setting, the addition of antibodies against VEGF or EGFR to standard adjuvant FOLFOX chemotherapy in patients with lymph node-positive, i.e., stage III, colon cancer has failed to improve outcome in randomized trials 21–23. Accordingly, there is no role for targeted therapy at this time in the treatment of non-metastatic CRC. In patients with stage III disease, micrometastases are responsible for disease relapse and the reasons for failure of targeted agents to improve their eradication awaits further study.
KRAS and BRAF oncogenes
KRAS and NRAS mutation status predict the efficacy of anti-EGFR antibodies with clinical benefit restricted to patient tumors with non-mutated KRAS or NRAS genes 16–18. More than one-third of CRCs carry mutations in exon 2 of KRAS, and an additional 15% of tumors were found to carry mutations at exons 3 and 4 of KRAS and exons 2, 3 and 4 at NRAS that predict resistance to anti-EGFR antibody therapy 16, 17, 24, 25. It is now recommended that expanded RAS mutation testing be performed for all patients being considered for anti-EGFR mAb treatment. In addition to RAS, mutations in the phosphoinositide 3-kinase catalytic subunit alpha (PIK3CA; exon 20 vs exon 9), which is part of the EGFR signaling pathway, may confer resistance to anti-EGFR therapies although further study is needed 25. The association of mutations in KRAS with prognosis in patients with CRC has produced conflicting data 26–28, yet recent data from adjuvant studies in node-positive, i.e., stage III, colon cancer have shown that KRAS exon 2 mutations are associated with poor clinical outcome 29, 30. Furthermore, the adverse impact of KRAS mutations on prognosis appears to be stronger in the distal compared to the proximal colon cancers 31.
A subset (~8%) of CRCs carry a point mutation (V600E) in the BRAF oncogene that is mutually exclusive with mutation in KRAS 32. Patients whose tumors carry BRAFV600E mutations have been consistently shown to have a poor prognosis in the metastatic setting 33–36. In addition, data from clinical trials of adjuvant therapy in stage III disease also reveal an association between mutant BRAFV600E and adverse outcome 14, 26, 29. The adverse prognostic impact of mutant BRAFV600E is particularly evident for overall survival and this finding may be explained, in part, by poor survival after recurrence that was observed for BRAFV600E mutant colon cancers 37, 38. In contrast to patients with BRAFV600 mutant melanoma 39, CRCs that harbor BRAFV600E mutations were found to be resistant to inhibition of the BRAF/MEK/ERK signaling pathway by vemurafenib 40. Resistance to vemurafenib was later found to be due to feedback activation of EGFR when BRAF is inhibited41. This finding has led to ongoing clinical trials that investigate combinations of inhibitors of BRAF, EGFR, and MEK with or without chemotherapy42. In a recent report, the combination of a BRAF inhibitor plus a MEK/ERK inhibitor demonstrated modest activity in a subset of patients with BRAFV600E mutant metastatic CRC 43.
DNA Mismatch Repair/Microsatellite Instability (MMR/MSI)
BRAFV600E mutated vs non mutated tumors are associated with poorer clinical outcome in dMMR and proficient (p) MMR tumors, with its effect being somewhat attenuated in dMMR cancers37.
Studies have consistently shown that CRCs with dMMR/MSI-H have a more favorable prognosis compared to those tumors with pMMR/ MSS 15, 25, 37, 44, 45. These studies include clinical trials comparing surgery alone with 5-FU-based chemotherapy 25, 44, and trials comparing 5-FU-based regimens 15, 37, 46. Of note, the survival benefit of dMMR/MSI-H status appears to be stronger among colon cancers with earlier stage vs later stage disease 46. Patients with dMMR/MSI-H stage II colon cancers have an excellent prognosis and do not receive benefit from 5-FU-based adjuvant therapy 47, 48. In stage III patients, a pooled analysis of two adjuvant chemotherapy trials that evaluated FOLFOX alone or combined with cetuximab showed that patients with dMMR vs pMMR tumors had significantly better patient DFS and OS that was limited to the FOLFOX arm49. Given that dMMR/MSI-H testing evaluates patients for LS and provides prognostic information, NCCN guidelines recommend such testing for all newly diagnosed CRC cases. While conflicting reports exist for the association of CIMP-positive vs negative tumors with clinical outcome, a meta-analysis concluded that CIMP was associated with a poor prognosis in both MSI-H and MSS tumors 50, 51. In a recent study using only CIMP-positive tumors, overall survival was decreased in those lacking MLH1 methylation compared to those with MLH1 hypermethylation that is consistent with the close association between MLH1 hypermethylation and MSI-H and the reduction in significance after controlling for MSI status 52.
Tumors with dMMR/MSI-H are hypermutated and express abundant frameshift peptides that serve as neoantigens to elicit a brisk immune response characterized by abundant tumor infiltrating lymphocytes (TILs) 53. CRCs with dMMR/MSI show increased expression of checkpoint proteins (PD-1, PD-L1, CTLA-4, LAG-3, IDO) compared to pMMR/MSS tumors that help enable them to evade immune destruction 54. In a recent phase II study, metastatic CRCs with MSI-H were treated with an anti-programmed death ligand-1 (PD-1) antibody (permbrolizumab) which achieved a 62% objective response rate compared to 0% for pMMR/MSS CRCs that did not respond to this therapy 55. Progression-free survival has not yet been reached for the treatment arm of this small patient cohort that included sporadic dMMR/MSI-H and Lynch Syndrome cases as well as some extracolonic malignancies associated with this syndrome. While only ~4% of metastatic CRCs are dMMR/MSI-H indicating that the target population is relatively small 56, the potential clinical benefit appears to be quite substantial and may potentially apply to earlier stage patients.
Classification by Molecular Subtype
While single gene analysis has been shown to provide valuable predictive and prognostic information, analyses from multiple studies support the feasibility of classifying CRCs by combining molecular markers or using gene expression profiling. The identification of CRC subtypes that link molecular features to clinical outcome can guide patient stratification and inform the rational design of drugs targeting specific pathways. In participants in an adjuvant chemotherapy trial, the combination of KRAS and BRAFV600E mutations with the status of the DNA MMR system was shown to identify colon cancer subtypes with distinct clinicopathological features and prognoses. These data were then validated in an independent patient cohort (Fig. 1)14. Evaluation of a similar molecular classifier that included CIMP status yielded similar results in a large population-based cohort of individuals with varying stages of CRC 51, suggesting the clinical relevance of this classifier. Several studies have utilized gene expression profiling to develop expression signatures that can identify distinct tumor subgroups 3–7. Using an unsupervised classification strategy involving over 1,100 individuals with colon cancer, three main molecular subtypes were identified based upon genetic, epigenetic and clinical features 4. These colon cancer subtypes (CCS) included CCS1 tumors that were characterized by mutations in KRAS and TP53 genes, displayed increased Wnt signaling, and showed evidence of marked CIN. CCS2 cancers were strongly enriched for CIMP-positive tumors, showed enhanced immune cell infiltration, and were frequently located in the right colon. CCS3 tumors consisted of both MSI-H and CIN tumors, but were enriched for BRAFV600E and PIK3CA mutations and displayed a mesenchymal phenotype. A similar molecular classification developed by other investigators included at least three major subtypes that were largely based on three biological hallmarks of the tumor: epithelial-to-mesenchymal transition, deficiency in MMR genes and MSI-H, and cellular proliferation 6. In these classifications, tumors with dMMR/MSI-H had the best prognosis whereas tumors with a mesenchymal expression phenotype had the worst outcome. These molecular subtypes show overlap with previous classification systems that include CIN, MSI, CIMP, or KRAS/ BRAFV600E mutation status, but cannot be identified solely based on single mutations or epigenetic features.
Figure 1.

Categorization of stage III colon cancers into 5 subtypes based on mutations in BRAFV600E and KRAS (exon 2) and MMR status (top) identifies subtypes with distinct clinicopathological features and prognoses 14. DFS is shown for individual molecular subtypes in an independent cohort of stage III colon cancer patients used for external validation of the subtype classifier (bottom). (Reprinted with permission from Sinicrope FA, et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology 2015;148:88–99.)
Current classifiers show superficial similarities, yet important differences exist that are likely related to differences in data processing and algorithms applied to diverse patient cohorts, sample preparation methods, and gene expression platforms. To address this issue, a CRC Subtyping Consortium was established to perform a cross-comparison of subtype assignments obtained by the various approaches on a common set of tumor samples 57. This effort identified four molecular subtypes (CMS) with distinguishing features: CMS1 [MSI immune, 14%] showing hypermutation; CMS2 (canonical, 37%) that is epithelial with marked WNT and MYC signaling activation; CMS3 (metabolic, 13%) that is epithelial with metabolic dysregulation; and CMS4 (mesenchymal, 23%) showing prominent transforming growth factor (TGF)-β activation, stromal invasion and angiogenesis. While validation in independent cohorts preferably from clinical trials is needed, the CMS groups can be considered to be the most robust classification system currently available for CRC with a clear biological basis that may inform future clinical stratification and subtype-based interventions.
Translation
Prognostic biomarkers can explain some of the variability in outcomes seen among CRC patients, inform patient management and follow-up strategies, and serve as stratification factors. Predictive biomarkers can enable the selection of patients for targeted therapy and are individualizing oncologic practice. The prime example is expanded RAS testing to select metastatic CRC patients for anti-EGFR therapy. There remains, however, a critical need to identify additional predictive biomarkers to enhance therapeutic outcomes. Several biomarkers provide both prognostic and predictive information. One such example is dMMR/MSI-H which has been consistently associated with favorable patient survival rates 25, 37, 44, 45, and evidence suggests that it can predict for lack of benefit from adjuvant 5-fluorouracil 47, 48. While data are limited, patients with dMMR/MSI-H CRC appear to receive benefit from standard FOLFOX as adjuvant chemotherapy as do pMMR/MSS cases49, 58. A potentially practice-changing development is the therapeutic benefit from immune checkpoint inhibitors in observed in patients with metastatic CRC 55. To advance the personalized care of patients with CRC, guidelines from the NCCN and a forthcoming consensus guideline authored by the several professional organizations recommend that that dMMR/MSI testing be performed in all newly diagnosed CRC patients for the identification of Lynch Syndrome. In addition, it is recommended that BRAFV600 mutation analysis be performed in conjunction with dMMR/ MSI testing for prognostic stratification, and that expanded RAS testing be performed to identify candidates for anti-EGFR therapy.
Roadblocks and limitations
Despite advances in the molecular profiling of CRCs, progress in identifying predictive markers beyond RAS status remains limited, with a prime example being lack of predictive biomarkers for anti-angiogenesis drugs. This issue is complicated by whether the target is the tumor vs the tumor microenvironment or both. CRCs are molecularly heterogeneous tumors and differences can also be found between the primary tumor and its metastases. For biomarker studies, important issues include standardization, reproducibility, and interpretation that must be ensured before use in the clinical setting. Given the limitations of single gene analysis, gene expression profiling has identified intrinsic tumor subtypes that are associated with clinical outcome 3–7, yet further study is needed to establish the consistency of these signatures and to perform external validation. In addition to analysis of pretreatment tumors, expression profiling at the time of treatment resistance and progression may further inform therapeutic decision-making. Recent advances in genomic technology enable comprehensive tumor characterization using whole genome or whole exome analyses which are being increasing utilized due to evolving ease of performance and declining costs. Obstacles exist including the need for sophisticated bioinformatic analysis and the need to perform testing/analysis in a time frame that is realistic for use in the clinic.
Conclusions
Advances in the molecular characterization of CRC have led to the personalized treatment of this malignancy which is evolving at a rapid pace. In patients with metastatic CRC, expanded RAS testing for patients who are being considered for anti-EGFR therapy is routine and current guidelines recommend BRAFV600E mutational analysis in conjunction with MMR testing for prognostic stratification. Universal testing of all newly diagnosed CRCs for KRAS/BRAF and MSI/MMR will have the added benefit of increasing the detection of patients with Lynch Syndrome. Biomarker combinations of KRAS, BRAF and MMR status with/without CIMP can identify colon cancer subtypes with distinct prognoses. Moreover, gene expression profiling can identifies colon cancer subtypes that show some overlap with biomarker-based classifiers, and can enable prognostic stratification with the potential for subtype-based targeted interventions. Advances in genomic technology enable more comprehensive tumor characterization using whole genome or whole exome sequencing, yet pose new challenges for data interpretation and clinical application.
Fig. 2.
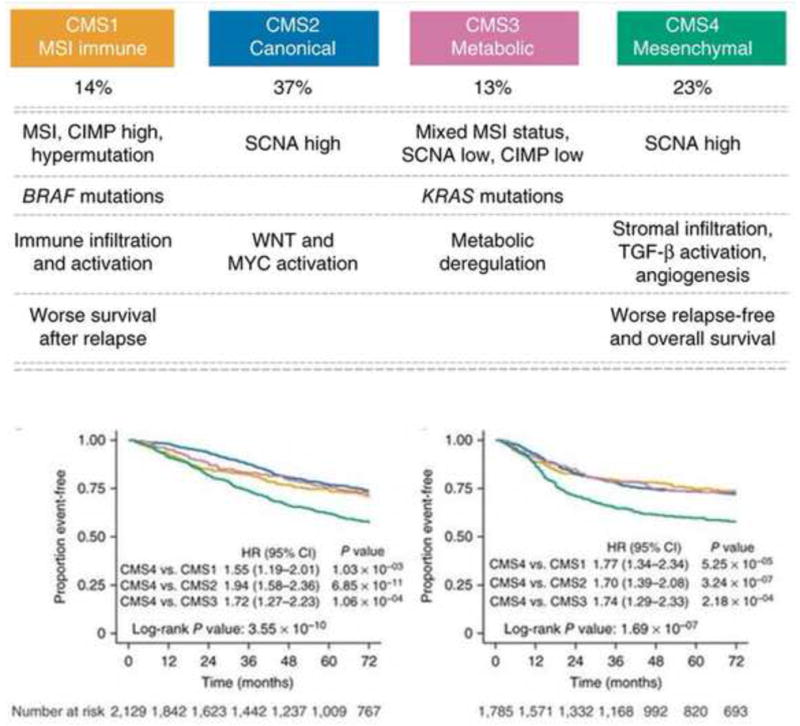
Proposed taxonomy of colorectal cancer reflecting significant biological differences in the gene expression-based consensus molecular subtypes (CMS). Prognostic value of CMS1 (yellow), CMS2 (blue), CMS3 (pink) and CMS4 (green) with Kaplan-Meier survival analysis is shown in the aggregated cohort for overall survival (n = 2,129) [left] and for relapse-free survival (n = 1,785)[right]. (Reprinted with permission from Macmillan Publishers Ltd on behalf of Cancer Research UK: Nature Medicine, doi: 10.1038/nm.3967. Epub 2015 Oct 12. Guinney J, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6.)
Footnotes
Conflicts of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–86. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tie J, Kinde I, Wang Y, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol. 2015;26:1715–22. doi: 10.1093/annonc/mdv177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Budinska E, Popovici V, Tejpar S, et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol. 2013;231:63–76. doi: 10.1002/path.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Sousa EMF, Wang X, Jansen M, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19:614–8. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 5.Marisa L, de Reynies A, Duval A, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roepman P, Schlicker A, Tabernero J, et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer. 2014;134:552–62. doi: 10.1002/ijc.28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–25. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–6. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 11.Hawkins N, Norrie M, Cheong K, et al. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology. 2002;122:1376–87. doi: 10.1053/gast.2002.32997. [DOI] [PubMed] [Google Scholar]
- 12.Ogino S, Kawasaki T, Kirkner GJ, et al. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J Mol Diagn. 2007;9:305–14. doi: 10.2353/jmoldx.2007.060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domingo E, Espin E, Armengol M, et al. Activated BRAF targets proximal colon tumors with mismatch repair deficiency and MLH1 inactivation. Genes Chromosomes Cancer. 2004;39:138–42. doi: 10.1002/gcc.10310. [DOI] [PubMed] [Google Scholar]
- 14.Sinicrope FA, Shi Q, Smyrk TC, et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2015;148:88–99. doi: 10.1053/j.gastro.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinicrope FA, Sargent DJ. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clin Cancer Res. 2012;18:1506–12. doi: 10.1158/1078-0432.CCR-11-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 17.Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–75. doi: 10.1016/S1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- 18.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 19.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 20.Fakih M. Biologic therapies in colorectal cancer: indications and contraindications. Am Soc Clin Oncol Educ Book. 2015;35:e197–206. doi: 10.14694/EdBook_AM.2015.35.e197. [DOI] [PubMed] [Google Scholar]
- 21.Alberts SR, Sargent DJ, Nair S, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA. 2012;307:1383–93. doi: 10.1001/jama.2012.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allegra CJ, Yothers G, O’Connell MJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol. 2013;31:359–64. doi: 10.1200/JCO.2012.44.4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taieb J, Tabernero J, Mini E, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:862–73. doi: 10.1016/S1470-2045(14)70227-X. [DOI] [PubMed] [Google Scholar]
- 24.Bokemeyer C, Bondarenko I, Hartmann JT, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22:1535–46. doi: 10.1093/annonc/mdq632. [DOI] [PubMed] [Google Scholar]
- 25.De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 26.Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60–00 trial. J Clin Oncol. 2010;28:466–74. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 27.Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29:1261–70. doi: 10.1200/JCO.2010.30.1366. [DOI] [PubMed] [Google Scholar]
- 28.Mouradov D, Domingo E, Gibbs P, et al. Survival in stage II/III colorectal cancer is independently predicted by chromosomal and microsatellite instability, but not by specific driver mutations. Am J Gastroenterol. 2013;108:1785–93. doi: 10.1038/ajg.2013.292. [DOI] [PubMed] [Google Scholar]
- 29.Blons H, Emile JF, Le Malicot K, et al. Prognostic value of KRAS mutations in stage III colon cancer: post hoc analysis of the PETACC8 phase III trial dataset. Ann Oncol. 2014;25:2378–85. doi: 10.1093/annonc/mdu464. [DOI] [PubMed] [Google Scholar]
- 30.Yoon HH, Tougeron D, Shi Q, et al. KRAS codon 12 and 13 mutations in relation to disease-free survival in BRAF-wild-type stage III colon cancers from an adjuvant chemotherapy trial (N0147 alliance) Clin Cancer Res. 2014;20:3033–43. doi: 10.1158/1078-0432.CCR-13-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinicrope FA, Mahoney MR, Yoon HH, et al. Analysis of Molecular Markers by Anatomic Tumor Site in Stage III Colon Carcinomas from Adjuvant Chemotherapy Trial NCCTG N0147 (Alliance) Clin Cancer Res. 2015;21:5294–304. doi: 10.1158/1078-0432.CCR-15-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajagopalan H, Bardelli A, Lengauer C, et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 33.Maughan TS, Adams RA, Smith CG, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–14. doi: 10.1016/S0140-6736(11)60613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sobrero AF, Maurel J, Fehrenbacher L, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:2311–9. doi: 10.1200/JCO.2007.13.1193. [DOI] [PubMed] [Google Scholar]
- 35.Tveit KM, Guren T, Glimelius B, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30:1755–62. doi: 10.1200/JCO.2011.38.0915. [DOI] [PubMed] [Google Scholar]
- 36.Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 37.Gavin PG, Colangelo LH, Fumagalli D, et al. Mutation profiling and microsatellite instability in stage II and III colon cancer: an assessment of their prognostic and oxaliplatin predictive value. Clin Cancer Res. 2012;18:6531–41. doi: 10.1158/1078-0432.CCR-12-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogino S, Shima K, Meyerhardt JA, et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clin Cancer Res. 2012;18:890–900. doi: 10.1158/1078-0432.CCR-11-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323–32. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kopetz S, Chang GJ, Overman MJ, et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol. 2009;27:3677–83. doi: 10.1200/JCO.2008.20.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lito P, Pratilas CA, Joseph EW, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–82. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yaeger R, Cercek A, O’Reilly EM, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–20. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J Clin Oncol. 2015;33:4023–31. doi: 10.1200/JCO.2015.63.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–57. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sinicrope FA, Foster NR, Thibodeau SN, et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst. 2011;103:863–75. doi: 10.1093/jnci/djr153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klingbiel D, Saridaki Z, Roth AD, et al. Prognosis of stage II and III colon cancer treated with adjuvant 5-fluorouracil or FOLFIRI in relation to microsatellite status: results of the PETACC-3 trial. Ann Oncol. 2015;26:126–32. doi: 10.1093/annonc/mdu499. [DOI] [PubMed] [Google Scholar]
- 47.Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–26. doi: 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sargent DJ, Shi Q, Yothers G, et al. Prognostic impact of deficient mismatch repair (dMMR) in 7,803 stage II/III colon cancer (CC) patients (pts): A pooled individual pt data analysis of 17 adjuvant trials in the ACCENT database. J Clin Oncol. 2014;32:3507. [Google Scholar]
- 49.Zaanan A, Shi Q, Taieb J, et al. Analysis of DNA mismatch repair (MMR) and clinical outcome in stage III colon cancers from patients (pts) treated with adjuvant FOLFOX +/− cetuximab in the PETACC8 and NCCTG N0147 adjuvant trials. J Clin Oncol. 2015;33:3506. [Google Scholar]
- 50.Juo YY, Johnston FM, Zhang DY, et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol. 2014;25:2314–27. doi: 10.1093/annonc/mdu149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phipps AI, Limburg PJ, Baron JA, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148:77–87. e2. doi: 10.1053/j.gastro.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine AJ, Phipps AI, Baron JA, et al. Clinicopathological risk factor distributions for MLH1 promoter region methylation in CIMP positive tumors. Cancer Epidemiol Biomarkers Prev. 2015 doi: 10.1158/1055-9965.EPI-15-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwitalle Y, Kloor M, Eiermann S, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134:988–97. doi: 10.1053/j.gastro.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 54.Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koopman M, Kortman GA, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100:266–73. doi: 10.1038/sj.bjc.6604867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andre T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: updated 10-year survival and outcomes according to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol. 2015;33:4176–87. doi: 10.1200/JCO.2015.63.4238. [DOI] [PubMed] [Google Scholar]